

Abstract
The incidence of focal segmental glomerulosclerosis (FSGS) is approximately 10% in children <6 years, 20% in adolescents, and 20-25% in adults. A retrospective observational study was done to document clinicopathological correlation, treatment response, and risk factors in the progression of chronic kidney disease (CKD) of primary FSGS in adults and adolescents. A total of 170 patients were studied with a mean follow-up of 4.32 ± 1.2 years. FSGS not otherwise specified was the most common subtype (56%) followed by tip variant (24%). About 32% had complete remission (CR) at a mean time of 6.4 months, 23% had partial remission (PR) at a mean time of 5.7 months, and 45% had no response to steroids. Persistent nephrotic proteinuria at 3rd and 6th month and presence of interstitial fibrosis and tubular atrophy >30% in renal biopsy are the independent predictors of poor response to treatment. Presence of anemia, interstitial fibrosis, and tubular atrophy of >30% in renal biopsy and the absence of remission after treatment were the independent predictors of CKD progression. Overall renal survival was 78% at 3 years and 54% at 5 years. Renal survival difference with or without nephrotic proteinuria at onset was 39% and 69% at 5 years. Renal survival was higher in patients with normal renal function (66%) compared with those who had renal failure (42%) at 5 years. Renal survival at 5 years for CR was 69%, PR was 49%, and no remission was 42%.
Key words: Chronic kidney disease, primary focal segmental glomerulosclerosis, remission, risk factors, steroids
Introduction
Focal segmental glomerulosclerosis (FSGS) is a common cause of nephrotic syndrome, accounting for 20-25% of nephrotic syndrome in adults.[1] Around 50% of patients reach end-stage renal disease (ESRD) at 8 years, accounting for 20% of dialysis patients. Kitiyakara et al. reported an 11-fold increase in FSGS among dialysis patients older than 21 years.[2] Remission rates of 80% can be achieved with longer treatment, and is an independent predictor of renal survival.[3] In adults, responsiveness to steroids usually takes up to 16 weeks, and can be slowly tapered over 3-6 months. Our aim was to study the epidemiological profile and clinicopathologic correlation of primary FSGS in adults and adolescents. Treatment response and risk factors in the progression of chronic kidney disease (CKD) was also studied.
Subjects and Methods
This was a retrospective observational study in adult and adolescent patients (age 13-60 years) with biopsy proven FSGS during the period of January 2007 to December 2013. All patients treated with oral prednisolone for 24 weeks with at least 6 months follow-up were included. We excluded patients with secondary FSGS, patients who did not receive steroid therapy, poor compliance with drugs, and follow-up with <6 months.
Demographic profile, clinical examination, laboratory parameters viz urine analysis, 24 h urine protein, serum creatinine, blood urea, serum albumin, and lipid profile at the onset of disease and renal biopsy details were analyzed. The diagnosis of FSGS was made when at least one glomerulus shows a typical sclerotic lesion in a segment in light microscopy. Subtyping of FSGS was performed in according to Columbia classification described by D'Agati.[4] All patients were started on oral prednisolone 1 mg/kg/day and continued for 6 months, tapered and stopped within 1-month. If there was no response to steroids at 6 months, they were started on second-line drugs that include oral cyclophosphamide, cyclosporine A (CSA), tacrolimus, or mycophenolate mofetil (MMF). All received a maximal tolerable dose of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers along with supportive treatment and statins.
Definitions
Complete remission (CR) – proteinuria <0.3 g/day with a stable serum creatinine (<50% increase from baseline)
Partial remission (PR) – proteinuria between 0.3 and 3.5 g/day with at least 50% reduction in proteinuria from the baseline and a stable serum creatinine
Relapse – proteinuria >3.5 g/24 hr after remission
CKD – CrCl <60 ml/min/1.73 m2 after 3 months
ESRD – CrCl <15 ml/min/1.73 m2 or the need for dialysis or renal transplantation.[1,5]
Statistical analysis
All variables were expressed in mean ± standard deviation or percentage. Univariate analysis was done by Fisher's exact test and Chi-square test for categorical variables and analysis of variance was used for continuous variables. Statistical significant variables by univariate analysis were subjected to linear regression analysis to assess multivariable association. Renal survival rate was determined by the Kaplan–Meier method. The log-rank test was used to evaluate the significance of the difference between survival mean values in subgroups. Statistical analysis was done with SPSS software 16 (SPSS Inc., Chicago, IL). In all analytic procedures, P < 0.05 was considered as significant. Institutional Ethics Committee approval was obtained.
Results
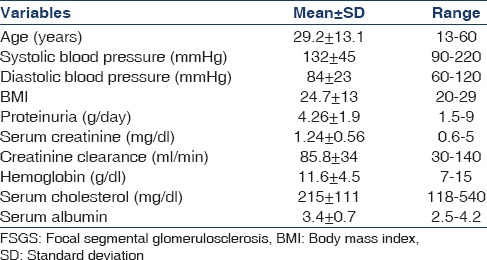
A total of 170 patients were included with a mean follow-up of 4.32 ± 1.2 years. About 65% were males (male: female ratio − 1.9:1). The predominant age group was between 21 and 40 years accounting for 54% of total patients. Baseline patient characteristics at the time of biopsy are shown in Table 1. The most common symptom was edema (98%), followed by nephrotic proteinuria (79%), hypertension (41%), microhematuria (30%), sub-nephrotic proteinuria (21%), and renal failure (20%). Venous thrombosis and cellulitis due to anasarca were occurred as complications of disease process. Infection is the most common complication followed by cushingoid features due to drugs. Two patients suffered from glaucoma and eight patients had cataract due to steroid therapy.
Table 1.
Baseline characteristics of patients with primary FSGS
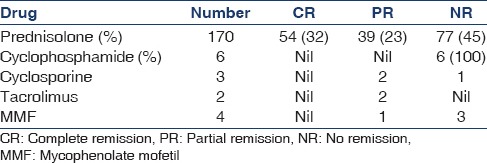
Response to treatment as defined previously is expressed as CR, PR, and NR, and the details of immunosuppression therapy are described in Table 2. About 49% of the patients progressed to CKD at mean follow-up. Incidence of ESRD is 17% at a mean time of 4.32 years and two patients died due to uremia at a mean time of 2.4 years. During follow-up, 13 patients out of 93 who achieved remission (CR or PR) had a relapse at a mean duration of 2.8 years. Eighty percentage of them had prior PR only.
Table 2.
Treatment response
Not otherwise specified (NOS) was the most common lesion, present in 96 (56%), followed by tip variant in 41 (24%), perihilar type in 16 (10%), and cellular 15 (9%). Only two (1%) patients had collapsing FSGS and reached ESRD in 2.2 years. Mesangial hypercellularity and intra-glomerular foam cells were present in 11% and 26%, respectively. Significant interstitial fibrosis and tubular atrophy was present (>30% of cortical parenchyma) in 29% of patients. Hyaline arteriosclerosis was seen in 94 patients (55%). Around 90 patients (53%) showed IgM positivity and 56 patients (33%) had C3 positivity in immunofluorescence.
Among subtypes, perihilar variant was present with less microhematuria, nephrotic proteinuria compared to NOS (P < 0.001), and cellular variety (P < 0.001). Cellular variant was present more with renal failure (P < 0.05) at presentation versus tip variant and more arterial hyalinosis in renal biopsy(P < 0.05) compared to the perihilar lesion. Hypoalbuminemia (P 0.001) was commonly seen in tip lesion and hypertension in perihilar variant (P = 0.007) compared to other groups. Interstitial fibrosis and tubular atrophy were seen more in NOS (P = 0.007) versus cellular variant. CR was seen more in tip variant (P 0.001) when compared to others. Less remission and progression to CKD was increasingly noted in NOS type compared to tip lesion (P = 0.003 and P = 0.009, respectively).
Predictors of poor response to treatment and progression to CKD are given Table 3. Overall renal survival was 78% at 3 years and 54% at 5 years. Renal survival was significantly higher in patients presented with normal renal function compared with those with renal failure at presentation with 66% versus 42% at 5 years. Renal survival difference with or without nephrotic proteinuria at onset was 39% versus 69% at 5 years [Figure 1]. Renal survival by Kaplan-Meier analysis at 5 years with CR was 69%, PR was 49%, and NR was 42% [Figure 2]. There was no significant difference between those achieve PR and nil response.
Table 3.
Predictors of poor treatment response and CKD progression (univariate analysis)

Figure 1.
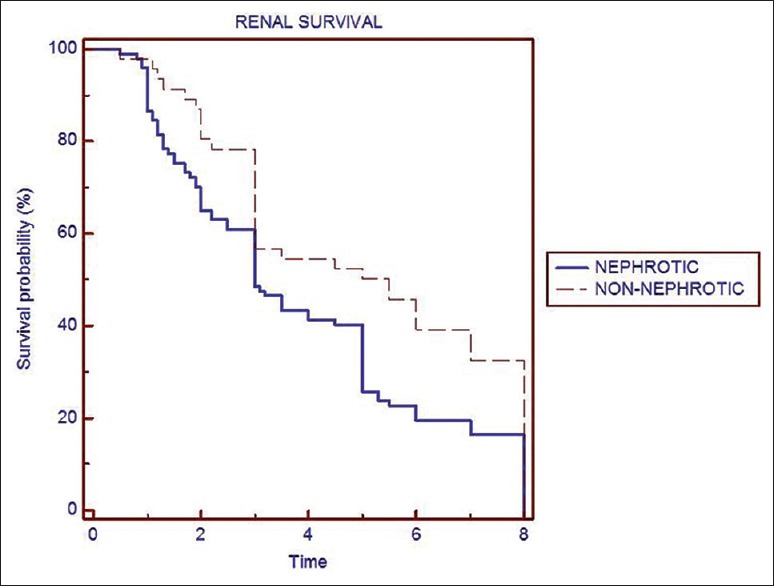
Renal survival at 5-year nephrotic versus nonnephrotic proteinuria
Figure 2.
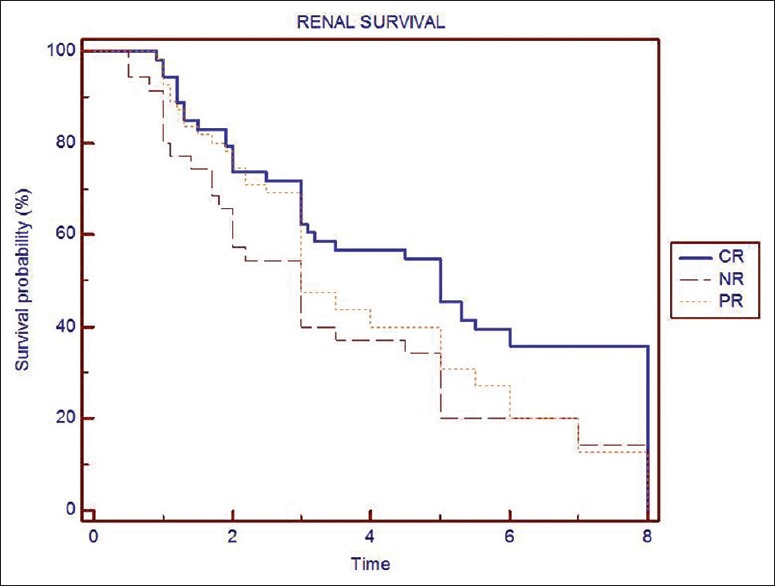
Survival analysis by Kaplan–Meier method
Discussion
FSGS is characterized by marked proteinuria, variable steroid responsiveness hypertension, and a risk of progression to CKD.[5] Classification of FSGS include primary, genetic, and secondary. Primary (idiopathic) occurs without known inciting injury.[4] A working group has defined five subtypes of FSGS[6] based on light microscopy (Columbia classification) which include collapsing FSGS, tip lesion, cellular, perihilar variant, and NOS.
The most common symptom was edema (98%), followed by nephrotic proteinuria (79%), hypertension (41%), and renal insufficiency (20%), while Singh et al.[7] showed hypertension in 42.2%, gross hematuria in 36.4%, micro-hematuria in 63.6%, and azotemia in 37.5%. Mean proteinuria was 5.53 ± 2.89 g/day while in our study it was 4.26 ± 1.9 g/day. Taheri et al.[8] reported mean systolic blood pressure of 121.19 mmHg, diastolic blood pressure of 77.52 mmHg, serum creatinine of 1.18 mg/dl, plasma albumin of 3.29 g/dl, and glomerular filtration rate of 87.18 ml/min, while in our study it was 132 mmHg, 84 mmHg, 1.24 mg/dl, 3.4 mg/dl, and 85.8 ml/min, respectively.
On steroid therapy, PR occurred earlier in those who eventually achieved CR. Hence, early partial response is the predictor of CR. Miyata et al.[9] reviewed 32 FSGS patients with steroids alone in which 44% had CR, 12% had PR, and 44% had nil response. The prospective Regional Glomerulonephritis Registry Study[10] followed 95 adult and pediatric patients with FSGS, for a mean period of 61 months and showed a remission rate of 39% in adults after prolonged therapy with steroids.
Relapse occurred in 14% cases at a mean time of 2.8 years who achieved remission. In other studies where relapse rate was reported as high as 40%. Males (80%) had more relapse than females. CSA is the only agent tested by randomized controlled trials in adults with FSGS.[11] The main drawback of CSA is that 40–75% patients experience a relapse within 2 months of stopping or tapering the drug.[12] In a recent study of 25 patients with steroid-resistant FSGS, who were treated with tacrolimus (0.15 mg/kg/day) and full dose prednisone for at least 8 weeks, CR was achieved in 40% and PR in 8%.[13] Briggs et al.[14] reported a remission rate of 66% with MMF in steroid resistant FSGS and is an alternative to CSA in patients with progressive renal failure due to its negligible side effects on renal hemodynamics and metabolic profile. Lanewala et al.[15] showed steroid-dependent nephrotic syndrome in 63.8% and steroid resistance in 35% of children with idiopathic FSGS. They also showed that majority of them could achieve high rates of sustained remission with second- and third-line immunosuppressive drugs.
NOS was the most common subtype (56%), followed by tip variant (24%), perihilar (10%), cellular (9%), and collapsing (1%). In a study by Deegens et al., tip lesion was the most common (37%), followed by NOS (32%), perihilar (26%), and collapsing (5%).[16] Schwartz and Lewis[17] showed IgM positivity in 17% and mesangial cellularity in 16%. Presence of mesangial hypercellularity and hyalinosis was suggested as predictors for lower remission rate.[18] The tip variant was associated with highest remission rate in our study of 70% while other studies also showed higher remission rate of 57–60%. A study of primary FSGS in children <17 years from Pakistan showed NOS as the highly prevalent variant.[19]
By multivariate analysis, persistent nephrotic proteinuria at 3rd and 6th month and presence of interstitial fibrosis and tubular atrophy >30% in renal biopsy were the independent predictors of poor renal response to treatment. Risk factors such as male gender, nephrotic proteinuria at onset, persistent nephrotic proteinuria at 3 and 6 months, renal failure at onset, persistent renal failure at 3 and 6 months, presence of hypertension, anemia, interstitial fibrosis and tubular atrophy of >30% in renal biopsy, and NR after treatment predicted the progression to CKD at mean follow-up. Of this, last three factors were the independent predictors. Absence of remission was the single strongest predictor for the progression of CKD. Most studies demonstrated that presence of significant interstitial fibrosis was the only independent predictive factor for response to treatment.[20,21] Korbet[1] showed that serum creatinine >1.3 mg/dl, range of proteinuria, and interstitial fibrosis >20% were positive predictors of the occurrence of ESRD.
Overall renal survival by Kaplan-Meier survival analysis was 78% at 3 years and 54% at 5 years. Renal survival was significantly higher in patients presented with normal renal function (66%) compared with those with renal failure (42%) at 5 years. Renal survival difference with or without nephrotic proteinuria at onset was 39% versus 69% at 5 years. Renal survival at 5 years for CR was 69%, for PR 49%, and for NR 42%. There was no significant difference between those achieve PR and nil response. Therefore, attaining CR significantly improves renal survival. Renal survival rates were 68.6% and 27.3% at 5 and 10 years, respectively, in a study by Singh et al.[7] Cameron[22] reported 5-year and 10-year survival rates as 70% and 40%, respectively.
This study involves significant number of patients with longer duration of follow-up from India and validates the observation that nonremission of proteinuria is the strongest risk factor for CKD progression. The limitations include retrospective analysis and the role of immunosuppressants other than steroids was not studied due to less number.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Korbet SM. Primary focal segmental glomerulosclerosis. J Am Soc Nephrol. 1998;9:1333–40. doi: 10.1681/ASN.V971333. [DOI] [PubMed] [Google Scholar]
- 2.Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol. 2003;23:172–82. doi: 10.1053/snep.2003.50025. [DOI] [PubMed] [Google Scholar]
- 3.Banfi G, Moriggi M, Sabadini E, Fellin G, D’Amico G, Ponticelli C. The impact of prolonged immunosuppression on the outcome of idiopathic focal-segmental glomerulosclerosis with nephrotic syndrome in adults. A collaborative retrospective study. Clin Nephrol. 1991;36:53–9. [PubMed] [Google Scholar]
- 4.D’Agati V. The many masks of focal segmental glomerulosclerosis. Kidney Int. 1994;46:1223–41. doi: 10.1038/ki.1994.388. [DOI] [PubMed] [Google Scholar]
- 5.Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC. Toronto Glomerulonephritis Registry Group. Focal and segmental glomerulosclerosis: Definition and relevance of a partial remission. J Am Soc Nephrol. 2005;16:1061–8. doi: 10.1681/ASN.2004070593. [DOI] [PubMed] [Google Scholar]
- 6.D’Agati VD. The spectrum of focal segmental glomerulosclerosis: New insights. Curr Opin Nephrol Hypertens. 2008;17:271–81. doi: 10.1097/MNH.0b013e3282f94a96. [DOI] [PubMed] [Google Scholar]
- 7.Singh RG, Agarwal DK, Usha, Johny KV, Jaiprakash Ten years follow-up of idiopathic focal and segmental glomerulosclerosis. Saudi J Kidney Dis Transpl. 1994;5:354–8. [PubMed] [Google Scholar]
- 8.Taheri D, Chehrei A, Samanianpour P, Hassanzadeh A, Sadrarhami S, Seyrafian S. Correlation of kidney biopsy findings and clinical manifestations of primary focal and segmental glomerulosclerosis. Saudi J Kidney Dis Transpl. 2009;20:417–23. [PubMed] [Google Scholar]
- 9.Miyata J, Takebayashi S, Taguchi T, Naito S, Harada T. Evaluation and correlation of clinical and histological features of focal segmental glomerulosclerosis. Nephron. 1986;44:115–20. doi: 10.1159/000183978. [DOI] [PubMed] [Google Scholar]
- 10.Pei Y, Cattran D, Delmore T, Katz A, Lang A, Rance P. Evidence suggesting under-treatment in adults with idiopathic focal segmental glomerulosclerosis. Regional Glomerulonephritis Registry Study. Am J Med. 1987;82:938–44. doi: 10.1016/0002-9343(87)90155-0. [DOI] [PubMed] [Google Scholar]
- 11.Frassinetti Castelo Branco Camurça Fernandes P, Bezerra Da Silva G, Jr, De Sousa Barros FA, Costa Oliveira CM, Kubrusly M, Evangelista JB., Jr Treatment of steroid-resistant nephrotic syndrome with cyclosporine: Study of 17 cases and a literature review. J Nephrol. 2005;18:711–20. [PubMed] [Google Scholar]
- 12.Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, et al. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int. 1999;56:2220–6. doi: 10.1046/j.1523-1755.1999.00778.x. [DOI] [PubMed] [Google Scholar]
- 13.Duncan N, Dhaygude A, Owen J, Cairns TD, Griffith M, McLean AG, et al. Treatment of focal and segmental glomerulosclerosis in adults with tacrolimus monotherapy. Nephrol Dial Transplant. 2004;19:3062–7. doi: 10.1093/ndt/gfh536. [DOI] [PubMed] [Google Scholar]
- 14.Briggs WA, Choi MJ, Scheel PJ., Jr Successful mycophenolate mofetil treatment of glomerular disease. Am J Kidney Dis. 1998;31:213–7. doi: 10.1053/ajkd.1998.v31.pm9469489. [DOI] [PubMed] [Google Scholar]
- 15.Lanewala A, Mubarak M, Kazi JI, Akhter F, Sher A, Fayyaz A, et al. A clinicopathologic study of primary focal segmental glomerulosclerosis in children. Saudi J Kidney Dis Transpl. 2012;23:513–20. [PubMed] [Google Scholar]
- 16.Deegens JK, Steenbergen EJ, Borm GF, Wetzels JF. Pathological variants of focal segmental glomerulosclerosis in an adult Dutch population – Epidemiology and outcome. Nephrol Dial Transplant. 2008;23:186–92. doi: 10.1093/ndt/gfm523. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz MM, Lewis EJ. Focal segmental glomerular sclerosis: The cellular lesion. Kidney Int. 1985;28:968–74. doi: 10.1038/ki.1985.225. [DOI] [PubMed] [Google Scholar]
- 18.Das P, Sharma A, Gupta R, Agarwal SK, Bagga A, Dinda AK. Histomorphological classification of focal segmental glomerulosclerosis: A critical evaluation of clinical, histologic and morphometric features. Saudi J Kidney Dis Transpl. 2012;23:1008–16. doi: 10.4103/1319-2442.100883. [DOI] [PubMed] [Google Scholar]
- 19.Shakeel S, Mubarak M, Kazi JI. Frequency and clinicopathological correlations of histopathological variants of pediatric idiopathic focal segmental glomerulosclerosis. Indian J Nephrol. 2014;24:148–53. doi: 10.4103/0971-4065.132003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rydel JJ, Korbet SM, Borok RZ, Schwartz MM. Focal segmental glomerular sclerosis in adults: Presentation, course, and response to treatment. Am J Kidney Dis. 1995;25:534–42. doi: 10.1016/0272-6386(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 21.Wehrmann M, Bohle A, Held H, Schumm G, Kendziorra H, Pressler H. Long-term prognosis of focal sclerosing glomerulonephritis. An analysis of 250 cases with particular regard to tubulointerstitial changes. Clin Nephrol. 1990;33:115–22. [PubMed] [Google Scholar]
- 22.Cameron JS. Focal segmental glomerulosclerosis in adults. Nephrol Dial Transplant. 2003;18(Suppl 6):vi45–51. doi: 10.1093/ndt/gfg1058. [DOI] [PubMed] [Google Scholar]