

Abstract
Background
We previously have shown that glibenclamide (GBC), a sulfonylurea receptor 1–transient receptor potential M4 (SUR1‐TRPM4) channel inhibitor, improves survival and neurological outcome after asphyxial cardiac arrest and cardiopulmonary resuscitation (ACA/CPR). Here, we further compare the efficacy of GBC with target temperature management (TTM) and determine whether the efficacy of GBC is affected by TTM.
Methods and Results
Male Sprague‐Dawley rats (n=213) subjected to 10‐minute ACA/CPR were randomized to 4 groups after return of spontaneous circulation (ROSC): normothermia control (NT); GBC; TTM; and TTM+GBC. Survival, neurodeficit scores, histological injury, as well as the expressions of SUR1 and TRPM4 were evaluated. The 7‐day survival rate was 34.4% (11 of 32) in the NT group, 65% (13 of 20) in the GBC group, 50% (10 of 20) in the TTM group, and 70% (14 of 20) in the TTM+GBC group. Rats that received either GBC, TTM alone, or in combination showed less neurological deficit than NT control at 24, 48, and 72 hours and 7 days after ROSC. Moreover, TTM or GBC ameliorated neuronal degeneration and glial activation in the hippocampal CA1 region with similar efficacy, whereas the combination of them had a trend toward better effect. The subunits of SUR1‐TRPM4 heterodimers were both strongly upregulated after ACA/CPR and expressed in multiple types of brain cells, but partly suppressed by TTM.
Conclusions
GBC is comparable to TTM in improving survival and neurological outcome after ACA/CPR. When GBC is given along with TTM, less histological injury tended to be achieved.
Keywords: cardiopulmonary resuscitation, glibenclamide, heart arrest, sulfonylurea receptor 1‐transient receptor potential M4 channel, target temperature management
Subject Categories: Cardiopulmonary Arrest, Cardiopulmonary Resuscitation and Emergency Cardiac Care
Introduction
Survivors from out‐of‐hospital cardiac arrest (OHCA) are still facing high risks of death and disability.1 Target temperature management (TTM), also known as therapeutic hypothermia, is the only approach proven to improve outcome after ventricular fibrillation‐induced OHCA.2, 3, 4 Despite that, the efficacy of TTM on comatose adult patients whose initial rhythms are nonshockable requires further confirmation.5, 6 In addition, the adverse effects of hypothermia, the increased nursing labor, and costs for management of hypothermia are also issues of concern.7, 8 Overall, TTM is still underutilized.9 Development of alternative or additive therapeutic approaches are required to favor post‐CA (cardiac arrest) patients.
Glibenclamide (US adopted name, glyburide, glibenclamide [GBC]) is a US Food and Drug Administration (FDA)‐approved sulfonylurea for treating type 2 diabetes mellitus for decades. In recent years, GBC has been proved to be neuroprotective in several disease models, including cerebral infraction, traumatic brain injury, subarachnoid hemorrhage, hemorrhagic encephalopathy of prematurity, spinal cord injury, hepatic encephalopathy, and multiple sclerosis.10, 11, 12, 13, 14, 15, 16 Moreover, the use of sulfonylureas (to treat diabetes) around the time of acute ischemic stroke has been associated with less neurological deficit and lower risk of hemorrhagic conversion.17, 18 Two clinical trials (ClinicalTrials.gov identifier: NCT01454154 and NCT01794182) testing the efficacy of GBC on traumatic brain injury as well as malignant edema and stroke are underway. Although the mechanisms are not fully uncovered, it is suggested that GBC blocks the activity of the transient receptor potential M4 (TRPM4) through interfering with the sulfonylurea receptor 1 (SUR1) subunit and thus ameliorates the resulting injuries to the neurovascular unit.19
In our previous study, post‐CA treatment with GBC improved survival and neurological outcome in a rat model of 8‐minute asphyxial cardiac arrest and cardiopulmonary resuscitation (ACA/CPR).20 In this study, we further compared the efficacy of GBC with TTM in a more‐severe model of ACA/CPR. From a viewpoint of clinical translation, we also examined whether the efficacy of GBC was affected by TTM.
Methods
Animal Preparation
This study was approved by the Animal Care and Use Committee of the Nanfang Hospital, Southern Medical University (Guangzhou, China) and followed the National Guidelines for Animal Experimentation. Male Sprague‐Dawley rats weighing between 300 and 350 g were obtained from the Experimental Animal Center of Southern Medical University and housed on a 12‐hour light/dark cycle with free access to water and food.
Rat Model of Asphyxial Cardiac Arrest
A rat model of 10‐minute ACA/CPR was established with modification (Figure 1A).20 In brief, rats were anesthetized with isoflurane (4% for induction and 2% for maintenance; RWD, Shenzhen, China), orotracheally intubated, and connected to a mechanical ventilator (RWD). Intravascular catheters (PE50; Smiths Medical, Ashford, UK) were inserted into the right femoral artery and vein for dynamic artery blood pressure monitoring and drug administration. Electrocardiogram was recorded by subcutaneous needle electrodes. After 10‐minute stabilization, rats were chemically paralyzed by IV vecuronium (2 mg/kg) and inspired gas was changed to room air. Then, the ventilator was disconnected for 10 minutes, leading to circulatory arrest within 4 minutes, which was confirmed by cessation of arterial pulse and reduction of mean arterial pressure (MAP) to below 20 mm Hg. At the end of 10‐minute asphyxia, CPR was initiated by reconnecting the ventilator and thoracic compressions were performed by a custom‐made mechanical chest compressor at a rate of 400 compressions per minute. To prolong the duration of CPR, epinephrine (0.01 mg/kg) was administered after 1 minute of thoracic compressions and repeated at 3‐minute intervals (0.02 mg/kg), if necessary. Return of spontaneous circulation (ROSC) was defined as increase of MAP beyond 60 mm Hg for 10 minutes. Rats failed to ROSC within 5 minutes were excluded from the continuing experiments.
Figure 1.
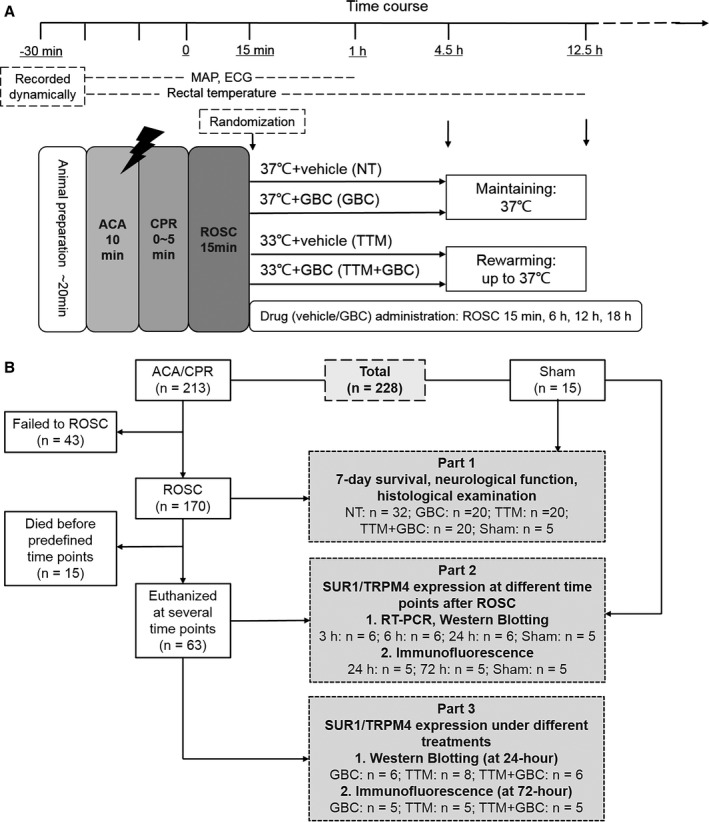
Experimental procedure and flow diagram of the study. A, Experimental procedure and measurements during baseline, asphyxial cardiac arrest and cardiopulmonary resuscitation (ACA/CPR), and return of spontaneous circulation (ROSC). B, Flow diagram of the experiment. The whole study consisted of 3 parts. In part 2, rats were euthanized at 3, 6, 24, or 72 hours after ROSC. ECG indicates electrocardiogram; GBC, glibenclamide; MAP, mean arterial pressure; NT, normothermia; TTM, target temperature management.
Core temperature was monitored by a rectal temperature probe (RC‐4; Elitech, Xuzhou, China) and maintained with a temperature feedback system (RWD) as appropriate temperature management. Brain temperature was monitored in a subgroup of animals by placing a temperature probe (Physitemp Instruments, Inc., Clifton, NJ) in the right brain cortex (3 mm posterior and 3 mm lateral to the bregma and 4 mm below the skull surface). Blood glucose was detected intermittently using a glucometer (Optium, Abbott, CA). After spontaneous breath started, animals were weaned from the ventilator. After 1‐hour observation, all catheters were removed and surgical wounds were sutured. At the end of each experimental period, rats were returned to their cages with easily accessible food and water and were observed in a room of constant temperature (22°C). Saline (20 mL/kg) was administered daily to rats until they were able to feed themselves.
Study Protocol
Fifteen minutes after ROSC, rats were block randomized to 4 treatment groups by using a random number table (Figure 1A): normothermia and vehicle (NT); normothermia and GBC (GBC); TTM and vehicle (TTM); and TTM and GBC (TTM+GBC). Rats in the GBC groups were intraperitoneally administered GBC (Sigma‐Aldrich, St. Louis, MO) with a loading dose of 10 μg/kg at randomization and 4 maintenance doses of 1.2 μg per 6 hours after ROSC, whereas rats in the vehicle groups received an equal volume of DMSO and saline.20 In the TTM groups, rapid cooling was initiated after randomization. By using ice packs and an electric fan, a target temperature of 33°C was reached within 15 minutes and then maintained for 4 hours, followed by slow rewarming to 37°C at a rate of 0.5°C/h. Normothermic rats were constantly maintained at 37°C. Rats that underwent all procedures except ACA and CPR were used as sham control.
The present study includes 3 parts (Figure 1B). In part 1, 92 rats that successfully resuscitated were followed up for 7 days and survival, neurological outcome ,and histological injury were evaluated. In parts 2 and 3, expressions of SUR1 and TRPM4 at various time points and under different treatments were examined.
Survival Study and Neurological Function Evaluation
Survival was recorded daily. Neurological function was assessed by 2 investigators unaware of animal grouping at 24, 48, and 72 hours and 7 days after ROSC, using the neurodeficit score (NDS),21 a previously validated scale consisted of 7 components: general behavioral deficit; brainstem function; motor assessment; sensory assessment; motor behavior; behavior; and seizures. A total NDS of 80 was considered normal, whereas 0 was brain death. To avoid the confounding brought by mortality, NDSs were analyzed in survived rats only.
Histological Examination
After 7‐day follow‐up, rats were euthanized and coronal brain sections (6‐μm thickness) located at 3.5 mm posterior to bregma were obtained (Leica CM1800; Leica Microsystems GmbH, Heidelberg, Germany). Immunohistochemistry was performed with antibodies against neuronal nuclei (NeuN; Abcam, Cambridge, UK) for detection of neuron, microtubule‐associated protein 2 (MAP2; Sigma‐Aldrich) for dendrite, glial fibrillary acidic protein (GFAP, Abcam) for astrocyte, and ionized calcium‐binding adapter molecule‐1 (Iba‐1; Wako, Osaka, Japan) for microglia, using the protocol suggested by the manufacturer (ZSGB‐BIO, Beijing, China). In each brain section, 2 slide fields were randomly examined.22 The number of NeuN‐positive cells and the relative intensity of MAP2, Iba‐1, and GFAP staining was determined with ImageJ software (1.49v; National Institutes of Health [NIH], Bethesda, MD).
For immunofluorescence, brain sections were incubated with primary antibodies directed against SUR1 (Sigma‐Aldrich), TRPM4 (Abcam), NeuN, von Willebrand factor (vWF; Abcam), GFAP, and Iba‐1. Bound primary antibodies were detected with Alexa Fluor dye of donkey antimouse or antirabbit (Abcam) and observed with a confocal microscope (FluoView; Olympus, Tokyo, Japan). The total number of SUR1‐ and TRPM4‐positive cells as well as the percentages of SUR1‐staining in different types of cells were calculated by ImageJ (NIH).
Western Blotting
Western blot was routinely performed as previously reported.20 Antibodies against SUR1, TRPM4, and β‐actin (CWBIO, Beijing, China) were used. The densities of protein blots were quantified by using ImageJ software (NIH) and normalized to level of β‐actin.
Measurement of Gene Expression
mRNA levels of Abcc8 (encoding SUR1), Trpm4, and Gapdh were routinely measured by quantitative real‐time polymerase chain reaction.20 Relative changes of mRNA expressions were normalized to levels of Gapdh.
Statistical Analysis
To calculate the sample size in survival analysis, a power analysis was performed based on our preliminary experiment. The survival rate of the NT group was estimated at 30% and the GBC group at 70%. Assuming an α error of 0.05 with a power of 0.85, a ratio of 1.6 for the NT‐to‐GBC group, we obtained a necessary sample size of 32 in the NT group and 20 in the GBC group to show a significant effect. The ratio of 1.6 for the NT‐to‐GBC group was determined based on estimated survival and to ensure sufficient brain samples for histological examination.
Data are presented as mean±SD. For multiple comparisons, one‐way ANOVAs were used, followed by least‐significant difference tests between multiple groups. Physiological variables were examined by repeated‐measures analyses comprising treatment group, time, and treatment‐by‐time interaction. NDSs are presented as medians and 25th to 75th percentiles and were analyzed by Kruskal–Wallis with Mann–Whitney U analyses between multiple groups. Difference in survival rate was analyzed by Kaplan–Meier analysis with log‐rank test. SPSS (version 20.0; IBM Corp, Armonk, NY) and GraphPad Prism software (version 5.0; GraphPad Software Inc., La Jolla, CA) were used for statistical analyses. P<0.05 was considered statistically significant.
Results
GBC was Comparable to TTM in Improving Survival and Neurological Outcome After ACA/CPR
A total of 228 rats were prepared for the study (Figure 1B). Of 170 rats that successfully resuscitated, 15 died (3 in the 24 hours group, 2 in the 72 hours group, 3 in the GBC group, 5 in the TTM group, and 2 in the TTM+GBC group) before predefined time points were excluded. There were no significant differences in body weight, time from asphyxia to CA, time required for ROSC, and epinephrine usage among the 4 experimental groups (Table 1). There were also no differences among the 4 experimental groups in MAP, heart rate, blood gases, or serum lactate at baseline or during post‐CA care after ROSC (Table 2). Rectal temperature in hypothermic animals reduced rapidly to 33.0°C at 30 minutes after ROSC, whereas normothermic animals were maintained around 37.0°C during the whole period (Figure 2A). Brain temperature dropped faster than rectal temperature during asphyxia, but revealed linear trend with rectal temperature after ROSC (Figure 2B and 2C). Blood glucose levels in the GBC and TTM+GBC groups seemed lower than those in the NT and TTM groups, but no hypoglycemia (<3.9 mmol/L) was detected (Figure 2D).
Table 1.
Baseline Characteristics Among the 4 Experimental Groups
| Parameters | NT (n=32) | GBC (n=20) | TTM (n=20) | TTM+GBC (n=20) |
|---|---|---|---|---|
| Body weight, g | 327±23 | 332±20 | 332±38 | 331±34 |
| Time from asphyxia to CA, sec | 212±26 | 218±24 | 225±21 | 222±18 |
| Time required for ROSC, sec | 194±65 | 173±72 | 179±65 | 167±73 |
| Total dose of epinephrine, μg | 7.3±3.2 | 6.3±3.3 | 6.6±3.4 | 5.3±3.2 |
CA indicates cardiac arrest; GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
Table 2.
Physiological Parameters and Laboratory Results of the 4 Experimental Groups at Baseline and During Post‐CA Care After ROSC
| Parameters | Time Points | NT (n=32) | GBC (n=20) | TTM (n=20) | TTM+GBC (n=20) |
|---|---|---|---|---|---|
| MAP, mm Hg | Baseline | 108±9 | 112±8 | 108±9 | 104±7 |
| 10 minutes | 114±9 | 116±11 | 123±9 | 119±5 | |
| 30 minutes | 87±7 | 86±8 | 87±5 | 88±9 | |
| 60 minutes | 87±5 | 82±12 | 80±7 | 85±11 | |
| Heart rate, bpm | Baseline | 405±9 | 399±11 | 398±9 | 402±15 |
| 10 minutes | 436±20 | 425±16 | 417±20 | 421±17 | |
| 30 minutes | 386±14 | 385±13 | 392±9 | 378±14 | |
| 60 minutes | 388±7 | 379±14 | 376±11 | 376±15 | |
| PH | Baseline | 7.41±0.01 | 7.40±0.04 | 7.38±0.03 | 7.39±0.03 |
| 10 minutes | 7.16±0.03 | 7.15±0.03 | 7.14±0.05 | 7.14±0.06 | |
| 30 minutes | 7.20±0.05 | 7.19±0.03 | 7.19±0.03 | 7.17±0.07 | |
| 60 minutes | 7.25±0.05 | 7.23±0.02 | 7.23±0.06 | 7.22±0.05 | |
| PaO2, mm Hg | Baseline | 85±5 | 88±4 | 86±5 | 86±7 |
| 10 minutes | 105±7 | 109±6 | 112±7 | 111±6 | |
| 30 minutes | 112±8 | 118±6 | 117±10 | 115±9 | |
| 60 minutes | 99±4 | 96±7 | 99±7 | 100±8 | |
| PaCO2, mm Hg | Baseline | 40±4 | 40±4 | 42±4 | 41±2 |
| 10 minutes | 59±7 | 65±10 | 66±6 | 69±8 | |
| 30 minutes | 56±5 | 55±5 | 54±5 | 57±7 | |
| 60 minutes | 51±3 | 46±3 | 48±4 | 51±6 | |
| Lactate, mmol/L | Baseline | 1.20±0.37 | 1.28±0.37 | 1.34±0.26 | 1.34±0.26 |
| 10 minutes | 5.52±1.78 | 4.44±1.25 | 4.82±1.53 | 4.62±1.14 | |
| 30 minutes | 4.00±0.42 | 3.78±0.89 | 4.24±0.52 | 3.98±0.72 | |
| 60 minutes | 3.08±0.80 | 3.22±0.61 | 3.26±0.50 | 2.64±0.33 |
All values were expressed as mean±SD. There were no significant differences for treatment by time among the four groups by mixed‐effects model for repeated‐measures analyses. bpm indicates beats per minute; CA, cardiac arrest; GBC, glibenclamide; MAP, mean arterial pressure; NT, normothermia; PaCO2, arterial carbon dioxide pressure; PH, potential of hydrogen; ROSC, return of spontaneous circulation; TTM, target temperature management.
Figure 2.
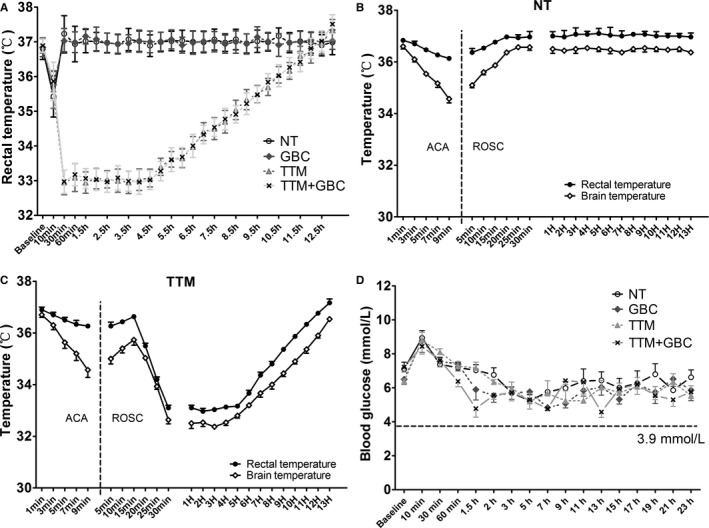
Core temperature and blood glucose monitoring at baseline and after ROSC. A, Rectal temperature monitoring of all rats in the experimental groups. B and C, Rectal temperature and brain temperature in the NT (n=3) and TTM (n=3) groups during ACA and after ROSC. D, Blood glucose monitoring at baseline and after ROSC. ACA indicates asphyxial cardiac arrest; GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
The 7‐day survival rate was 34.4% (11 of 32) in the NT group, 65% (13 of 20) in the GBC group, 50% (10 of 20) in the TTM group, and 70% (14 of 20) in the TTM+GBC group (Figure 3A). In addition, rats in the GBC, TTM, and TTM+GBC groups all showed higher NDSs than those in the NT group at 24, 48, and 72 hours and 7 days after ROSC (Figure 3B). These results suggest that GBC is comparable to, or even better than, TTM in improving survival and neurological outcome after 10‐minute ACA/CPR.
Figure 3.
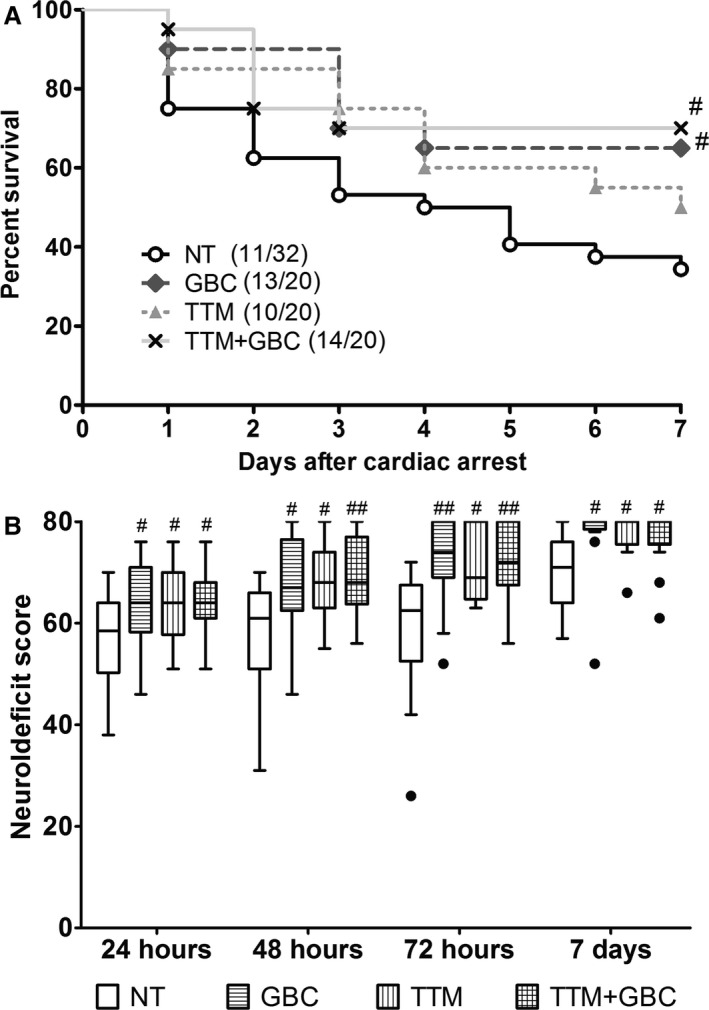
Effects of GBC and TTM on survival and neurological outcome after ACA/CPR. A, Kaplan–Meier analyses of cumulative survival during 7‐day follow‐up after ROSC. B, Neurodeficit scores (0=brain death; 80=normal) of survived rats at 24, 48, and 72 hours and at 7 days after ROSC. Filled circle indicates outlier. # P<0.05; ## P<0.01 versus NT group. ACA indicates asphyxial cardiac arrest; CPR, cardiopulmonary resuscitation; GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
GBC and TTM Ameliorated Histological Injury in the Hippocampal CA1 Region
Transient global cerebral ischemia could lead to delayed neuronal loss and activation of microglia and astrocytes in selectively vulnerable areas, such as the hippocampal CA1.22 As indicated in the histological analysis of the hippocampal CA1 region in Figure 4, TTM or GBC alone significantly recovered numbers of neurons by NeuN staining compared to NT treatment, with their combination resulted in a further recovery (Figure 4A and 4E). In addition, dendritic injury revealing by MAP2 staining was partly prevented by GBC or TTM treatment (Figure 4B and 4F), with better preservation by TTM+GBC than GBC alone. Moreover, there were significantly more Iba‐1‐positive microglia and GFAP‐positive astrocytes in the NT group than the other groups (Figure 4C, 4D, 4G and 4H). Together, these results imply that GBC is comparable to TTM in ameliorating histological injury after ACA/CPR, whereas their combination has a trend toward better effect.
Figure 4.
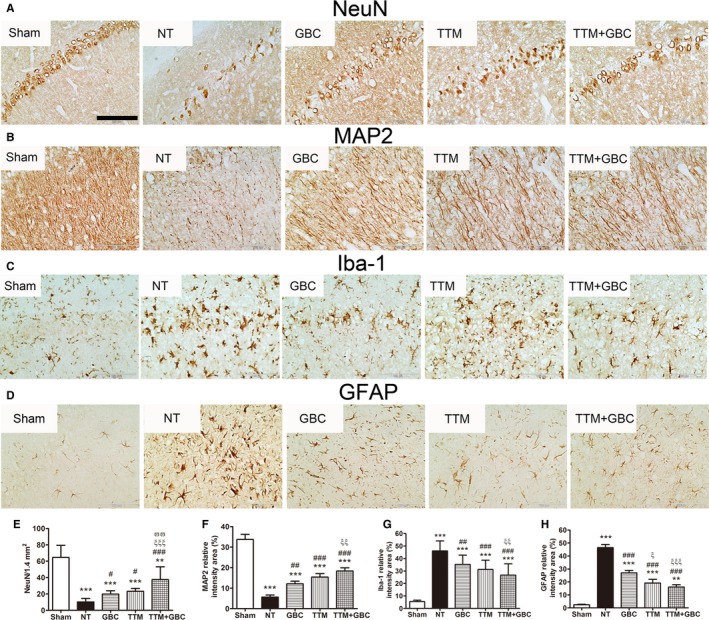
Effects of GBC and TTM on neuronal death and activation of microglia and astrocytes after ACA/CPR. A through D, Representative photomicrographs of immunohistochemistry for neuronal nuclei (NeuN), microtubule‐associated protein 2 (MAP2), ionized calcium‐binding adapter molecule 1 (Iba‐1), and glial fibrillary acidic protein (GFAP) in the hippocampal CA1 region of the sham‐operated and the experimental groups at 7 days after ROSC. Bar, 100 μm. E through H, Semiquantitative results of NeuN, MAP2, Iba‐1, and GFAP, respectively. **P<0.01 and ***P<0.001 versus the sham‐operated group; # P<0.05, ## P<0.01, and ### P<0.001 versus the NT group; ξ P<0.05, ξξ P<0.01, and ξξξ P<0.001 versus the GBC group; ϖϖ P<0.01 versus the TTM group. ACA indicates asphyxial cardiac arrest; CPR, cardiopulmonary resuscitation; GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
TTM, but Not GBC, Inhibited the Upregulation of SUR1 and TRPM4 Induced by ACA/CPR
Given that the SUR1‐TRPM4 channel is the putative therapeutic target of GBC,10, 23 we then examined expressions of this channel subunits before and after ACA/CPR. Results showed that both mRNA levels and protein levels of SUR1 and TRPM4 were significantly upregulated at 6 hours after ACA/CPR and reached a higher level at 24 hours (Figure 5A through 5C). Immunofluorescent labeling also revealed more SUR1‐ and TRPM4‐postive cells in the brain cortex and hippocampal CA1 region in post‐CA rats (Figure 5D). Moreover, TTM, but not GBC, reduced the levels of SUR1 and TRPM4 (Figure 5E and 5F).
Figure 5.
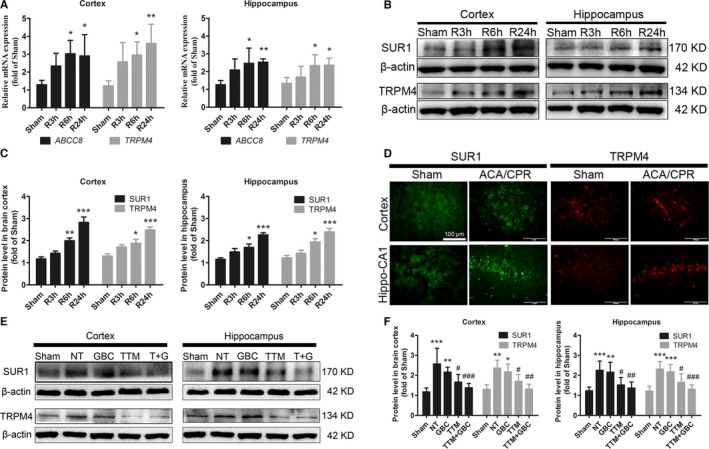
Effects of GBC and TTM on expressions of sulfonylurea receptor 1 (SUR1) and transient receptor potential M4 (TRPM4) after ACA/CPR. A, Quantitative real‐time polymerase chain reaction results of message RNA expressions of Abcc8 (encoding SUR1) and Trpm4 in sham‐operated rats and postcardiac arrest (post‐CA) rats that euthanized at 3, 6, and 24 hours after ROSC. B and C, Western blot results of SUR1 and TRPM4 in sham‐operated and post‐CA rats that euthanized at different time points after ROSC. D, Representative photomicrographs of the immunofluorescence labeled with SUR1 (green) and TRPM4 (red) in brain cortex and hippocampal CA1 region from sham‐operated and post‐CA rats. Bar, 100 μm. E and F, Western blot findings of SUR1 and TRPM4 under different treatments detected at 24‐hour after ROSC. T+GBC, TTM, and GBC. *P<0.05, **P<0.01, and ***P<0.001 versus the sham‐operated group; # P<0.05, ## P<0.01, and ### P<0.001 versus the NT group. ACA indicates asphyxial cardiac arrest; CPR, cardiopulmonary resuscitation; DAPI, 4′,6‐diamidino‐2‐phenylindole; GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
At 72 hours after ROSC, SUR1 was expressed in multiple types of brain cells, including neuron (NeuN‐positive), endothelial cell (vWF‐positive), microglia (Iba‐1‐positive), and astrocytes (GFAP‐positive; Figure 6A). Besides, SUR1 colocalized closely with TRPM4, in agreement with the fact that SUR1 and TRPM4 form a heterodimer.24 TTM and TTM+GBC reduced the total number of SUR1‐ and TRPM4‐positive cells (Figure 6B), without interfering the cellular distribution of the SUR1 (Figure 6C).
Figure 6.
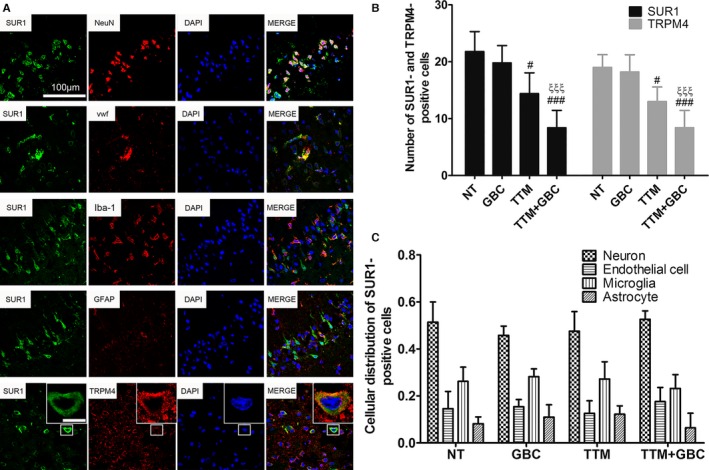
Cellular distribution of SUR1 in the brain at 72‐hour after ROSC. A, Representative photomicrographs observed under confocal microscope are shown. SUR1 (green) colocalizes with multiple cellular markers (red), including neuronal nuclei (NeuN), von Willebrand factor (vWF), ionized calcium‐binding adapter molecule 1 (Iba‐1), and glial fibrillary acidic protein (GFAP). SUR1 also colocalizes with TRPM4 (red). Bar, 100 μm. Inserted bar, 5 μm. B, Number of SUR1‐ and TRPM4‐positive cells in each group. C, Cellular distribution of SUR1in different groups. All n=5. # P<0.05 and ### P<0.001 versus the NT group; ξξξ P<0.001 versus the GBC group. GBC, glibenclamide; NT, normothermia; ROSC, return of spontaneous circulation; TTM, target temperature management.
Discussion
GBC has been shown to improve survival and neurological outcome in a rat model of 8‐minute ACA/CPR.20 In this study, we demonstrated that GBC was comparable to TTM in improving survival and neurological outcome, attenuating neuronal injury and glial activation in a rat model of 10‐minute ACA/CPR. When GBC was combined with TTM, there was a trend toward less histological injury than either of them alone. We also found paralleled upregulation of SUR1 and TRPM4 after ACA/CPR, which were inhibited by TTM.
TTM is the cornerstone of post‐CA care.2 However, there is a dispute on the target temperature, especially after the findings presented by Nielsen et al.,25 that hypothermia at a temperature of 33°C in unconscious survivors of OHCA did not confer a benefit as compared to 36°C. Although the lack of benefit at the lower temperature might be ascribed to the improvement in critical care or diversity of illness severity, the concept of TTM, aiming to prevent hyperthermia, seems to have become an established paradigm of postresuscitative care.26, 27 The current American Heart Association guideline recommends comatose adult patients with ROSC after CA to have TTM, by selecting and maintaining a constant temperature between 32°C and 36°C.2 In this study, 33°C was selected and maintained for all TTM.
Besides the depth of hypothermia, the initiation, duration, and rate of rewarming are all crucial factors to optimize TTM.28 In humans, TTM is suggested to be maintained for at least 24 hours after achieving target temperature.2 However, shorter duration of TTM was commonly utilized in animal studies, because of the high metabolic rate of rodents and shorter time in reaching target temperature.22, 29, 30 Ye et al.31 reported that 2‐hour duration of mild hypothermia induced rapidly after ROSC improved survival, myocardial and cerebral functions as well as prolonged duration of hypothermia. As for rewarming, it is recommended to be conducted at a rate of 0.25 to 0.5°C/h.1 A rate of rewarming at 2°C/h may abolish the beneficial effects of hypothermia and, at 4°C/h, can adversely affect the brain.29, 32 Taking these factors into account, we initiated TTM at 15 minutes after ROSC, maintained it for 4 hours after reaching 33°C, and followed by a rewarming rate at 0.5°C/h. As expected, TTM alleviated neurological deficit and histological injury after 10‐minute ACA/CPR.
From the viewpoint of clinical translation, it is also important to note that the combined therapy of TTM and GBC had a trend of better effect than TTM alone in improving survival and alleviating histological injury. TTM moderates a variety of destructive processes after CA/CPR, but the exact mechanism is still unknown.33 Given that the subunits of the SUR1‐TRPM4 channel were upregulated after ACA/CPR and targeting this channel by GBC or gene manipulation have been shown to provide benefit in many neuropathological circumstances,10, 23 we checked here whether TTM would regulate this channel. As indicated in Figure 5E and 5F, TTM inhibited the upregulations of SUR1 and TRPM4 induced by ACA/CPR. Although the causal relationship between the SUR1‐TRPM4 channel and post‐CA brain injury requires to be further determined, targeting the SUR1‐TRPM4 channel might be one of the mechanism that leads to the neuroprotection of TTM.
The SUR1‐TRPM4 channel is a heterodimer that exhibits pharmacological properties of SUR1 and biophysical properties of TRPM4.24 Once it has been overactivated upon ischemia, hypoxia, trauma, or inflammation, the SUR1‐TRPM4 channel could cause unchecked influx of extracellular sodium and water, leading to consequential cell swelling, cell death, release of inflammatory cytokines, and breakdown of the blood–brain barrier.10, 11, 12, 13 GBC is well documented to bind to the SUR1 subunit and increase the probability of long closed states of the TRPM4 channel,16, 19, 34 without altering their structural expressions.20 Consistent with our previous data,20 we showed paralleled upregulations of SUR1 and TRPM4 after ACA/CPR. However, because of technological limitation, we have no direct evidence for the blocking of this channel by GBC and thus are not able to rule out other possible mechanisms that might contribute to the salutary effect of GBC. Therefore, our findings suggest a possible role of the SUR1‐TRPM4 channel in post‐CA brain injury, but further studies are required to prove their causal relationship.
In histological examination, we demonstrated that not only neurons, but also dendrites were salvaged by GBC treatment. GBC also suppressed the activation of microglia and astrocytes, which could either be a reaction to tissue damage or a key contributor to the pathophysiology of brain ischemic injuries, such as delayed neuronal loss.22, 35 Similar to that found in ischemic stroke,10 the upregulated SUR1 was evident in neurons, astrocytes, and endothelial cells after ACA/CPR, with paralleled upregulation of TRPM4. Moreover, the delayed upregulation of SUR1 and TRPM4 from 6 to 24 hours or even 72 hours post‐ROSC might suggest a broader window for GBC therapy in ACA/CPR and better effects brought by longer repetitive application of GBC.34, 36
It is worthy of note that no hypoglycemia was detected after GBC treatment, either alone or in combination with TTM. In 2 previous studies, GBC in triple dosage (33 μg/kg) of that used here did not induce a sharp reduction of serum glucose either, even when applied under hypothermia.37, 38 Therefore, the findings from our present and previous studies may have demonstrated the effectiveness and safety of GBC in different degrees of ACA/CPR. Although the SUR1‐TRPM4 channel was supposed to be the therapeutic target of GBC, as stated above, our experiments did not prove a causal relationship here, which is the key limitation of this study.
Conclusion
GBC is comparable to TTM in improving survival and neurological outcome in a rat model of 10‐minute ACA/CPR. When GBC is applied along with TTM, a trend toward better effects in ameliorating neuronal injury and activation of microglia and astrocytes is observed. As an FDA‐approved medication, GBC merits further exploration in ACA for clinical translation.
Sources of Funding
This work is supported by the National Natural Science Foundation of China (Nos. 81271521 and 81471339).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003465 doi: 10.1161/JAHA.116.003465)
References
- 1. Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT Jr, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez‐Nunez A, Sellke FW, Spaulding C, Sunde K, Vanden Hoek T. Post‐cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008;118:2452–2483. [DOI] [PubMed] [Google Scholar]
- 2. Callaway CW, Donnino MW, Fink EL, Geocadin RG, Golan E, Kern KB, Leary M, Meurer WJ, Peberdy MA, Thompson TM, Zimmerman JL. Part 8: post‐cardiac arrest care: 2015 American Heart Association Guidelines Update for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2015;132:S465–S482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Hypothermia After Cardiac Arrest Study Group . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. [DOI] [PubMed] [Google Scholar]
- 4. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out‐of‐hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. [DOI] [PubMed] [Google Scholar]
- 5. Kim Y, Yim H, Jeong S, Klem ML, Callaway CW. Does therapeutic hypothermia benefit adult cardiac arrest patients presenting with non‐shockable initial rhythms? A systematic review and meta‐analysis of randomized and non‐randomized studies. Resuscitation. 2012;83:188–196. [DOI] [PubMed] [Google Scholar]
- 6. Perman SM, Grossestreuer AV, Wiebe DJ, Carr BG, Abella BS, Gaieski DF. The utility of therapeutic hypothermia for post–cardiac arrest syndrome patients with an initial nonshockable rhythm. Circulation. 2015;132:2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Varon J, Marik PE, Einav S. Therapeutic hypothermia: a state‐of‐the‐art emergency medicine perspective. Am J Emerg Med. 2012;30:800–810. [DOI] [PubMed] [Google Scholar]
- 8. Nichol G, Aufderheide TP, Eigel B, Neumar RW, Lurie KG, Bufalino VJ, Callaway CW, Menon V, Bass RR, Abella BS, Sayre M, Dougherty CM, Racht EM, Kleinman ME, O'Connor RE, Reilly JP, Ossmann EW, Peterson E. Regional systems of care for out‐of‐hospital cardiac arrest: a policy statement from the American Heart Association. Circulation. 2010;121:709–729. [DOI] [PubMed] [Google Scholar]
- 9. Merchant RM, Soar J, Skrifvars MB, Silfvast T, Edelson DP, Ahmad F, Huang KN, Khan M, Vanden HT, Becker LB, Abella BS. Therapeutic hypothermia utilization among physicians after resuscitation from cardiac arrest. Crit Care Med. 2006;34:1935–1940. [DOI] [PubMed] [Google Scholar]
- 10. Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1‐regulated NC(Ca‐ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simard JM, Kilbourne M, Tsymbalyuk O, Tosun C, Caridi J, Ivanova S, Keledjian K, Bochicchio G, Gerzanich V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma. 2009;26:2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V. Glibenclamide reduces inflammation, vasogenic edema, and caspase‐3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tosun C, Kurland DB, Mehta R, Castellani RJ, Dejong JL, Kwon MS, Woo SK, Gerzanich V, Simard JM. The protective effect of glibenclamide in a model of hemorrhagic encephalopathy of prematurity. Brain Sci. 2013;3:215–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z, Woo SK, Gerzanich V. Endothelial sulfonylurea receptor 1‐regulated NCCa‐ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007;117:2105–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jayakumar AR, Valdes V, Tong XY, Shamaladevi N, Gonzalez W, Norenberg MD. Sulfonylurea receptor 1 contributes to the astrocyte swelling and brain edema in acute liver failure. Transl Stroke Res. 2014;5:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makar TK, Gerzanich V, Nimmagadda VKC, Jain R, Lam K, Mubariz F, Trisler D, Ivanova S, Woo SK, Kwon MS, Bryan J, Bever CT, Simard JM. Silencing of Abcc8 or inhibition of newly upregulated Sur1‐Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2015;12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunte H, Busch MA, Trostdorf K, Vollnberg B, Harms L, Mehta RI, Castellani RJ, Mandava P, Kent TA, Simard JM. Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann Neurol. 2012;72:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kunte H, Schmidt S, Eliasziw M, Del Zoppo GJ, Simard JM, Masuhr F, Weih M, Dirnagl U. Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke. Stroke. 2007;38:2526–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simard JM, Sheth KN, Kimberly WT, Stern BJ, del Zoppo GJ, Jacobson S, Gerzanich V. Glibenclamide in cerebral ischemia and stroke. Neurocrit Care. 2014;20:319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang K, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X, Xie Z, Pan S. Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit Care Med. 2015;43:e341–e349. [DOI] [PubMed] [Google Scholar]
- 21. Geocadin RG, Ghodadra R, Kimura T, Lei H, Sherman DL, Hanley DF, Thakor NV. A novel quantitative EEG injury measure of global cerebral ischemia. Clin Neurophysiol. 2000;111:1779–1787. [DOI] [PubMed] [Google Scholar]
- 22. Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Ohta S, Fukuda K, Hori S. Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest, independent of targeted temperature management. Circulation. 2014;130:2173–2180. [DOI] [PubMed] [Google Scholar]
- 23. Tosun C, Kurland DB, Mehta R, Castellani RJ, Dejong JL, Kwon MS, Woo SK, Gerzanich V, Simard JM. Inhibition of the Sur1‐Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke. 2013;44:3522–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. The sulfonylurea receptor 1 (Sur1)‐transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem. 2013;288:3655–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, Pellis T, Stammet P, Wanscher M, Wise MP, Åneman A, Al‐Subaie N, Boesgaard S, Bro‐Jeppesen J, Brunetti I, Bugge JF, Hingston CD, Juffermans NP, Koopmans M, Køber L, Langørgen J, Lilja G, Møller JE, Rundgren M, Rylander C, Smid O, Werer C, Winkel P, Friberg H; TTM Trial Investigators . Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. N Engl J Med. 2013;369:2197–2206. [DOI] [PubMed] [Google Scholar]
- 26. Rittenberger JC, Callaway CW. Temperature management and modern post‐cardiac arrest care. N Engl J Med. 2013;369:2262–2263. [DOI] [PubMed] [Google Scholar]
- 27. Drabek T, Kochanek PM. Improving outcomes from resuscitation: from hypertension and hemodilution to therapeutic hypothermia to H2. Circulation. 2014;130:2133–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, Gabrielli A, Silvers SM, Zaritsky AL, Merchant R, Vanden Hoek TL, Kronick SL; American Heart Association . Part 9: post‐cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S768–S786. [DOI] [PubMed] [Google Scholar]
- 29. Lu X, Ma L, Sun S, Xu J, Zhu C, Tang W. The effects of the rate of postresuscitation rewarming following hypothermia on outcomes of cardiopulmonary resuscitation in a rat model. Crit Care Med. 2014;42:e106–e113. [DOI] [PubMed] [Google Scholar]
- 30. Jia X, Koenig MA, Shin HC, Zhen G, Pardo CA, Hanley DF, Thakor NV, Geocadin RG. Improving neurological outcomes post‐cardiac arrest in a rat model: immediate hypothermia and quantitative EEG monitoring. Resuscitation. 2008;76:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ye S, Weng Y, Sun S, Chen W, Wu X, Li Z, Weil MH, Tang W. Comparison of the durations of mild therapeutic hypothermia on outcome after cardiopulmonary resuscitation in the rat. Circulation. 2012;125:123–129. [DOI] [PubMed] [Google Scholar]
- 32. Wang B, Armstrong JS, Lee JH, Bhalala U, Kulikowicz E, Zhang H, Reyes M, Moy N, Spicer D, Zhu J, Yang ZJ, Koehler RC, Martin LJ, Lee JK. Rewarming from therapeutic hypothermia induces cortical neuron apoptosis in a swine model of neonatal hypoxic‐ischemic encephalopathy. J Cereb Blood Flow Metab. 2015;35:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–S202. [DOI] [PubMed] [Google Scholar]
- 34. Simard JM, Woo SK, Bhatta S, Gerzanich V. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol. 2008;8:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simard JM, Woo SK, Tsymbalyuk N, Voloshyn O, Yurovsky V, Ivanova S, Lee R, Gerzanich V. Glibenclamide‐10‐h treatment window in a clinically relevant model of stroke. Transl Stroke Res. 2012;3:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V. Protective effect of delayed treatment with low‐dose glibenclamide in three models of ischemic stroke. Stroke. 2009;40:604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li X, Ji Z, Gu Y, Hu Y, Huang K, Pan S. Mild hypothermia decreases the total clearance of glibenclamide after low dose administration in rats. Neurosci Lett. 2015;614:55–59. [DOI] [PubMed] [Google Scholar]