

Abstract
Background
Mitochondria‐mediated cell death plays a critical role in myocardial ischemia‐reperfusion (IR) injury. We hypothesized that nanoparticle‐mediated drug delivery of mitochondrial division inhibitor 1 (Mdivi1) protects hearts from IR injury through inhibition of mitochondria outer membrane permeabilization (MOMP), which causes mitochondrial‐mediated cell death.
Methods and Results
We formulated poly (lactic‐co‐glycolic acid) nanoparticles containing Mdivi1 (Mdivi1‐NP). We recently demonstrated that these nanoparticles could be successfully delivered to the cytosol and mitochondria of cardiomyocytes under H2O2‐induced oxidative stress that mimicked IR injury. Pretreatment with Mdivi1‐NP ameliorated H2O2‐induced cell death in rat neonatal cardiomyocytes more potently than Mdivi1 alone, as indicated by a lower estimated half‐maximal effective concentration and greater maximal effect on cell survival. Mdivi1‐NP treatment of Langendorff‐perfused mouse hearts through the coronary arteries at the time of reperfusion reduced infarct size after IR injury more effectively than Mdivi1 alone. Mdivi1‐NP treatment also inhibited Drp1‐mediated Bax translocation to the mitochondria and subsequent cytochrome c leakage into the cytosol, namely, MOMP, in mouse IR hearts. MOMP inhibition was also observed in cyclophilin D knockout (CypD‐KO) mice, which lack the mitochondrial permeability transition pore (MPTP) opening. Intravenous Mdivi1‐NP treatment in vivo at the time of reperfusion reduced IR injury in wild‐type and CypD‐KO mice, but not Bax‐KO mice.
Conclusions
Mdivi1‐NP treatment reduced IR injury through inhibition of MOMP, even in the absence of a CypD/MPTP opening. Thus, nanoparticle‐mediated drug delivery of Mdivi1 may be a novel treatment strategy for IR injury.
Keywords: drug delivery system, ischemia‐reperfusion injury, mitochondria, myocardial infarction, nanoparticle
Subject Categories: Basic Science Research, Myocardial Infarction
Introduction
Coronary heart disease is the leading cause of morbidity and mortality worldwide.1 Myocardial infarction (MI) size is a major determinant of clinical outcome/prognosis in patients with acute MI.2 MI size has been reduced using early reperfusion therapy with thrombolytic drugs and/or percutaneous coronary intervention in these patients. However, restoration of blood supply to the ischemic myocardium induces ischemia‐reperfusion (IR) injury, which may limit the therapeutic effects of early reperfusion and lead to a devastatingly poor prognosis for acute MI patients.3, 4, 5, 6 Various pharmacological agents have shown cardioprotective effects against IR injury in animals, but none of them has achieved sufficient cardioprotective effects for IR injury in clinical practice.4, 7, 8 Therefore, innovative therapeutic strategies to protect the heart from IR injury are needed to adequately reduce MI size and improve the clinical outcomes of these patients.
Accumulating evidence suggests that mitochondria‐mediated cell death plays a key role in the pathogenesis of myocardial IR injury. The opening of the mitochondrial permeability transition pore (MPTP) is an established mechanism of mitochondria‐mediated cell death during myocardial IR.7, 9, 10 Genetic ablation of cyclophilin D (CypD), a key regulatory molecule for the MPTP opening, was shown to reduce IR injury in mice.11, 12 In a recent clinical trial, however, intravenous administration of cyclosporine A (CsA), an inhibitor of CypD, did not reduce heart failure or left ventricular remodeling after acute MI,13, 14 possibly because blockade of the MPTP opening alone is not sufficient to improve clinical prognosis. These findings prompted us to explore another mechanism of mitochondria‐mediated cell death, apoptotic cell death induced by mitochondria outer membrane permeabilization (MOMP). Myocardial IR induces Bcl2‐associated X protein (Bax) recruitment from the cytosol and oligomerization on the mitochondrial outer membrane, which causes MOMP.15 MOMP induces cytochrome c release from the mitochondrial intermembrane space to the cytosol, followed by activation of caspases and apoptotic cell death.15, 16, 17
Cassidy‐Stone et al. described mitochondria division inhibitor 1 (Mdivi1) as a chemical inhibitor of dynamin‐related protein (Drp1) that regulates MOMP.18 Drp1 regulates mitochondrial fission and contributes to mitochondrial quality control in concert with mitochondrial fusion.19, 20 Drp1 also regulates Bax/Bak‐dependent MOMP, leading to cytochrome c release from mitochondria.18, 21 In animal models, Mdivi1 has been shown to protect the heart,22, 23 kidneys,24 and brain25, 26 from IR injury, although it has not been determined whether this protection is dependent on regulation of mitochondrial dynamics or MOMP inhibition.18, 22, 23, 24, 25, 26
Using a mouse model of myocardial IR injury, Ong et al.22 reported that Mdivi1 was effective only when administered before induction of ischemia, but not at the time of reperfusion, which makes it unsuitable for clinical settings. The timing of this effect may be attributed to an insufficient local Mdivi1 concentration at the ischemic myocardium—the therapeutic target of Mdivi1—when it is administered at the time of reperfusion. Another possible limitation of the clinical application of Mdivi1, which achieves pharmacological inhibition of Drp1, is the adverse effect of Mdivi1 on off‐target organs; because genetic deletion of Drp1 causes organ dysfunction.27, 28 Therefore, from a clinical perspective, Mdivi1 must be delivered specifically to the IR myocardium to achieve immediate elevation of the local concentration of Mdivi1.
We recently reported that a drug delivery system (DDS) used to administer bioabsorbable poly (lactic acid/glycolic acid) (PLGA) nanoparticles in rat and mouse IR injury models preferentially delivered to the myocardium in the ischemic risk area. This lowered the effective dose of cardioprotective agents, such as a statin or a cyclosporine (eg, CsA), for reduction of myocardial IR injury.29, 30 Indeed, in an IR mouse model, intravenous administration of CsA‐NP at the time of reperfusion increased the local concentration of CsA in the ischemic area of the IR myocardium during an early phase (5 minutes) after reperfusion.30 We thought that a deeper understanding of Mdivi1's cardioprotective mechanisms might show that PLGA‐NP targeting Mdivi1 into the ischemic myocardium can be developed as a clinically feasible therapeutic modality.
In the present study, we tested the hypotheses that (1) application of a PLGA‐NP‐mediated DDS could enhance Mdivi1‐induced cardioprotection and (2) Mdivi1‐NPs have cardioprotective effect against IR injury through inhibition of Drp1‐mediated Bax translocation to the mitochondria, namely, through MOMP, even in mice lacking a CypD/MPTP opening.
Methods
Preparation of PLGA Nanoparticles
PLGA, with an average molecular weight of 20 000 and a copolymer ratio of lactide to glycolide of 75:25 (Wako Pure Chemical Industries Ltd, Osaka, Japan), was used as a matrix for the nanoparticles, and polyvinylalcohol (PVA‐403; Kuraray, Osaka, Japan) was used as a dispersing agent. PLGA nanoparticles incorporating the fluorescent marker, FITC (FITC‐NP; Dojin Chemical, Tokyo, Japan), Mdivi1 (Mdivi1‐NP; Sigma‐Aldrich, St. Louis, MO), or CsA (CsA‐NP; Sigma‐Aldrich) were prepared using an emulsion solvent diffusion method in purified water, as previously described.29, 31, 32 The FITC‐NP, Mdivi1‐NP, and CsA‐NP contained 4.0% (wt/wt) FITC, 3.4% (wt/wt) Mdivi1, and 2.67% (wt/wt) CsA, respectively. A sample of the nanoparticle suspension in distilled water was used to determine particle size. The diameters of the FITC‐NP, Mdivi1‐NP, and CsA‐NP were 223, 194, and 221 nm, respectively. Surface charge (zeta potential) was analyzed using a Zetasizer Nano (Sysmex, Hyogo, Japan) and was found to be anionic (−24.6 mV [FITC‐NP], −21.4 mV [Mdivi1‐NP], and −20.2 mV [CsA‐NP]).
Experimental Animals
The study protocol was reviewed and approved by the committee on the ethics of animal experiments, Kyushu University Faculty of Medicines, and all experiments were conducted according to the guidelines of the American Physiological Society.
Mice
Male C57BL/6J (wild‐type; WT) and cyclophilin D knockout (CypD‐KO) mice were purchased from The Jackson Laboratory (Stock No.: 009071; Bar Harbor, ME). Conditional Bax knockout (Bax‐KO) mice were generated by crossing Bax floxed mice (Baxflox/flox) with transgenic mice expressing Cre recombinase. Transgenic mice expressing Cre under control of the alpha‐myocin heavy chain (Myh6) promoter were used to produce cardiac‐specific (CS) Bax‐KO mice (Baxflox/flox; Myh6‐Cre). Bax floxed mice were kindly provided by Prof Osamu Takeuchi, Kyoto University.33 Animals were maintained on a 12‐hour light/dark cycle with free access to normal chow and water. We totally used 95 WT mice, 33 CypD‐KO mice, and 17 CS‐Bax‐KO mice in the experiments.
Cardiomyocyte Preparation and Culture
Primary rat neonatal ventricular cardiomyocytes (RNVMs) cultures were prepared using neonatal Sprague‐Dawley (SD) rat ventricles, as previously described.30 Briefly, neonatal SD rats were euthanized by decapitation under isoflurane anesthesia, and the hearts were rapidly excised and digested with trypsin (Wako Chemicals USA, Inc., Cambridge, MA) and collagenase type 2 (Worthington, Lakewood, NJ), and cells were cultured in DMEM (Sigma‐Aldrich) containing 10% FBS (Thermo Scientific, Waltham, MA), 1% penicillin (Invitrogen, Carlsbad, CA), and streptomycin (Invitrogen). Cells were plated twice in 100‐mm culture dishes (Cellstar; Greiner Bio‐One, Kremsmünster, Austria) for 70 minutes each to reduce the number of nonmyocytes. Nonadherent cells were plated in culture dishes (Primaria, Corning, NY) at an appropriate density for each experiment. Cardiomyocytes were maintained at 37°C in humidified air with 5% CO2 for 36 hours after plating to culture dishes. We have used these cardiomyocytes after confirming the beating under a microscope.
Cellular Uptake of FITC‐NP
RNVMs were washed with HBSS. RNVMs were treated with FITC alone (100 μmol/L) or FITC‐NP (100 μmol/L) for 1 hour under control conditions or treatment with H2O2 (250 μmol/L). RNVMs were also loaded with 10 μmol/L of Hoechst 33342 (Dojin Chemical Co., Kumamoto, Japan) for 20 minutes and washed with PBS. RNVMs were fixed with methanol at −20°C for 20 minutes and observed using a fluorescence microscope. FITC signal intensity (excitation wavelength, 498 nm; emitted fluorescent wavelength, 522 nm) was measured using a fluorescence plate reader (Infinite 200; PLATE manager V5; Wako) and corrected by the amount of protein.
RNVM Viability
RNVMs were pretreated with Mdivi1 alone (1, 5, 10, 50, 100, and 1000 μmol/L) or Mdivi1‐NP (1, 5, 10, 50, 100, and 1000 μmol/L) for 10 minutes. RNVMs were loaded with 250 μmol/L of H2O2 (Wako), which mimics oxidative stress during IR, in culture medium for 3 hours. RNVM viability was assessed using Cell Titer Blue assays (Promega, Madison, WI) in accord with the supplier's protocol.
Mitochondrial Morphology
RNVMs were pretreated with Mdivi1 alone (50 μmol/L) or Mdivi1‐NP (5 μmol/L) for 10 minutes. RNVMs were loaded with 250 nmol/L of Mito Tracker Orange CMTMRos (Invitrogen) and 10 μmol/L of Hoechst 33342 (Dojin Chemical) 3 hours after oxidative stress. RNVMs were washed and fixed with methanol at −20°C for 20 minutes.
Western Blot Analysis
Homogenates of RNVMs and mouse myocardium were analyzed using immunoblotting. Mitochondrial and cytosolic fractions were isolated from RNVMs in accord with the manufacturer's protocol (Abcam, Cambridge, MA) 3 hours after H2O2‐induced oxidative stress. Samples were solubilized in lysis buffer. Proteins (mitochondria: 2.0 μg, cytosol: 5.0 μg) were separated on 7.5% or 12% SDS/polyacrylamide gels and blotted to PVDF membranes. The following antibodies were used as primary antibodies: Drp1 (1:500, sc‐32898; Santa Cruz Biotechnology, Dallas, TX); Bax (1:1000, #2772; Cell Signaling Technology, Danvers, MA); cytochrome c (1:1000, A8 sc‐20357; Santa Cruz Biotechnology), GAPDH (1:1000, sc‐20357; Santa Cruz Biotechnology); and voltage‐dependent anion channel protein (VDAC; 1:1000, #4661S; Cell Signaling Technology). Densitometry analyses were performed using Image Quant TL (General Electric, Fairfield, CT).
Evaluation of Apoptosis
DNA fragmentation was detected using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; DeadEnd Fluorometric TUNEL system; Promega) 3 hours after H2O2‐induced (250 μmol/L) oxidative stress, in accord with the manufacturer's protocol. Nuclear density was determined by manual counting of Hoechst 33342 (Dojin Chemical) in 4 fields for each group, and TUNEL‐positive nuclei were counted by examining the entire section.
Adenovirus Transduction
Adenovirus harboring shRNA for Drp1 (shDrp1) was kindly provided by Prof Junichi Sadoshima, Rutgers New Jersey Medical School.34 We transduced shDrp1 or adenovirus harboring LacZ (control) to RNVMs for 48 hours.
Evaluation of Viability and Mitochondrial Drp1 and Bax on shDrp1‐Transduced RNVMs
We performed viability assays, mitochondrial isolation, and western blot analysis on RNVMs transduced with shDrp1 or Ad‐LacZ 3 hours after H2O2‐induced oxidative stress.
Langendorff‐Perfused Mouse Heart IR Model
The murine model for Langendorff myocardial IR injury was based on previously described methods.35, 36 Adult male mice (age, 10–12 weeks; body weight [BW], 20–30 g) were anesthetized by an intraperitoneal injection of sodium pentobarbital (60 mg/kg), which was maintained using 1% isoflurane and heparin (1000 IU/kg) intravenously. Mouse hearts were rapidly excised. Aortae were cannulated using shortened and blunted 23‐gauge needles and retrogradely perfused at a constant pressure (80 mm Hg) with modified KH buffer (Sigma‐Aldrich) equilibrated with 95% O2/5% CO2 at a pH of 7.4 at 37°C. We monitored coronary flow rate using a tubing flow module (Transonic, Ithaca, NY), and heart rate was monitored by electrocardiogram. To induce IR injury, Langendorff mouse hearts were subjected to 45 minutes of ischemia induced by cessation of perfusion 20 minutes after equilibration. Hearts were then reperfused with KH buffer.
Experimental Protocols for Langendorff‐Perfused Hearts
Experimental protocol 1
At the time of reperfusion, WT mouse hearts were divided into 3 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer), (2) FITC alone (KH buffer with 5 μmol/L of FITC), or (3) FITC‐NP (KH buffer with PLGA that contained 5 μmol/L of FITC). FITC fluorescence in the myocardium and FITC signals in subcellular mitochondrial and cytosolic fractions were examined 120 minutes after reperfusion (Figure 1A).
Figure 1.
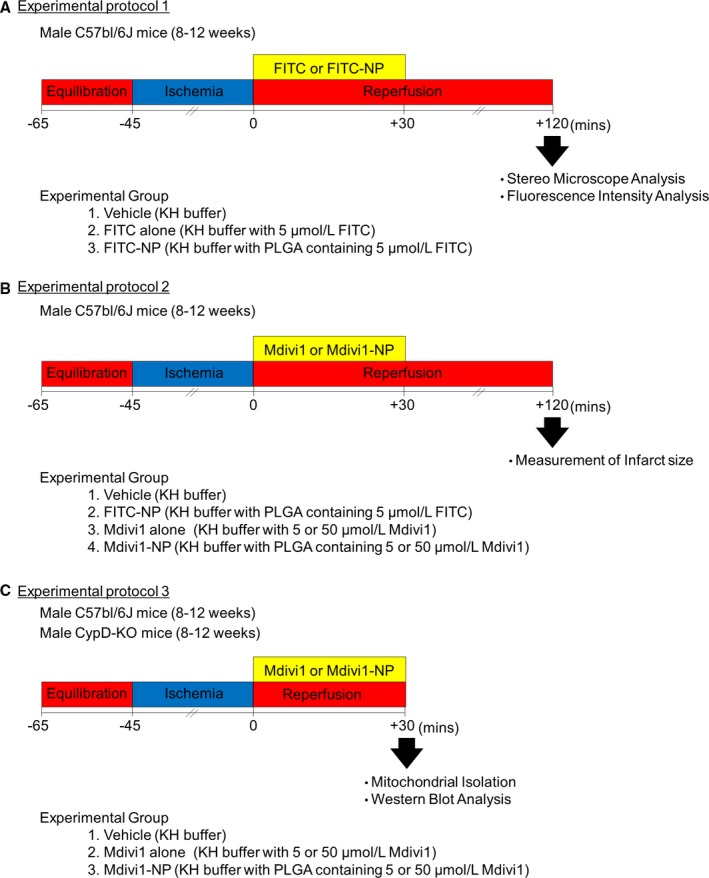
Experimental protocols for Langendorff‐perfused hearts. Experimental protocol 1: At the time of reperfusion, WT mouse hearts were divided into 3 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer), (2) FITC alone (KH buffer with 5 μmol/L of FITC), or (3) FITC‐NP (KH buffer with PLGA that contained 5 μmol/L of FITC). FITC fluorescence in the myocardium and FITC signals in subcellular mitochondrial and cytosolic fractions were examined 120 minutes after reperfusion. A, Experimental protocol 2: At the time of reperfusion, WT mouse hearts were divided into 4 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer), (2) FITC‐NP (KH buffer with PLGA that contained 5 μmol/L of FITC), (3) Mdivi1 alone (KH buffer with 5 or 50 μmol/L of Mdivi1), or (4) Mdivi1‐NP (KH buffer with PLGA that contained 5 or 50 μmol/L of Mdivi1). MI size was determined 2 hours after reperfusion with 1% 2,3,5‐triphenyltetrazolium chloride (TTC; Sigma‐Aldrich). B, Experimental protocol 3: At the time of reperfusion, WT mouse hearts and CypD‐KO mouse hearts were divided into 2 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer) or (2) Mdivi1‐NP (KH buffer with PLGA that contained 5 μmol/L of Mdivi1). Isolation of ischemic mouse heart mitochondria and western blot analysis were performed 30 minutes after reperfusion. CypD‐KO indicates cyclophilin D knockout; Mdivi1, mitochondrial division inhibitor 1; MI, myocardial infarction; NP, nanoparticles; PLGA, poly (lactic acid/glycolic acid); WT, wild type.
Experimental protocol 2
At the time of reperfusion, WT mouse hearts were divided into 4 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer), (2) FITC‐NP (KH buffer with PLGA that contained 5 μmol/L of FITC), (3) Mdivi1 alone (KH buffer with 5 or 50 μmol/L of Mdivi1), or (4) Mdivi1‐NP (KH buffer with PLGA that contained 5 or 50 μmol/L of Mdivi1). MI size was determined 2 hours after reperfusion with 1% 2,3,5‐triphenyltetrazolium chloride (TTC; Sigma‐Aldrich) (Figure 1B).
Experimental protocol 3
At the time of reperfusion, WT mouse hearts and CypD‐KO mouse hearts were divided into 2 groups and perfused for 30 minutes with KH buffer containing (1) vehicle (KH buffer) or (2) Mdivi1‐NP (KH buffer with PLGA that contained 5 μmol/L of Mdivi1). Isolation of ischemic mouse heart mitochondria and western blot analysis were performed 30 minutes after reperfusion (Figure 1C).
Mouse Myocardial IR Model
The murine model for myocardial IR injury was based on previously described methods.30 Briefly, adult male mice (age, 10–12 weeks; BW, 20–30 g) were anesthetized by an intraperitoneal injection of sodium pentobarbital (60 mg/kg) and maintained using 1% isoflurane with a ventilator after intubation. The heart was exposed by a left thoracotomy on a heated board. Transient myocardial ischemia was produced by ligation of the anterior descending branch of the left coronary artery (LAD) using an 8‐0 silk suture with a silicon tube placed alongside the LAD. After reperfusion, the chest was closed, and animals were allowed to recover from surgery.
Experimental Protocols for the In Vivo Mouse IR Model
Experimental protocol 4
WT mice were subjected to 30 minutes of ischemia followed by reperfusion. At the time of reperfusion, WT mice were divided into the 3 groups and received intravenous injections of (1) vehicle (5.0 mL/kg of saline), (2) Mdivi1 alone (1.2 mg/kg in 5.0 mL/kg of saline), or (3) Mdivi1‐NP (PLGA that contained 0.24, 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline). Mice were euthanized, and MI size was measured 24 hours after reperfusion (Figure 2A).
Figure 2.
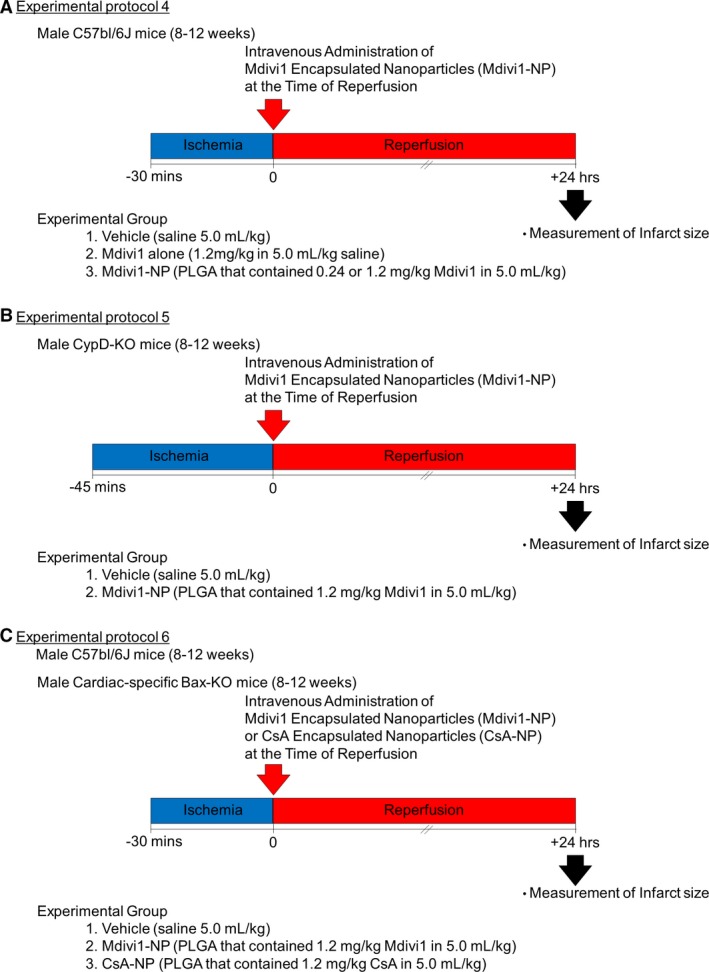
Experimental protocols for the in vivo mouse IR model. A, Experimental protocol 4: WT mice were subjected to 30 minutes of ischemia followed by reperfusion. At the time of reperfusion, WT mice were divided into the 3 groups and received intravenous injections of (1) vehicle (5.0 mL/kg of saline), (2) Mdivi1 alone (1.2 mg/kg in 5.0 mL/kg saline), or (3) Mdivi1‐NP (PLGA that contained 0.24, 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline). Mice were euthanized, and MI size was measured 24 hours after reperfusion. B, Experimental protocol 5: CypD‐KO mice were subjected to 45 minutes of ischemia followed by reperfusion. At the time of reperfusion, CypD‐KO mice were divided into 2 groups and received intravenous injections of (1) vehicle (5.0 mL/kg saline) or (2) Mdivi1‐NP (PLGA that contained 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline). Mice were euthanized, and MI size was measured 24 hours after reperfusion. C, Experimental protocol 6: CS‐Bax‐KO mice and control WT mice were subjected to 30 minutes of ischemia followed by reperfusion. At the time of reperfusion, CS‐Bax‐KO mice were divided into 3 groups and received intravenous injections of (1) vehicle (5.0 mL/kg of saline) or (2) Mdivi1‐NP (PLGA that contained 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline) (3) CsA‐NP (PLGA that contained 1.2 mg/kg of CsA in 5.0 mL/kg of saline). Mice were euthanized, and the infarct was measured 24 hours after reperfusion. CsA indicates cyclosporine A; CypD‐KO, cyclophilin D knockout; IR, ischemia‐reperfusion; Mdivi1, mitochondrial division inhibitor 1; MI, myocardial infarction; NP, nanoparticles; PLGA, poly (lactic acid/glycolic acid); WT, wild type.
Experimental protocol 5
CypD‐KO mice were subjected to 45 minutes of ischemia followed by reperfusion. At the time of reperfusion, CypD‐KO mice were divided into 2 groups and received intravenous injections of (1) vehicle (5.0 mL/kg of saline) or (2) Mdivi1‐NP (PLGA that contained 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline). Mice were euthanized, and MI size was measured 24 hours after reperfusion (Figure 2B).
Experimental protocol 6
CS‐Bax‐KO mice and control WT mice were subjected to 30 minutes of ischemia followed by reperfusion. At the time of reperfusion, CS‐Bax‐KO mice were divided into 3 groups and received intravenous injections of (1) vehicle (5.0 mL/kg of saline) or (2) Mdivi1‐NP (PLGA that contained 1.2 mg/kg of Mdivi1 in 5.0 mL/kg of saline) (3) CsA‐NP (PLGA that contained 1.2 mg/kg of CsA in 5.0 mL/kg of saline). Mice were euthanized, and the infarct was measured 24 hours after reperfusion (Figure 2C).
Statistical Analysis
Data are expressed as the means±SD. We analyzed differences between the 2 groups using the Mann–Whitney U test. Differences between 3 or more groups were assessed using an ANOVA and post‐hoc Bonferroni multiple comparison tests or Kruskal–Wallis test with post‐hoc Dunn multiple comparisons test. When we compare samples with independent variables, we analyzed data using 2‐way ANOVA and post‐hoc Bonferroni multiple comparison tests. In these independent variables, silencing effects of shDrp1 were differently affected by each adenovirus amount, whereas other independent variables have no interaction effect, respectively. Dose‐response curve to Mdivi1 alone or Mdivi1‐NP in terms of viability of RNVMs was evaluated by Probit analysis, and results were compared using the extra sum‐of‐squares F test. Mitochondrial morphology was compared using chi‐square tests with the Bonferroni correction. P<0.05 were considered statistically significant. Data were assessed with GraphPad Prism Software (version 6.0; GraphPad Software Inc., La Jolla, CA).
Results
Mdivi1‐NP Protects Cardiomyocytes Against Cell Death Under Oxidative Stress
We first examined the cellular uptake of FITC alone or FITC‐NP in cultured RNVMs. Neither FITC alone nor FITC‐NP was taken up by RNVMs under control conditions. FITC‐NP, but not FITC alone, accumulated in RNVMs after treatment with H2O2 (250 μmol/L, 1 hour), which represented oxidative stress during IR (Figure 3A). An ≈6‐fold higher FITC signal was detected in oxidative‐stressed RNVMs in the FITC‐NP treatment group compared with RNVMs without oxidative stress (Figure 3B). These results suggest that PLGA‐NPs accumulated in damaged cardiomyocytes and that nanoparticles may serve as a feasible DDS for the targeting of cardiomyocytes injured by oxidative stress under conditions mimicking IR injury.
Figure 3.
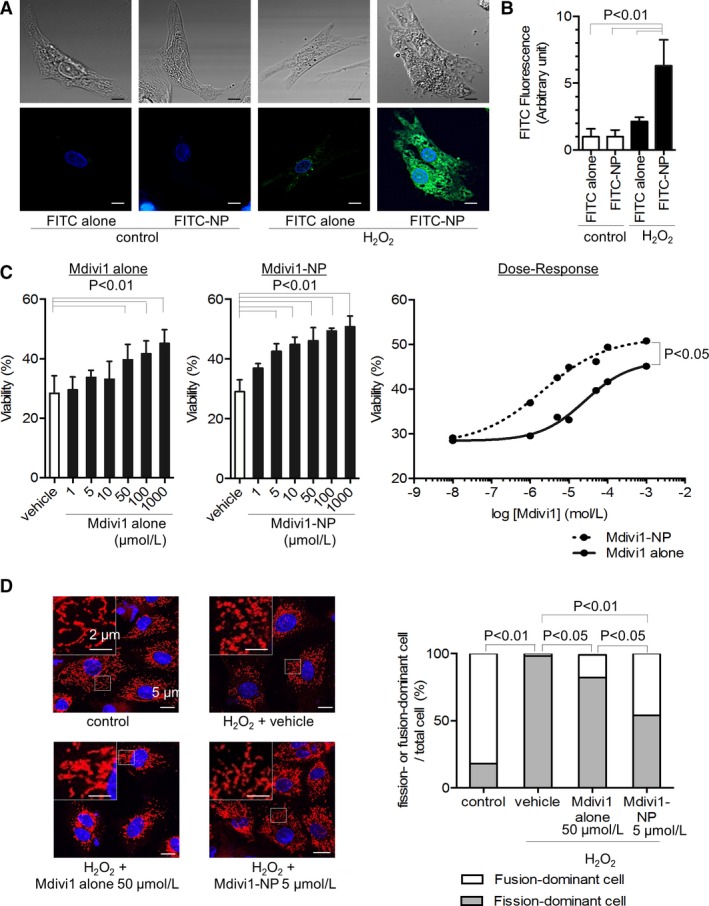
Distribution of nanoparticles in cardiomyocytes under H2O2‐induced oxidative stress and effects of Mdivi1‐NP on cardiomyocytes under oxidative stress. A, Representative confocal micrographs of rat neonatal ventricular cardiomyocytes (RNVMs) under control conditions or H2O2‐induced oxidative stress (H2O2 250 μmol/L, 1 hour). RNVMs were treated with either FITC alone or FITC‐NP. Differential interference contrast microscopic images are shown in the upper panels. Fluorescence microscopic images are shown in the lower panels. FITC is shown in green, nuclei in blue (Hoechst; Dojin Chemical Co., Kumamoto, Japan). Scale bar: 5 μm. B, Quantification of FITC fluorescence intensity in RNVMs under control conditions or oxidative stress (H2O2 250 μmol/L, 1 hour). RNVMs were treated with FITC alone or FITC‐NP (N=6 per group). C, Viability of RNVMs under H2O2‐induced oxidative stress (H2O2 250 μmol/L, 3 hours) after 10 minutes of pretreatment with either Mdivi1 alone (left) or Mdivi1‐NP (center) evaluated using the Cell Titer Blue assay (N=6 per group; Promega, Madison, WI). Dose‐response curve to Mdivi1 alone or Mdivi1‐NP in terms of viability of RNVMs is shown in right figure. D, Assessment of mitochondrial morphology under H2O2‐induced oxidative stress (H2O2 250 μmol/L, 3 hours) and pretreatment with Mdivi1 alone or Mdivi1‐NP (right). Gray bar: fission‐dominant cell/total cell; white bar: fusion‐dominant cell/total cell (N=22–63 cells per group). Representative confocal micrographs of each group (left). Mitochondria are shown in red (Mito Tracker Orange; Invitrogen, Carlsbad, CA), nuclei in blue (Hoechst). Scale bar: 5 μm (right bottom), 2 μm (left upper). Mdivi1 indicates mitochondrial division inhibitor 1; NP, nanoparticles.
We next examined the protective effect of Mdivi1 alone and Mdivi1‐NP on H2O2‐induced cell death in RNVMs. Oxidative stress induced by H2O2 (250 μmol/L, 3 hours) caused 71±4% cell death, and 10‐minute pretreatment with Mdivi1 alone or Mdivi1‐NP improved viability under H2O2‐induced oxidative stress in a dose‐dependent manner. The minimum therapeutic dose of Mdivi1‐NP was lower than that of Mdivi1 alone (Figure 3C). Importantly, pretreatment with Mdivi1‐NP improved the viability of RNVMs under H2O2‐induced oxidative stress more efficiently than pretreatment with Mdivi1 alone, as indicated by a lower estimated EC50 (2.0 vs 25 μmol/L; P<0.01) and greater maximal effect on cell survival (50.8% vs 45.1%; P<0.05; (Figure 3C). These results suggest that Mdivi1‐NP inhibits cell death more efficiently than Mdivi1 alone.
Because Mdivi1 is an inhibitor of Drp1, we examined mitochondrial morphology in RNVMs using mitochondrial staining. H2O2‐induced oxidative stress increased the percentage of fission‐dominant cells, which possess predominantly fractioned (<2 μm in length) mitochondria,22 whereas pretreatment with Mdivi1 alone (50 μmol/L; P<0.05) or Mdivi1‐NP (5 μmol/L; P<0.01) inhibited this increase (Figure 3D). These data suggested that nanoparticulation effectively lowered the minimal effective dose of Mdivi1 to inhibit H2O2‐induced mitochondrial fission.
Mdivi1‐NP Inhibits Drp1 and Subsequent Bax Translocation and MOMP in RNVMs Under Oxidative Stress
Because Drp1 is reported to colocalize with Bax during MOMP,37 we examined the effect of Mdivi1‐NP on Bax translocation to mitochondria during oxidative stress that induces MOMP. We isolated protein from the mitochondrial and cytosolic fractions of RNVMs 3 hours after oxidative stress. Drp1 and Bax were increased in the mitochondrial fraction after oxidative stress, whereas pretreatment with Mdivi1‐NP (5 μmol/L) inhibited this increase. Drp1 and Bax in the cytosolic fraction were not altered after oxidative stress (Figure 4A). These findings suggest that Mdivi1‐NP inhibited Drp1 and Bax recruitment to the mitochondria after H2O2‐induced oxidative stress in RNVMs. Bax‐mediated MOMP induces cytochrome c leakage from mitochondria to the cytosol, causing apoptotic cell death.16, 38 Therefore, we examined the effect of Mdivi1‐NP on apoptotic cell death and found that pretreatment with Mdivi1‐NP (5 μmol/L) reduced TUNEL‐positive apoptotic RNVMs under H2O2‐induced oxidative stress (Figure 4B). These results suggest that Mdivi1‐NP inhibits Drp1 and Bax translocation to the mitochondria and subsequent apoptotic cell death.
Figure 4.
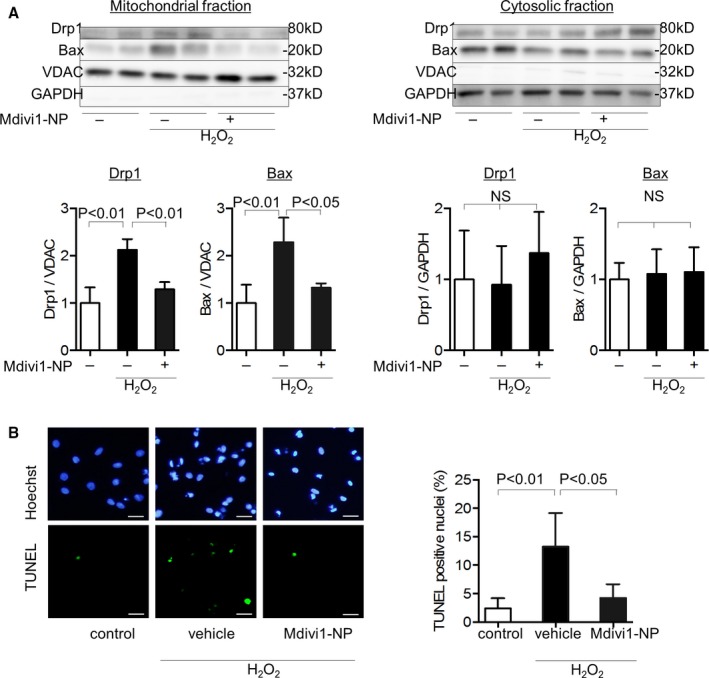
Effects of Mdivi1‐NP on Drp1 and subsequent Bax translocation to the mitochondria and MOMP in rat neonatal ventricular cardiomyocytes (RNVMs) under oxidative stress. A, Effect of Mdivi1‐NP (5 μmol/L) on Drp1 and Bax in mitochondrial fractions (left) and cytosolic fractions (right) of RNVMs under H2O2‐induced oxidative stress (H2O2 250 μmol/L, 3 hours) assessed using western blot analysis (N=4 per group). B, Effect of Mdivi1‐NP (5 μmol/L) on the proportions of TUNEL‐positive nuclei under oxidative stress (H2O2 250 μmol/L, 3 hours). In fluorescence microscopic images (left), apoptotic cells were identified using TUNEL (green; lower), and nuclei were identified using Hoechst (blue; upper). Quantification of TUNEL‐positive nuclei (right) by counting the number of TUNEL‐positive cells in fluorescence microscopic images (N=4 images per group). Scale bar: 20 μm. Bax indicates Bcl2‐associated X protein; Drp1, dynamin‐related protein 1; Mdivi1, mitochondrial division inhibitor 1; MOMP, mitochondria outer membrane permeabilization; NP, nanoparticles; NS, not significant; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling; VDAC, voltage‐dependent anion channel protein.
Drp1 shRNA Reduces BAX Translocation and Cell Death Under Oxidative Stress
To elucidate the cause‐and‐effect relationship between Mdivi1‐NP‐induced inhibition of Drp1 and Bax translocation and cell death, we transduced adenovirus harboring Drp1 shRNA (shDrp1) to RNVMs. Transduction of shDrp1 at 15 and 50 multiplicities of infection (MOIs) decreased Drp1 protein levels by ≈50% in RNVMs (Figure 5A), and 50‐MOI adenovirus treatment reduced RNVM viability (data not shown).
Figure 5.
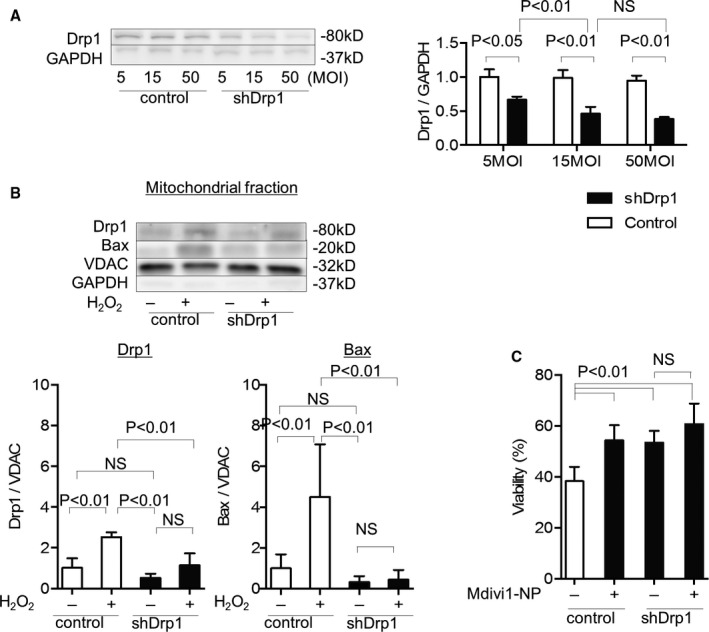
Effects of shDrp1 on Bax translocation and cell death under oxidative stress. A, Western blot analysis of Drp1 in rat neonatal ventricular cardiomyocytes (RNVMs) transduced with adenovirus harboring Drp1 shRNA (shDrp1) or adenovirus harboring LacZ (control) at 5, 15, and 50 MOIs (N=3 per group). B, Effects of shDrp1 (15 MOIs) on mitochondrial Drp1 and Bax under H2O2‐induced oxidative stress (H2O2 250 μmol/L, 3 hours) evaluated using western blot analysis of mitochondrial fractions from RNVMs (N=7 per group). C, Effects of shDrp1 (15 MOIs) on RNVM viability under H2O2‐induced oxidative stress (H2O2 250 μmol/L, 3 hours) and effect of Mdivi1‐NP (5 μmol/L) on shDrp1‐transduced RNVMs viability under oxidative stress evaluated using the Cell Titer Blue assay (N=7 each group; Promega, Madison, WI). Bax indicates Bcl2‐associated X protein; Drp1, dynamin‐related protein 1; MOI, multiplicity of infection; NS, not significant; VDAC, voltage‐dependent anion channel protein.
We isolated mitochondria from LacZ or shDrp1 (15 MOIs)‐transduced RNVMs and performed western blotting analysis of the mitochondrial fraction. H2O2‐induced oxidative stress caused Drp1 and Bax translocation to the mitochondria in LacZ‐transduced RNVMs (control), whereas shDrp1 transduction suppressed the oxidative‐stress–induced translocation of Drp1 and Bax in RNVMs (Figure 5B).
shDrp1 (15 MOIs) transduction improved the viability of RNVMs under H2O2‐induced oxidative stress (38.3% vs 53.4%; P<0.01), whereas Mdivi1‐NP did not effectively improve viability of RNVMs (53.4% vs 60.7%; P=0.218; Figure 5C). These results suggest that Drp1 regulates Bax translocation to the mitochondria under oxidative stress in RNVMs and that Mdivi1‐NP exerts cytoprotective effects under oxidative stress by Drp1 inhibition, which may reduce MOMP‐mediated cell death.
Mdivi1‐NP Protects Langendorff‐Perfused Hearts From IR Injury
Langendorff mouse hearts were subjected to 45 minutes of ischemia induced by cessation of the perfusion. We examined the distribution of FITC in Langendorff hearts reperfused for 30 minutes with KH buffer containing FITC alone or FITC‐NP (Figure 1A). FITC signals were detected in the cross‐sections of hearts perfused with FITC‐NP, but not the hearts perfused with saline or FITC alone (Figure 6A). We next isolated the IR myocardium and detected significant FITC signals in both the cytosolic and mitochondrial fractions from the hearts perfused with FITC‐NP (Figure 6B). These results suggest an enhancement of FITC delivery by PLGA‐NP into the mitochondria and cytosol of IR myocardium.
Figure 6.
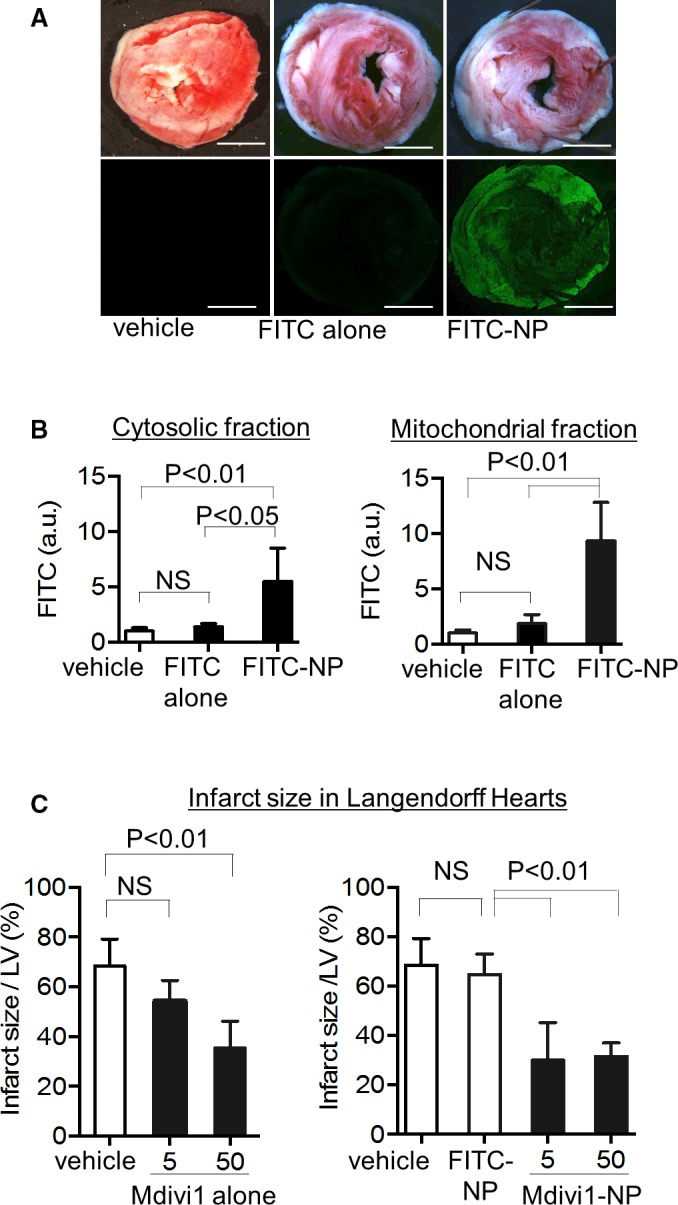
>Distribution of PLGA‐NPs in the myocardium and effects of Mdivi1‐NP on the Langendorff‐perfused mouse heart IR model. A, Representative light (upper) and fluorescence (lower) stereomicrographs of cross‐sections of IR hearts treated with saline (vehicle), FITC alone, or FITC‐NP. In the light images, the hearts were stained with 2,3,5‐triphenyltetrazolium chloride (TTC) to determine the infarct area. Scale bar: 2 mm. B, Quantification of FITC fluorescence intensity in the cytosolic fraction (left) and mitochondrial fraction (right) from whole IR hearts 30 minutes after reperfusion. Hearts were treated with saline (vehicle), FITC alone, or FITC‐NP (N=5 per group). C, Effects of Mdivi1 alone and Mdivi1‐NP on infarct size after IR injury in Langendorff‐perfused mouse hearts (N=5 each group). a.u. indicates arbitrary units; IR, ischemia‐reperfusion; LV, left ventricle; Mdivi1, mitochondrial division inhibitor 1; NP, nanoparticles; NS, not significant.
We then examined the infarct‐limiting effects of Mdivi1 alone or Mdivi1‐NP in Langendorff hearts. We reperfused the Langendorff hearts with KH buffer containing Mdivi1 alone or Mdivi1‐NP for 30 minutes after 45 minutes of ischemia (Figure 1B). Mdivi1 alone (5 μmol/L) did not reduce infarct size compared with vehicle (68.4% vs 54.4%; P=0.62), although a higher dose of Mdivi1 alone (50 μmol/L) had a therapeutic effect (68.4% vs 35.4%; P<0.01). By contrast, Mdivi1‐NP containing 5 μmol/L of Mdivi1 significantly reduced infarct size compared with FITC‐NP (64.7% vs 30.0%; P<0.01; Figure 6C). Treatment with Mdivi1 alone or Mdivi1‐NP did not affect heart rate or flow rate during perfusion (Tables 1 and 2).
Table 1.
Effects of Mdivi1‐NP on HR in Langendorff‐Perfused Mouse
| Groups | μmol/L | HR (bpm) | ||
|---|---|---|---|---|
| Equilibration | Reperfusion | |||
| 5 Minutes | 120 Minutes | |||
| Vehicle | 193±40 | 158±25 | 194±26 | |
| FITC‐NP | 5 | 212±21 | 197±47 | 201±17 |
| Mdivi1 alone | 5 | 208±29 | 193±39 | 190±25 |
| 50 | 234±43 | 190±55 | 212±29 | |
| Mdivi1‐NP | 5 | 224±53 | 182±23 | 176±35 |
| 50 | 216±40 | 183±19 | 188±30 | |
Data are expressed means±SD (N=5 each). bpm indicates beats per minute; HR, heart rate; Mdivi1, mitochondrial division inhibitor 1; NP, nanoparticles.
Table 2.
Effects of Mdivi1‐NP on Coronary Arteries Flow Rate in Langendorff‐Perfused Mouse Hearts
| Groups | μmol/L | Flow Rate (mL/min) | ||
|---|---|---|---|---|
| Equilibration | Reperfusion | |||
| 5 Minutes | 120 Minutes | |||
| Vehicle | 6.7±1.7 | 6.4±1.6 | 6.6±1.5 | |
| FITC‐NP | 5 | 7.7±1.0 | 7.3±1.8 | 6.5±1.2 |
| Mdivi1 alone | 5 | 7.6±2.1 | 5.8±1.5 | 6.0±1.2 |
| 50 | 5.5±0.9 | 6.8±1.7 | 7.0±1.2 | |
| Mdivi1‐NP | 5 | 6.9±0.9 | 7.5±1.6 | 7.5±1.2 |
| 50 | 7.7±1.9 | 6.5±1.2 | 5.7±30 | |
Data are expressed means±SD (N=5 each). Mdivi1, mitochondrial division inhibitor 1; NP, nanoparticles.
Mdivi1‐NP Inhibits Cytochrome c Leakage by MOMP During IR
To investigate the mechanism by which Mdivi1‐NP reduces myocardial IR injury, we isolated the mitochondrial and cytosolic fractions of Langendorff IR hearts 30 minutes after reperfusion and examined the translocation of cytochrome c, Drp1, and Bax (Figure 1C). IR caused cytochrome c leakage into the cytosol in WT mice, whereas Mdivi1‐NP containing 5 μmol/L of Mdivi1 inhibited this leakage. CypD deficiency reduced cytochrome c leakage. Importantly, Mdivi1‐NP further inhibited cytochrome c leakage in CypD‐KO mice. These results suggest the role of an MPTP‐independent mechanism in cytochrome c leakage, namely, MOMP, during myocardial IR injury (Figure 7A). Under the same experimental conditions, IR increased Drp1 and Bax in the mitochondrial fractions in WT mice, whereas Mdivi1‐NP inhibited this increase (P<0.01). These results suggest that Bax translocation to the mitochondria is dependent on Drp1 (Figure 7B). Moreover, inhibition of Bax translocation by Mdivi1‐NP was observed in CypD‐KO mice, which suggests that Mdivi1‐NP reduces cytochrome c leakage by inhibition of MOMP in CypD‐KO mice.
Figure 7.
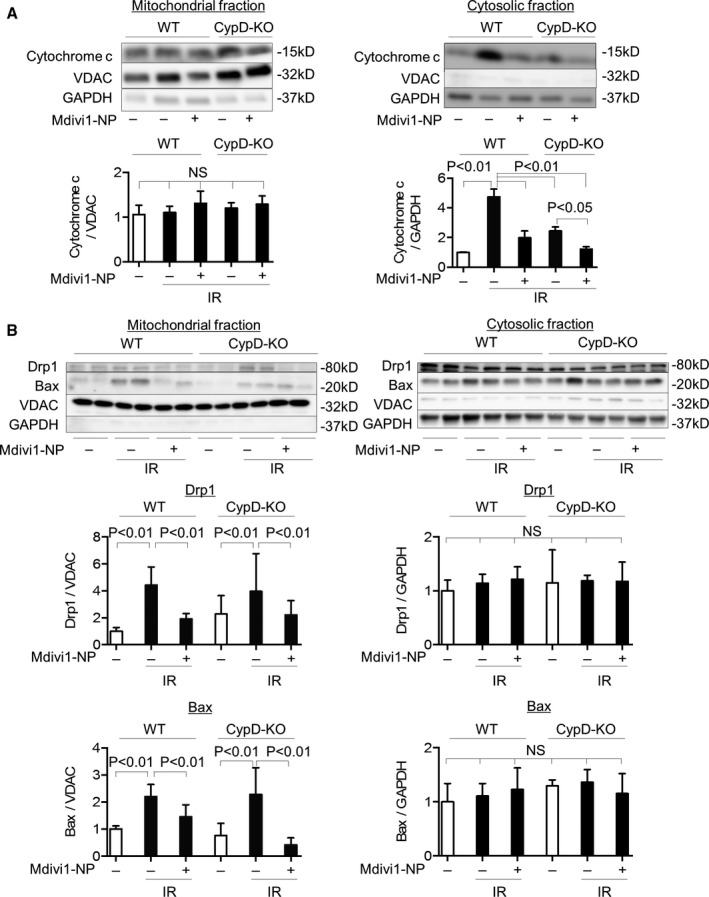
Effects of Mdivi1‐NP on Drp1/Bax translocation and cytochrome c leakage in IR hearts of WT and CypD‐KO mice. A, Effects of Mdivi1‐NP (5 μmol/L) on cytochrome c in the mitochondrial fraction (left) and cytosolic fraction (right) of IR myocardium from WT mice or CypD‐KO mice, as assessed using western blot analysis (N=3 per group). B, Effects of Mdivi1‐NP (5 μmol/L) on Drp1 and Bax in the mitochondrial fraction (left) and cytosolic fraction (right) of IR myocardium from WT mice or CypD‐KO mice (data for Drp1 N=4–9 per group; data for Bax N=4–5 per group). Bax indicates Bcl2‐associated X protein; CypD‐KO, cyclophilin D knockout; Drp1, dynamin‐related protein 1; IR, ischemia‐reperfusion; Mdivi1, mitochondrial division inhibitor 1; NP, nanoparticles; NS, not significant; VDAC, voltage‐dependent anion channel protein; WT, wild type.
Mdivi1‐NP Reduces Infarct Size in IR Hearts by CypD‐Independent, but Bax‐Dependent, Mechanisms
We next investigated whether Mdivi1‐NP would reduce infarct size in an in vivo myocardial IR injury model (Figure 2). Treatment with Mdivi1 alone—1.2 mg/kg, equivalent to the in vitro concentration of 50 μmol/L, similar to the dose used in a previous study22—at the time of reperfusion did not reduce infarct size (Figure 8A), which is consistent with the previous study.22 Intravenous treatment with Mdivi1‐NP containing 0.24 or 1.2 mg/kg of Mdivi1, corresponding to in vitro concentrations of 10 and 50 μmol/L, respectively,22 at the time of reperfusion reduced infarct size 24 hours after reperfusion (62.9% vs 34.0%; P<0.01; 62.9% vs 21.3%; P<0.01; Figure 8A). Of note, intravenous treatment with Mdivi1‐NP (1.2 mg/kg) at the time of reperfusion significantly reduced MI size in CypD‐KO mice (40.3% vs 22.2%; P<0.01) 24 hours after reperfusion (Figure 8B), suggesting Mdivi1‐NP protects IR hearts by inhibition of MOMP in myocardial IR injury in vivo.
Figure 8.
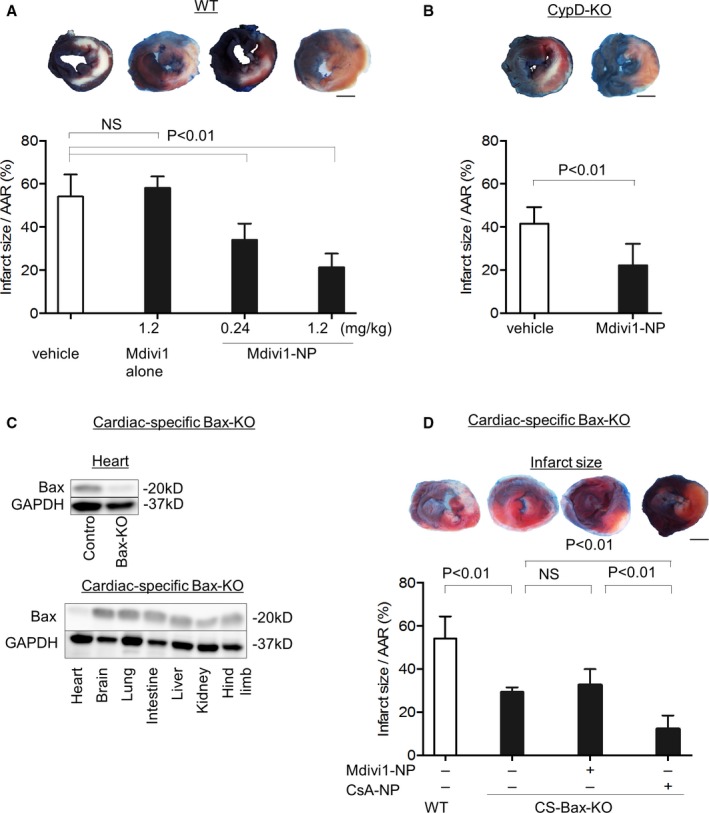
Effects of intravenous administration of Mdivi1‐NP on infarct size in vivo in IR hearts of WT mice, CypD‐KO mice, and cardiac‐specific Bax‐KO mice. A, Effects of Mdivi1 alone (1.2 mg/kg) or Mdivi1‐NP (0.24 and 1.2 mg/kg) on infarct size after intravenous administration at the time of reperfusion in WT mice (N=7–8 per group). Scale bar: 2 mm. B, Effects of Mdivi1‐NP (1.2 mg/kg) on infarct size in CypD‐KO mice (N=7–8 per group). Scale bar: 2 mm. C, Confirmation of deletion of Bax protein expression in cardiac‐specific Bax‐KO mice heart. In the left figure, we compared to control mice heart. In the right figure, we compared with other organs in cardiac‐specific Bax KO mice. D, Effects of Mdivi1‐NP (1.2 mg/kg) on infarct size in cardiac‐specific Bax‐KO mice (left; N=4 per group). The area at risk is shown as a percentage of the left ventricle (N=5 per group). Scale bar: 2 mm. AAR indicates area at risk; Bax, Bcl2‐associated X protein; CS, cardiac‐specific; CsA, cyclosporine A; CypD‐KO, cyclophilin D knockout; IR, ischemia‐reperfusion; Mdivi1, mitochondrial division inhibitor 1; NP, nanoparticles; NS, not significant; WT, wild type.
We also manufactured CS Bax‐KO mice by combining the myh6‐Cre transgene with floxed Bax alleles. We confirmed deletion of Bax protein expression in the heart (Figure 8C). Infarct size after IR was reduced in CS‐Bax‐KO mice, which revealed an important role of Bax‐mediated MOMP in myocardial IR injury. However, treatment with Mdivi1‐NP did not reduce infarct size in CS‐Bax‐KO mice (29.3% vs 32.6%; Figure 8D), which suggests that the therapeutic effect of Mdivi1‐NP was dependent on Bax. On the other hand, treatment with CsA‐NP reduced infarct size in CS‐Bax‐KO mice, suggesting that MOMP causes IR injury independently, at least in part, of MPTP opening.
Discussion
In the present study, we made the following novel findings: (1) PLGA‐NP‐mediated delivery of Mdivi1 (Mdivi1‐NP) enhanced Mdivi1‐induced cardioprotection against IR injury and (2) Mdivi1‐NP protected hearts from IR injury by inhibition of Drp1‐dependent Bax translocation to the mitochondria, which causes MOMP, even in the absence of CypD/MPTP opening. We characterized the role of MOMP as a therapeutic target in myocardial IR injury. The lower degree of myocardial IR injury in CS‐Bax‐KO mice strongly supports this hypothesis (Figure 8D). Drp1 suppression by shRNA reduced Bax translocation under oxidative stress in RNVMs (Figure 5B), confirming that Drp1 regulates Bax translocation to mitochondria in RNVMs under conditions of oxidative stress mimicking IR injury. These findings support our strategy to employ the Drp1 inhibitor, Mdivil, to reduce MOMP‐mediated myocardial IR injury.
MPTP Opening/MOMP Interaction
Ong et al.22 reported that Mdivi1‐induced cardioprotection was associated with the preservation of mitochondrial membrane potential, which is regulated by MPTP. Karch et al.39 recently demonstrated that inhibition of MOMP by Bax and Bak deficiency prevented mitochondrial swelling and rupture, an effect that is attributable to MPTP opening. These findings suggest that MOMP plays a role in MPTP opening‐dependent cardiomyocyte deaths, although the molecular mechanisms are unknown (Figure 9). In the present study, Mdivi1‐NP inhibited the translocation of Drp1 and Bax to mitochondria in a Langendorff‐perfused IR model established in CypD‐KO mice (Figure 7B) and reduced infarct size in IR models in CypD‐KO mice (Figure 8B). In contrast, treatment with CsA‐NP reduced MI size after IR injury in Bax‐KO mice (Figure 8D). These results suggest that MOMP causes IR injury independently, at least in part, of MPTP opening. There are several reports on signaling pathways that converge to mitochondrial cell deaths constituting reperfusion injury, such as the reperfusion injury salvage kinases pathway, most of which were associated with MPTP opening.40 Our results indicate that Mdivi1‐NP can target not only MPTP, but also MOMP, another type of mitochondria cell death inducer, by inhibiting Drp1 and Bax recruitment to mitochondria during IR. Mdivi1‐NP may exert additive cardioprotection over CyPD/MPTP inhibition (Figures 5, 6 through 7). Further examination is necessary to elucidate detailed molecular mechanisms and relative importance of the Drp1/Bax‐mediated pathway in the complex molecular networks which regulate cardioprotective signaling.
Figure 9.
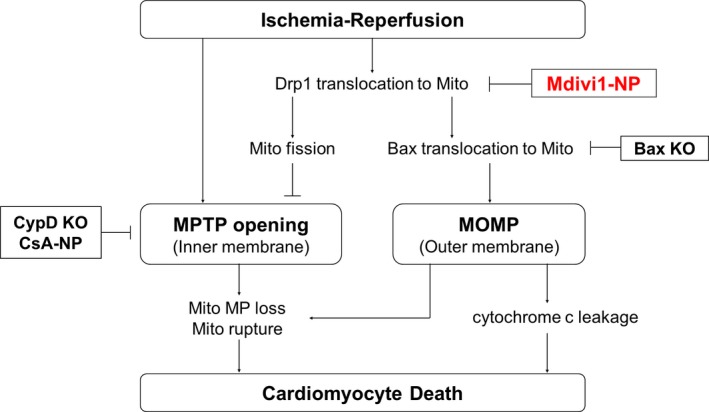
Schematic mechanistic pathway of mitochondrial cell death and mitochondrial dynamics after IR and how Mdivi1‐NP protects the hearts after IR. Mdivi1‐NP inhibited the translocation of Drp1 to mitochondria during IR. This inhibition of Drp1 translocation suppresses MPTP opening and Bax translocation followed by MOMP. Inhibition of MOMP could suppress mitochondrial membrane potential loss and mitochondrial rupture, though molecular mechanisms are uncovered so far. CsA‐NP indicates cyclosporine A‐nanoparticle; CypD, cyclophilin D; Bax indicates Bcl2‐associated X protein; Drp1, dynamin‐related protein 1; IR, ischemia‐reperfusion; KO, knockout; Mdivi1, mitochondrial division inhibitor 1; Mito, mitochondria; MOMP, mitochondria outer membrane permeabilization; MP, membrane potential; MTPT, mitochondrial permeability transition pore; NP, nanoparticles.
Mitochondrial Dynamics
Mitochondrial dynamics are linked to mitochondrial quality control by autophagy of dysfunctional fractions of mitochondria (ie, mitophagy).19, 41 Several studies have demonstrated that genetic (either conditional or inducible) deletion of Drp1 causes cardiac dysfunction and heart failure.27, 28, 34 Ikeda et al.34 reported that CS‐Drp1‐KO mice are susceptible to myocardial IR injury. Although these results are somewhat discrepant with our findings, the effect of Drp1 inhibition may depend on the time course. For instance, chronic inhibition of mitochondrial fission through genetic deletion induces cardiac dysfunction through failure of mitochondrial quality control. However, our findings indicate that acute inhibition of Drp1 at the time of reperfusion may be protective against IR injury. Other investigators have also shown that acute inhibition of Drp1, by either Mdivi1,22, 23, 34 P110,42 or a dominant‐negative Drp1 (Drp1K38A),22 protects the heart against IR injury. However, these studies have not clarified whether the therapeutic effects of acute Drp1 inhibition are dependent on regulation of mitochondrial dynamics. Whelan et al.43 reported that in mouse embryonic fibroblasts, mitochondrial fusion by Mdivi1/Drp1 inhibition makes mitochondria susceptible to the opening of MPTP and exacerbates myocardial IR injury.42 Our study decisively demonstrated that the cardioprotective effect of Mdivi1‐NP was dependent on Bax (Figure 8D) and excludes the potentially harmful effect of Mdivi1‐NP induced through inhibition of mitochondrial fission, because the mitochondrial fusion and fission rates in the adult mouse heart are extremely slow.19 Acute Drp1 inhibition in a mouse myocardial IR model may not affect mitochondrial dynamics.
PLGA Nanoparticle as DDS
In this study, we used a PLGA‐NP‐mediated DDS to deliver Mdivi1‐NP to achieve higher local concentrations of Mdivi1 in the IR myocardium immediately after administration. The intracellular uptake of PLGA‐NP was increased in RNVMs injured by oxidative stress in in vitro experiments (Figure 3A and 3B), and PLGA‐NP was increased in the IR myocardium in the cytosol and mitochondria in ex vivo experiments (Figure 6A and 6B). We have previously shown that endocytosis is a major mechanism of intracellular uptake of PLGA nanoparticles in vascular smooth muscle cells.44 In cardiomyocytes, Lin et al.45 reported that a transient MPTP opening, observed in the early phase after IR in vivo and in cardiomyocytes after hypoxia/reoxygenation in vitro, causes extensive and unconventional endocytic responses, which result in increased intracellular uptake of nanoparticles. Very recently, we have demonstrated that a loss of mitochondrial membrane potential causes the delivery of PLGA‐NP to the vicinity of mitochondria in cardiomyocytes.30 Drp1 localizes primarily in the cytosol and translocates to the mitochondria during IR, which causes mitochondrial membrane potential loss. Mdivi1‐NP, which is delivered to the vicinity of mitochondria during IR, may interfere Drp1/Bax translocation to mitochondria more efficiently than Mdivi1 alone. Indeed, PLGA‐NP lowered the therapeutic dose of Mdivi1 in vitro (Figure 3C), and Mdivi1‐NP treatment reduced infarct size after IR even when it was administered at the time of reperfusion, whereas treatment with Mdivi1 alone at the time of reperfusion did not exert a cardioprotective effect (Figure 8A). Therefore, the preferential delivery of PLGA‐NP into the IR myocardium may underpin the cardioprotective effect of Mdivi1‐NP. PLGA polymers as drug carriers have received biosafety approval for human use by the US Food and Drug Administration, the European Medicine Agency, and the Japanese regulatory agency. We adopted good manufacturing practices for our PLGA‐NP‐mediated DDS and produced PLGA‐NPs incorporating a statin.29, 31 Statin‐incorporated PLGA‐NPs underwent a phase I clinical trial at Kyushu University Hospital (clinical trial registry identifier: UMIN000014940 and UMIN000019189) to investigate the safety of a single administration or 7 days of repeated intravenous infusion. This nanoparticle‐based technology may represent an innovative therapeutic modality for coronary artery and other vascular diseases.
Limitations of the Study
There are several limitations in the present study. At first, we have used isolated RNVMs to examine molecular mechanisms of Drp1‐mediated cardiomyocytes death. Therefore, a possibility cannot be ruled out that cardiomyocytes were modified/dedifferentiated during the isolation procedure, though RNVMs are well known and generally used material for in vitro experiments.46 Second, there are some reports showing a possibility that cardioprotective signaling is different between species, especially between mice and larger mammals including humans.7 Therefore, further examination using large mammals is necessary to translate our results into humans.
Conclusions
In conclusion, nanoparticle‐mediated delivery of Mdivi1 to the ischemic myocardium protected the heart from IR injury through inhibition of Drp1/Bax‐mediated MOMP. The present study indicates that Mdivi1‐NP as a monotherapy, or in combination with cyclosporine, may exert cardioprotection against IR injury. Mdivi1‐NP may represent a novel therapeutic strategy to improve the prognosis of patients with acute myocardial infarction.
Sources of Funding
This study was supported by grants from the Ministry of Education, Science, and Culture, Tokyo, Japan (Grants‐in‐Aid for Scientific Research 25461135 to Matoba and 23790863 and 23790861 to Egashira), the Ministry of Health Labor and Welfare, Tokyo, Japan (Health Science Research Grants, Research on Translational Research, Intractable Diseases, and Nanomedicine to Egashira) and intractable diseases overcome research project from the Japan Agency for Medical Research and Development, AMED (to Egashira).
Disclosures
Dr Egashira holds a patent on the results reported in the present study. The remaining authors report no conflicts of interest.
Acknowledgments
We thank Eiko Iwata, Satomi Abe, and Shizuka Egusa for their excellent technical assistance.
(J Am Heart Assoc. 2016;5:e003872 doi: 10.1161/JAHA.116.003872)
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J‐P, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 2. Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JMO, Warnica JW, Flaker GC, Braunwald E, Pfeffer MA. Predictors of late development of heart failure in stable survivors of myocardial infarction. J Am Coll Cardiol. 2003;42:1446–1453. [DOI] [PubMed] [Google Scholar]
- 3. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 4. Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113:451–463. [DOI] [PubMed] [Google Scholar]
- 5. Ibáñez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. [DOI] [PubMed] [Google Scholar]
- 6. Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia‐Dorado D, Hausenloy DJ, Heusch G, Vinten‐Johansen J, Yellon DM, Schulz R; Working Group of Cellular Biology of Heart of European Society of Cardiology . Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406–423. [DOI] [PubMed] [Google Scholar]
- 7. Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008;103:501–513. [DOI] [PubMed] [Google Scholar]
- 9. Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32:1552–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. [DOI] [PubMed] [Google Scholar]
- 11. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. [DOI] [PubMed] [Google Scholar]
- 12. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- 13. Cung T‐T, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D, Bonnefoy‐Cudraz E, Guérin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit J‐F, Jouve B, Motreff P, Tron C, Labeque J‐N, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, De Poli F, Morice M‐C, Ider O, Dubois‐Randé JL, Unterseeh T, Le Breton H, Béard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. [DOI] [PubMed] [Google Scholar]
- 14. Ottani F, Latini R, Staszewsky L, La Vecchia L, Locuratolo N, Sicuro M, Masson S, Barlera S, Milani V, Lombardi M, Costalunga A, Mollichelli N, Santarelli A, De Cesare N, Sganzerla P, Boi A, Maggioni AP, Limbruno U. Cyclosporine A in reperfused myocardial infarction. J Am Coll Cardiol. 2016;67:365–374. [DOI] [PubMed] [Google Scholar]
- 15. Gustafsson AB, Gottlieb RA. Bcl‐2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2006;292:C45–C51. [DOI] [PubMed] [Google Scholar]
- 16. Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. [DOI] [PubMed] [Google Scholar]
- 18. Cassidy‐Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak‐dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dorn GW, Kitsis RN. The mitochondrial dynamism‐mitophagy‐cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res. 2015;116:167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ong S‐B, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010;88:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Montessuit S, Somasekharan SP, Terrones O, Lucken‐Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy‐Wetzel E, Basañez G, Meda P, Martinou J‐C. Membrane remodeling induced by the dynamin‐related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. [DOI] [PubMed] [Google Scholar]
- 23. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin‐related protein 1 (Drp1)‐mediated diastolic dysfunction in myocardial ischemia‐reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brooks C, Wei Q, Cho S‐G, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Y‐X, Cui M, Chen S‐F, Dong Q, Liu X‐Y. Amelioration of ischemic mitochondrial injury and Bax‐dependent outer membrane permeabilization by Mdivi‐1. CNS Neurosci Ther. 2014;20:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J, Wang P, Li S, Wang S, Li Y, Liang N, Wang M. Mdivi‐1 prevents apoptosis induced by ischemia‐reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species‐activated mitochondrial pathway. J Stroke Cerebrovasc Dis. 2014;23:1491–1499. [DOI] [PubMed] [Google Scholar]
- 27. Ishihara T, Ban‐Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, Mizushima N, Mihara K, Ishihara N. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol. 2014;35:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagaoka K, Matoba T, Mao Y, Nakano Y, Ikeda G, Egusa S, Tokutome M, Nagahama R, Nakano K, Sunagawa K, Egashira K. A new therapeutic modality for acute myocardial infarction: nanoparticle‐mediated delivery of pitavastatin induces cardioprotection from ischemia‐reperfusion injury via activation of PI3K/Akt pathway and anti‐inflammation in a rat model. PLoS One. 2015;10:e0132451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikeda G, Matoba T, Nakano Y, Nagaoka K, Ishikita A, Nakano K, Funamoto D, Sunagawa K, Egashira K. Nanoparticle‐mediated targeting of cyclosporine A enhances cardioprotection against ischemia‐reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci Rep. 2016;6:20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oda S, Nagahama R, Nakano K, Matoba T, Kubo M, Sunagawa K, Tominaga R, Egashira K. Nanoparticle‐mediated endothelial cell‐selective delivery of pitavastatin induces functional collateral arteries (therapeutic arteriogenesis) in a rabbit model of chronic hind limb ischemia. J Vasc Surg. 2010;52:412–420. [DOI] [PubMed] [Google Scholar]
- 32. Chen L, Nakano K, Kimura S, Matoba T, Iwata E, Miyagawa M, Tsujimoto H, Nagaoka K, Kishimoto J, Sunagawa K, Egashira K. Nanoparticle‐mediated delivery of pitavastatin into lungs ameliorates the development and induces regression of monocrotaline‐induced pulmonary artery hypertension. Hypertension. 2011;57:343–350. [DOI] [PubMed] [Google Scholar]
- 33. Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX, BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci USA. 2005;102:11272–11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. [DOI] [PubMed] [Google Scholar]
- 35. Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings B, Kass DA, Champion HC, Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Onitsuka K, Ide T, Arai S, Hata Y, Murayama Y, Hosokawa K, Sakamoto T, Tobushi T, Sakamoto K, Fujino T, Sunagawa K. Cardiac phase‐targeted dynamic load on left ventricle differentially regulates phase‐sensitive gene expressions and pathway activation. J Mol Cell Cardiol. 2013;64:30–38. [DOI] [PubMed] [Google Scholar]
- 37. Karbowski M, Lee Y‐J, Gaume B, Jeong S‐Y, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez‐Caballero S, Osinska H, Cheng EH‐Y, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2:e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre‐, post‐, and remote conditioning. Circ Res. 2015;116:674–699. [DOI] [PubMed] [Google Scholar]
- 41. Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Disatnik M‐H, Ferreira JCB, Campos JC, Gomes KS, Dourado PMM, Qi X, Mochly‐Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long‐term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461 doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Whelan RS, Konstantinidis K, Wei A‐C, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW, O'Rourke B, Kitsis RN. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA. 2012;109:6566–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimura S, Egashira K, Nakano K, Iwata E, Miyagawa M, Tsujimoto H, Hara K, Kawashima Y, Tominaga R, Sunagawa K. Local delivery of imatinib mesylate (STI571)‐incorporated nanoparticle ex vivo suppresses vein graft neointima formation. Circulation. 2008;118:S65–S70. [DOI] [PubMed] [Google Scholar]
- 45. Lin M‐J, Fine M, Lu J‐Y, Hofmann SL, Frazier G, Hilgemann DW. Massive palmitoylation‐dependent endocytosis during reoxygenation of anoxic cardiac muscle. Elife. 2013;2:e01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chlopcíková S, Psotová J, Miketová P. Neonatal rat cardiomyocytes‐a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2001;145:49–55. [PubMed] [Google Scholar]