

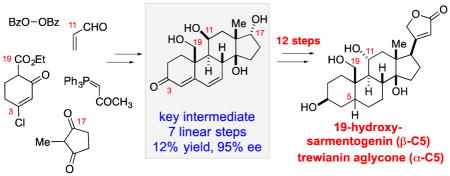
Abstract
The expedient and scalable approach to cardiotonic steroids carrying oxygenation at the C11- and C19- positions has been developed and applied to the total asymmetric synthesis of steroids 19-hydroxysarmentogenin and trewianin aglycone as well as to the assembly of the panogenin core. This new approach features enantioselective organocatalytic oxidation of an aldehyde, diastereoselective Cu(OTf)2-catalyzed Michael reaction/tandem aldol cyclizations and one-pot reduction/transposition reactions allowing a rapid (7 linear steps) assembly of a functionalized cardenolide skeleton. The ability to quickly set this steroidal core with preinstalled functional handles and diversity elements eliminates the need for difficult downstream functionalizations and substantially improves the accessibility to the entire class of cardenolides and their derivatives for biological evaluation.
Keywords: Steroids, cardenolides, enantioselective synthesis
Graphical Abstract
INTRODUCTION
Steroids are among the most universally pervasive, naturally occurring small molecules. Despite having a common structural core, steroids with differing peripheral substitutions or stereochemical relationships can confer an incredible range of valuable physiological effects.1 Two particularly useful classes of steroids are the plant-derived cardenolides, typified by ouabagenin (1A) and the animal-derived bufadienolides (e.g. 1B) (Figure 1A).2 These polyoxygenated steroids have characteristic β-C14 alcohol, β-C17 heterocyclic ring, and an unusual cis-C/D ring junction pattern, which imparts a rigid “U” shape to the molecule. These features help make the cardenolides potent inhibitors of the Na+/K+ATPase pumps on cardiac myocytes.3 Inhibition of the sodium pump leads to downstream retention of Ca2+ in the myocytes and results in a slowing of the heart rate and a positive inotropic response, directly counteracting the effects of congestive heart failure. Accordingly, the cardenolide steroids have been used for centuries to treat congestive heart failure, an ailment resulting from the heart becoming a less efficient pump, and as such have become some of the most ingested drugs in medicine.4 In addition, recent studies have demonstrated that cardenolides and the related bufadienolides possess a wide spectrum of biological activities including anti-cancer, immunoregulatory, anti-inflammatory and anti-hypertensive properties.5 Due to these valuable medicinal properties, the drug-oriented design and synthesis of the cardenolides has received considerable attention and many landmark efforts have been accomplished.
Figure 1.

Examples of bioactive cardiotonic steroids (A), and proposed synthetic approach (B)
Among such studies, the approaches based on partial synthesis starting with androstane/pregnane derivatives have been by far the most explored tactics.2b,6–8 However, these routes have significant limitation as the available androstanes and pregnanes lack oxygenation at the key positions of the steroid skeleton (i.e. at the C1, C11, C14, and C19 positions). While the presence of the C3 and C17 oxygenation often permits straightforward oxygenation of the C1, C5, and C14 positions, the oxidation of the C11 and C19 positions represents a longstanding challenge in steroid synthesis. The recent studies on ouabagenin (1A)8 and 19-hydroxysarmentogenin (2)7 by the Deslongchamps, Baran and Inoue groups provided elegant and clever solutions to the aforementioned problems. However, a general and flexible synthetic approach from non-steroidal precursors allowing control over the oxygenation patterns (i.e. 1A, 1C, and 2) and absolute and relative stereochemistry (i.e. 1D, 2, and 3) is highly desirable.
Our group recently described a new approach to oxygenated steroids based on sequential enantioselective bis(oxazoline) copper(II)-catalyzed Michael addition/intramolecular aldol reactions.9 This approach enabled one of the most concise (4–5 step) enantioselective syntheses of 9 oxygenated steroid analogs. However, the lack of oxygenation at the C3 and C11 positions typically present in natural cardenolides impeded application of this strategy to the synthesis of natural cardiotonic steroids (i.e. 1–3). To address this drawback, and, in particular, the necessity for the C3 oxygenation, we have expanded on that work and developed a new strategy, which allows rapid and scalable access to fully functionalized chiral steroid intermediate 4 (Figure 1B). This new approach features enantioselective organocatalytic oxidation of an aldehyde to form enone 8, which undergoes diastereoselective Cu(OTf)2-catalyzed Michael reaction to adduct 6 that is cyclized to steroid 5. Subsequent one-pot reduction/transposition reaction of 5 is the major element of the design that allows a rapid (7 linear steps, 9 total steps) and scalable assembly of fully functionalized intermediate 4. Steroid 4 is the key building block in the synthesis of various natural cardenolides (i.e. 2 and 3). The utility of this building block is demonstrated in the synthesis 19-hydroxysarmentogenin (2) and trewianin aglycone (3), cardenolides epimeric at the C5 position. These syntheses are among the most concise totally synthetic enantioselective approaches (19 linear steps, 21 total steps) to the C11/C19 oxygenated cardenolides reported to date.
RESULTS AND DISCUSSION
One of the major challenges in the strategy outlined in Figure 1B is achieving a stereoselective Michael reaction between 7 and 8. Not only will the Michael reaction resulting in vicinal quaternary and tertiary stereocenters be retarded by the steric hindrance, but also one would expect even further rate retardation upon introduction of an additional OBz group. Finally, the presence of the -OBz group on 8 introduces an additional challenge as the relative configuration of the C9 and C10 stereocenters has to be controlled. Consequently, to test the prospects of this Michael reaction, the model study for the reaction of racemic enone 8 and commercially available ketoester 9 was investigated (Scheme 1). Remarkably, it was discovered that the presence of OBz-containing γ-stereocenter results in excellent levels of stereocontrol, which allows a diastereoselective formation of the corresponding Michael adduct (>20:1 dr) even if Cu(OTf)2 (20 mol%) without chiral ligand is employed as the catalyst. While γ-alkoxide containing stereocenters are known to have a strong influence on the stereochemical outcome of intramolecular Michael reactions,8a,10 stereoselective intermolecular variants of such reactions (i.e. Michael addition in Scheme 1) have not been described.11 Considering that the vinylogous polar Felkin-type model has been invoked to describe the selectivity of the cuprate addition to γ-alkoxy enones,12 we propose a similar model (i.e. 11) to explain the diastereoselectivity of the Michael reaction of 9 and 8. The subsequent cyclization of the resulting Michael adduct with DBU provided steroid 10, and the relative configuration was then elucidated by X-ray crystallographic analysis. Importantly, the product 10 contained the desired trans-C9/C10 configuration, the feature that is essential for the approach depicted in Figure 1B. It should be noted that the DBU-promoted cyclization resulted in the unnatural α-configuration of the CD-ring junction of 10. At the same time, our prior studies suggest that the proper stereochemistry of the C13 and C14 stereocenters can be established under equilibrating conditions.9
Scheme 1.

Model studies of diastereoselective Cu(II)-catalyzed Michael reaction
The asymmetric synthesis of intermediate 4 was investigated next (Scheme 2). Our studies commenced with the Michael reaction of commercially available diketone 12 and acrolein. The resulting aldehyde product was subjected to an organocatalytic oxidation catalyzed by commercially available L-prolinol-based catalyst 13 in 79% yield.13 The resultant aldehyde was then subjected to a Wittig reaction with the commercially available 1-(tripheylphosphoranylidene)-2-propanone to provide chiral enone 8 (57% yield, 95% ee, 3 steps).
Scheme 2.

Scalable enantio- and diastereoselective synthesis of cardenolides core 5a
aReagents and conditions: (a) acrolein, H2O, 12 h, 97% yield; (b) 13 (10 mol%), (BzO)2, hydroquinone, THF, H2O, 1.5 h, 79% yield; (c) 1-(tripheylphosphoranylidene)-2-propanone, toluene, 75% yield, 95% ee; (d) 7, Cu(OTf)2 (50 mol%), CH2Cl2, 9 h, 70% yield (6), 6:1 dr; 20% yield (6a); (e) p-TSA, CH3CN, 55 °C, 48 h, 68% yield from 6; 56% yield from 6a; (f) NaHMDS, toluene, −78 to 42 °C, 1.5 h, 48% yield (68% BRSM).
Importantly, this sequence is amenable to scale up and 8 was conveniently generated via this 3-step procedure in 11.5 g batches without erosion in yield or selectivity. With the chiral enone 8 in hand, the key Cu(II)-catalyzed diastereoselective Michael reaction was attempted. The early attempts to utilize β-ketoesters 7a and 7b failed as these ketoesters were found to be unreactive with enone 8. Gratifyingly, easily available (3 steps, 44% yield, 19 g scale) chlorinated ketoester 7 was found to be significantly more reactive, and diastereomer 6 was generated from 7 and 8 in 6.4 g scale batches (70% yield, 6:1 dr) along with its monocyclized product 6a (20% yield). Increased loadings of Cu(OTf)2 (i.e. 50 mol%) were employed to enhance the reaction times; however, lower catalysts loading (i.e. 20 mol%) can be used without significant erosion in yield or selectivity. The diastereoselectivity of this reaction was consistent with the prior observations made for 10 (Scheme 1). This Michael adduct (6) was converted to steroid 14 upon its exposure to p-TSA (68% yield, >20:1 dr) to produce pure 14 in 5.3 g batches. Sideproduct 6a, can also be converted to steroid 14 upon exposure to p-TSA (56% yield, >20:1 dr). The NMR analysis of 14 (cf. SI) indicated that it possessed the unnatural α-configuration of the C13/C14 stereocenters. However, our prior studies revealed that steroids with unsaturation at the C5 position prefer the natural β-C13/β-C14 configuration under equilibrating conditions. Correspondingly, steroid 14 was subjected to epimerization with NaHMDS, which presumably proceeded through 14a to provide the desired steroid 5 with the natural β–CD ring junction. This transformation was accomplished on 2.0 g batches of 14 and resulted in 0.9 g of 5 (48% yield) along with 0.4 g of recovered 14 (68% yield brsm).
At this point, 5 had to be transformed into the key intermediate 4. We envisioned this could be accomplished with a global reduction to generate the C7 alcohol followed by an acid catalyzed transposition. Importantly, the C3 chlorine provided a means of trapping the initial transposition product as a ketone so as to avoid complication from diastereo- and regioselectivity during the reaction.14 Finally, we sought for the concomitant reduction of the C19 ester and C11-OBz group to minimize the number of redox manipulations. This transformation required careful optimization of the reaction conditions, and initial evaluation of various reducing agents (LiAlH4, LiBH4, etc.) did not result in clean reduction. However, switching the reducing agent to DIBAL-H helped to circumvent this problem. Remarkably, the resulting reaction mixture can be quenched with water and formic acid without the isolation of 15 and warmed up to 85 °C for 1 h to provide the desired intermediate 4 in a single step (72% yield, 8:1 dr, 0.28 g scale).
With an established 7 linear step route to 4, we next demonstrated that this intermediate could indeed be elaborated to diastereomeric cardenolides with cis- and trans- configuration of the AB ring junction found in steroids 2, 3 and 1D (Scheme 4). Thus, 4 can be subjected to a 3–4 step sequences to provide diastereomeric scaffolds 16, 17 and 18, which are precursors to 2, 1D and 3, correspondingly. The C19 hydroxyl-directed hydrogenation of 4 to establish the cis-AB ring junction in 16 was accomplished under basic conditions (Pd/C, KOH, quinoline) with excellent diastereoselectivity (84% yield, >20:1 dr).15 Protection at C19 with TIPS and oxidation at C11 and C17 with DMP resulted in 16. Trans-reductions of steroid-derived A-ring α,β-unsaturated enones are typically conducted using dissolving metal conditions as the hydrogenation reactions are typically cis- selective. However, based on the trends observed by Knox et al.16 we argued that the presence of the C19 hydroxyl would allow overturning this selectivity trends if a bulky protecting group is introduced at this position. Thus, the C19 and C11 alcohol functionalities of intermediate 4 can be protected as acetonide (69% yield). This product can be subjected to hydrogenation reaction (H2, Pd/C) to provide a crude mixture of the products epimeric at the C5 position (2.5:1 dr) upon filtration. This mixture can be selectively reduced with LiAl(OtBu)3H and separated to provide panogenin core 17 (59% yield, 2 steps) as the major diastereomer. Trewianin core 18 can be generated in a fashion similar to 17. Thus, substrate 4 was subjected to selective TIPS protection of the C19 hydroxyl, followed by Dess-Martin oxidation of the remaining alcohol functionalities. The resulting product was subjected to diastereoselective steric-controlled hydrogenation to provide exclusive formation of the trans- product 18 (61% yield, >20:1 dr, 3 steps).
Scheme 4.

Elaboration of intermediate 4 into precursors of 19-hydroxysarmentogenin (2), panogenin (1D) and trewianin (3)a
aReagents and conditions: (a) H2, 10% Pd/C (25% w/w loading), KOH (1% w/v), quinoline (1% v/v), MeOH, 84% yield, >20:1 dr; (b) TIPSCl, ImH, DMF, 6 h, 82% yield; (c) DMP, Py, CH2,Cl2, 2 h, 89% yield; (d) 2,2′-dimethoxypropane, (+)-CSA, DMF, 69% yield; (e) DMP, Py, CH2Cl2, 1.5 h, 90% yield; (f) H2, Pd/C (25% w/w), EtOAc/MeOH/CH2Cl2 (3:1:1), 20 min, 2.5:1 dr; (g) LiAl(OtBu)3H, THF, −78 to −40 °C; 2.5 h, 59% yield, >20:1 dr (2 steps); (h) TIPSCl, ImH, DMF, rt, 6 h, 81% yield; (i) DMP, Py, CH2Cl2, 2 h, 91% yield; (j) H2, Pd/C, MeOH, Py, 4 h; 83% yield, >20:1 dr.
With these results in hand, we demonstrated that the outlined strategy is applicable to the synthesis of fully functionalized cardiotonic steroids by elaborating 16 and 18 into 2 and 3 (Scheme 5). The C3 ketone of 16 was selectively reduced with K-selectride, and the resulting product was subjected to protection (TBSOTf, Et3N) to provide 19 (73% yield, 2 steps). With the exception of the C19 protecting group (TIPS vs TBS), 19 is identical to the corresponding intermediate previously synthesized by the Inoue group in their studies toward 19-hydroxysarmentogenin (2).7 The C11-ketone moiety of 19 was subjected to the reduction and the resulting α–C11 alcohol-containing product was converted to iodide 20 via a 3-step sequence (66% yield). Iodide 20 was then cross-coupled with the known stannane 21 to provide protected cardenolide 22 in 92% yield. With this key intermediate, the remaining β–C17 stereocenter was installed by global TMS-protection of 22 followed by hydrogenation over Pd/C (2.7:1 dr). Finally, the complete removal of the protecting groups was accomplished with hydrofluoric acid to provide 19-hydroxysarmentogenin (2) in 27% yield from 22 (3 steps).
Scheme 5.

Stereodivergent conversion of intermediates 16 and 18 into (+)-19-hydroxysarmentogenin (2) and (+)-trewianin aglycone (3)
aReagents and conditions: (a) K-selectride, THF, −78 to −30 °C, 1.5 h, 77% yield, >20:1 dr; (b) TBSOTf, Et3N, CH2Cl2, −78 to −30 °C, 1.5 h, 95% yield; (c) Li, NH3, THF, −78 °C, 30 min; (d) TBAF, THF, −78 °C, 5 min, 73% yield (2 steps); (e) N2H4•H2O, Et3N, EtOH, 50 °C, 6 h then I2, Et3N, THF, rt, 1 h, 90% yield; (f) 21, Pd(PPh3)4, CuCl, LiCl, DMSO, 50 °C, 1 h, 92% yield; (g) TMSOTf, 2,6-lutidine, CH2Cl2, −78 °C to rt, 2h, then SiO2 (dry), 10 h, 64% yield; (h) H2, Pd/C, EtOAc, 30 min, 2.7:1 dr (β-C17: α–C17); (i) HF in CH3CN/H2O/CH2Cl2, rt, 3 days, 42% yield, 2 steps; (j) LiAlH(OtBu)3, THF, −78 to −40 °C, 4 h, 96% yield, >20:1 dr; (k) TBSOTf, Et3N, CH2Cl2, −78 to −30 °C, 1.5 h, 87% yield; (l) Li, NH3, THF, −78 °C, 30 min; (m) TBAF, THF, −78 °C, 5 min, 91% yield (2 steps); (n) N2H4•H2O, Et3N, EtOH, 50 °C, 6 h then I2, Et3N, THF, rt, 1 h, 83% yield; (o) 21, Pd(PPh3)4, CuCl, LiCl, DMSO, 50 °C, 1 h, 93% yield; (p) TMSOTf, 2,6-lutidine, CH2Cl2, −78 °C to rt, 2h, then SiO2 (dry), 10 h, 79% yield; (q) H2, Pd/C, EtOAc, 30 min; 54% yield of β-C17 and 22% of α-C17; (r) HF in CH3CN/H2O/CH2Cl2, 3 days, 91% yield.
The 1H and 13C NMR spectral data as well as other physical properties of 2 were identical in all respects to the corresponding data previously reported by the Inoue group.7 In addition, the obtained specific optical rotation value for 2 (i.e. [α]D23= 13, c = 0.1, MeOH) was in good agreement with the reported value ([α]D23= 17, c = 0.45, MeOH).
Similarly, trans-AB ring-containing intermediate 18 was subjected to highly diastereoselective reduction with LiAlH(OtBu)3 followed by TBS protection to provide 23 in 84% yield (2 steps). The subsequent reduction with Li/NH3 followed by deprotection with TBAF and installation of the vinyl iodide moiety (N2H4/I2) resulted in vinyl iodide 24 (76% yield, 3 steps). The cross-coupling of 24 and 21 resulted in efficient formation of 25 (93% yield), which was subjected to a 3-step protection/reduction/deprotection sequence to provide 3 (39% yield, 3 steps). Similar to the 19-hydroxysarmentogenin (2) case, the hydrogenation of protected 25 resulted in a 2.5:1 mixture of the β–C17: α–C17 diastereomeric products, and the major diastereomer (β–C17) was isolated prior to the deprotection in 54% yield. The spectroscopic data for 3 were in good agreement with the spectroscopic data obtained for trewianin17 and 19-hydroxysarmentogenin (2).
CONCLUSION
In summary, a concise enantioselective approach to C11- and C19- oxygenated cardenolides has been developed. This approach features a rapid (7 linear steps, 9 total steps) enantioselective synthesis of functionalized intermediate 4, which is the key building block in the synthesis of various natural cardenolides. The utility of this building block is demonstrated in the synthesis 19-hydroxysarmentogenin (2) and trewianin aglycone (3), cardenolides epimeric at the C5 position. The studies on the further diversification of 4 and conversion of it to C1- and C5- oxygenated cardenolides and bufadienolides are ongoing in our laboratory.
Supplementary Material
Scheme 3.

Reduction/transposition for the synthesis of intermediate 4a
aReagents and conditions: (a) DIBALH, THF, −78 to 60 °C, 12 h then HCOOH, H2O, 85 °C, 1 h, 72% yield, 8:1 dr.
Acknowledgments
Funding Sources and Acknowledgement
This manuscript is dedicated to Professor Samuel J. Danishefsky on the occasion of his 80th birthday. This work was supported by NIGMS R01 grant (1R01GM111476-01). PN is the Sloan Foundation and Amgen Young Investigator Fellow. WK is an AFPE and University of Michigan CBI program fellow. We acknowledge funding from NSF grant CHE-0840456 for X-ray instrumentation.
Footnotes
Supporting Information Available.
Experimental procedures, 1H and 13C NMR spectra, and X-ray data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Corey EJ, Czako B, Kurti L. Molecules and medicine. Hoboken, NJ: Wiley; 2007. [Google Scholar]; (b) Newman DJ, Cragg GM. J Nat Prod. 2012;75:311. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Wiesner K, Tsai TYR. Pure & Appl Chem. 1986;58:799. [Google Scholar]; (b) Heasley B. Chem Eur J. 2012;18:3092. doi: 10.1002/chem.201103733. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bukharovich IF, Kukin M. Prog Cardiovasc Dis. 2006;48:372. doi: 10.1016/j.pcad.2005.11.002. [DOI] [PubMed] [Google Scholar]; (b) Schoner W, Scheiner-Bobis G. Am J Physiol Cell Physiol. 2007;293:C509. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]; (c) Laursen M, Yatime L, Nissen P, Fedosova NU. Proc Nat Acad Sci, USA. 2013;110:10958. doi: 10.1073/pnas.1222308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Wasserstrom JA, Aistrup GL. Am J Physiol Heart Circ Physiol. 2005;289:H1781. doi: 10.1152/ajpheart.00707.2004. [DOI] [PubMed] [Google Scholar]; (b) Bauman JL, Di-Domenico RJ, Galanter WL. Am J Cardiovasc Drugs. 2006;6:77. doi: 10.2165/00129784-200606020-00002. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zhao M, Bai L, Wang L, Toki A, Hasegawa T, Kikuchi m, Abe M, Sakai J, Hasegawa R, Bai Y, Mitsui T, Ogura H, Kataoka T, Oka S, Tsushima H, Kiuchi M, Kirose K, Tomida A, Tsuruo T, Ando M. J Nat Prod. 2007;70:1098. doi: 10.1021/np068066g. [DOI] [PubMed] [Google Scholar]; (b) Platz EA, Yegnasubramanian S, Liu JO, Chong CR, Shim JS, Kenfield SA, Stampfer MJ, Willett WC, Giovannucci E, Nelson WG. Cancer Dis. 2011;1:68. doi: 10.1158/2159-8274.CD-10-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Banuls LMY, Urban E, Gelbcke M, Dufrasne F, Kopp B, Kiss R, Zehl M. J Nat Prod. 2013;76:1078. doi: 10.1021/np400034d. [DOI] [PubMed] [Google Scholar]; (d) Ferrari P. Biochim Biophys Acta. 2010;12:1254. doi: 10.1016/j.bbadis.2010.01.009. [DOI] [PubMed] [Google Scholar]; (e) Terness P, Navolan D, Duter C, Kopp B, Opelz G. Int Immunopharmacol. 2001;1:119. doi: 10.1016/s0162-3109(00)00264-2. [DOI] [PubMed] [Google Scholar]
- 6.Selected approaches to cardenolides: Yoshii E, Oribe T, Tumura K, Koizumi T. J Org Chem. 1978;43:3946.Lavallee JF, Deslongchamps P. Tetrahedron Lett. 1988;29:6033.Deng W, Jensen MS, Overman LE, Rucker PV, Vionnet JP. J Org Chem. 1996;61:6760. doi: 10.1021/jo961209r.Stork G, West F, Lee HY, Isaacs RCA, Manabe S. J Am Chem Soc. 1996;118:10660.Overman LE, Rucker PV. Tetrahedron Lett. 1998;39:4643.Jung ME, Davidov P. Angew Chem Int Ed. 2002;41:4125. doi: 10.1002/1521-3773(20021104)41:21<4125::AID-ANIE4125>3.0.CO;2-E.Jung ME, Yoo D. Org Lett. 2011;13:2698. doi: 10.1021/ol200796r.Honma M, Nakada M. Tetrahedron Lett. 2007;48:1541.
- 7.Synthesis of 19-hydroxysarmentogenin (2): Mukai K, Urabe D, Kasuya S, Aoki N, Inoue M. Angew Chem Int Ed. 2013;52:5300. doi: 10.1002/anie.201302067.
- 8.Ouabagenin (1A) synthesis: Zhang H, Reddy MS, Phoenix S, Deslongchamps P. Angew Chem Int Ed. 2008;47:1272. doi: 10.1002/anie.200704959.Reddy MS, Zhang H, Phoenix S, Deslongchamps P. Chem-Asian J. 2009;4:725. doi: 10.1002/asia.200800429.Renata H, Zhou Q, Baran PS. Science. 2013;339:59. doi: 10.1126/science.1230631.Renata H, Zhou Q, Dunstl G, Felding J, Merchant RR, Chien-Hung Y, Baran PS. J Am Chem Soc. 2015;137:1330. doi: 10.1021/ja512022r.Mukai K, Kasuya S, Nakagawa Y, Urabe D, Inoue M. Chem Sci. 2015;6:3387. doi: 10.1039/c5sc00212e.
- 9.Cichowicz N, Kaplan W, Khomutnik I, Bhattarai B, Sun Z, Nagorny P. J Am Chem Soc. 2015;137:14341. doi: 10.1021/jacs.5b08528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Kwan EE, Scheerer JR, Evans DA. J Org Chem. 2013;78:175. doi: 10.1021/jo302138z. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Scheerer JR. Angew Chem Int Ed. 2005;44:6038. doi: 10.1002/anie.200502296. [DOI] [PubMed] [Google Scholar]
- 11.Selected reviews on substrate-directed reactions: Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307.Rousseau G. Breit, B Angew Chem Int Ed. 2011;50:2450. doi: 10.1002/anie.201006139.Micalizio GC, Hale SB. Acc Chem Res. 2015;48:663. doi: 10.1021/ar500408e.
- 12.Roush WR, Michaelides MR, Tai DF, Lesur BM, Chong WKM, Harris DJ. J Am Chem Soc. 1989;111:2984. [Google Scholar]
- 13.(a) Keno T, Mil H, Maruoka K. J Am Chem Soc. 2009;131:3450. doi: 10.1021/ja809963s. [DOI] [PubMed] [Google Scholar]; (b) Vaismaa MJP, Yau SC, Tomkinson NCO. Tetrahedron Lett. 2009;50:3625. [Google Scholar]
- 14.While the examples of Stork-Danheiser rearrangement of 3-chlorovinyl alcohols are known, to our knowledge, the vinylogous variant of this transformation is unprecedented: Liotta D, Zima G, Saindane M. J Org Chem. 1982;47:1258.Demeke D, Forsyth CJ. Org Lett. 2000;2:3177. doi: 10.1021/ol006376z.Gan P, Smith MW, Braffman NR, Snyder SA. Angew Chem Int Ed. 2016;55:3625. doi: 10.1002/anie.201510520.
- 15.Bach G, Capitaine J, Engel ChR. Can J Chem. 1968;46:733. [Google Scholar]
- 16.Knox LH, Blossey E, Caprio H, Cervantes L, Crabbe P, Velarde E, Edwards JA. J Org Chem. 1995;30:2198. [Google Scholar]
- 17.Shilpi JA, Gray AI, Seidel V. Filoterapia. 2010;81:536. doi: 10.1016/j.fitote.2010.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.