

Abstract
Background
GlycA, a novel protein glycan biomarker of N‐acetyl side chains of acute‐phase proteins, was recently associated with incident cardiovascular disease (CVD) in healthy women. Whether GlycA predicts CVD events in the setting of statin therapy in men and women without CVD but with evidence of chronic inflammation is unknown.
Methods and Results
In the Justfication for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial (NCT00239681), participants with low‐density lipoprotein cholesterol <130 mg/dL and high‐sensitivity C‐reactive protein (hsCRP) ≥2 mg/L were randomized to rosuvastatin 20 mg/day or placebo. GlycA was quantified by nuclear magnetic resonance spectroscopy in 12 527 before randomization and 10 039 participants at 1 year. A total of 310 first primary CVD events occurred during maximum follow‐up of 5.0 years (median, 1.9). GlycA changed minimally after 1 year on study treatment: 6.8% and 4.7% decrease in the rosuvastatin and placebo groups, respectively. Overall, baseline GlycA levels were associated with increased risk of CVD: multivariable‐adjusted hazard ratio (HR) per SD increment, 1.20 (95% CI, 1.08–1.34; P=0.0006). After additionally adjusting for hsCRP, this was slightly attenuated (HR, 1.18; 95% CI, 1.04–1.35; P=0.01). On‐treatment GlycA levels were also associated with CVD; corresponding multivariable‐adjusted HRs per SD before and after additionally adjusting for hsCRP: 1.27 (95% CI, 1.13–1.42; P<0.0001) and 1.24 (95% CI, 1.07–1.44; P=0.004), respectively. Tests for heterogeneity by treatment arm were not significant (P for interaction, >0.20).
Conclusion
In the JUPITER trial, increased levels of GlycA were associated with an increased risk of CVD events independent of traditional risk factors and hsCRP.
Clinical Trials Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00239681.
Keywords: cardiovascular disease, epidemiology, glycoprotein, metabolomics, statin intervention
Subject Categories: Epidemiology, Primary Prevention, Inflammation, Basic Science Research
Introduction
Although statins are effective agents in preventing cardiovascular disease (CVD) in combination with optimal lifestyle behaviors, cardiovascular events remain the leading cause of morbidity and mortality worldwide and still commonly occur during statin therapy.1 Recent research has also found that specific glycan changes facilitate the course of atherosclerosis and also track with cardiometabolic risk factors.2, 3 In addition, glycan attachments have been known to functionally modify cytokines and other inflammatory mediators implicated in atherosclerosis.4 Yet, the potential role of glycans in CVD prevention has not been well explored. Considerable technological advances for glycan analysis are now facilitating the investigation of glycans and their side chains as novel diagnostic and pharmacotherapeutic targets for various conditions, such as infectious diseases and cancers.5
To this effect, we recently reported that GlycA, a novel proton nuclear magnetic resonance (NMR) biomarker that identifies a consensus sequence of glycans common to a host of acute phase glycoproteins,6 was associated with the risk of incident CVD in initially healthy middle‐aged and older women in a manner similar to high‐sensitivity C‐reactive protein (hsCRP)7 despite the negligible contribution of hsCRP to the GlycA signal.6 Nonetheless, glycan structures change in response to the phenotypic and metabolic state of a cell, and it is unknown whether the glycan sequence present in GlycA predicts CVD in a high‐risk population recruited on the basis of elevated hsCRP. Furthermore, although GlycA was correlated with modifiable CVD risk factors,7, 8 the effect of statin therapy on GlycA and its association with CVD events during treatment with high‐intensity statin therapy is unknown.
Therefore, in the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER),9 we measured baseline and on‐treatment GlycA levels in order to assess: (1) the effect of 1 year of rosuvastatin treatment on GlycA; (2) the association of baseline and on‐statin GlycA with incident CVD; and (3) whether the CVD relative risk reduction attributable to rosuvastatin treatment in JUPITER was modified by GlycA levels.
Methods
Study Population
JUPITER (ClinicalTrial.gov No.: NCT00239681) was a double‐blind, placebo‐controlled trial that evaluated rosuvastatin 20 mg daily versus placebo in the primary prevention of first major CVD events among 17 802 apparently healthy men ≥50 years and women ≥60 years with low low‐density lipoprotein cholesterol (LDL‐C; <130 mg/dL), but who were at increased risk of cardiovascular events on the basis of elevated hsCRP (≥2 mg/L).9 Key exclusion criteria for JUPITER included previous or current use of lipid‐lowering therapy, current use of postmenopausal hormonal therapy, diabetes mellitus, and inflammatory conditions, such as severe arthritis, lupus or inflammatory bowel disease, or treatment with immunosuppressant medications. The trial protocol required study participants to provide a baseline blood sample before randomization and after 1 year on study treatment. Study participants were also requested, but not required, to provide samples for additional phenotyping. For this study, we analyzed a total of 12 527 participants who provided a sufficient blood sample at baseline for NMR GlycA measurements; and of these, 10 039 participants had a sufficient blood sample at both baseline and at 1 year. The JUPITER trial protocol was approved by the local institutional review board at each participating center, and all study participants provided written informed consent.
Laboratory Methods
Standard lipids, apolipoprotein‐B, and hsCRP were assayed immediately after blood collection in a core laboratory as previously described.10 For the current analysis, GlycA was measured for each participant from baseline and 1‐year samples that were paired together and stored in liquid nitrogen until the time for assay. GlycA signals were quantified from signal amplitudes generated from the automated proton (H1) NMR LipoProfile test at the CLIA‐certified LipoScience, Inc. (now LabCorp, Raleigh, NC) clinical laboratory.6 The GlycA signal is centered at 2.00±0.01 ppm in H1 NMR spectra of plasma, and only N‐acetylglucosamine with specific glycosidic linkage, that is, β (1→2) or β (1→6) with a preceding mannose residue, contributes to the GlycA signal. NMR signal amplitudes originating from the N‐acetyl methyl group protons of the N‐acetylglucosamine moieties located on the bi‐, tri‐, and tetra‐antennary branches of specific serum proteins (mainly α1‐acid glycoprotein, haptoglobin, α1‐antitrypsin, α1‐antichymotrypsin, and transferrin) were used to calculate the concentrations of GlycA expressed in μmol/L of N‐acetyl methyl groups.7
Outcomes
The primary endpoint of the current study is the trial primary endpoint, defined as nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, arterial revascularization, or cardiovascular death. We additionally examined the expanded endpoint of the primary endpoint and all‐cause death, consistent with previous biomarker analyses in JUPITER.11 All components of the primary endpoint occurring through the end of the JUPITER trial (March 30, 2008) were adjudicated in a blinded fashion by an independent endpoint committee and were confirmed using standardized diagnostic criteria.9 Only deaths adjudicated to be clearly attributed to a cardiovascular or cerebrovascular cause were included in the primary endpoint. For the expanded outcome, all deaths were included regardless of whether data were available to confirm the cause of death.
Statistical Analyses
Statistical analyses were performed with SAS software (version 9.3; SAS Institute Inc., Cary, NC). Baseline characteristics are expressed as medians (25th–75th percentiles) or percentages. Spearman coefficients were used to compare correlations of GlycA with risk factors. Wilcoxon signed‐rank tests compared baseline and 1‐year GlycA levels by randomization arm. Changes in GlycA after 1 year of randomization are expressed as percentages within each arm. The effect of 1 year of rosuvastatin therapy versus placebo on GlycA levels was assessed by the Wilcoxon rank‐sum tests. We performed a similar analysis for hsCRP.
Person‐time of follow‐up was assessed from the time of randomization to the first occurrence of a primary endpoint or the date of death, last study visit, withdrawal from study, loss to follow‐up, or trial completion, whichever came first. Absolute event rates were calculated per 100 person‐years. Consistent with previous published analyses examining on‐treatment hsCRP and LDL‐C in relation to cardiovascular outcomes,12 on‐treatment GlycA levels were defined as the values obtained after the first year of treatment. A cumulative incidence plot and log‐rank statistics were used to compared the unadjusted primary event rates between baseline quartiles of GlycA. Hazard ratios (HRs) and 95% CIs for the association of baseline and on‐treatment GlycA with cardiovascular outcomes were quantified using Cox proportional hazards regression according to quartiles of GlycA and per SD. The P value for linear trend was computed by fitting a continuous variable that assigned the median value for each quartile in regression models. We used 3 models that sequentially adjusted for demographic variables (age, sex, race, and randomization treatment assignment), traditional CVD risk factors (smoking, blood pressure, body mass index, fasting glucose, LDL‐C, high‐density lipoprotein cholesterol [HDL‐C], triglycerides, and family history of premature coronary disease), and natural log‐transformed hsCRP. We tested for treatment interaction by including a cross‐product term between GlycA and randomized treatment and by stratifying the analysis according to randomization arm. We evaluated the incremental predictive value of GlycA by calculating the likelihood ratio χ2 statistic and corresponding P value comparing models with and without the addition of GlycA We performed additional analyses to examine whether the association of GlycA with events was modified by sex (male or female), age (≤65 or >65 years), smoking (yes or no), family history of coronary heart disease (CHD; yes or no), metabolic syndrome (yes or no), time to event (≤24 or >24 months), or fasting glucose (<100 or ≥100 mg/dL). Finally, we examined whether the efficacy of rosuvastatin therapy on the primary outcome was differential in subgroups defined by quartiles of baseline GlycA levels. All probability values were 2‐tailed, with values <0.05 considered statistically significant.
Results
Baseline Characteristics
Baseline characteristics of study participants with measurable GlycA levels were similar to that of the entire JUPITER cohort except for more white participants in the current study (Table 1). GlycA was normally distributed, with a mean (SD) of 411 (70) μmol/L and a median value of 404 μmol/L (25th–75th percentile: 364–449 μmol/L); corresponding median value in men was 395 μmol/L (356–439) whereas that in women was 419 μmol/L (381–466). GlycA correlated positively in increasing magnitude with apolipoprotein B, triglycerides, hemoglobin A1c, and hsCRP (Spearman r=0.13–0.46), but not substantively with fasting glucose, LDL‐C, body mass index, and apolipoprotein B (Spearman, r<0.07; Table 2).
Table 1.
Baseline Characteristics
| Characteristics | Current Study n=12 527 | Not in Current Study n=5275 | Overall Study n=17 802 |
|---|---|---|---|
| Age, y | 66 (60–71) | 66 (61–71) | 66 (60–71) |
| Female | 4542 (36.3) | 2259 (42.8) | 6801 (38.2) |
| Rosuvastatin | 6182 (49.4) | 2719 (51.6) | 8901 (50.0) |
| Race/ethnicity | |||
| White | 10 251 (81.8) | 2432 (46.1) | 12 683 (71.3) |
| Black | 886 (7.1) | 1338 (25.4) | 2224 (12.5) |
| Asian | 184 (1.5) | 99 (1.9) | 283 (1.6) |
| Hispanic | 1105 (8.8) | 1156 (21.9) | 2261 (12.7) |
| Other/unknown | 99 (0.8) | 250 (4.7) | 349 (2.0) |
| Body mass index, kg/m2 | 28.4 (25.5–32.0) | 28.1 (25.0–32.0) | 28.4 (25.3–32.) |
| Systolic blood pressure, mmHg | 134 (124–146) | 134 (125–144) | 134 (124–145) |
| Diastolic blood pressure, mmHg | 80 (75–86) | 80 (76–89) | 80 (75–87) |
| Current smoker | 1883 (15.0) | 937 (17.8) | 2820 (15.9) |
| Family history of premature coronary disease | 1575 (12.6) | 470 (9.0) | 2045 (11.5) |
| Metabolic syndrome | 4987 (40.4) | 2329 (44.5) | 7316 (41.6) |
| Fasting glucose, mg/dL | 95 (88–102) | 93 (86–101) | 94 (88–102) |
| Hemoglobin A1c, % | 5.7 (5.4–5.9) | 5.7 (5.5–6.0) | 5.7 (5.5–5.9) |
| High‐sensitivity C‐reactive protein, mg/L | 4.1 (2.8–6.9) | 4.7 (3.0–7.9) | 4.3 (2.9–7.1) |
| LDL cholesterol, mg/dL | 109 (95–119) | 107 (91–118) | 108 (94–119) |
| Total cholesterol, mg/dL | 186 (170–200) | 183 (165–198) | 185 (169–200) |
| Triglycerides, mg/dL | 116 (84–167) | 122 (88–175) | 118 (85–169) |
| HDL cholesterol, mg/dL | 49 (41–60) | 47 (39–58) | 49 (40–60) |
Values stated are median (25th–75th percentile) or n (%). Percentages may not add up because of rounding off. HDL indicates high‐density lipoprotein; LDL, low‐density lipoprotein.
Table 2.
Spearman Correlation Coefficients
| GlycA | |
|---|---|
| hsCRP, mg/L | 0.46 |
| Total cholesterol, mg/dL | 0.09 |
| LDL cholesterol, mg/dL | −0.02 |
| Apolipoprotein B, mg/dL | 0.13 |
| HDL cholesterol, mg/dL | −0.03 |
| Apolipoprotein A‐1, mg/dL | 0.01a |
| Triglycerides, mg/dL | 0.20 |
| Body mass index, kg/m2 | 0.07 |
| Fasting glucose, mg/dL | 0.01a |
| Hemoglobin A1c, % | 0.21 |
| 1‐Year Change | ||
|---|---|---|
| GlycAb | ||
| Placebo | Rosuvastatin | |
| hsCRP, mg/Lb | 0.47 | 0.44 |
| Total cholesterol, mg/dLb | −0.05 | 0.06 |
| LDL cholesterol, mg/dLb | −0.06 | 0.05 |
| Apolipoprotein B, mg/dLb | 0.06 | 0.13 |
| HDL cholesterol, mg/dLb | −0.08 | −0.07 |
| Apolipoprotein A‐1, mg/dLb | −0.10 | −0.08 |
| Triglycerides, mg/dLb | 0.06 | 0.10 |
HDL indicates high‐density lipoprotein; hsCRP, high‐sensitivity C‐reactive protein; LDL‐C, low density lipoprotein cholesterol.
Nonsignificant P values for Spearman correlations; otherwise, P values are <0.01.
Denotes change at 1 year; otherwise, biomarkers represent baseline measurements.
Effect of Rosuvastatin on GlycA
After 1 year of study treatment, GlycA levels were minimally decreased in both placebo and rosuvastatin treatment groups to median values of 383 (345–427) and 375 μmol/L (337–419), respectively; P<0.0001 for 1‐year vs baseline values in each group). The resulting median percent change (25th–75th percentile) was −4.7% (−12% to 2.7%) in the placebo group and −6.8% (−13.8% to 0.8%) in the rosuvastatin group (P comparing changes between treatment groups <0.0001; Table 3 and Figure 1). The corresponding percent changes in hsCRP were −20% (−49.2% to 20%) and −48% (−68.8% to −15.2%), respectively. Similar changes were observed in analyses stratified by age, sex, metabolic syndrome, family history of premature coronary disease, and current smoking (data not shown).
Table 3.
Median (25th–75th Percentile) Concentrations of GlycA and High‐Sensitivity C‐Reactive Protein by Randomized Treatment Group
| Baseline | Year 1a | Changeb | % Change | |
|---|---|---|---|---|
| GlycA, μmol/L | ||||
| Placebo | 404 (363, 446) | 383 (345, 427) | −19 (−48, 10) | −4.7 (−11.7, 2.7) |
| Rosuvastatin | 404 (364, 448) | 375 (337, 419) | −27 (−57, 3) | −6.8 (−13.8, 0.8) |
| hsCRP, mg/L | ||||
| Placebo | 4.1 (2.8, 6.9) | 3.4 (1.9, 5.9) | −0.7 (−2.3, 0.7) | −20 (−49.2, 20) |
| Rosuvastatin | 4.1 (2.8, 6.7) | 2.1 (1.2, 4.2) | −1.7 (−3.4, −0.5) | −48 (−68.8, −15.2) |
Values obtained from individuals with both baseline and 1‐year measurements (n=10 039). hsCRP indicates high‐sensitivity C‐reactive protein.
P values from the Wilcoxon signed‐rank test comparing baseline and year 1 values were statistically significant (P<0.0001).
P values from the Wilcoxon rank‐sum test comparing the change among the rosuvastatin group with the change among the placebo group were <0.0001.
Figure 1.
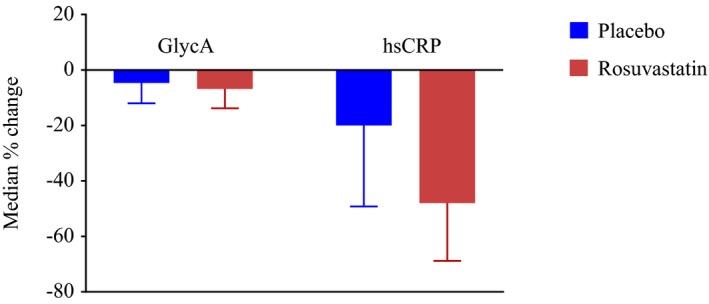
Baseline to 1‐year median percent change in GlycA with error bar indicating limit of 75th percentile according to randomized treatment, among individuals with both baseline and 1‐year measurements (n=10 039). P values from the Wilcoxon signed‐rank test comparing baseline and year 1 values were P<0.0001. P values from the Wilcoxon rank‐sum test comparing the change among the rosuvastatin group with the change among the placebo group were <0.0001. hsCRP indicates high‐sensitivity C‐reactive protein.
In the rosuvastatin treatment group, 1‐year change in GlycA correlated positively with 1‐year change in hsCRP (Spearmen, r=0.44), but not substantively with 1‐year changes in LDL‐C, apolipoprotein B, and triglycerides (Spearmen, r<0.13; Table 2). Generally, similar correlations were noted in the placebo group.
Association of Baseline and On‐Treatment GlycA With CVD
Among 12 527 JUPITER participants with baseline GlycA levels, the study primary outcome was confirmed in 310 participants over a maximum follow‐up of 5.0 years (median, 1.9). Of these, 201 (1.6%) primary events occurred in the placebo group and 109 (0.87%) in the rosuvastatin group, the proportion of these were similar to those observed in the overall JUPITER trial.9 The cumulative primary event rates diverged increasingly according to quartiles of GlycA with the lowest and highest events rates found in quartiles 1 and 4, respectively (log‐rank, P<0.0001; Figure 2 and Table 4). In a multivariable model that included age, race, sex, randomized treatment assignment, smoking, blood pressure, body mass index, fasting glucose, LDL‐C, HDL‐C, triglycerides, and family history of premature coronary disease, HRs for the primary endpoint for quartiles 1 to 4 of baseline GlycA were 1.00, 1.05 (95% CI, 0.74–1.49), 1.23 (95% CI, 0.88–1.72), and 1.57 (95% CI, 1.12–2.18; P linear trend=0.004). This association was slightly attenuated, but remained statistically significant, after adjustment for hsCRP: HR for quartile 4 versus 1 was 1.45 (95% CI, 1.01–2.10; P linear trend=0.03). Risk estimates per SD increase in baseline GlycA levels were 1.20 (95% CI, 1.08–1.34; P=0.0006) and 1.18 (95% CI, 1.04–1.35; P=0.01) in the corresponding multivariable adjusted models (Table 4). Results were similar when baseline GlycA was examined in relation to the expanded primary endpoint that included all‐cause death (528 events); the HR in the fully adjusted model for quartile 4 versus 1 was 1.83 (95% CI, 1.38–2.42; P linear trend<0.0001), with a corresponding HR per SD increase in baseline GlycA of 1.25 (95% CI, 1.15–1.37; P<0.0001; Table 4).
Figure 2.
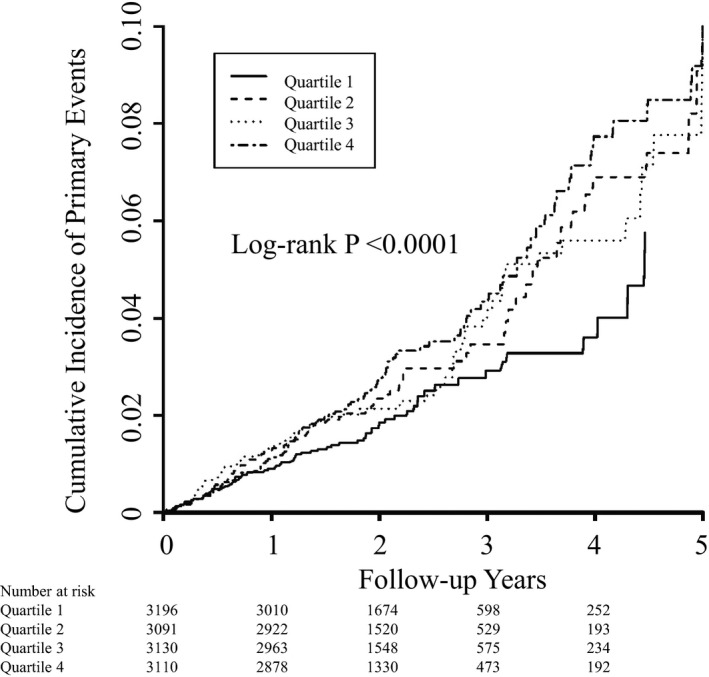
Cumulative incidence rates of the primary endpoint according to baseline quartiles of GlycA.
Table 4.
Baseline GlycA in Relation to Incident Events, Overall and by Treatment Group
| Quartile 1 ≤364 μmol/L | Quartile 2 365 to 404 μmol/L | Quartile 3 405 to 449 μmol/L | Quartile 4 >449 μmol/L | P Linear trend | HR (95% CI, P Values) Per SD | P LRT a | |
|---|---|---|---|---|---|---|---|
| Cardiovascular events | |||||||
| Overall (No. of events/n:310/12 527)b | |||||||
| Incidence rate (95% CI) | 0.95 (0.75–1.21) | 0.98 (0.77–1.25) | 1.16 (0.93–1.44) | 1.55 (1.28–1.89) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.13 (0.80–1.59) | 1.35 (0.98–1.88) | 1.85 (1.35–2.54) | <0.0001 | 1.27 (1.15–1.40, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.05 (0.74–1.49) | 1.23 (0.88–1.72) | 1.57 (1.12–2.18) | 0.004 | 1.20 (1.08–1.34, 0.0006) | 0.001 |
| Model 3 | 1 | 1.04 (0.73–1.47) | 1.19 (0.84–1.68) | 1.45 (1.01–2.10) | 0.03 | 1.18 (1.04–1.35, 0.01) | 0.01 |
| Placebo (No. of events/n:201/6345) | |||||||
| Incidence rate (95% CI) | 1.18 (0.88–1.59) | 1.25 (0.92–1.68) | 1.47 (1.12–1.93) | 2.02 (1.59–2.57) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.13 (0.74–1.73) | 1.40 (0.93–2.11) | 1.94 (1.30–2.87) | 0.0004 | 1.28 (1.13–1.44, <0.0001) | 0.0002 |
| Model 2 | 1 | 1.02 (0.66–1.57) | 1.23 (0.81–1.87) | 1.55 (1.03–2.34) | 0.02 | 1.20 (1.06–1.36, 0.006) | 0.008 |
| Model 3 | 1 | 1.03 (0.67–1.59) | 1.26 (0.82–1.93) | 1.63 (1.03–2.57) | 0.02 | 1.26 (1.07–1.48, 0.005) | 0.006 |
| Rosuvastatin (No. of events/n:109/6182) | |||||||
| Incidence rate (95% CI) | 0.71 (0.48–1.10) | 0.70 (0.47–1.06) | 0.83 (0.58–1.21) | 1.07 (0.77–1.50) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.14 (0.64–2.02) | 1.23 (0.71–2.13) | 1.68 (0.99–2.87) | 0.048 | 1.24 (1.04–1.47, 0.02) | 0.02 |
| Model 2 | 1 | 1.13 (0.62–2.03) | 1.19 (0.67–2.09) | 1.56 (0.89–2.73) | 0.11 | 1.20 (1.002–1.44, 0.047) | 0.06 |
| Model 3 | 1 | 1.04 (0.57–1.89) | 1.04 (0.58–1.86) | 1.16 (0.62–2.17) | 0.64 | 1.07 (0.86–1.33, 0.56) | 0.56 |
| Cardiovascular events or all‐cause death | |||||||
| Overall (No. of events/n:528/12 527)b | |||||||
| Incidence rate (95% CI) | 1.48 (1.22–1.79) | 1.55 (1.28–1.88) | 1.82 (1.53–2.16) | 3.09 (2.69–3.54) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.16 (0.88–1.52) | 1.37 (1.05–1.78) | 2.33 (1.83–2.97) | <0.0001 | 1.37 (1.28–1.46, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.15 (0.87–1.51) | 1.34 (1.02–1.74) | 2.08 (1.61–2.67) | <0.0001 | 1.30 (1.21–1.39, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.12 (0.84–1.47) | 1.26 (0.96–1.66) | 1.83 (1.38–2.42) | <0.0001 | 1.25 (1.15–1.37, <0.0001) | <0.0001 |
| Placebo (No. of events/n:322/6345) | |||||||
| Incidence rate (95% CI) | 1.65 (1.28–2.12) | 1.96 (1.54–2.48) | 2.08 (1.65–2.61) | 3.85 (3.24–4.85) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.28 (0.90–1.81) | 1.44 (1.02–2.03) | 2.59 (1.89–3.55) | <0.0001 | 1.36 (1.25–1.48, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.24 (0.87–1.76) | 1.37 (0.97–1.95) | 2.18 (1.57–3.03) | <0.0001 | 1.28 (1.17–1.40, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.23 (0.86–1.75) | 1.35 (0.94–1.93) | 2.10 (1.46–3.02) | <0.0001 | 1.28 (1.14–1.44, <0.0001) | <0.0001 |
| Rosuvastatin (No. of events/n:197/3110) | |||||||
| Incidence rate (95% CI) | 1.30 (0.97–1.74) | 1.13 (0.82–1.56) | 1.55 (1.18–2.03) | 2.30 (1.84–2.89) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 0.99 (0.64–1.54) | 1.26 (0.84–1.88) | 1.98 (1.35–2.90) | 0.0001 | 1.37 (1.22–1.53, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.03 (0.66–1.60) | 1.27 (0.84–1.93) | 1.90 (1.28–2.82) | 0.0005 | 1.32 (1.18–1.48, <0.0001) | <0.0001 |
| Model 3 | 1 | 0.96 (0.61–1.51) | 1.13 (0.74–1.73) | 1.47 (0.95–2.29) | 0.054 | 1.22 (1.06–1.41, 0.005) | 0.007 |
Model 1 is adjusted for age, race, and sex. Model 2 is model 1 plus smoking, blood pressure, body mass index, fasting glucose, LDL‐C, high‐density lipoprotein cholesterol, triglycerides, and family history of premature coronary disease. Model 3 is model 2 plus high‐sensitivity C‐reactive protein. SD was 70.28 μmol/L. Incidence rates are reported per 100 person‐years. HR indicates hazard ratio; LDL‐C, low‐density lipoprotein cholesterol.
P LRT likelihood ratio χ2 comparing model with and without GlycA as a continuous variable.
Additionally adjusted for randomized treatment assignment.
Somewhat stronger magnitudes of associations were observed among the 10 039 participants with on‐treatment levels of GlycA: the HRs for quartile 4 versus 1 of on‐treatment GlycA with the primary endpoint (224 events) in multivariable adjusted models with and without hsCRP were 2.25 (95% CI, 1.48–3.42; P linear trend<0.0001) and 2.05 (95% CI, 1.28–3.28; P linear trend=0.002; Table 5). The corresponding HR per SD increase in on‐treatment GlycA levels were 1.27 (95% CI, 1.13–1.42; P<0.0001) and 1.24 (95% CI, 1.07–1.44; P=0.0004), respectively. In analyses examining on‐treatment GlycA levels with the expanded primary endpoint that included all‐cause death (315 events), the HR in the fully adjusted model for quartile 4 versus 1 was 1.98 (95% CI, 1.33–2.94; P linear trend=0.0002), with a corresponding HR per SD increase in on‐treatment GlycA of 1.37 (95% CI, 1.22–1.54; P<0.0001; Table 5).
Table 5.
On‐Treatment GlycA in Relation to Incident Events, Overall and by Treatment Group
| Quartile 1 ≤341 μmol/L | Quartile 2 342 to 379 μmol/L | Quartile 3 380 to 423 μmol/L | Quartile 4 >423 μmol/L | P Linear trend | HR (95% CI, P Values) Per SD | P LRT a | |
|---|---|---|---|---|---|---|---|
| Cardiovascular events | |||||||
| Overall (No. of events/n:224/10 039)b | |||||||
| Incidence rate (95% CI) | 0.62 (0.45–0.86) | 0.83 (0.62–1.11) | 1.18 (0.93–1.50) | 1.33 (1.06–1.67) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.46 (0.94–2.25) | 2.10 (1.40–3.16) | 2.47 (1.65–3.70) | <0.0001 | 1.32 (1.18–1.47, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.36 (0.87–2.14) | 2.03 (1.34–3.08) | 2.25 (1.48–3.42) | <0.0001 | 1.27 (1.13–1.42, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.33 (0.85–2.09) | 1.94 (1.26–2.98) | 2.05 (1.28–3.28) | 0.002 | 1.24 (1.07–1.44, 0.004) | 0.006 |
| Placebo (No. of events/n: 143/5113) | |||||||
| Incidence rate (95% CI) | 0.66 (0.41–1.04) | 1.00 (0.70–1.43) | 1.56 (1.17–2.08) | 1.63 (1.24–2.14) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.73 (0.96–3.12) | 2.73 (1.58–4.72) | 3.10 (1.80–5.35) | <0.0001 | 1.37 (1.20–1.57, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.62 (0.89–2.95) | 2.55 (1.46–4.45) | 2.67 (1.53–4.69) | 0.0003 | 1.27 (1.11–1.45, 0.0005) | 0.001 |
| Model 3 | 1 | 1.65 (0.90–3.02) | 2.65 (1.49–4.70) | 2.88 (1.55–5.35) | 0.0005 | 1.35 (1.13–1.62, 0.001) | 0.002 |
| Rosuvastatin (No. of events/n:81/4926) | |||||||
| Incidence rate (95% CI) | 0.59 (0.37–0.93) | 0.65 (0.41–1.02) | 0.77 (0.51–1.19) | 0.97 (0.65–1.44) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.19 (0.62–2.29) | 1.46 (0.78–2.76) | 1.81 (0.97–3.36) | 0.048 | 1.21 (0.996–1.47, 0.054) | 0.069 |
| Model 2 | 1 | 1.09 (0.55–2.17) | 1.46 (0.76–2.80) | 1.72 (0.90–3.30) | 0.07 | 1.18 (0.96–1.44, 0.11) | 0.13 |
| Model 3 | 1 | 1.02 (0.51–2.04) | 1.29 (0.66–2.54) | 1.34 (0.63–2.86) | 0.38 | 1.06 (0.82–1.37, 0.67) | 0.67 |
| Cardiovascular events or all‐cause death | |||||||
| Overall (No. of events/n:315/10 039)b | |||||||
| Incidence rate (95% CI) | 0.84 (0.64–1.12) | 1.04 (0.80–1.34) | 1.59 (1.30–1.95) | 2.10 (1.76–2.52) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.35 (0.93–1.98) | 2.06 (1.45–2.92) | 2.79 (1.99–3.91) | <0.0001 | 1.47 (1.36–1.60, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.28 (0.87–1.89) | 1.99 (1.40–2.85) | 2.52 (1.77–3.58) | <0.0001 | 1.42 (1.30–1.54, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.20 (0.81–1.78) | 1.76 (1.22–2.55) | 1.99 (1.33–2.94) | 0.0002 | 1.37 (1.22–1.54, <0.0001) | <0.0001 |
| Placebo (No. of events/n:195/5113) | |||||||
| Incidence rate (95% CI) | 0.91 (0.62–1.35) | 1.17 (0.84–1.64) | 2.03 (1.58–2.61) | 2.47 (1.98–3.09) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.48 (0.88–2.49) | 2.55 (1.60–4.08) | 3.35 (2.11–5.30) | <0.0001 | 1.49 (1.35–1.65, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.43 (0.84–2.42) | 2.49 (1.55–4.01) | 3.01 (1.88–4.82) | <0.0001 | 1.39 (1.25–1.54, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.38 (0.81–2.35) | 2.34 (1.44–3.82) | 2.67 (1.58–4.52) | <0.0001 | 1.41 (1.21–1.64, <0.0001) | <0.0001 |
| Rosuvastatin (No. of events/n:120/4926) | |||||||
| Incidence rate (95% CI) | 0.78 (0.53–1.17) | 0.90 (0.61–1.33) | 1.11 (0.78–1.58) | 1.65 (1.22–2.24) | |||
| Hazard ratio (95% CI) | |||||||
| Model 1 | 1 | 1.23 (0.70–2.15) | 1.52 (0.89–2.62) | 2.17 (1.30–3.64) | 0.002 | 1.43 (1.25–1.63, <0.0001) | <0.0001 |
| Model 2 | 1 | 1.11 (0.62–1.98) | 1.44 (0.83–2.51) | 1.91 (1.12–3.27) | 0.009 | 1.37 (1.19–1.58, <0.0001) | <0.0001 |
| Model 3 | 1 | 1.02 (0.56–1.83) | 1.25 (0.70–2.21) | 1.42 (0.77–2.63) | 0.21 | 1.30 (1.09–1.56, 0.004) | 0.007 |
Model 1 is adjusted for age, race, and sex. Model 2 is model 1 plus smoking, blood pressure, body mass index, fasting glucose, LDL‐C, high‐density lipoprotein cholesterol, triglycerides, and family history of premature coronary disease. Model 3 is model 2 plus high‐sensitivity C‐reactive protein. SD was 70.28 μmol/L. Incidence rates are reported per 100 person‐years. HR indicates hazard ratio; LDL‐C, low‐density lipoprotein cholesterol.
P LRT likelihood ratio χ2 comparing model with and without GlycA as a continuous variable.
Additionally adjusted for randomized treatment assignment.
We found no evidence of statistical interaction by GlycA across varied profiles of CVD risk factors when assessed at baseline (Figure 3A) or on‐treatment (Figure 3B; P>0.05).
Figure 3.
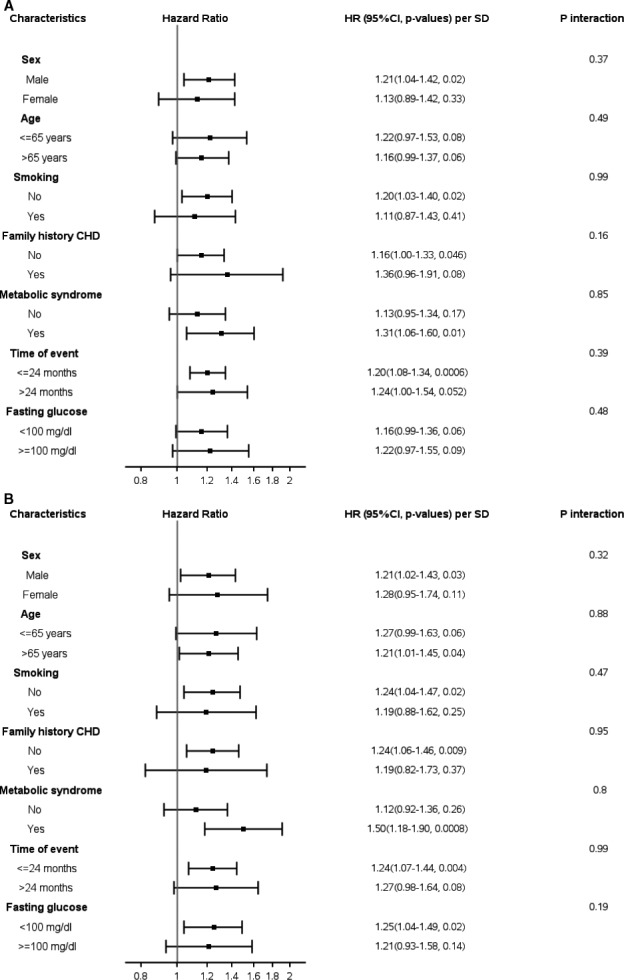
A, Stratified hazard ratios (95% CIs) per SD of baseline GlycA with the primary endpoint adjusted for age, race, sex, randomization treatment assignment, smoking, blood pressure, body mass index, fasting glucose, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, triglycerides, family history of premature coronary disease, and high‐sensitivity C‐reactive protein. B, Stratified hazard ratios (95% CIs) per SD of on‐treatment GlycA with the primary endpoint adjusted for age, race, sex, randomization treatment assignment, smoking, blood pressure, body mass index, fasting glucose, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, triglycerides, family history of premature coronary disease, and high‐sensitivity C‐reactive protein. CHD indicates coronary heart disease; HR, hazard ratio.
Association of Baseline and On‐Treatment GlycA With CVD in the Rosuvastatin and Placebo Groups
Risk estimates observed for the placebo subgroups when baseline and on‐treatment GlycA were examined with the primary endpoint were similar to that for the overall study population; P linear trend for baseline and on‐treatment GlycA in the fully adjusted model were 0.02 and 0.0005 respectively, the corresponding HRs per SD of GlycA were 1.26 (95% CI, 1.07–1.48; P=0.005) and 1.35 (95% CI, 1.13–1.62; P=0.001), respectively. Similar magnitudes of effect were observed among the rosuvastatin subgroup, although the associations did not reach statistical significance; corresponding P linear trend were 0.64 and 0.38 while HRs per SD of GlycA were 1.07 (95% CI, 0.86–1.33; P=0.56) and 1.06 (95% CI, 0.82–1.37; P=0.67), respectively. There was no statistically significant interaction between either baseline or on‐treatment GlycA levels with rosuvastatin therapy on the occurrence of the primary events (P for interaction >0.05 in all models).
Stronger associations were observed in analysis examining the expanded primary endpoint that included all‐cause death in the placebo subgroup; P linear trend for baseline and on‐treatment GlycA in the fully adjusted model were <0.0001 and <0.0001, respectively, the corresponding HRs per SD of GlycA were 1.28 (95% CI, 1.14–1.44; P<0.0001) and 1.41 (95% CI, 1.21–1.64; P<0.0001). Likewise, results in the rosuvastatin subgroup trended toward statistical significance; corresponding P linear trend were 0.054 and 0.21, whereas HRs per SD increase in GlycA were 1.22 (95% CI, 1.06–1.41; P=0.005) and 1.30 (95% CI, 1.09–1.56; P=0.004), respectively. Similarly, there was no evidence of statistical interaction by rosuvastatin therapy (P for interaction >0.05 in all models; Tables 4 and 5).
Efficacy of Rosuvastatin According to Baseline GlycA
In an analysis that examined participants based on 8 categories that took both treatment assignment and GlycA levels according to quartiles into account, those who were on placebo and had GlycA levels ≤364 μmol/L (first quartile) were considered as the referent. For both placebo and rosuvaststin, there was a suggested trend of increasing risk with increasing baseline GlycA levels; however, at each level, the rosuvastatin group had lower risk (Figure 4A). Consequently, rosuvastatin therapy had similar efficacy regardless of GlycA levels (Figure 4B), with estimates centered around that reported in the original JUPITER trial (HR, 0.56; 95% CI, 0.46–0.69).
Figure 4.
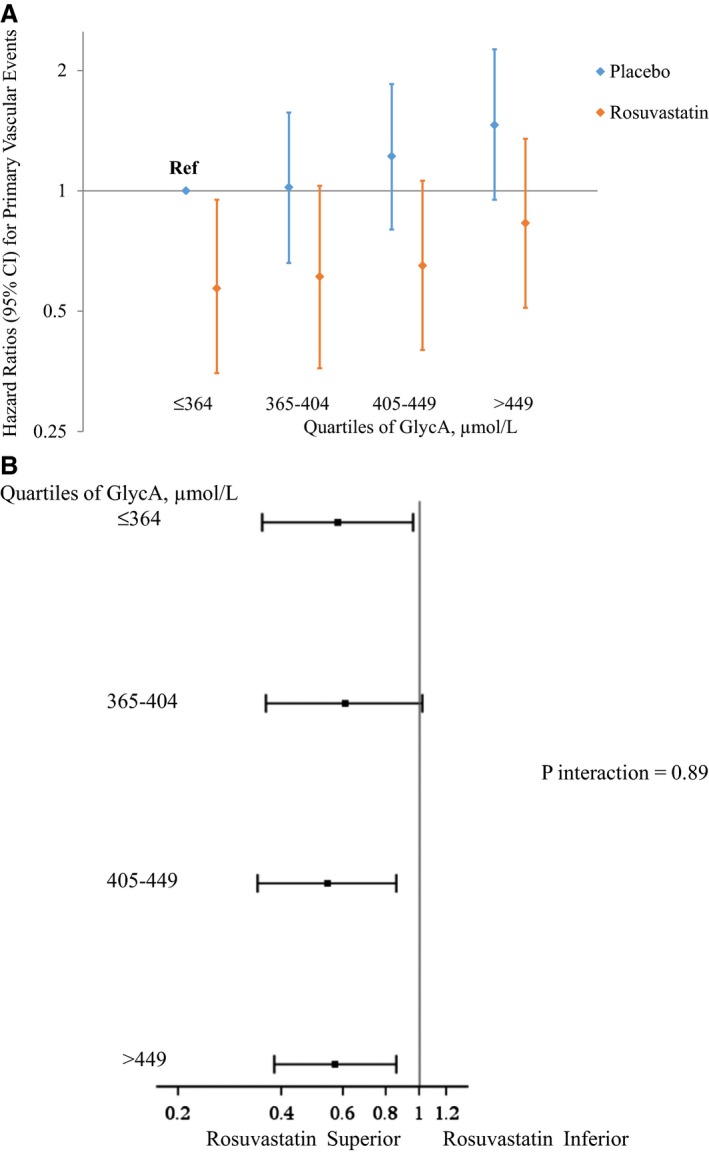
A, Hazard ratios (95% CIs) for the primary endpoint according to baseline GlycA levels events in relation to randomized treatment assignment, adjusted for age, race, sex, smoking, blood pressure, body mass index, fasting glucose, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, triglycerides, high‐sensitivity C‐reactive protein, and family history of premature coronary disease. B, Efficacy of rosuvastatin for the primary endpoint according to baseline GlycA levels adjusted for age, race, sex, smoking, blood pressure, body mass index, fasting glucose, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, triglycerides, high‐sensitivity C‐reactive protein, and family history of premature coronary disease.
Discussion
In the JUPITER trial, baseline and on‐treatment levels of GlycA, a recently characterized biomarker of circulating glycan side chains common to a host of acute‐phase glycoproteins, were positively associated with the first occurrence of CVD independent of traditional risk factors and hsCRP. Importantly, there was no evidence of effect heterogeneity by rosuvastatin treatment, and the association did not vary by levels of traditional CVD risk factors.
Among CVD biomarkers assessed at baseline and on‐treatment in the current study, GlycA correlated positively with hsCRP (r=0.5), similar to the magnitude of correlation of the change in GlycA with change in hsCRP (r=0.5), consistent with an inflammatory origin of GlycA. Nonetheless, while rosuvastatin therapy was associated with a meaningful reduction in hsCRP levels compared to placebo, the effect of rosuvastatin on GlycA levels was minimal (median percent decrease of ≈2%) and may partly relate to the negligibly low contribution of hsCRP to GlycA.7 Statins may reduce GlycA by inhibiting expression of the acute‐phase proteins contributing to GlycA or act more directly by repressing synthesis of the glycan side chains that represent GlycA. Consistent with our results, a prior study found that atorvastatin lowered levels of alpha‐1‐acid glycoprotein, an acute‐phase protein that is rich in the glycan detected by the GlycA signal, by a magnitude that was much less that than observed for hsCRP.13 A subsequent study found that rosuvastatin downregulated alpha‐1‐acid glycoprotein expression.14 Whether or not treatment with targeted anti‐inflammatory agents reduces circulating levels of GlycA remains to be evaluated.
The prognostic potential of GlycA for CVD risk assessment was recently documented in the Women's Health Study, where baseline GlycA levels were positively associated with incident CVD events, but that association was attenuated after adjusting for hsCRP over a 17‐year period.7 That study also demonstrated a significant time interaction whereby GlycA was independent of hsCRP in the short term (up to 6 years of follow‐up).7 Consistent with this short‐term observation, the present analysis of JUPITER, which included men and women from an international clinical trial with a maximum follow‐up of 5 years, found the association of GlycA with incident CVD to be independent of hsCRP. Several acute‐phase proteins contribute to the GlycA signal with major contribution from α1‐acid glycoprotein, haptoglobin, α1‐antitrypsin, α1‐antichymotrypsin, and transferrin, but not from CRP.7 Bell et al.15 first characterized the proton NMR signal of chemically distinct glycan conjugates linked to several acute‐phase proteins and showed that the intensity of the NMR signal was proportional to the degree of inflammation.15 GlycA may, in fact, represent a global burden of low‐grade systemic inflammation, given that its unique molecular signature arises from a host of acute‐phase proteins.
Although enzymatic glycosylation serve to modify protein function, the glycan conjugates on the acute‐phase proteins contributing to GlycA may have been synthesized cotranslationally or posttranslationally. In this context, the degree by which the glycoconjugates nested within GlycA impacts the native function of the contributory acute‐phase proteins, particularly with respect to their role in atherosclerosis, is unknown. However, the inflammatory origin of GlycA suggests a pathogenic role for these glycoconjugates. Nevertheless, we observed that the association of GlycA with clinically relevant CVD was independent of hsCRP. GlycA may therefore reflect inflammatory burden that is not completely accounted for by hsCRP.7
The correlation of GlycA with atherogenic lipids mostly targeted by statins ranged from negligible for LDL‐C (r=−0.02) to weak for apolipoprotein B (r=0.13). Furthermore, the lack of effect heterogeneity by randomization treatment on the occurrence of events suggests that GlycA, a novel inflammatory biomarker, is a determinant of residual CVD risk in JUPITER. Although effect sizes did not reach statistical significance in the rosuvastatin‐treated subgroup for the primary endpoint, they were significant for the expanded primary endpoint that included all‐cause death and had a greater number of events. Within this context, the lack of interaction between GlycA levels and rosuvastatin therapy on CVD risk and the stable estimates of rosuvastatin efficacy across varying levels of GlycA suggest that GlycA may mediate residual vascular risk through a pathway that, if targeted, may complement statin therapy for managing the risk of CVD. Given that specific glycan chains unique to a host of acute‐phase reactants are captured by GlycA, and presumably the contributing acute‐phase proteins bear glycan sequence that do not contribute GlycA, a better understanding of the holo‐glycan sequence for each contributing protein may shed light on the potential utility of GlycA for developing diagnostics, and perhaps therapy, that address CVD through inflammation. Research endeavors exploiting glycan biology are already making gains in virology and cancer‐preventive research.5, 16, 17 It is worth noting that heparin, which is broadly indicated in the management of coronary events, is one of the oldest known anticoagulants and was later discovered to be a glycan‐based therapeutic.18
Merits and limitations of the present analysis warrant mentioning. The present study includes a large number of male and female participants who were recruited internationally and randomly allocated to a potent statin or placebo, had both baseline and on‐treatment measurement of GlycA assessed, and were extensively phenotyed for CVD risk factors. Limitations of our study includes lack of generalizability given the entry criteria of JUPITER, which excluded participants with low hsCRP, high LDL‐C, high triglycerides, known CVD, or diabetes and a limited follow‐up duration. Finally, we expect that missing data on GlycA at 1 year occurred at random in a nondifferential manner that would, if anything, bias our results toward the null.
To conclude, this post‐hoc analysis of JUPITER found that baseline and on‐treatment GlycA levels were directly associated with the risk of future CVD independent of traditional CVD risk factors, rosuvastatin therapy, and hsCRP. These findings support the contribution of low‐grade systemic inflammation to the occurrence of CVD in statin‐treated individuals and, importantly, provide a significant biological illustration on the potential role of glycan‐based research to address prevailing CVD risk burden.
Sources of Funding
JUPITER was financially supported by AstraZeneca, which collected trial data and monitored sites but had no role in the design or conduct of the present study, including data analysis or interpretation, drafting or editing of this report, or preparation, review, or the decision to submit the manuscript for publication. LipoScience Inc., Raleigh, NC (now LabCorp, Burlington, NC) absorbed the cost of performing the GlycA measurements and performed them in a blinded manner. Research reported in this publication was supported, in part, by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL117861 to Dr Mora and a charitable gift from the Molino Family Trust. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Dr Akinkuolie was supported by the National Heart, Lung, and Blood Institute (T32 HL007575).
Disclosures
Dr Ridker has received research grant support from AstraZeneca, Novartis, Amgen, and the National Heart, Lung, and Blood Institute and has served as a consultant to Genzyme, Janssen, Aegerion, ISIS, Vascular Biogenics, Boehringer Ingelheim, Pfizer, and Merck. Dr Ridker is listed as a convector on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in CVD that have been licensed to AstraZeneca and Siemens. Dr Mora has received institutional research support from AstraZeneca, Atherotech Diagnostics, and NHLBI; served as a consultant to Genzyme, Quest Diagnostics, and Cerenis Therapeutics; and received speaker honoraria from AstraZeneca and the National Lipid Association for educational (nonpromotional) activities. Dr Mora has a patent application on the use of NMR‐measured GlycA for predicting risk of colorectal cancer. The other authors report no conflicts.
(J Am Heart Assoc. 2016;5:e003822 doi: 10.1161/JAHA.116.003822)
References
- 1. Minino AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep. 2011;59:1–126. [PubMed] [Google Scholar]
- 2. Scott DW, Chen J, Chacko BK, Traylor JG Jr, Orr AW, Patel RP. Role of endothelial N‐glycan mannose residues in monocyte recruitment during atherogenesis. Arterioscler Thromb Vasc Biol. 2012;32:e51–e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischer K, Kettunen J, Wurtz P, Haller T, Havulinna AS, Kangas AJ, Soininen P, Esko T, Tammesoo ML, Magi R, Smit S, Palotie A, Ripatti S, Salomaa V, Ala‐Korpela M, Perola M, Metspalu A. Biomarker profiling by nuclear magnetic resonance spectroscopy for the prediction of all‐cause mortality: an observational study of 17,345 persons. PLoS Med. 2014;11:e1001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varki A, Freeze HH. Glycans in acquired human diseases In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, eds. Essentials of Glycobiology. New York, NY: Cold Spring Harbor; 2009:601–616. [PubMed] [Google Scholar]
- 5. Shriver Z, Raguram S, Sasisekharan R. Glycomics: a pathway to a class of new and improved therapeutics. Nat Rev Drug Discov. 2004;3:863–873. [DOI] [PubMed] [Google Scholar]
- 6. Otvos JD, Shalaurova I, Wolak‐Dinsmore J, Connelly MA, Mackey RH, Stein JH, Tracy RP. Glyca: a composite nuclear magnetic resonance biomarker of systemic inflammation. Clin Chem. 2015;61:714–723. [DOI] [PubMed] [Google Scholar]
- 7. Akinkuolie AO, Buring JE, Ridker PM, Mora S. A novel protein glycan biomarker and future cardiovascular disease events. J Am Heart Assoc. 2014;3:e001221 doi: 10.1161/JAHA.114.001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akinkuolie AO, Pradhan AD, Buring JE, Ridker PM, Mora S. Novel protein glycan side‐chain biomarker and risk of incident type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2015;35:1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; Group JS . Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 10. Ridker PM, Genest J, Boekholdt SM, Libby P, Gotto AM, Nordestgaard BG, Mora S, MacFadyen JG, Glynn RJ, Kastelein JJ; Group JTS . HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet. 2010;376:333–339. [DOI] [PubMed] [Google Scholar]
- 11. Mora S, Glynn RJ, Ridker PM. High‐density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation. 2013;128:1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; Group JTS . Reduction in C‐reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. [DOI] [PubMed] [Google Scholar]
- 13. Nakamura K, Masuda H, Kariyazono H, Arima J, Iguro Y, Yamada K, Sakata R. Effects of atorvastatin and aspirin combined therapy on inflammatory responses in patients undergoing coronary artery bypass grafting. Cytokine. 2006;36:201–210. [DOI] [PubMed] [Google Scholar]
- 14. Schmidt WM, Spiel AO, Jilma B, Wolzt M, Muller M. In‐vivo effects of simvastatin and rosuvastatin on global gene expression in peripheral blood leucocytes in a human inflammation model. Pharmacogenet Genomics. 2008;18:109–120. [DOI] [PubMed] [Google Scholar]
- 15. Bell JD, Brown JC, Nicholson JK, Sadler PJ. Assignment of resonances for ‘acute‐phase’ glycoproteins in high resolution proton NMR spectra of human blood plasma. FEBS Lett. 1987;215:311–315. [DOI] [PubMed] [Google Scholar]
- 16. Brown JR, Crawford BE, Esko JD. Glycan antagonists and inhibitors: a fount for drug discovery. Crit Rev Biochem Mol Biol. 2007;42:481–515. [DOI] [PubMed] [Google Scholar]
- 17. Arnold JN, Saldova R, Hamid UM, Rudd PM. Evaluation of the serum N‐linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics. 2008;8:3284–3293. [DOI] [PubMed] [Google Scholar]
- 18. Jorpes E. The chemistry of heparin. Biochem J. 1935;29:1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]