

Abstract
Background
Phenotypic overlap of type 3 long QT syndrome (LQT3), Brugada syndrome (BrS), cardiac conduction disease (CCD), and sinus node dysfunction (SND) is observed with SCN5A mutations. SCN5A‐E1784K is the most common mutation associated with BrS and LQTS3. The present study examines the genotype–phenotype relationship in a large family carrying SCN5A‐E1784K and SCN5A‐H558R polymorphism.
Methods and Results
Clinical work‐up, follow‐up, and genetic analysis were performed in 35 family members. Seventeen were SCN5A‐E1784K positive. They also displayed QTc prolongation, and either BrS, CCD, or both. One carrier exhibited SND. The presence of SCN5A‐H558R did not significantly alter the phenotype of SCN5A‐E1784K carriers. Fourteen SCN5A‐E1784K patients underwent implantable cardioverter‐defibrillator (ICD) implantation; 4 developed VF and received appropriate ICD shocks after 8±3 months of follow‐up. One patient without ICD also developed VF after 6.7 years. These 5 cases carried both SCN5A‐E1784K and SCN5A‐H558R. Functional characterization was achieved by expressing SCN5A variants in TSA201 cells. Peak (INa,P) or late (INa,L) sodium currents were recorded using whole‐cell patch‐clamp techniques. Co‐expression of SCN5A‐E1784K and SCN5A‐WT reduced INa,P to 70.03% of WT, shifted steady‐state inactivation by −11.03 mV, and increased INa,L from 0.14% to 1.86% of INa,P. Similar changes were observed when SCN5A‐E1784K was co‐expressed with SCN5A‐H558R.
Conclusions
We demonstrate a strong genotype‐phenotype correlation with complete penetrance for BrS, LQTS, or CCD in the largest family harboring SCN5A‐E1784K mutation described so far. Phenotype of LQTS is present during all decades of life, whereas CCD develops with increasing age. Phenotypic overlap may explain the high event rate in carriers.
Keywords: Brugada syndrome, channelopathies, conduction defect, electrophysiology, genetics, long QT syndrome
Subject Categories: Catheter Ablation and Implantable Cardioverter-Defibrillator, Sudden Cardiac Death, Ion Channels/Membrane Transport, Genetics, Functional Genomics
Introduction
Mutations in the SCN5A gene have been associated with long QT syndrome (LQTS) type 3, Brugada syndrome (BrS), sick sinus syndrome, and cardiac conduction disease (CCD).1, 2, 3 Some of these mutations can cause an overlapping electrocardiographic phenotype.4, 5 SCN5A‐E1784K was first described in clinical cases presenting with mild bradycardia, LQTS, and sudden cardiac death (SCD).6 Recently, this mutation was associated with the phenotype of BrS, LQTS, and sick sinus syndrome. The SCN5A‐E1784K mutation has been recognized as the most common LQT3 mutation and accounts for up to 34% of LQT3 cases.5 In vitro studies suggest that this common SCN5A mutation causes the mixed phenotype by enhanced sodium channel inactivation, a negative shift of steady‐state sodium channel inactivation, and enhanced tonic block in response to sodium channel blockers.5
Polymorphisms have been shown to modulate SCN5A mutations in the sense of either aggravation of the channelopathy or rescue of the pathophysiologic effect of the mutation.7, 8, 9, 10 SCN5A‐H558R is a common polymorphism and has been shown to restore normal sodium channel function in the case of specific mutations and to enhance sodium channel dysfunction in others.8, 11, 12
The objective of the present study was to elucidate clinical spectrum and electrocardiographic phenotype in a large European family with familial SCD and carrying the SCN5A‐E1784K mutation and the common polymorphism SCN5A‐H558R. Furthermore, the electrophysiologic effects of the SCN5A‐H558R polymorphism on the SCN5A‐E1784K mutation were studied both in vitro and in vivo.
Methods
Clinical Analysis
The European family studied consisted of 76 family members (Figure 1). All members were encouraged to undergo a complete clinical work‐up and follow‐up. The work‐up included medical and family history, physical examination, 12‐lead baseline and stress ECG, and echocardiogram. ECGs were analyzed by 2 independent cardiologists blended to genotype status. In case of abnormal findings, invasive cardiologic investigations were performed. All members were invited to participate in an ajmaline challenge test and genetic screening for causative mutations. The clinical and genetic studies were approved by the local human ethics committees and performed after written informed consent was obtained from all participants. All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Figure 1.
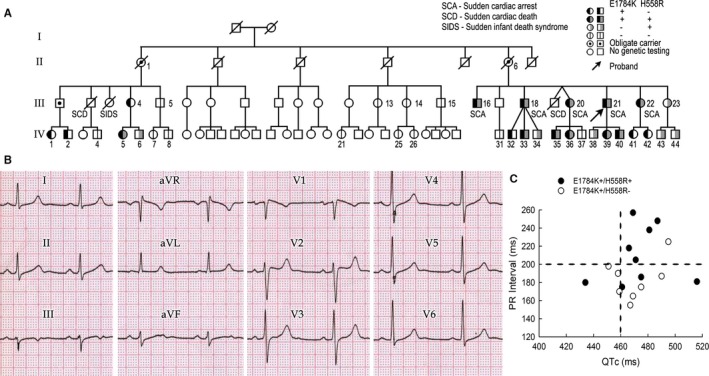
Clinical summary of the affected family. A, Pedigree of the family. B, Baseline ECG of the index patient (proband), who is positive for SCN5A‐E1784K and H558R. C, PR interval vs QTc among SCN5A‐E1784K carriers. Upper limit of normal values is indicated by dotted lines.
Genetic Analysis
Genomic DNA was prepared from peripheral blood lymphocytes of the patient. Genomic DNA was extracted from peripheral blood lymphocytes using a commercial kit (Gentra System; Puregene, Valencia, CA). All exons and intron borders of the susceptibility genes (SCN5A, SCN1‐4B, GPD1L, CACNA1C, CACNB2b, CACNA2D1, KCNE3‐5, KCNJ8, ABCC9), including the alternative splice variants, were amplified and analyzed by direct sequencing. Polymerase chain reaction products were purified with a commercial enzyme (ExoSAP‐ IT; USB, Cleveland, OH) and directly sequenced from both directions using an Applied Biosystems 3100 Genetic Analyzer (Applied Biosystem, Foster City, CA). Comprehensive open‐reading frame/splice site mutational analysis was then performed.
Cellular Electrophysiologic Analysis
The appropriate nucleotide changes were engineered into human cardiac voltage‐dependent sodium channel SCN5A/hNav1.5 in the pcDNA3.1 vector (Invitrogen, Carlsbad, CA) as reported previously.13, 14 Wild‐type (WT) and mutant channels were expressed transiently in TSA201 cells for functional study.13 The WT SCN1B cloned in pRC‐CMV were co‐expressed in each experimental group. CD8 beads were used for identification.
Macroscopic INa was measured using a standard whole‐cell patch clamp method at a temperature of 22°C to 24°C. The extracellular (bath) solution contained (in mmol/L): 140 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 2.8 Na acetate, 10 HEPES, and 10 glucose (pH 7.3 with NaOH). Tetraethylammonium (5 mmol/L) was added to the buffer. The pipette solution contained (in mmol/L): 5 NaCl, 5 KCl, 130 CsF, 1.0 MgCl2, 5 EGTA, and 10 HEPES (pH 7.2 with CsOH). Pipettes had resistances between 0.8 and 2.8 MΩ when filled with recording solution. The data were acquired using pClamp 9.2 (Axon Instruments Inc., Union City, CA) and analyzed using Clampfit (Axon Instruments Inc.). The standard voltage clamp protocols and other details are presented with the data and as described in detail previously.15 Late INa was recorded in bath solution without and with 25 μmol/L tetrodotoxin. To ensure the accuracy of the value of late INa, we only included cells with peak INa>1nA.
Statistical Analysis
Clinical and electrophysiological data were presented as mean±SD and mean±SE, respectively. Before choosing the appropriate statistical test, we checked for normality (Kolmogorov–Smirnov test) and assumption of equal variance (Levene's test). In case of a non‐normality or unequal variance the Wilcoxon signed‐rank test, the Mann–Whitney U test, and the Kruskal–Wallis test were used. Continuous variables among multiple subgroups were analyzed by ANOVA coupled with a Student‐Newman‐Keuls (SNK) test for electrophysiological study. Chi‐square test and Fisher's exact test were used for comparison of categorical variables. Kaplan–Meier survival curves were generated to compare the outcome according to the underlying genotype. Receiver operating characteristic curves were used to analyze ECG parameters in predicting a positive genotype with the corresponding sensitivity and specificity. A P<0.05 was considered statistically significant. All calculations were performed using SPSS 21 (SPSS Inc, Chicago, IL).
Results
Clinical Analysis
The proband (III,21), a 47‐year‐old male of European descent, presented with aborted SCD (or sudden cardiac arrest [SCA]) due to ventricular fibrillation (VF). His basal ECG showed QTc prolongation of 465 ms, first‐degree atrioventricular block (AVB), and ST elevation in the right precordial leads with a type‐I and type‐II Brugada pattern (Figure 1B). He received an implantable cardioverter‐defibrillator (ICD).
The family consisted of 76 family members (Figure 1A). The family history revealed aborted SCA of proband's elder brother (III,18) 3 years ago. The brother exhibited sinus bradycardia, first‐degree AVB (PR, 225 ms), and QTc interval of 469 ms without ST‐segment elevation in the right precordial leads. He received an ICD and subsequently experienced 4 appropriate ICD shocks due to VF. Furthermore, 3 other family members suffered SCD at the age of 1 year (III,3), 22 years (III,19), and 45 years of age (III,2).
Of the 76 family members, 28 refused genetic analysis and 13 patients died before they could be evaluated. A total of 35 family members (19 males, mean age 29.8±15.9 years) were clinically and genetically evaluated. None of them received Class III anti‐arrhythmic drug as medication.
Genetic Analysis
Of the 35 genotyped family members, 17 (8 males/47%; mean age 30.6±15.5 years) were carriers of the SCN5A‐E1784K mutation. Eighteen members (11 males/61%; mean age 29±16.7 years) were negative for the mutation. No significant differences with respect to age and sex were observed.
The SCN5A‐H558R polymorphism was identified in a total of 18 members. Nine patients (5 males, mean age 35.1±16.3 years) were positive for both SCN5A‐E1784K mutation and SCN5A‐H558R polymorphism, whereas 8 patients (3 males; mean age 25.7±13.9 years) carried only the mutation, but not the polymorphism. Common single nucleotide polymorphisms, V1869M (22.9%) and L1868P (33.1%) in CACNA1c, were also found in the proband, which have never been reported as disease‐causing or disease‐modulating variants.
Out of 18 patients negative for SCN5A‐E1784K, 9 patients (7 males/78%; mean age 17.6±9.4 years) were carriers of H558R polymorphism. Nine members (4 males/44%; mean age 40.5±14.7 years) were neither carrier of the mutation nor the polymorphism. Patients carrying the polymorphism without mutation were significantly younger than family members negative for both (P=0.002).
Electrocardiographic Analysis
SCN5A‐E1784K positive versus SCN5A‐E1784K negative
Electrocardiographic measurements of SCN5A‐E1784K carriers versus noncarriers are shown in Table 1. Patients positive for SCN5A‐E1784K exhibited significantly longer PR interval, QRS duration, and QTc interval compared to noncarriers (P<0.0001, P=0.03, P<0.0001, respectively). Ajmaline challenge was positive in 87% of patients positive for SCN5A‐E1784K. In 2 patients, ajmaline administration had to be stopped due to QRS widening >130% and the occurrence of premature ventricular contractions (PVCs). The response to intravenous ajmaline was negative in all SCN5A‐E1784K noncarriers.
Table 1.
Electrocardiographic Parameters of SCN5A‐E1784K Carriers Versus Noncarriers
| E1784K+ n=17 | E1784K− n=18 | P Value | |
|---|---|---|---|
| RR, ms | 920±258 | 865±168 | 0.46 |
| PR, ms | 196±31 | 162±17 | <0.0001 |
| QRS, ms | 100±20 | 87±13 | 0.03 |
| QTc, ms | 467±16 | 408±17 | <0.0001 |
| ST elevation V1, mV | 0.04±0.07 | 0.01±0.03 | 0.11 |
| ST elevation V2, mV | 0.04±0.07 | 0.01±0.03 | 0.08 |
| ST elevation V3, mV | 0.02±0.05 | 0.04±0.02 | 0.2 |
| Brugada type II/III | 7 | 0 | 0.008 |
| Ajmaline positive | 13/15 (87%)a | 0/11 (0%) | <0.0001 |
Two had to be stopped due to QRS widening >130% and the occurrence of premature ventricular contractions.
In the receiver operating characteristic analysis, QRS interval was not found to be predictive for the identification of an SCN5A‐E1784K carrier (area under the curve, 0.692). There was good correlation between PR interval and SCN5A‐E1784K mutation carrier (area under the curve 0.842). Using a PR interval of 176 ms, sensitivity and specificity for identification of a SCN5A‐E1784K carrier was 76.5% and 72.2%. The best correlation was found for QTc interval. The cut‐off value of QTc=445 ms identified an SCN5A‐E1784K mutation carrier with a sensitivity of 88% and a specificity of 100% (area under the curve 0.993, Figure 2A).
Figure 2.
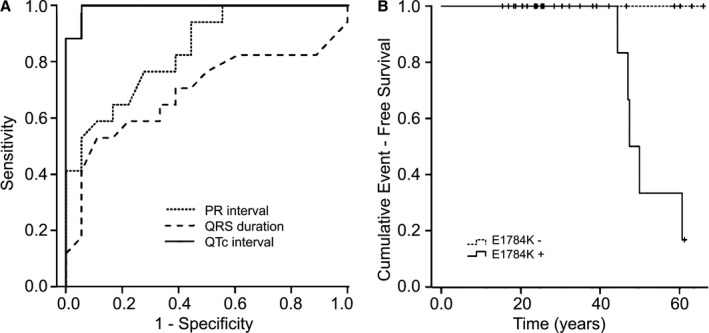
A, Receiver operating characteristic curve for PR interval, QRS duration, and QTc interval for the identification of SCN5A‐E1784K carriers vs noncarriers. The best correlation was found for QTc interval (area under the curve 0.993). B, Kaplan–Meier event‐free survival curves for SCN5A‐E1784K mutation carrier vs noncarriers are displayed. Noncarriers had no cardiac events. Four mutation carriers developed sudden cardiac arrest and ventricular fibrillation beyond the age of 40 years.
SCN5A‐E1784K and SCN5A‐H558R
Electrocardiographic parameters with respect to mutation and polymorphism are represented in Table 2 and Figure 1C. There were no significant differences within SCN5A‐E1784K mutation carriers positive or negative for SCN5A‐H558R. Carriers with the mutation and polymorphism exhibited a Brugada saddle‐back type ECG (55%) more often than patients without the polymorphism (25%). The response to ajmaline administration was positive in 75% (E1784K+/H558R+) and 100% (E1784K+/H558R−), respectively. QRS widening over 130% and PVCs during ajmaline administration occurred in 2 patients with E1784K+/H558R+, which led to the interruption of the test.
Table 2.
Demographic and Electrocardiographic Parameters with Respect to Mutation and Polymorphism
| E1784K+ n=17 | E1784K− n=18 | |||||
|---|---|---|---|---|---|---|
| H558R+ n=9 | H558R− n=8 | P Value | H558R+ n=9 | H558R− n=9 | P Value | |
| Male sex (%) | 5 (56%) | 3 (38%) | 0.46 | 7 (78%) | 4 (44%) | 0.15 |
| Age, y | 35.1±16.3 | 25.7±13.9 | 0.22 | 17.6±9.4 | 40.4±14.6 | 0.002 |
| RR, ms | 911±215 | 930±315 | 0.88 | 822±196 | 909±132 | 0.29 |
| PR, ms | 208±34 | 183±22 | 0.94 | 154±15 | 170±16 | 0.04 |
| QRS, ms | 102±24 | 98±16 | 0.78 | 80±8 | 96±12 | 0.005 |
| QTc, ms | 464±17 | 470±15 | 0.42 | 412±15 | 404±20 | 0.34 |
| QT dispersion, ms | 44±20 | 36±24 | 0.74 | 29±14 | 28±16 | 0.95 |
| QTp‐e dispersion, ms | 43±24 | 51±25 | 0.52 | 38±18 | 34±15 | 0.44 |
| ST elevation in V1, mV | 0.05±0.08 | 0.04±0.07 | 0.74 | 0.11±0.25 | 0.11±0.33 | 1.0 |
| ST elevation in V2, mV | 0.03±0.05 | 0.06±0.09 | 0.46 | 0 | 0.02±0.03 | 0.35 |
| ST elevation in V3, mV | 0.01±0.03 | 0.03±0.06 | 0.42 | 0.01±0.02 | 0.02±0.04 | 0.33 |
| Brugada type II/III | 5 (55%) | 2 (25%) | 0.2 | 0 | 0 | 1.0 |
| Ajmaline positive | 6/8 (75%) | 7/7 (100%) | 0.2 | 0/7 | 0/4 | 1.0 |
In patients negative for SCN5A‐E1784K mutation, carriers of the polymorphism had a significantly shorter PR interval and QRS duration (P=0.04 and 0.005, respectively). However, patients positive for the polymorphism were significantly younger than patients without the polymorphism (17.6±9.4 versus 40.5±14.7 y/o; P=0.002). In all patients without mutation, ajmaline challenge tests were negative.
Age and mutation
The impact of age on the electrocardiographic presentation of the SCN5A‐E1784K carriers versus noncarriers is presented in Table 3. Patients were divided in subgroups according to age (age <40 years [mean age 20.4 years] and ≥40 years [mean age 49.3 years]) and presence or absence of SCN5A‐E1784K. Among patients positive for the SCN5A‐E1784K mutation, RR‐interval, PR interval, and QRS duration were significantly longer in patients ≥40 years of age (P=0.007, 0.0001, 0.04). All mutation carriers ≥40 years exhibited a first‐degree AVB with a mean PR interval of 231±19 ms. QTc interval was not significantly different between young (<40 years of age) and old (≥40 years of age) mutation carriers (465±15 ms versus 470±19 ms; P=0.55). In noncarriers there were no significant differences between patients in the subgroups <40 and ≥40 years, although an increase of PR interval and QRS duration was observed.
Table 3.
Demographic and Electrocardiographic Parameters with Respect to Mutation and Age
| E1784K+ n=17 | E1784K− n=18 | |||||
|---|---|---|---|---|---|---|
| <40 n=11 | ≥40 n=6 | P Value | <40 n=14 | ≥40 n=4 | P Value | |
| Male sex | 5 (45%) | 3 (50%) | 1.0 | 9 (64%) | 2 (50%) | 1.0 |
| Age, y | 20.4±7.3 | 49.3±4.1 | 0.0001 | 21.7±10.0 | 55.1±3.6 | 0.001 |
| RR, ms | 804±219 | 1132±182 | 0.007 | 837±164 | 960±166 | 0.19 |
| PR, ms | 176±12 | 231±19 | 0.0001 | 166±10 | 181±12 | 0.02 |
| QRS, ms | 91±14 | 117±21 | 0.03 | 84±11 | 94±7 | 0.1 |
| QTc, ms | 465±15 | 470±19 | 0.55 | 408±18 | 407±17 | 0.96 |
| QT dispersion, ms | 36±21 | 48±22 | 0.29 | 25±11 | 39±15 | 0.13 |
| QTp‐e dispersion, ms | 45±26 | 50±21 | 0.70 | 32±14 | 39±16 | 0.32 |
| Max. ST, mV | 0.03±0.06 | 0.13±0.08 | 0.02 | 0.01±0.03 | 0.02±0.05 | 0.63 |
| Brugada type II/III | 0 | 5 (83%) | 0.001 | 0 | 0 | 1.0 |
| Ajmaline positive | 7/9 (78%) | 6/6 (100%) | 0.2 | 0/9 | 0/2 | 1.0 |
In comparison of patients <40 years with respect to the absence or presence of SCN5A‐E1784K, significant differences were found for PR interval and QTc interval. Mutation carriers <40 years had significantly longer PR and QTc intervals compared to noncarriers at the same age (P=0.03 and 0.0001). There was a trend of longer QRS duration in mutation carriers (P=0.07). In older patients (≥40 years), mutation carriers exhibited significantly longer PR‐, QTc interval, and maximal ST elevation in the right precordial leads (P=0.001, 0.001, and 0.032, respectively). In addition, when SCN5A‐H558R was taken into consideration, there is no difference among those with SCN5A‐E1784K−, no matter in which age subgroup (<40 or ≥40 years, Table 4). When comparing electrocardiographic parameters of SCN5A‐E1784K+ carriers, the presence or absence of SCN5A‐H558R also had a similar distribution with age, except the SCN5A‐H558R+ group showed a lower heart rate in the older‐age subgroup (≥40 years), and the SCN5A‐H558R− group showed a higher ST elevation in the younger‐age subgroup (<40 years).
Table 4.
Electrocardiographic and Demographic Parameters with Respect to Genotype and Age
| E1784K+ | E1784K− | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H558R+ | H558R− | H558R+ | H558R− | |||||||||
| <40 n=4 | ≥40 n=5 | P Value | <40 n=7 | ≥40 n=1 | P Value | <40 n=8 | ≥40 n=1 | P Value | <40 n=5 | ≥40 n=4 | P Value | |
| Male sex | 2 | 3 | 1.0 | 3 | 0 | 1.0 | 7 | 0 | — | 3 | 2 | 1.0 |
| Age, y | 18.5±5.6 | 48.3±3.7 | 0.0001 | 21.6±8.3 | 54.4 | 0.25 | 14.9±5.1 | 42.1 | 0.02 | 28.6±5.1 | 55.1±3.6 | 0.22 |
| RR, ms | 718±154 | 1065±89 | 0.02 | 854±246 | 1467 | 0.25 | 818±109 | 845 | 0.56 | 867±95 | 960±166 | 1.0 |
| PR, ms | 176±8 | 233±21 | 0.02 | 177±15 | 225 | 0.25 | 154±15 | 160 | 0.7 | 161±14.7 | 182±12 | 1.0 |
| QRS, ms | 89±17 | 111±25 | 0.286 | 98 ±16 | 106 | 05 | 79±7 | 73 | 1.0 | 92±12 | 101±13 | 0.22 |
| QTc, ms | 461±19 | 465±17 | 1.0 | 467±13 | 495 | 0.25 | 411±15 | 417 | 0.73 | 400±23 | 407±18 | 0.67 |
| QT dispersion, ms | 32±15 | 53±19 | 0.06 | 38±25 | 22 | 0.75 | 25± | 41 | 0.11 | 19±10 | 39±15 | 0.22 |
| QTp‐e dispersion, ms | 34±24 | 50±23 | 0.29 | 51±27 | 50 | 1.0 | 34±4 | 45 | 0.19 | 26±12 | 39±14 | 0.44 |
| ST elevation in V1, mV | 0 | 0.09±0.09 | 0.19 | 0.01±0.04 | 0.2 | 0.25 | 0.01±0.03 | 0 | 0.56 | 0 | 0.02±0.05 | 1.0 |
| ST elevation in V2, mV | 0 | 0.05±0.05 | 0.19 | 0.03±0.07 | 0.2 | 0.25 | 0 | 0 | 0.56 | 0 | 0.02±0.07 | 1.0 |
| ST elevation in V3, mV | 0 | 0.02±0.04 | 0.73 | 0.01±0.04 | 0.15 | 0.25 | 0 | 0 | 0.56 | 0 | 0.02±0.04 | 1.0 |
| Brugada type II/III | 0 | 4 (80%) | 0.02 | 0 | 1 | 0.005 | 0 | 0 | 1.0 | 0 | 0 | 1.0 |
| Ajmaline positive | 1/3 | 5/5 | 0.17 | 6/6 | 1/1 | 1.0 | 0/6 | 0/1 | 1.0 | 0/2 | 0/2 | 1.0 |
Patient II,6, the mother of the proband, was an obligate mutation carrier. Repetitive ECG recordings at the age of 64, 75, 77, 78, and 81 were available (Figure 3). She developed progressive conduction disease with increasing age. At the age of 75, the patient showed first‐degree AVB and 3 years later second‐degree AVB. Consecutively, a DDD pacemaker was implanted due to symptomatic bradycardia due to sinus node dysfunction (SND) and second‐degree AVB. Furthermore, intermittent ST‐segment elevation in lead V1 (typical saddle‐back type for the BrS), was visible (Figure 3B).
Figure 3.
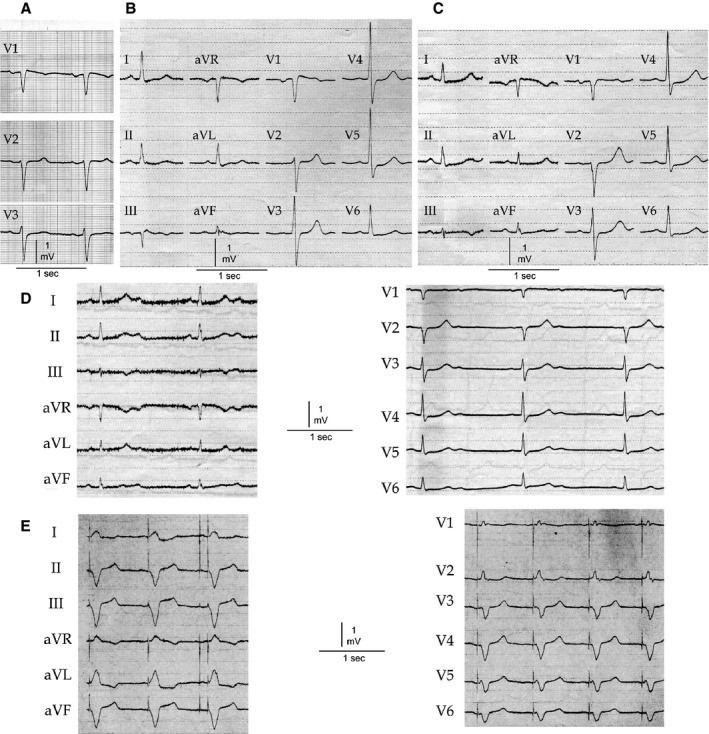
Basal ECGs of patient II,6. The ECGs show QTc prolongation, intermittent saddle‐back type Brugada ECG in the right precordial leads, and progressive conduction disease. The patient developed second‐degree atrioventricular block at the age of 78 years. A, Age 64: PR 193 ms, QRS 85 ms, QTc 461 ms. B, Age 75: PR 205 ms, QRS 94 ms, QTc 467 ms. C, Age 77: PR 229 ms, QRS 95 ms, QTc 472 ms. D, Age 78: Second‐degree AV block, QRS 104 ms, QTc 465 ms. E, Age 81: DDD pacemaker ECG.
Treatment and follow‐up
The index patient (III,21) and his brother (III,18) received an ICD for secondary prophylaxis after successful resuscitation. The asymptomatic sisters of the proband (III,20 and III,22) with the electrocardiographic phenotype of LQTS and BrS were implanted with an ICD for primary prophylaxis after diagnosis due to the strong family history of SCD. All patients with a positive genotype were advised to avoid medication and circumstances, including fever, that might aggravate the phenotype and increase the risk for cardiac events according to www.crediblemeds.org and www.brugadadrugs.org.16
A total of 14 patients (82%) positive for SCN5A‐E1784K mutation and the clinical phenotype of LQTS underwent ICD implantation. Three patients refused prophylactic ICD implantation. Including the index patient, 4 family members had VF and SCA and received adequate ICD shocks (2 males; mean age 47.3±3 years) after a mean follow‐up of 8±3 months (Figure 4). Clinical characteristics of patients with adequate ICD therapies or SCA during follow‐up are presented in Table 5. One male patient (III,16), who refused an ICD implantation, also developed VF and SCA after 6.7 years of follow‐up. All 5 patients were ≥40 years of age, had positive ajmaline challenge, first‐degree AVB, and prolonged QTc interval. SCN5A‐E1784K mutation and SCN5A‐H558R polymorphism were found in each of them. Patients negative for SCN5A‐E1784K mutations revealed no cardiac events (Figure 2B). A total of 6 stored intracardiac electrograms of adequate ICD therapies were analyzed in 4 cases. Patient III,18 had 2 episodes of VF. Mean basic cycle length before the inducing PVC was 770 and 576 ms. The VF‐inducing PVC was coupled with 86% and 89% of the previous basic cycle length. Patients III,20 and III,22 had 1 and 2 adequate ICD shocks during follow‐up, respectively. Basic cycle lengths were 888 ms in patient III,20 and 776 and 754 ms in patient III,22. Coupling interval of the VF‐triggering PVC was 52% in these 2 cases. The proband (III,21) also developed VF during follow‐up. In his case, basic cycle length was 720 ms and the inducing PVC was short‐coupled with 290 ms (40%).
Figure 4.
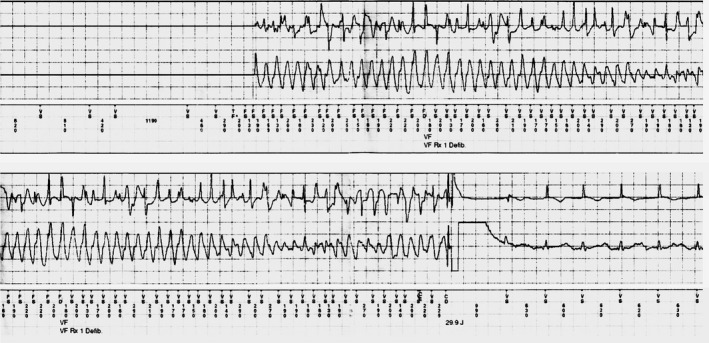
ECG from the ICD of patient III,22 positive for E1784K mutation and H558R polymorphism. Nine months after ICD implantation for primary prevention, the patient developed ventricular fibrillation, which was terminated by a single ICD shock. ICD indicates implantable cardioverter‐defibrillator.
Table 5.
Clinical and Demographic Data of Patients with Sudden Cardiac Arrest or Ventricular Fibrillation during Follow‐Up
| Patient | Sex | Event | Age With Event (y) | E1784K | H558R | PR | QRS | QTc | Ajmaline Test | ICD Implantation | CI (ms) | CI/Basic CL (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| III,16 | Male | SCA, VF | 60.8 | + | + | 248 | 132 | 487 | Positive | Refused | N/A | N/A |
| III,18 | Male | SCA, VF | 47.1 | + | + | 256 | 136 | 468 | Positive | Yes | 663 | 86.1% |
| 515 | 89.4% | |||||||||||
| III,20 | Female | SCA, VF | 50 | + | + | 238 | 105 | 456 | Positive | Yes | 460 | 51.8% |
| III,21 (proband) | Male | SCA, VF | 47.5 | + | + | 205 | 108 | 465 | Positive | Yes | 290 | 40.3% |
| III,22 | Female | SCA, VF | 44.5 | + | + | 218 | 107 | 469 | Positive | Yes | 410 | 52.8% |
| 390 | 51.7% |
CI indicates coupling interval of the first beat of polymorphic VT; CL, cycle length; ICD, implantable cardioverter‐defibrillator; N/A, not available; SCA, sudden cardiac arrest; VF, ventricular fibrillation.
Electrophysiology Study
Peak sodium current of SCN5A‐E1784K
The SCN5A‐E1784K mutation and the SCN5A‐H558R polymorphism were reproduced in vitro on the SCN5A (hH1c) background in order to study the functional consequences of the mutation both with and without the presence of the polymorphism. We expressed WT+WT, E1784K+WT, and E1784K+H558R channels in TSA201 cells for whole‐cell voltage‐clamp measurements. Figure 5A represents a typical Na+ current. Compared to the WT+WT group, the current amplitude was moderately but significantly reduced for E1784K+WT. When H558R co‐expressed with E1784K, the loss of function was slightly more prominent (Figure 5B). The maximum current amplitude was 665.14±87.60 pA/pF for WT+WT, 465.82±50.60 pA/pF for E1784K+WT, and 375.82±65.50 pA/pF for E1784K+H558R (Figure 5C). The voltage of peak sodium current was similar, and there was no difference of activation among the different groups (Figure 5D). Steady‐state inactivation was shifted to much more negative potentials by the mutation, but were not further shifted in the presence of the H558R polymorphism (Figure 5E); V 1/2=−92.48±0.82 mV and k=5.98±0.30 mV for WT+WT; V 1/2=−103.51±1.34 mV and k=7.14±0.46 mV for E1784K+WT; and V 1/2=−106.54±1.78 mV and k=6.89±0.65 mV for E1784K+H558R. P≤0.001 is for difference in V 1/2 between WT and the other 2 groups. No significant difference in k was observed among all groups (P=0.134).
Figure 5.
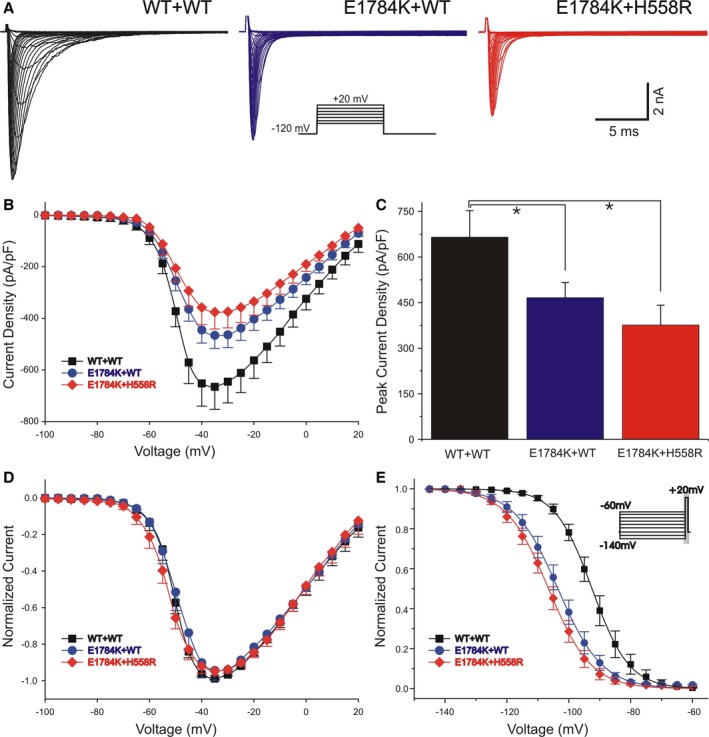
Functional expression studies evaluating the characteristics of IN a for WT+WT, E1784K+WT, and E1784K+H558R. A, Whole‐cell current traces from representative experiments of 3 groups. The voltage‐clamp protocol is shown as the inset. IN a was elicited by depolarizing pulses ranging from −90 mV to +20 mV in 5‐mV increments with a holding potential of −120 mV. B, Current–voltage relationship for WT+WT (squares, n=33), E1784K+WT (circles, n=18), and E1784K+H558R (diamonds, n=11). C, Summary of maximum peak IN a density of the 3 groups (*P<0.05). Peak E1784K+WT and E1784K+H558R current at −35 mV was smaller by 29.97% and 43.50% relative to WT+WT, respectively (P=0.043 and P=0.032). D, Normalized peak current–voltage relationships of the 3 groups. E, Steady‐state inactivation for WT+WT (squares, n=22), E1784K+WT (circles, n=7), and E1784K+H558R (diamonds, n=12) using the standard protocol indicated in the diagram. Peak current was normalized to their respective maximum values and plotted against the conditioning potential. Data were fitted by Boltzman function. For values see text.
Late sodium current of SCN5A‐E1784K
E1784K and H558R channels were also evaluated for late INa (Figure 6A). TTX‐sensitive currents were compared at the end of a 300‐ms depolarization, and values were obtained by subtracting the values before and after TTX. Summary data for late INa, presented as percent of peak INa, were significantly higher when E1784K was expressed (1.86±0.23% co‐expressed with WT, n=7; 1.89±0.25% co‐expressed with H558R, n=10), compared with the WT group (0.14±0.02%, n=22; Figure 6B). Moreover, late INa amplitude recorded from the E1784K channel, with WT or H558R, was also obviously larger than in WT. Late INa density was 1.30±0.29 pA/pF (n=22) for WT+WT channels, 9.91±2.48 pA/pF (n=7) for E1784+WT and 8.53±1.63 pA/pF (n=10) for E1784+H558R mutant channels (Figure 6C). H558R variant did not significantly alter increased late INa produced by SCN5A‐E1784K mutation.
Figure 6.
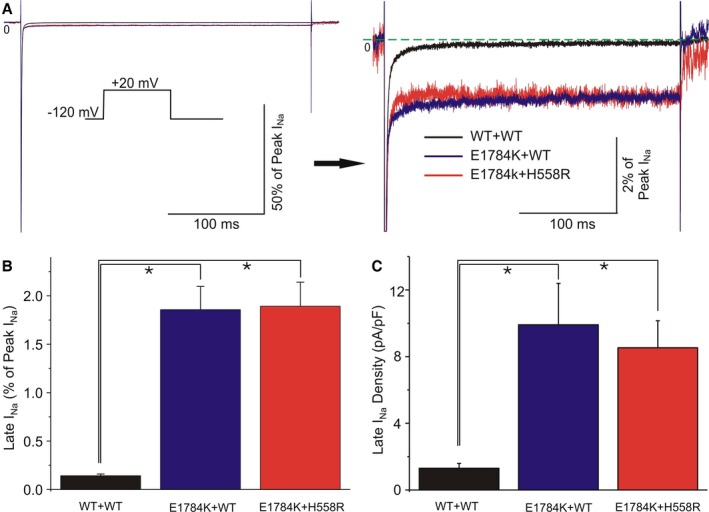
Functional expression studies evaluating the characteristics of late IN a for WT+WT, E1784K+WT, and E1784K+H558R. A, Representative TTX‐sensitive sodium current traces obtained by subtraction, recorded during a 300‐ms depolarization to −20 mV from −120 mV (left panel). Amplified traces showing late INa (right panel). B and C, Bar graph of relative late INa (% of peak INa) and late INa density (pA/pF) among 3 groups. Statistically significant differences (*P<0.05) were observed between E1784K+WT/E1784K+H558R and WT+WT in both panels (n=7, 10, 22 for each group). No statistically significant difference was observed between E1784K+WT and E1784K+H558R. For values see text.
Discussion
SCN5A‐E1784K is a frequently reported mutation in patients with LQT3 syndrome. It was first described as a congenital LQTS mutation caused by a defect in sodium channel inactivation giving rise to a small, persistent current.6 These characteristics were confirmed by several subsequent studies.17, 18 In 2000, Priori and co‐workers reported that flecainide not only shortens the QT interval, but can also induce a BrS phenotype in SCN5A‐E1784K carriers.19 Two years later, it was confirmed by the same group that SCN5A‐E1784K is associated with BrS in a study of 200 subjects diagnosed with BrS.20 Recently, it has been reported as a mutation causing phenotypic overlap.5, 21 Of 41 SCN5A‐E1784K carriers, 93% had LQT3, 22% had BrS, and 39% had SND.5
Here, we report on the clinical and electrocardiographic phenotypes of a large white family carrying the SCN5A‐E1784K mutation and examine the modulatory effect of the common polymorphism H558R. Carriers of SCN5A‐E1784K showed a strong genotype–phenotype correlation in this family, with a penetrance of 100% with respect to a prolonged QT interval. Additionally, 13 of 15 patients (87%) presented with the Brugada phenotype and 7% with SND. We demonstrate, for the first time, an association of SCN5A‐E1784K with progressive CCD in mutation carriers above the age of 40 years. These electrocardiographic features are consistent with our finding of a loss of function of INa,P and a gain of function of INa,L.
Genotype–Phenotype Correlation of SCN5A‐E1784K
Compared to previous studies, patients positive for SCN5A‐E1784K were found to exhibit the clinical phenotype of LQT 3 syndrome.5, 6 In the present cohort, the QTc cut‐off of 445 ms was able to differentiate carriers from noncarriers with a sensitivity of 88% and a corresponding specificity of 100%. These findings are in line with in vitro studies showing a gain of function of late INa. In the present study, SCN5A‐E1784K was associated with a 7.6‐fold higher late INa density of E1784+WT compared with WT+WT channels. These electrophysiologic findings are comparable to those of previous studies.5, 6, 17
Priori et al reported overlapping clinical phenotypes of LQT3 syndrome and BrS. Thirteen patients with SCN5A mutations and LQTS received intravenous flecainide.19 Three of 13 patients were carriers of SCN5A‐E1784K. Following flecainide administration, 6 of 13 patients developed a Brugada phenotype; 1 of the 3 patients (33%) was an SCN5A‐E1784K carrier. In a larger study focusing on SCN5A‐E1784K, Makita et al described an overlap of LQT3 and BrS in 9 of 41 mutation carriers.5 Only 1 SCN5A‐E1784K carrier showed spontaneous Brugada type 1 ECG. In the present study, the overlap of LQT3 and Brugada phenotype was much greater. At baseline, 7/17 SCN5A‐E1784K carriers revealed a saddle‐back‐type ECG pattern. Following intravenous ajmaline, a diagnostic Brugada type‐I ECG could be unmasked in 13/15 mutation carriers, whereas none of the SCN5A‐E1784K‐negative family members displayed a Brugada pattern. In our study the overlap of LQT3 and Brugada phenotype was significantly higher than in the study by Makita et al (87% versus 22%, respectively). The main difference between these 2 studies was the sodium channel blocker used. In previous studies, flecainide and pilsicainide were used to unmask the Brugada ECG pattern. In the present study, ajmaline was used. We have previously shown that ajmaline is significantly more sensitive than flecainide in provoking the Brugada phenotype. This may explain the higher overlap of Brugada and LQT3 phenotypes in the present study.22
Late sodium channel blockers, including mexiletine, flecainide, ranolazine, and GS‐6615 have been used in patients with LQT3 for primary and secondary prevention. These agents reduced QTc and are expected to lower the arrhythmogenic risk.23, 24, 25 Due to the high proportion of Brugada phenotypes in SCN5A‐E1784K carriers, the Class IC sodium channel blockers, such as flecainide, should be avoided.19 Whether ranolazine or GS‐6615 are safe with respect to provocation of a Brugada phenotype in SCN5A‐E1784K carriers is currently under investigation.26, 27
SND has previously been associated with SCN5A mutations.28 Makita et al recently reported that 16 of 41 SCN5A‐E1784K carriers presented with SND.5 In our family, only 1 patient (III,18; male; 47 years old) presented with SND characterized by persistent sinus bradycardia and chronotropic incompetence. This patient also carried SCN5A‐H588R as well. His mother (II,6), an obligate carrier of SCN5A‐E1784K and SCN5A‐H558R, developed SND with increasing age. The mechanism underlying the SCN5A‐E1784K‐mediated sick sinus syndrome is likely due to the negative shift of steady‐state inactivation (Figure 5) and the prolonged action potential duration caused by the increase in INa,L (Figure 6).
Clinical manifestation of progressive cardiac conduction disease characterized by reduced excitability and conduction slowing of the His‐Purkinje system results in prolongation of AV‐conduction and widening of the QRS. SCN5A mutations have been shown to cause progressive cardiac conduction disease.29, 30 Our study is the first to associate SCN5A‐E1784K with progressive cardiac conduction disease. SCN5A‐E1784K carriers presented with slowed AV‐nodal conduction and intraventricular conduction compared to noncarriers (PR: 196±31 ms versus 162±17 ms, P<0.0001; QRS: 100±20 ms versus 87±13 ms, P=0.03). Prolongation of AV and intraventricular conduction was age dependent. SCN5A‐E1784K carriers below the age of 40 (mean age 20±7 years) displayed significantly shorter RR, PR, and QRS intervals than mutation carriers (average age, 49±4 years, Table 3). QTc interval was not significantly different between young and old mutation carriers. In the present large family, cardiac events occurred mainly in adulthood, above the age of 40. Thus, the addition of progressive CCD to prolonged QTc intervals and the Brugada phenotype may be responsible for the increased risk of SCD in this family.
Modulating Polymorphism—SCN5A‐H558R
Single nucleotide polymorphisms have been shown to modify SCN5A mutations by either aggravation of the channelopathy or rescue of the pathophysiologic effect of the mutation. H558R is one of the most common SCN5A‐polymorphisms, with an allelic frequency of 20% to 30% within individuals of white ethnicity.7 In the general population, the presence of SCN5A‐H558R (without any SCN5A mutation) has been reported to be associated with increased heart rate, longer QTc intervals, and an increased susceptibility to atrial fibrillation in population studies.31, 32, 33, 34, 35 In the absence of sodium channel mutations, there is no functional effect of H588R detectable in heterologous expression systems in vitro.35, 36
In patients with SCN5A mutations, the presence of H558R had diverse effects on the cellular and clinical phenotype. H558R has been shown to restore defective sodium channel function and/or trafficking of specific mutations (ie, R282H, T512I, D1275N, D1690N, G1748D, M1766L, V1951L, and P2006A) by modifying INa,P and/or INa,L.8, 10, 37, 38, 39, 40 It is reported to improve ECG characteristics (QRS duration, J‐point elevation, and “aVR sign”) and clinical phenotype of BrS among carriers of SCN5A mutations.12 In contrast, the sodium channel defects of other mutants (ie, E161K, R222Q, G400A, A572D, P1298L, R1632H, and I1835T) are aggravated in the SCN5A‐H558R background.13, 36, 38, 41 In the case of other SCN5A mutations, including the missense and deletion mutants (ie, L212P, T220I, F1617Del, T187I, R878C, and G1408R), as well as the truncated mutants (ie, W1421X, K1578fs/52, R1623X), sodium currents are not significantly affected by H558R.38
In the present study, we evaluated the potential effect of SCN5A‐H558R both in vitro and in vivo. When comparing electrocardiographic parameters of SCN5A‐E1784K carriers, the presence or absence of SCN5A‐H558R had no significant effect on PR, QRS, and QTc or the expression of a Brugada phenotype. In the in vitro study, co‐expression of SCN5A‐E1784K with H558R produced no significant effect on peak or late INa, although there was a tendency to a further reduction of INa,P. Above all, this kind of discrepancy of in vitro and in vivo study may result from several possibilities. First, there are some other underlying mechanisms or modulating background among carriers, which are not present in the cell system used for experimental study. If we had been aware of those factors, we could conduct further investigation. Secondly, the intermittent severe phenotype, such as VT/VF or SCA, likely to be induced or triggered by certain acute conditions (ischemia, stress, fever, hypoxia, change of pH, etc), will not be duplicated in the normal in vitro study. Lastly, despite the chance being very small, there is still the possibility of coincidence. One of the evidences to support this assumption is that patients with SCA or VF during follow‐up are the oldest carriers of SCN5A‐E1784K, so the effect of aging on the mutant carriers could not be totally excluded.
Aging and Sodium Channel
There is some experimental evidence showing that sodium channels may decline with age. Huang et al demonstrated interesting results indicating an age‐dependent alteration in expression and function of sodium current in rat heart.42 Atrial fibrillation, a main arrhythmia significantly contributing to morbidity and mortality in elderly patients, has been correlated with enhanced age‐dependent atrial fibrosis. Henry and his group showed decreased Nav1.5 expression and slowed conduction velocity by reduction of sodium channel expression and augmentation of fibrosis among an aged rat atrial fibrillation model. The effect was reversed by relaxin treatment in all aged animals with atrial fibrillation.43 Although the role of aging on genetic variants in SCN5A remains unclear, it is reasonable to speculate that decreased sodium channel and increased fibrosis caused by aging will lead to a more serious phenotype for SCN5A mutation, such as what we observed in our SCN5A‐E1784K family.
Risk of SCD
All 5 patients with SCA carried both the SCN5A‐E1784K mutation and H558R polymorphism, and their phenotype presented as a combination of long QT interval, CCD, and type I Brugada ECG following ajmaline challenge, suggesting that SCN5A‐H558R aggravated the SCN5A‐E1784K−‐associated phenotype under certain conditions. Interestingly, the mechanism of induction of ventricular tachyarrhythmias was different in the 4 patients, whose intracardiac electrograms were available. In 1 male patient, the coupling interval that triggered VF was 90% of the mean previous cycle length. This late coupled PVC suggests that prolonged QT interval is the underlying mechanism for SCA. However, in 2 female patients and the proband the coupling interval was short (40–52%), which indicates BrS as the responsible pathology for sudden death. From these findings, we cannot conclude which phenotype is the responsible one for SCA in these patients. Possibly, both mechanisms are responsible within the same patient; thus, circumstances and drugs provoking either phenotype in these patients should be avoided.
Conclusions
We demonstrate high penetrance for LQTS, BrS, and CCD in a large family harboring the SCN5A‐E1784K mutation. Prolonged QT interval was present at all ages, whereas CCD developed after the fourth decade of life. Moreover, at least 87% of SCN5A‐E1784K carriers displayed ST‐segment elevation diagnostic of BrS. This reveals one of the highest penetrances in a family with BrS syndrome reported by far. SCD/SCA cases occurred mainly after the fourth decade of life in carriers of both E1784K and H558R, displaying overlapping phenotypes of LQTS, BrS, and CCD. In vivo and in vitro studies showed no statistically significant differences in the electrocardiographic phenotypes or INa characteristics between E1784K with and without H588R.
Author Contributions
Hu, Veltmann, and Antzelevitch designed the study. Veltmann, Hu, Borggrefe, Wolpert, Schimpf, coordinated and collected the clinical evaluations. Hu and Antzelevitch supervised and coordinated the genetic and electrophysiology study. Pfeiffer and Cáceres did the genetic screening. Burashnikov provided the plasmids. Hu and Barajas‐Martinez performed the electrophysiology laboratory work. Hu and Barajas‐Martinez organized and summarized the genetic database. Veltmann, Hu, and Barajas‐Martinez analyzed the data. Veltmann, Hu, and Antzelevitch wrote the manuscript. All co‐authors contributed to editing of the manuscript.
Sources of Funding
This work was supported by NIH grant HL47678 (Antzelevitch), NYSTEM grant #C026424 (Antzelevitch), CONACYT #FM201866 (Hu and Barajas‐Martinez), NYS, Florida, Massachusetts, and Connecticut Masons and DZHK #FKZ81Z5500251 (Veltmann).
Disclosures
All authors have no financial or other considerations to disclose. All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Acknowledgments
The authors are grateful to Judy Hefferon for technical assistance.
(J Am Heart Assoc. 2016;5:e003379 doi: 10.1161/JAHA.116.003379)
References
- 1. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz‐Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze‐Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. [DOI] [PubMed] [Google Scholar]
- 2. Jiang C, Atkinson D, Towbin JA, Splawski I, Lehmann MH, Li H, Timothy K, Taggart RT, Schwartz PJ, Vincent GM. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994;8:141–147. [DOI] [PubMed] [Google Scholar]
- 3. Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. [DOI] [PubMed] [Google Scholar]
- 4. Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan‐Sindhunata G, Bink‐Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long‐QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. [DOI] [PubMed] [Google Scholar]
- 5. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, Schulze‐Bahr E, Fukuhara S, Mochizuki N, Makiyama T, Itoh H, Christiansen M, McKeown P, Miyamoto K, Kamakura S, Tsutsui H, Schwartz PJ, George AL Jr, Roden DM. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei J, Wang DW, Alings M, Fish F, Wathen M, Roden DM, George AL. Congenital long‐QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation. 1999;99:3165–3171. [DOI] [PubMed] [Google Scholar]
- 7. Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, Keating MT, Jones G, Chadha M, Burrow CR, Stephens JC, Xu C, Judson R, Curran ME. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. [DOI] [PubMed] [Google Scholar]
- 8. Poelzing S, Forleo C, Samodell M, Dudash L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R, Guida P, Chahine M, Pitzalis M, Deschênes I. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation. 2006;114:368–376. [DOI] [PubMed] [Google Scholar]
- 9. Tan B‐H, Valdivia CR, Song C, Makielski JC. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am J Physiol Heart Circ Physiol. 2006;291:H1822–H1828. [DOI] [PubMed] [Google Scholar]
- 10. Ye B, Valdivia CR, Ackerman MJ, Makielski JC. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics. 2003;12:187–193. [DOI] [PubMed] [Google Scholar]
- 11. Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest. 2003;111:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lizotte E, Junttila MJ, Dube MP, Hong K, Benito B, De Zutter M, Henkens S, Sarkozy A, Huikuri HV, Towbin J, Vatta M, Brugada P, Brugada J, Brugada R. Genetic modulation of Brugada syndrome by a common polymorphism. J Cardiovasc Electrophysiol. 2009;20:1137–1141. [DOI] [PubMed] [Google Scholar]
- 13. Hu D, Viskin S, Oliva A, Carrier T, Cordeiro JM, Barajas‐Martínez H, Wu Y, Burashnikov E, Sicouri S, Brugada R, Rosso R, Guerchicoff A, Pollevick GD, Antzelevitch C. Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm. 2007;4:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu D, Barajas‐Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C, Schimpf R, Cai JJ, Nam G‐B, Deshmukh P, Scheinman M, Preminger M, Steinberg J, López‐Izquierdo A, Ponce‐Balbuena D, Wolpert C, Haïssaguerre M, Sánchez‐Chapula JA, Antzelevitch C. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu D, Barajas‐Martínez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, Priori SG, Tan HL, Hiraoka M, Brugada J, Wilde AAM. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up‐to‐date website (www.brugadadrugs.org). Heart Rhythm. 2009;6:1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deschênes I, Baroudi G, Berthet M, Barde I, Chalvidan T, Denjoy I, Guicheney P, Chahine M. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. [DOI] [PubMed] [Google Scholar]
- 18. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. [DOI] [PubMed] [Google Scholar]
- 19. Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome: the role of flecainide challenge. Circulation. 2000;102:945–947. [DOI] [PubMed] [Google Scholar]
- 20. Priori SG, Napolitano C, Gasparini M, Pappone C, Bella Della P, Giordano U, Bloise R, Giustetto C, De Nardis R, Grillo M, Ronchetti E, Faggiano G, Nastoli J. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–1347. [DOI] [PubMed] [Google Scholar]
- 21. Nakajima T, Kaneko Y, Saito A, Irie T, Tange S, Iso T, Kurabayashi M. Identification of six novel SCN5A mutations in Japanese patients with Brugada syndrome. Int Heart J. 2011;52:27–31. [DOI] [PubMed] [Google Scholar]
- 22. Wolpert C, Echternach C, Veltmann C, Antzelevitch C, Thomas GP, Spehl S, Streitner F, Kuschyk J, Schimpf R, Haase KK, Borggrefe M. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type‐3 long‐QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS. Safety and efficacy of flecainide in subjects with Long QT‐3 syndrome (DeltaKPQ mutation): a randomized, double‐blind, placebo‐controlled clinical trial. Ann Noninvasive Electrocardiol. 2005;10:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long‐QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. [DOI] [PubMed] [Google Scholar]
- 26. Zareba W, Rosero S, Zeng DW, Moss AJ, Robinson JL, Couderc JP, Karwatowska‐Prokopczuk E, Layug B, Belardinelli L. QTc shortening by GS‐6615, a new late sodium current blocker, in LQT3 patients. Heart Rhythm. 2014;11:S81. [Google Scholar]
- 27. Hu D, Barajas‐Martínez H, Ruan L, Pfeiffer R, Burashnikov E, Caceres G, Belardinelli L, Antzelevitch C. Effects of ranolazine on long QT3 syndrome are mutation dependent. Circulation. 2011;124:A15739. [Google Scholar]
- 28. Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003;112:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan HL, Bink‐Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort‐Krol GC, van Tintelen PJ, van Den Berg MP, Wilde AA, Balser JR. A sodium‐channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. [DOI] [PubMed] [Google Scholar]
- 30. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. [DOI] [PubMed] [Google Scholar]
- 31. Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P; D.E.S.I.R. Study Group . Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–1222. [DOI] [PubMed] [Google Scholar]
- 32. Chen J‐Z, Xie X‐D, Wang X‐X, Tao M, Shang Y‐P, Guo X‐G. Single nucleotide polymorphisms of the SCN5A gene in Han Chinese and their relation with Brugada syndrome. Chin Med J. 2004;117:652–656. [PubMed] [Google Scholar]
- 33. Wilton SB, Anderson TJ, Parboosingh J, Bridge PJ, Exner DV, Forrest D, Duff HJ. Polymorphisms in multiple genes are associated with resting heart rate in a stepwise allele‐dependent manner. Heart Rhythm. 2008;5:694–700. [DOI] [PubMed] [Google Scholar]
- 34. Chen LY, Ballew JD, Herron KJ, Rodeheffer RJ, Olson TM. A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clin Pharmacol Ther. 2007;81:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen L, Zhang W, Fang C, Jiang S, Shu C, Cheng H, Li F, Li H. Polymorphism H558R in the human cardiac sodium channel SCN5A gene is associated with atrial fibrillation. J Int Med Res. 2011;39:1908–1916. [DOI] [PubMed] [Google Scholar]
- 36. Tester DJ, Valdivia C, Harris‐Kerr C, Alders M, Salisbury BA, Wilde AAM, Makielski JC, Ackerman MJ. Epidemiologic, molecular, and functional evidence suggest A572D‐SCN5A should not be considered an independent LQT3‐susceptibility mutation. Heart Rhythm. 2010;7:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shinlapawittayatorn K, Dudash LA, Du XX, Heller L, Poelzing S, Ficker E, Deschênes I. A novel strategy using cardiac sodium channel polymorphic fragments to rescue trafficking‐deficient SCN5A mutations. Circ Cardiovasc Genet. 2011;4:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gui J, Wang T, Trump D, Zimmer T, Lei M. Mutation‐specific effects of polymorphism H558R in SCN5A‐related sick sinus syndrome. J Cardiovasc Electrophysiol. 2010;21:564–573. [DOI] [PubMed] [Google Scholar]
- 39. Núñez L, Barana A, Amorós I, la Fuente de MG, Dolz‐Gaitón P, Gómez R, Rodríguez‐García I, Mosquera I, Monserrat L, Delpón E, Caballero R, Castro‐Beiras A, Tamargo J. p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm. 2013;10:264–272. [DOI] [PubMed] [Google Scholar]
- 40. Shinlapawittayatorn K, Du XX, Liu H, Ficker E, Kaufman ES, Deschênes I. A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm. 2011;8:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng J, Morales A, Siegfried JD, Li D, Norton N, Song J, Gonzalez‐Quintana J, Makielski JC, Hershberger RE. SCN5A rare variants in familial dilated cardiomyopathy decrease peak sodium current depending on the common polymorphism H558R and common splice variant Q1077del. Clin Transl Sci. 2010;3:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang X, Du Y, Yang P, Lin S, Xi Y, Yang Z, Ma A. Age‐dependent alterations of voltage‐gated Na(+) channel isoforms in rat sinoatrial node. Mech Ageing Dev. 2015;152:80–90. [DOI] [PubMed] [Google Scholar]
- 43. Henry BL, Gabris B, Li Q, Martin B, Giannini M, Parikh A, Patel D, Haney J, Schwartzman DS, Shroff SG, Salama G. Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na(+) channels. Heart Rhythm. 2016;13:983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]