

Abstract
Background
The pathogenesis of essential hypertension is multifactorial with different underlying mechanisms contributing to disease. We have recently shown that TNF superfamily member 14 LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14) induces hypertension when injected into mice. Research reported here was undertaken to examine the role of transglutaminase (TGase) in LIGHT‐induced hypertension.
Methods and Results
Initial experiments showed that plasma and kidney TGase activity was induced by LIGHT infusion (13.91±2.92 versus 6.75±1.92 mU/mL and 19.86±3.55 versus 12.00±0.97 mU/10 μg) and was accompanied with hypertension (169±7.16 versus 117.17±11.57 mm Hg at day 14) and renal impairment (proteinuria, 61.33±23.21 versus 20.38±9.01 μg/mg; osmolality, 879.57±93.02 versus 1407.2±308.04 mmol/kg). The increase in renal TGase activity corresponded to an increase in RNA for the tissue TGase isoform, termed TG2. Pharmacologically, we showed that LIGHT‐induced hypertension and renal impairment did not occur in the presence of cystamine, a well‐known competitive inhibitor of TGase activity. Genetically, we showed that LIGHT‐mediated induction of TGase, along with hypertension and renal impairment, was dependent on interleukin‐6 and endothelial hypoxia inducible factor‐1α. We also demonstrated that interleukin‐6, endothelial hypoxia inducible factor‐1α, and TGase are required for LIGHT‐induced production of angiotensin receptor agonistic autoantibodies.
Conclusions
Thus, LIGHT‐induced hypertension, renal impairment, and production of angiotensin receptor agonistic autoantibodies require TGase, most likely the TG2 isoform. Our findings establish TGase as a critical link between inflammation, hypertension, and autoimmunity.
Keywords: angiotensin receptor, AT1‐AA, hypertension, inflammation, transglutaminase
Subject Categories: Inflammation, Hypertension, Animal Models of Human Disease, Vascular Biology, Genetically Altered and Transgenic Models
Introduction
The pathogenesis of essential hypertension is multifactorial, with different underlying mechanisms contributing to the disease. In recent years, there has been an increased effort to determine the role of the immune system in the development of hypertension.1, 2, 3, 4, 5 Research concerning the immunological basis of hypertension has revealed that both the innate and adaptive arms of the immune system contribute to the pathophysiology of hypertension, features explored by research presented here. The inflammatory response that accompanies activation of the innate immune system has received considerable attention and has revealed a prominent role for proinflammatory cytokines in the development of hypertension.6, 7, 8 Human hypertension is associated with highly elevated levels of key proinflammatory markers, including C‐reactive protein, tumor necrosis factor (TNF), interleukin (IL)‐1β, IL‐6, and IL‐17, among others.6, 7, 8 The molecular mechanisms linking inflammation to hypertension are poorly understood.
We have examined the role of inflammatory cytokines in an experimental model of hypertension in mice.9 Specifically we have recently shown that TNF superfamily member 14, LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator [HVEM], a receptor expressed by T lymphocytes, also known as TNFSF14) is elevated in women with preeclampsia (a serious hypertensive condition of pregnancy) and induces hypertension when injected into pregnant or nonpregnant mice.9 LIGHT is produced by cells of the innate and adaptive immune system, including granulocytes, monocytes, macrophages, dendritic cells, and T cells.10, 11 It is initially present on the cell membrane as a homotrimer but can be released from the cell membrane via proteolytic cleavage and exist in a soluble form.12 Whether membrane bound or free, LIGHT can bind to two receptors, HVEM, which is present on T cells, and lymphotoxin β receptor, present on nonlymphoid hematopoietic cells including natural killer cells.13 LIGHT‐mediated activation of HVEM receptors on T cells results in a costimulatory response leading to T‐cell activation, cell proliferation, and cytokine production.11 LIGHT‐deficient mice have reduced T‐cell immunity associated with impaired allograph rejection.14 Thus, considerable evidence supports a role for LIGHT in adaptive T‐cell immunity and cytokine production. The molecular mechanisms responsible for LIGHT‐induced hypertension are not understood, a knowledge gap addressed by experiments reported here.
Transglutaminases (TGases) are a widely distributed family of enzymes that modify proteins through the hydrolytic deamination or transamidation of the carboxamide moiety of glutamine residues on proteins.15 Several lines of evidence link TGases with inflammation and hypertension.16, 17, 18, 19, 20, 21 Initial evidence that TGase contributes to hypertension came from studies showing that cystamine, a transglutaminase inhibitor, reduced blood pressure in the spontaneously hypertensive rat.22 More recently, we showed that circulating TGase activity is significantly elevated in women with preeclampsia and in an experimental model of preeclampsia in mice.23 A pathogenic role for TGase in preeclampsia is suggested by our experiments showing that the TGase inhibitor cystamine or small interfering RNA (siRNA) to tissue TGase (TG2, a member of the TGase family of enzymes) mRNA significantly attenuated the hypertension and proteinuria in this mouse model. Data linking TGase to inflammation are based on reports showing that Tgm2 gene expression (encodes TG2) is induced by transforming growth factor (TGF)‐β,24 TNF‐α,19 and IL‐6,20 proinflammatory cytokines that are highly elevated in hypertensive disease,6, 7, 8 including preeclampsia.25, 26, 27 Cytokine‐induced Tgm2 gene transcription is mediated by nuclear factor‐κB28, 29 and hypoxia inducible factor (HIF)‐1α.30 Experiments reported here test the hypothesis that TGase is a critical link between inflammation and hypertension.
Other factors potentially linking inflammation with hypertension are agonistic autoantibodies to the AT1 angiotensin receptor (AT1R) that are associated with hypertensive conditions in humans.31, 32, 33 These autoantibodies, termed AT1‐AAs, were initially identified in preeclampsia where they are present in the maternal circulation of a large majority of affected women.34, 35 These autoantibodies cause hypertension and proteinuria when introduced into pregnant or nonpregnant mice36 and presumably contribute to these features in the women with preeclampsia from whom they were obtained. In addition to preeclampsia, these pathogenic autoantibodies are also associated with hypertensive conditions outside of pregnancy including malignant hypertension,37, 38 refractory hypertension,39, 40, 41 and primary aldosteronism.42, 43, 44, 45 An interesting feature of these autoantibodies is that they uniformly recognize the same epitope (AFHYESQ) located on the second extracellular loop of AT1Rs. Evidence linking AT1‐AAs with inflammation is provided by experiments showing that proinflammatory cytokines (TNF, IL‐6, IL‐17, and LIGHT) induce their production in pregnant animals.46, 47, 48, 49 Proinflammatory cytokines also contribute hypertension in nonpregnant animals,9, 50 but the possibility of pathogenic autoantibody production in these cases has not been examined. We show here that LIGHT‐induced hypertension in nonpregnant mice is accompanied with the production of AT1‐AAs and that the production of these pathogenic autoantibodies required IL‐6 and endothelial HIF‐1α–dependent induction of TGase. Overall, our results show that TGase is a critical factor in cytokine‐induced hypertension and the production of AT1‐AAs, pathogenic autoantibodies associated with hypertension.
Materials and Methods
Animals
Wild‐type 8‐ to 10‐week‐old C57BL6 mice were purchased from Harlan Laboratories. IL‐6–deficient mice (IL‐6−/−) congenic with the C57BL6 background were generated and genotyped as described.51 We generated mice with specific endothelial HIF‐1α deficiency by mating floxed HIF‐1α mice (HIF‐1αf/f) with the VE‐cadherin‐Cre + mice containing a transgene expressing Cre recombinase only in the endothelial cells. Hif‐1αf/f mice and Hif‐1αf/f /VE‐cadherin cre + mice, also congenic with C57BL6 mice, were originally from Dr Holger Eltzchig's laboratory at the University of Colorado at Denver and have been described in a previous publication.52 Six to 8 mice for each group were infused with LIGHT via minipump. Mice were anesthetized with isoflurane (2%), and osmotic minipumps were implanted subcutaneously in the neck. Recombinant mouse LIGHT (R&D Systems) was delivered at a rate of 4 ng/d into mice for 14 days. Cystamine‐treated mice were provided drinking water containing 0.9 g/L cystamine dihydrochloride throughout the 14 days. Control mice were infused with saline. We collected urine on days 3, 5 and 14 and measured blood pressure at 0, 3, 5, 7, 10, and 14 days. After treatment for 14 days, mice were killed. All protocols involving animal research were reviewed and approved by the Institutional Animal Welfare Committee of the University of Texas Health Science Center at Houston. All animal procedures were in accordance with institutional guidelines.
Plasma TGase Activity
TGase activity in human and mouse samples was determined using in vitro TGase assay kits (Covalab; Sigma‐Aldrich) following the manufacturer's instructions. The increased TGase activity we see in LIGHT‐infused animals is unlikely to be factor XIIIa transglutaminase because the use of EDTA as an anticoagulant in plasma collection results in the cleavage and inactivation of factor XIIIa by thrombin.53
Real‐Time Polymerase Chain Reaction of Kidney RNA
RNA was extracted using Trizol reagent (Invitrogen). Transcript levels were quantified using real‐time quantitative reverse transcription–polymerase chain reaction. Syber green was used for analysis of Tgm2 and Gapdh mRNAs by using the following primers: mouse: Tgm2, forward: 5′‐ggtggcaggccattgacccc‐3′, reverse: 5′‐gccccacgaccaaggaacgg‐3′, Gapdh, forward: 5′‐tgacctcaactacatggtctaca‐3′, reverse: 5′‐cttcccattctcggccttg‐3′. Polymerase chain reaction was performed using an ABI Prism 7700 sequence Detector (Applied Biosystems). Each cDNA sample was run in triplicate. For data analysis, the ΔΔCt method was used. For each gene, the fold change was calculated as a difference in kidney gene expression.
IL‐6 ELISA of Mice
We detected IL‐6 levels in mice blood using a BD OptEIA™ Mouse IL‐6 ELISA Kit (BD Biosciences) following the instructions provided by the vendor.
Blood Pressure Measurement and Quantification of Urinary Protein
The systolic blood pressure was noninvasively measured by determining the tail blood volume with a volume pressure recording sensor and an occlusion tail‐cuff (CODA System; Kent Scientific). We routinely adapt the mice to blood pressure measurements by training them 3 or 4 times in the device before initiating the actual experiments and have successfully used this approach many times in recent years.9, 36, 54, 55, 56 Blood pressure was measured at the same time daily (±1 hour) while the mice were kept warm using a warming pad. In previous experiments, we have validated blood pressure measurements through invasive monitoring of intracaroitid arterial pressure in mice on the last day of tail‐cuff measurement.9 Further, published results show that this method is in good agreement with radiotelemetry measurements of blood pressure.57
Twenty‐four‐hour urine specimens were collected for analysis through the use of metabolic cages (Nalgene). Mice were trained in metabolic cages for 2 days before urine collection. All of the mice were killed on GD18.5 before delivery, when their serum and organs were collected. We quantified urinary albumin using ELISA (Exocell) and measured urinary creatinine with the use of a picric acid colorimetric assay (Exocell). We used the ratio of urinary albumin to urinary creatinine as an index of urinary protein. Urine osmolality was measured by using a vapor pressure osmometer (Wescor).
Measurement of AT1‐AAs
ELISA
AT1‐AAs were measured as previously described49 using a commercially available sandwich ELISA (CellTrend GmbH) that is now marketed by One Lambda. The ELISA from CellTrend was supplied to measure human AT1‐AAs. For measurement of murine AT1‐AAs, the HRP‐linked antihuman IgG that was supplied with the kit was replaced with horseradish peroxidase–linked antimouse IgG secondary antibody (Jackson ImmunoResearch Labs). Other steps in this measurement were carried out according to vendor instructions. We have recently described the use of the original assay to detect human AT1‐AAs and its modification to measure mouse AT1‐AAs.49
Peptide display
Plasma samples from mice were assayed for binding to the 7–amino acid epitope peptide by using an assay in which the epitope sequence AFHYESQ is displayed in a high‐avidity format on the bacterial cell surface.58 The assay detects only mouse IgG antibodies that bind to the 7–amino acid epitope peptide. This is because after incubation with mouse plasma (1:100 dilution), whatever binds to the 7–amino acid epitope peptide is detected with the use of biotinylated goat antimouse IgG antibody, diluted 1:200 (Vector Labs). The latter is detected with the use of a streptavidin‐conjugated R‐phycoerythrin (bright red‐orange fluorescence), diluted 1:333. All of these incubations are carried out with reagents diluted in PBS with 0.1% BSA. AT1‐AA binding activity is measured as fold fluorescence over background (a negative control consisting of bacteria with an empty scaffold, ie, no displayed peptide). Thus, this assay only detects mouse antibodies that bind to the empty scaffold and to the scaffold with the 7–amino acid epitope peptide displayed. Further, another step of preincubating the plasma with the negative control bacteria is also used to remove anything binding to bacteria with the empty scaffold. Antibody binding to the 7–amino acid epitope peptide is expressed as fold increase over background. This assay was initially described by Elliott et al58 and subsequently used by Liu et al.49 This methodology does not require purified antibodies as additional work on bacterial displayed peptide libraries has used diluted plasma with an Ig‐specific secondary reagent.59 Detecting plasma Ig with a secondary reagent is consistently used wth ELISA and protein microarray studies as well.60
Statistical Analyses
All data are expressed as mean±SEM. Measurements of blood pressure, urinary protein concentrations, and plasma concentrations of autoantibodies and enzymes typically approximated a normal distribution. Unpaired Student t tests were applied in 2‐group analysis. Differences between the means of multiple groups were compared by using 1‐way ANOVA, followed by Tukey's multiple comparisons test. Blood pressure measurements data are mean values taken at different time points (days 0, 3, 5, 7, 10, and 14). The longitudinal data were analyzed by using a repeated‐measures ANOVA followed by Tukey's multiple comparisons test. AT1‐AA epitope binding activity data were analyzed by using 1‐way ANOVA followed by Dunnett's multiple comparisons test. A value of P<0.05 was considered significant. Statistical programs were run with GraphPad Prism 5 software (GraphPad Software).
Results
LIGHT Infusion Results in an IL‐6– and Endothelial HIF‐1α–Dependent Increase in Plasma and Renal TGase Activity
We have previously shown that LIGHT induces hypertension in pregnant and nonpregnant animals.9 To determine if elevated TGase is also associated with LIGHT‐induced hypertension, we infused recombinant mouse LIGHT into mice for 5 or 14 days and examined mice for elevated TGase. The results show that plasma TGase activity (Figure 1A) was significantly increased after 14 days of LIGHT infusion. Kidney TGase activity showed a small increase at day 5 and a significantly greater increase at day 14 (Figure 1B). The increase in plasma and renal TGase activity was prevented with continuous administration of cystamine, a competitive inhibitor of TGase.
Figure 1.
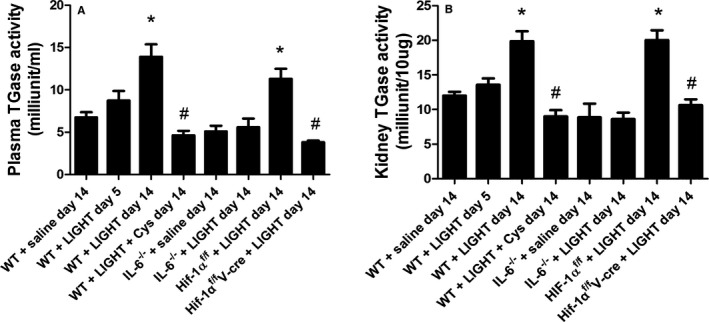
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14) infusion results in an interleukin (IL)‐6– and endothelial hypoxia inducible factor (HIF)‐1α–dependent increase in plasma and renal transglutaminase activity that is inhibited by cystamine (Cys). Wild‐type (WT) or mutant mice were infused with recombinant mouse LIGHT for up to 14 days in the presence or absence of Cys. Plasma transglutaminase (TGase) activity (A) and kidney TGase activity (B) were determined following 5 or 14 days of LIGHT infusion. Data are expressed as mean±SEM; *P<0.05 vs saline‐infused mice; # P<0.05 vs LIGHT‐infused mice. n=6 for all groups except for WT+saline day 14, WT+LIGHT day 14, where n=8 (V‐cre indicates VE‐cadherin‐cre + ).
To determine if the increased renal TGase was caused by increased transcriptional activity, we prepared kidney RNA from LIGHT‐infused animals and saline‐infused animals and determined the level of TGase mRNA through quantitative RT‐PCR. RNA was probed for the TG2 isoform of TGase, termed tissue TGase, that is the most abundant TGase isoform in the kidney. The results (Figure 2) show an increase in TG2 mRNA evident by day 5 of LIGHT infusion, reaching a significant increase by day 14. These results indicate that LIGHT infusion mediates increased transcription of the Tgm2 gene encoding TG2.
Figure 2.
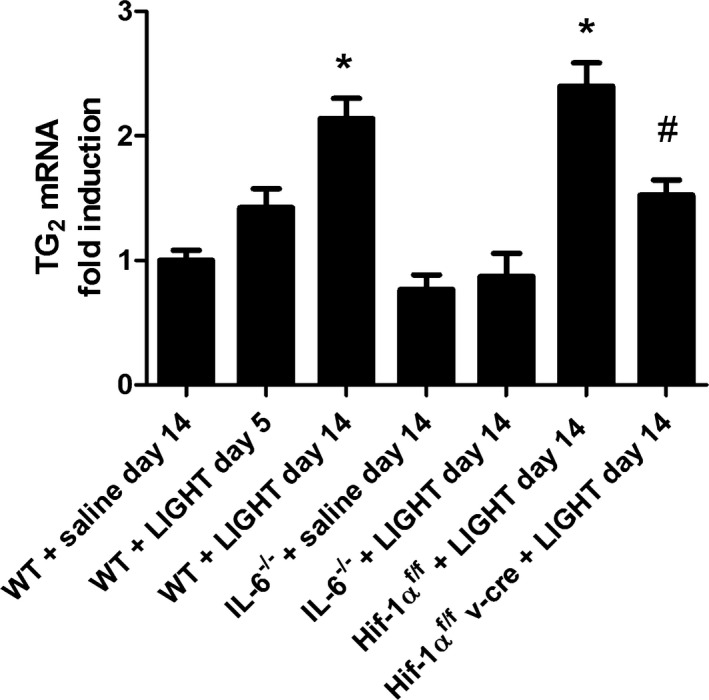
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14)‐induced TG2 mRNA in kidneys. Wild‐type mice were infused with recombinant mouse LIGHT for either 5 or 14 days. Kidneys were obtained, and RNA was extracted and analyzed by quantitative RT‐PCR for TG2 mRNA. Data are expressed as mean±SEM; *P<0.05 vs saline‐infused mice; # P<0.05 vs hypoxia inducible factor (HIF)‐1αf/f+LIGHT‐infused mice. n=6 for all groups except for WT+saline day 14, WT+LIGHT day 14, where n=8.
The Tgm2 gene is activated by inflammation and hypoxia and specifically includes regulatory elements that respond to IL‐620 and HIF‐1.30 To determine if IL‐6 or HIF‐1α is required for LIGHT induction of TG2, experiments were carried out using mice that are globally deficient in IL‐6 and mice that have an endothelial specific HIF‐1α deficiency (Hif‐1αf/f/ /VE‐cadherin cre +).52 The results (Figure 1A and 1B) show that LIGHT induction of plasma and renal TGase activity did not occur in these mice. Likewise, LIGHT‐mediated induction of renal TG2 mRNA was significantly inhibited in IL‐6–deficient mice or mice with an endothelial specific HIF‐1α deficiency (Figure 2). The requirement for IL‐6 is consistent with data showing that LIGHT infusion resulted in increased circulating IL‐6 (Figure 3). Overall, these results indicate that TG2, encoded by the Tgm2 gene TG2, is the TGase that is induced by LIGHT. Further, the results from the endothelial HIF‐1α–deficient mice suggest that the majority of the TG2 is derived from endothelial cells.
Figure 3.
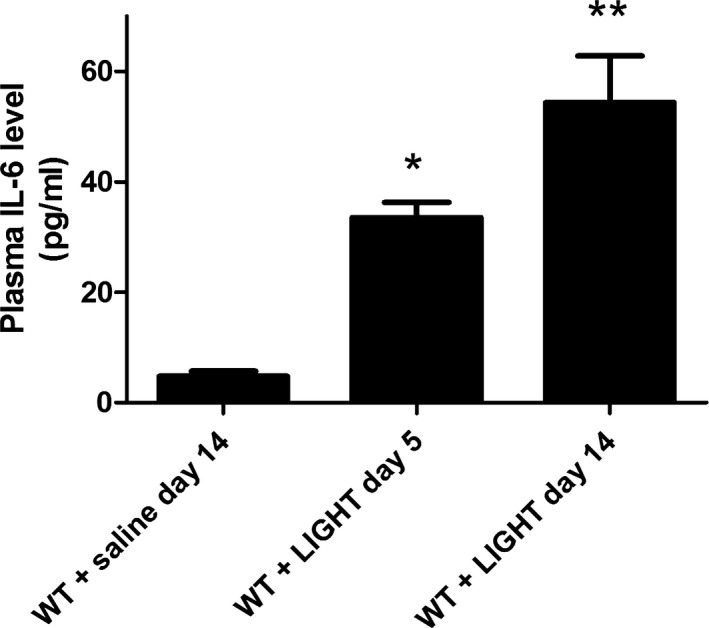
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14)‐induced interleukin (IL)‐6. Wild‐type mice were infused with recombinant mouse LIGHT for either 5 or 14 days. Plasma was obtained, and the concentration of IL‐6 was determined by ELISA. Data are expressed as mean±SEM; *P<0.05 vs saline‐infused mice; **P<0.01 vs saline‐infused mice. n=6 for WT + LIGHT day 5 and n=8 for WT+saline day 14 and WT+LIGHT day 14.
LIGHT‐Induced Hypertension Requires TGase and Is Dependent on IL‐6 and Endothelial HIF‐1α
To determine if elevated TGase is required for LIGHT‐induced hypertension, mice were continuously infused with LIGHT for 14 days in the presence or absence of cystamine. Blood pressure was monitored periodically from the day of injection through day 14 through the use of tail‐cuff plethysmography. We found (Figure 4A) that systolic blood pressure increased significantly by 3 days of LIGHT infusion and continued to increase throughout the 14‐day experiment. Blood pressure in the saline‐infused mice remained constant. The results (Figure 4A) also show that the presence of cystamine significantly inhibited LIGHT‐induced hypertension.
Figure 4.
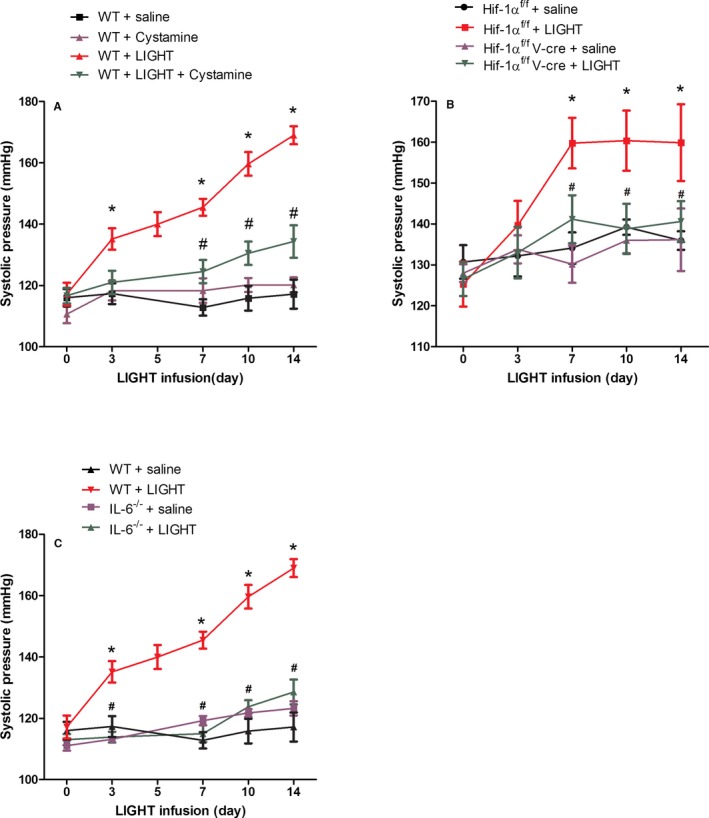
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14)‐induced hypertension is inhibited by cystamine and is dependent on interleukin (IL)‐6 and endothelial hypoxia inducible factor (HIF)‐1α. Wild‐type (WT) or mutant mice were infused with recombinant mouse LIGHT for up to 14 days in the presence or absence of cystamine (Cys). Systolic blood pressure was measured at periodic intervals by tail‐cuff plethysmography. Data are expressed as mean±SEM; *P<0.05 vs saline‐infused mice, # P<0.05 vs C57BL6+LIGHT (A and C) or HIF‐1αf/f+LIGHT (B). n=6 for all groups except for WT+saline day 14, WT+LIGHT day 14, where n=8 (V‐cre indicates VE‐cadherin‐cre +).
We also conducted LIGHT infusion experiments using IL‐6–deficient mice and mice with an endothelial cell–specific deficiency in HIF‐1α (Hif‐1αf/f /VE‐cadherin cre +). LIGHT‐mediated induction of hypertension did not occur in mice with an endothelial cell–specific deficiency in HIF‐1α (Figure 4B) or a global deficiency in IL‐6 (Figure 4C). As shown in Figure 1, LIGHT‐mediated induction of plasma and renal TGase did not occur in these mice. These results show that LIGHT infusion induces a robust increase in blood pressure that requires TGase and is dependent on IL‐6 and endothelial HIF‐1α.
LIGHT‐Induced Renal Impairment Requires TGase and Is Dependent on IL‐6 and Endothelial HIF‐1α
To determine if renal impairment was associated with LIGHT‐induced hypertension and the induction of TGase, we collected urine in metabolic cages on days 3, 5, and 14 for determination of proteinuria and urine osmolality. Saline‐infused animals were used as controls, and cystamine‐treated animals served to determine the contribution of TGase to any observed changes in proteinuria or urine osmolality. The results show that proteinuria (Figure 5A) began to increase by 3 and 5 days of LIGHT infusion, although the increase did not achieve statistical significance. However, by day 14, there was a significant LIGHT‐induced elevation in urinary protein (Figure 5A). We also observed a significant reduction in urine osmolality after 14 days of LIGHT infusion (Figure 5B). These LIGHT‐induced changes in renal physiology were prevented by cystamine treatment. Additionally, these changes did not occur in IL‐6–deficient mice or mice with an endothelial HIF‐1α deficiency. Overall, these results indicate that LIGHT‐induced renal impairment is dependent on IL‐6– and endothelial HIF‐1α–mediated induction of TG2.
Figure 5.
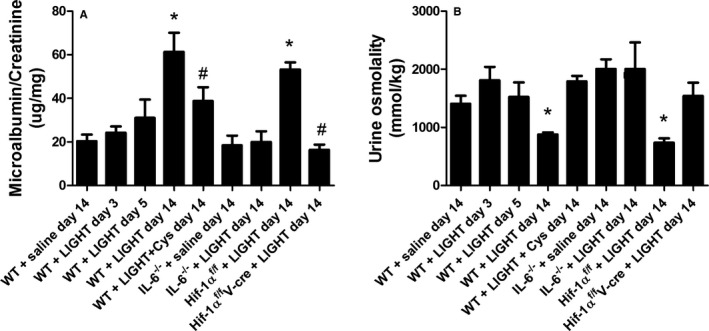
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14)‐induced proteinuria and urine osmolality is inhibited by cystamine and is dependent on interleukin (IL)‐6 and endothelial hypoxia inducible factor (HIF)‐1α. Wild‐type (WT) or mutant mice were infused with recombinant mouse LIGHT for up to 14 days in the presence or absence of cystamine (Cys). Twenty‐four‐hour urine specimens were collected at the times indicated, and the ratio of albumin to creatinine (A) and urine osmolality (B) was determined. Data are expressed as mean±SEM; *P<0.05 vs saline‐infused mice; # P<0.05 vs LIGHT‐infused mice. n=6 for all groups except for WT+saline day 14, WT+LIGHT day 14, where n=8 (V‐cre indicates VE‐cadherin‐cre +).
IL‐6– and Endothelial HIF‐1α–Dependent Induction of TGase Is Required for Production of AT1‐AAs in LIGHT‐Infused Mice
To determine if LIGHT‐induced hypertension and renal impairment were accompanied by the production of AT1‐AAs and to determine if TGase is required for cytokine‐induced AT1‐AA production, we carried out LIGHT infusion experiments in the presence or absence of cystamine, as described here earlier. Some mice were killed after 5 days of LIGHT infusion, while others were killed after 14 days of infusion. Blood was collected for determination of AT1‐AA titer. Autoantibody titers were determined by using a cell‐based ELISA, and a peptide display assay in which the epitope peptide recognized by human AT1‐AA (AFHYESQ) is displayed on the cell wall of specially engineered bacteria. The results from each assay (Figure 6A and 6B) are in good agreement and show that AT1‐AAs were slightly (though not significantly) elevated at day 5 of infusion and that autoantibody titers were significantly elevated following 14 days of LIGHT infusion. The results of the epitope peptide display assay indicates that the antibodies produced following LIGHT infusion bind to the same 7–amino acid epitope on the second extracellular loop of the AT1 receptor that is recognized by autoantibodies in women with preeclampsia. Additionally, the induction is blocked by the presence of cystamine, indicating that TGase activity is required for LIGHT‐induced autoantibody production. Mutant mice lacking IL‐6 or endothelial HIF‐1α also failed to show LIGHT‐induced autoantibody production (Figure 6A and 6B). Together, these results show that AT1‐AAs are produced in LIGHT‐infused mice by a process that depends on IL‐6– and endothelial HIF‐1α–dependent induction of TGase.
Figure 6.
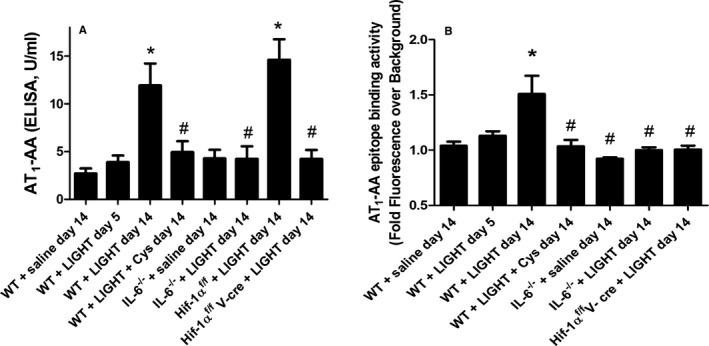
LIGHT (an acronym for homologous to lymphotoxins, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14)‐induced agonistic autoantibodies to the AT1 angiotensin receptor (AT1‐AAs) production is inhibited by cystamine (Cys) and is dependent on interleukin (IL)‐6 and endothelial hypoxia inducible factor (HIF)‐1α. Wild‐type (WT) or mutant mice were infused with recombinant mouse LIGHT for up to 14 days in the presence or absence of Cys. AT 1‐AA levels were determined by using a cell‐based ELISA (A) or a bacterial epitope peptide display assay (B). Data are expressed as mean±SEM; *P<0.05 vs saline‐infused WT mice; # P<0.05 vs LIGHT‐infused WT mice. n=6 for all groups except for WT+saline day 14, WT+LIGHT day 14, where n=8 (A). n=4 for WT+saline day 14, n=5 for WT+LIGHT day 5, n=8 for WT+LIGHT day 14, n=5 for WT+LIGHT+cys day 14, n=3 for IL‐6−/−+saline day 14 and IL‐6−/−+LIGHT day 14, n=6 for HIF‐1αf/ fV‐cre+LIGHT day 14 (B) (V‐cre indicates VE‐cadherin‐cre+).
Discussion
Research reported here was undertaken to identify factors that contribute to LIGHT‐induced hypertension.9 Using a combination of pharmacological and genetic approaches, we showed that elevated TGase activity was required for LIGHT‐induced hypertension and renal impairment. Pharmacologically, we showed that LIGHT‐induced hypertension and renal impairment did not occur in the presence of cystamine, a well‐known competitive inhibitor of TGase activity. Genetically, we showed that LIGHT‐induced hypertension, renal impairment and elevated TGase were dependent on IL‐6 and endothelial HIF‐1α. These results extend earlier observations that TGase inhibitors or siRNA to tissue TGase (TG2) can lower blood pressure in animal models of hypertensive disease.22, 23 We also show here that LIGHT induced the production of AT1‐AAs and that elevated TGase is required for autoantibody production. LIGHT‐induced AT1‐AA production also requires IL‐6 and endothelial HIF‐1α. Thus, our results show that LIGHT‐induced TGase, hypertension, renal impairment, and the production of AT1‐AAs are inhibited by cystamine and require IL‐6 and endothelial HIF‐1α.
Considerable evidence from human studies shows a strong association between hypertension and elevated inflammatory cytokines, including the inflammatory marker C‐reactive protein.61, 62, 63, 64, 65 These results have led some to hypothesize that hypertension is in part an inflammatory disorder. This hypothesis is supported by experimental studies in mice showing that TNF‐α, IL‐6, and IL‐17 are required for angiotensin II–induced hypertension in mice.66, 67, 68, 69, 70 Additional support comes from experiments showing that the introduction of IL‐17 or LIGHT, a TNF superfamily member, into mice results in hypertension.9, 50 Animal models, such as the one we describe here, that recapitulate the hypertensive consequences of elevated inflammatory cytokines provide critically important experimental opportunities to determine the mechanism of cytokine‐induced hypertension, a knowledge gap we have addressed by research reported here. Using our experimental model, we present data indicating that endothelial TG2 is required for LIGHT‐induced hypertension. Our research provides original evidence that TG2 is a previously unrecognized factor linking inflammation, autoimmunity, and hypertension.
Because AT1‐AAs are likely to be significant contributors to hypertension,32 it is important to understand the pathophysiological conditions and molecular mechanisms that initiate their production. Evidence linking inflammation with AT1‐AA production is provided by experiments showing that proinflammatory cytokines (TNF, IL‐6, IL‐17, and LIGHT) induce AT1‐AA production in pregnant animals.46, 47, 48 In these experiments, antibody was detected within 5 days of cytokine infusion or injection. However, proinflammatory cytokines also cause hypertension in nonpregnant animals,9, 50 but the possibility of pathogenic autoantibody production in these cases has not been thoroughly examined until now. Earlier studies failed to detect AT1‐AA production in nonpregnant rats within 5 days of initial infusion of proinflammatory cytokines TNF, IL‐6, and IL‐17.46, 47, 48 However, these investigators did not infuse nonpregnant animals for longer times and thus may have missed AT1‐AA production occurring after 5 days of cytokine infusion. We, too, did not observe a significant increase in AT1‐AA production following 5 days of cytokine infusion (Figure 6). However, by 14 days of LIGHT infusion, AT1‐AA production was readily apparent (Figure 6). Thus, AT1‐AAs appear within 5 days of cytokine injection in pregnant mice but require a longer time to appear in LIGHT‐injected nonpregnant mice. The cytokine infusion models of AT1‐AA production represent well‐defined and experimentally pliable model systems for identifying the molecular mechanisms that underlie autoantibody production.
A commonly considered mechanism for an autoimmune response is posttranslational modification, resulting in the creation of a neoantigen that is recognized as nonself by the immune system.71, 72 A large percentage of proteins in the body are posttranslationally modified, and it is now clear that some of these modifications create neoantigens that stimulate an autoimmune response.73, 74 One of the best examples of this is the posttranslational modification of arginine residues to citrulline, a process termed “citrullination” that contributes to autoimmune diseases such as rheumatoid arthritis and multiple sclerosis.72 Elevated TGase is commonly associated with inflammation and may contribute to autoimmunity by posttranslational modification of proteins, possibly by the addition of serotonin.75 We have recently reported that circulating TGase activity is elevated in women with preeclampsia and in an experimental mouse model of preeclampsia.23 Further, we have shown that TG2 modifies AT1 receptors in placentas of women with preeclampsia and in the placentas of a mouse model of preeclampsia by the addition of a small‐molecular‐weight molecule.23 We have shown here that LIGHT‐induced hypertension in nonpregnant mice is accompanied by the production of AT1‐AAs and that the production of these pathogenic autoantibodies is blocked by cystamine, a competitive inhibitor of TGase. An interesting feature of these autoantibodies is that they uniformly recognize the same epitope (AFHYESQ) located on the second extracellular loop of AT1 receptors. It is noteworthy that the epitope peptide contains a glutamine (Q) (1 of only 5 in the entire protein), a potential target of posttranslational modification by TGase. Thus, cytokine‐induced TG2 production in endothelial cells may result in posttranslational modification of AT1 receptors and autoimmune recognition of the TG2‐modified receptor. Our findings raise the interesting possibility that TG2‐mediated PTM of AT1 receptors may contribute to autoimmune recognition and autoantibody production (Figure 7), a possibility to be tested by future research.
Figure 7.
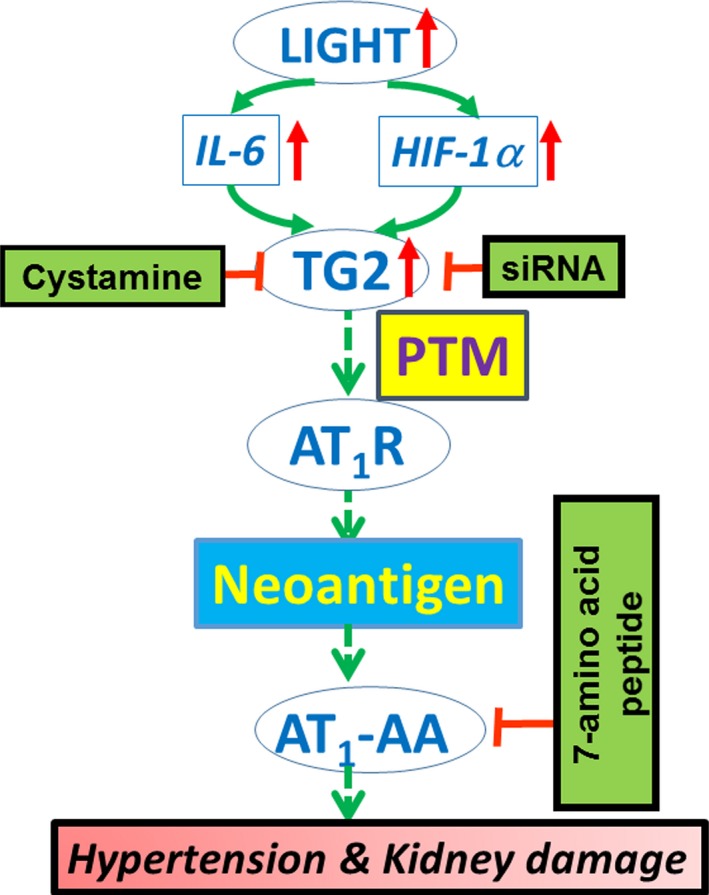
Hypothesis concerning cytokine‐induced hypertension. A model of hypertension involving LIGHT (an acronym for homologous to lymphotoxins, exhibits Inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes, also known as TNFSF14) induced tissue transglutaminase (TG2) and the posttranslational modification (PTM) of AT1 angiotensin receptor (AT1Rs) resulting in the creation of a neoantigen that underlies autoimmune production of agonistic autoantibodies to the AT1 angiotensin receptor (AT1‐AAs). The resulting autoantibodies contribute to hypertension by activation of AT 1Rs.
The mammalian TGase family includes 8 enzymatically active homologues (TG1‐7 and factor XIIIa) that catalyze the posttranslational modification of selected glutamine residues on proteins.76 Factor XIIIa is well known for its role in blood clotting and is located primarily in platelets. TG1 and TG3 are produced in keratinocytes of skin, and TG5 is produced in hair follicles. TG4 is predominantly found in the prostate. The expression patterns of TG6 and TG7 are not well characterized. TG2 (also called tissue TGase or endothelial TGase) is the most widely distributed member of the TGase family because of its presence in endothelial cells, smooth muscle cells, and fibroblasts. It is encoded by the Tgm2 gene and is the most studied.15 Previous studies have shown that TNF‐α and IL‐6 induce Tgm2 gene expression in cultured hepatocytes.19, 20 Recent studies have shown that Tgm2 expression is enhanced synergistically by interferon‐γ and TNF‐α in human small intestinal cells.18 We show here that LIGHT‐induced renal TGase and TG2 mRNA is prevented in IL‐6–deficient mice and mice with an endothelial specific deletion of HIF‐1α. IL‐6 and HIF‐1 are key transcriptional regulators of Tgm2 gene expression.15, 30, 77 Our results suggest that the majority of plasma and renal TGase activity induced by LIGHT is TG2 derived from endothelial cells. Further, the effect of the endothelial specific HIF‐1α knockout on LIGHT‐induced hypertension suggests that endothelial TG2 is a major contributor to cytokine‐induced hypertension. A role for TGase in blood pressure control was originally indicated by studies showing that cystamine treatment reduced blood pressure in the spontaneously hypertensive rat.22 In a different setting, work from our laboratory showed that the increase in blood pressure in a mouse model of preeclampsia (a hypertensive condition of pregnancy) was prevented by cystamine or by siRNA knockdown of tissue TGase (TG2) mRNA.23 Thus, TG2 is an attractive target for antihypertensive therapy. Because TG2‐deficient mice have no apparent developmental or physiological defects,78, 79 it is likely that TG2‐specific inhibitors will have no adverse side effects because of the inhibition of TG2.80, 81
Our results suggest that transglutaminase is an essential link between inflammation, autoimmunity, and hypertension. Genetic approaches presented here indicate that IL‐6 and endothelial HIF‐1α are required for LIGHT‐induced TGase gene expression and that transglutaminase is required for LIGHT‐induced hypertension, renal impairment, and AT1‐AA production. The results from endothelial HIF‐1α–deficient mice suggest that the LIGHT‐induced TGase is primarily from endothelial cells. Future research is required to determine the molecular mechanisms by which LIGHT induces Tgm2 gene expression, the role of IL‐6 in this process, and the mechanisms by which TG2 contributes to hypertension. Our results suggest that potent and specific TG2 inhibitors may be useful drugs for blood pressure control in some individuals.
Sources of Funding
This work was supported by National Institutes of Health grants HL19549 (to Dr. Xia), RC4HD067977 and HD34130 (to Drs. Xia and Kellems) and by China National Natural Science Foundation grant 81228044 (to Dr. Xia).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003730 doi: 10.1161/JAHA.116.003730)
References
- 1. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end‐organ damage. Circ Res. 2015;116:1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodriguez‐Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10:56–62. [DOI] [PubMed] [Google Scholar]
- 3. Rodriguez‐Iturbe B, Pons H, Quiroz Y, Johnson RJ. The immunological basis of hypertension. Am J Hypertens. 2014;27:1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trott DW, Harrison DG. The immune system in hypertension. Adv Physiol Educ. 2014;38:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc. 2014;125:130–138; discussion 138‐40. [PMC free article] [PubMed] [Google Scholar]
- 6. De Miguel C, Rudemiller NP, Abais JM, Mattson DL. Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep. 2015;17:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dinh QN, Drummond GR, Sobey CG, Chrissobolis S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int. 2014;2014:406960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang W, Parchim NF, Iriyama T, Luo R, Zhao C, Liu C, Irani RA, Zhang W, Ning C, Zhang Y, Blackwell SC, Chen L, Tao L, Hicks MJ, Kellems RE, Xia Y. Excess LIGHT contributes to placental impairment, increased secretion of vasoactive factors, hypertension, and proteinuria in preeclampsia. Hypertension. 2014;63:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. [DOI] [PubMed] [Google Scholar]
- 11. Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, Chen L. LIGHT, a TNF‐like molecule, costimulates T cell proliferation and is required for dendritic cell‐mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. [DOI] [PubMed] [Google Scholar]
- 12. Granger SW, Butrovich KD, Houshmand P, Edwards WR, Ware CF. Genomic characterization of LIGHT reveals linkage to an immune response locus on chromosome 19p13.3 and distinct isoforms generated by alternate splicing or proteolysis. J Immunol. 2001;167:5122–5128. [DOI] [PubMed] [Google Scholar]
- 13. Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, Jiang GW, Lu J, Tan J, Ugustus M, Carter K, Rojas L, Zhu F, Lincoln C, Endress G, Xing L, Wang S, Oh KO, Gentz R, Ruben S, Lippman ME, Hsieh SL, Yang D. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest. 1998;102:1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye Q, Fraser CC, Gao W, Wang L, Busfield SJ, Wang C, Qiu Y, Coyle AJ, Gutierrez‐Ramos JC, Hancock WW. Modulation of LIGHT‐HVEM costimulation prolongs cardiac allograft survival. J Exp Med. 2002;195:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta. 2012;1823:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sane DC, Kontos JL, Greenberg CS. Roles of transglutaminases in cardiac and vascular diseases. Front Biosci. 2007;12:2530–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim SY. Transglutaminase 2 in inflammation. Front Biosci. 2006;11:3026–3035. [DOI] [PubMed] [Google Scholar]
- 18. Bayardo M, Punzi F, Bondar C, Chopita N, Chirdo F. Transglutaminase 2 expression is enhanced synergistically by interferon‐gamma and tumour necrosis factor‐alpha in human small intestine. Clin Exp Immunol. 2012;168:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuncio GS, Tsyganskaya M, Zhu J, Liu SL, Nagy L, Thomazy V, Davies PJ, Zern MA. TNF‐alpha modulates expression of the tissue transglutaminase gene in liver cells. Am J Physiol. 1998;274:G240–G245. [DOI] [PubMed] [Google Scholar]
- 20. Suto N, Ikura K, Sasaki R. Expression induced by interleukin‐6 of tissue‐type transglutaminase in human hepatoblastoma HepG2 cells. J Biol Chem. 1993;268:7469–7473. [PubMed] [Google Scholar]
- 21. Elli L, Ciulla MM, Busca G, Roncoroni L, Maioli C, Ferrero S, Bardella MT, Bonura A, Paliotti R, Terrani C, Braidotti P. Beneficial effects of treatment with transglutaminase inhibitor cystamine on the severity of inflammation in a rat model of inflammatory bowel disease. Lab Invest. 2011;91:452–461. [DOI] [PubMed] [Google Scholar]
- 22. Engholm M, Eftekhari A, Chwatko G, Bald E, Mulvany MJ. Effect of cystamine on blood pressure and vascular characteristics in spontaneously hypertensive rats. J Vasc Res. 2011;48:476–484. [DOI] [PubMed] [Google Scholar]
- 23. Liu C, Wang W, Parchim N, Irani RA, Blackwell SC, Sibai B, Jin J, Kellems RE, Xia Y. Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination‐preventing isopeptide modification. Hypertension. 2014;63:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shweke N, Boulos N, Jouanneau C, Vandermeersch S, Melino G, Dussaule JC, Chatziantoniou C, Ronco P, Boffa JJ. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF‐beta activation and cell infiltration. Am J Pathol. 2008;173:631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borzychowski AM, Sargent IL, Redman CW. Inflammation and pre‐eclampsia. Semin Fetal Neonatal Med. 2006;11:309–316. [DOI] [PubMed] [Google Scholar]
- 26. Cotechini T, Graham CH. Aberrant maternal inflammation as a cause of pregnancy complications: a potential therapeutic target? Placenta. 2015;36:960–966. [DOI] [PubMed] [Google Scholar]
- 27. Staff AC, Johnsen GM, Dechend R, Redman CW. Preeclampsia and uteroplacental acute atherosis: immune and inflammatory factors. J Reprod Immunol. 2014;3:120–126. [DOI] [PubMed] [Google Scholar]
- 28. Lu S, Saydak M, Gentile V, Stein JP, Davies PJ. Isolation and characterization of the human tissue transglutaminase gene promoter. J Biol Chem. 1995;270:9748–9756. [DOI] [PubMed] [Google Scholar]
- 29. Mirza A, Liu SL, Frizell E, Zhu J, Maddukuri S, Martinez J, Davies P, Schwarting R, Norton P, Zern MA. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF‐kappaB. Am J Physiol. 1997;272:G281–G288. [DOI] [PubMed] [Google Scholar]
- 30. Jang GY, Jeon JH, Cho SY, Shin DM, Kim CW, Jeong EM, Bae HC, Kim TW, Lee SH, Choi Y, Lee DS, Park SC, Kim IG. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF‐kappaB activity in hypoxic tumor cells. Oncogene. 2010;29:356–367. [DOI] [PubMed] [Google Scholar]
- 31. Luft FC. Activating autoantibodies and cardiovascular disease. Physiology. 2013;28:254–261. [DOI] [PubMed] [Google Scholar]
- 32. Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circ Res. 2013;113:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wallukat G, Schimke I. Agonistic autoantibodies directed against G‐protein‐coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol. 2014;36:351–363. [DOI] [PubMed] [Google Scholar]
- 34. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension. 2010;55:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre‐eclampsia in pregnant mice. Nat Med. 2008;14:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjogren KG, Hjalmarson A, Muller‐Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18:945–953. [DOI] [PubMed] [Google Scholar]
- 38. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen‐Kelha M, Dechend R, Kintscher U, Rudolph B, Hoebeke J, Eckert D, Mazak I, Plehm R, Schonemann C, Unger T, Budde K, Neumayer HH, Luft FC, Wallukat G. Angiotensin II type 1‐receptor activating antibodies in renal‐allograft rejection. N Engl J Med. 2005;352:558–569. [DOI] [PubMed] [Google Scholar]
- 39. Wei F, Jia XJ, Yu SQ, Gu Y, Wang L, Guo XM, Wang M, Zhu F, Cheng X, Wei YM, Zhou ZH, Fu M, Liao YH; Group S‐AS . Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti‐AT1 receptor autoantibodies. Heart. 2011;97:479–484. [DOI] [PubMed] [Google Scholar]
- 40. Zhu F, Sun Y, Wang M, Ma S, Chen X, Cao A, Chen F, Qiu Y, Liao Y. Correlation between HLA‐DRB1, HLA‐DQB1 polymorphism and autoantibodies against angiotensin AT(1) receptors in Chinese patients with essential hypertension. Clin Cardiol. 2011;34:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1‐receptor and alpha1‐adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25:641–646. [DOI] [PubMed] [Google Scholar]
- 42. Rossitto G, Regolisti G, Rossi E, Negro A, Nicoli D, Casali B, Toniato A, Caroccia B, Seccia TM, Walther T, Rossi GP. Elevation of angiotensin‐II type‐1‐receptor autoantibodies titer in primary aldosteronism as a result of aldosterone‐producing adenoma. Hypertension. 2013;61:526–533. [DOI] [PubMed] [Google Scholar]
- 43. Kem DC, Li H, Velarde‐Miranda C, Liles C, Vanderlinde‐Wood M, Galloway A, Khan M, Zillner C, Benbrook A, Rao V, Gomez‐Sanchez CE, Cunningham MW, Yu X. Autoimmune mechanisms activating the angiotensin AT1 receptor in ‘primary’ aldosteronism. J Clin Endocrinol Metab. 2014;99:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H, Yu X, Cicala MV, Mantero F, Benbrook A, Veitla V, Cunningham MW, Kem DC. Prevalence of angiotensin II type 1 receptor (AT1R)‐activating autoantibodies in primary aldosteronism. J Am Soc Hypertens. 2015;9:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang J, Li L, Shang JY, Cai L, Song L, Zhang SL, Li H, Li X, Lau WB, Ma XL, Liu HR. Angiotensin II type 1 receptor autoantibody as a novel regulator of aldosterone independent of preeclampsia. J Hypertens. 2015;33:1046–1056. [DOI] [PubMed] [Google Scholar]
- 46. Parrish MR, Murphy SR, Rutland S, Wallace K, Wenzel K, Wallukat G, Keiser S, Ray LF, Dechend R, Martin JN, Granger JP, LaMarca B. The effect of immune factors, tumor necrosis factor‐alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms‐like tyrosine‐1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens. 2010;23:911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lamarca B, Speed J, Ray LF, Cockrell K, Wallukat G, Dechend R, Granger J. Hypertension in response to IL‐6 during pregnancy: role of AT1‐receptor activation. Int J Interferon Cytokine Mediat Res. 2011;2011:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J, Mosely J, Martin JN Jr, Dechend R, LaMarca B. IL‐17‐mediated oxidative stress is an important stimulator of AT1‐AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R353–R358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu C, Luo R, Elliott SE, Wang W, Parchim NF, Iriyama T, Daugherty PS, Blackwell SC, Sibai BM, Kellems RE, Xia Y. Elevated transglutaminase activity triggers angiotensin receptor activating autoantibody production and pathophysiology of preeclampsia. J Am Heart Assoc. 2015;4:e002323 doi: 10.1161/JAHA.115.002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G. Impaired immune and acute‐phase responses in interleukin‐6‐deficient mice. Nature. 1994;368:339–342. [DOI] [PubMed] [Google Scholar]
- 52. Luo R, Zhang W, Zhao C, Zhang Y, Wu H, Jin J, Zhang W, Grenz A, Eltzschig HK, Tao L, Kellems RE, Xia Y. Elevated endothelial hypoxia‐inducible factor‐1alpha contributes to glomerular injury and promotes hypertensive chronic kidney disease. Hypertension. 2015;66:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mary A, Achyuthan KE, Greenberg CS. The binding of divalent metal ions to platelet factor XIII modulates its proteolysis by trypsin and thrombin. Arch Biochem Biophys. 1988;261:112–121. [DOI] [PubMed] [Google Scholar]
- 54. Irani RA, Zhang Y, Blackwell SC, Zhou CC, Ramin SM, Kellems RE, Xia Y. The detrimental role of angiotensin receptor agonistic autoantibodies in intrauterine growth restriction seen in preeclampsia. J Exp Med. 2009;206:2809–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou CC, Irani RA, Zhang Y, Blackwell SC, Mi T, Wen J, Shelat H, Geng YJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody‐mediated tumor necrosis factor‐alpha induction contributes to increased soluble endoglin production in preeclampsia. Circulation. 2010;121:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iriyama T, Sun K, Parchim NF, Li J, Zhao C, Song A, Hart LA, Blackwell SC, Sibai BM, Chan LN, Chan TS, Hicks MJ, Blackburn MR, Kellems RE, Xia Y. Elevated placental adenosine signaling contributes to the pathogenesis of preeclampsia. Circulation. 2015;131:730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Feng M, Whitesall S, Zhang Y, Beibel M, D'Alecy L, DiPetrillo K. Validation of volume‐pressure recording tail‐cuff blood pressure measurements. Am J Hypertens. 2008;21:1288–1291. [DOI] [PubMed] [Google Scholar]
- 58. Elliott SE, Parchim NF, Liu C, Xia Y, Kellems RE, Soffici AR, Daugherty PS. Characterization of antibody specificities associated with preeclampsia. Hypertension. 2014;63:1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ballew JT, Murray JA, Collin P, Maki M, Kagnoff MF, Kaukinen K, Daugherty PS. Antibody biomarker discovery through in vitro directed evolution of consensus recognition epitopes. Proc Natl Acad Sci USA. 2013;110:19330–19335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HE, Skriner K, Hirschberg DL, Morris RI, Muller S, Pruijn GJ, van Venrooij WJ, Smolen JS, Brown PO, Steinman L, Utz PJ. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. [DOI] [PubMed] [Google Scholar]
- 61. Chamarthi B, Williams GH, Ricchiuti V, Srikumar N, Hopkins PN, Luther JM, Jeunemaitre X, Thomas A. Inflammation and hypertension: the interplay of interleukin‐6, dietary sodium, and the renin‐angiotensin system in humans. Am J Hypertens. 2011;24:1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C‐reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. [DOI] [PubMed] [Google Scholar]
- 63. Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C‐reactive protein, interleukin‐6, and TNF‐alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154. [DOI] [PubMed] [Google Scholar]
- 64. Bautista LE, Atwood JE, O'Malley PG, Taylor AJ. Association between C‐reactive protein and hypertension in healthy middle‐aged men and women. Coron Artery Dis. 2004;15:331–336. [DOI] [PubMed] [Google Scholar]
- 65. Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38:399–403. [DOI] [PubMed] [Google Scholar]
- 66. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin‐6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–H940. [DOI] [PubMed] [Google Scholar]
- 69. Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor‐alpha produced in the kidney contributes to angiotensin II‐dependent hypertension. Hypertension. 2014;64:1275–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II‐induced hypertension and chronic renal damage. Hypertension. 2012;59:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Doyle HA, Mamula MJ. Posttranslational protein modifications: new flavors in the menu of autoantigens. Curr Opin Rheumatol. 2002;14:244–249. [DOI] [PubMed] [Google Scholar]
- 72. Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. 2014;73:483–491. [DOI] [PubMed] [Google Scholar]
- 73. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, Gomez‐Tourino I, Arif S, Peakman M, Drijfhout JW, Roep BO. Posttranslational modification of HLA‐DQ binding islet autoantigens in type 1 diabetes. Diabetes. 2014;63:237–247. [DOI] [PubMed] [Google Scholar]
- 74. Dunne JL, Overbergh L, Purcell AW, Mathieu C. Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes. 2012;61:1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Penumatsa KC, Fanburg BL. Transglutaminase 2‐mediated serotonylation in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;306:L309–L315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. [DOI] [PubMed] [Google Scholar]
- 77. Penumatsa KC, Toksoz D, Warburton RR, Hilmer AJ, Liu T, Khosla C, Comhair SA, Fanburg BL. Role of hypoxia‐induced transglutaminase 2 in pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2014;307:L576–L585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. [DOI] [PubMed] [Google Scholar]
- 80. Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Badarau E, Collighan RJ, Griffin M. Recent advances in the development of tissue transglutaminase (TG2) inhibitors. Amino Acids. 2013;44:119–127. [DOI] [PubMed] [Google Scholar]