

Abstract
Post-tetanic potentiation (PTP) is a widespread form of short-term synaptic plasticity in which a period of elevated presynaptic activation leads to synaptic enhancement that lasts tens of seconds to minutes. A leading hypothesis for the mechanism of PTP is that tetanic stimulation elevates presynaptic calcium that in turn activates calcium-dependent protein kinase C (PKC) isoforms to phosphorylate targets and enhance neurotransmitter release. Previous pharmacological studies have implicated this mechanism in PTP at hippocampal synapses, but the results are controversial. Here we combine genetic and pharmacological approaches to determine the role of classic PKC isoforms in PTP. We find that PTP is unchanged in PKC triple knock-out (TKO) mice in which all calcium-dependent PKC isoforms have been eliminated (PKCα, PKCβ, and PKCγ). We confirm previous studies and find that in wild-type mice 10 μm of the PKC inhibitor GF109203 eliminates PTP and the PKC activator PDBu enhances neurotransmitter release and occludes PTP. However, we find that the same concentrations of GF109203 and PDBu have similar effects in TKO animals. We also show that 2 μm GF109203 does not abolish PTP even though it inhibits the PDBu-dependent phosphorylation of PKC substrates. We conclude that at the CA3 to CA1 synapse Ca2+-dependent PKC isoforms do not serve as calcium sensors to mediate PTP.
SIGNIFICANCE STATEMENT Neurons dynamically regulate neurotransmitter release through many processes known collectively as synaptic plasticity. Post-tetanic potentiation (PTP) is a widespread form of synaptic plasticity that lasts for tens of seconds that may have important computational roles and contribute to short-term memory. According to a leading mechanism, presynaptic calcium activates protein kinase C (PKC) to increase neurotransmitter release. Pharmacological studies have also implicated this mechanism at hippocampal CA3 to CA1 synapses, but there are concerns about the specificity of PKC activators and inhibitors. We therefore used a molecular genetic approach and found that PTP was unaffected when all calcium-dependent PKC isozymes were eliminated. We conclude that PKC isozymes are not the calcium sensors that mediate PTP at the CA3 to CA1 synapse.
Keywords: post-tetanic potentiation, protein kinase C, synaptic plasticity
Introduction
Post-tetanic potentiation (PTP) is a widespread form of short-term synaptic plasticity in which high-frequency presynaptic activity leads to synaptic enhancement that lasts for tens of seconds to minutes (Zucker and Regehr, 2002; Fioravante and Regehr, 2011; Regehr, 2012). It is thought that PTP is a consequence of calcium accumulation in presynaptic boutons during high-frequency activity that activates a calcium-dependent target to enhance neurotransmitter release (Xu et al., 2007; Regehr, 2012). Even though PTP has been studied for many decades, the specific mechanisms underlying PTP are poorly understood at most synapses. This has limited the ability to manipulate PTP in vivo to determine its functional and behavioral significance.
Pharmacological studies have implicated numerous calcium-sensitive proteins in PTP (Chapman et al., 1995; Rosahl et al., 1995; Wang and Maler, 1998; Alle et al., 2001; Brager et al., 2003; Fiumara et al., 2007; Lee et al., 2008), but most recent attention has been focused on the role of protein kinase C (PKC) in PTP. PKC inhibitors suppress PTP at the hippocampal CA3 to CA1 synapse (Brager et al., 2003), mossy fiber to hilar interneurons (Alle et al., 2001; Lee et al., 2007), the cerebellar granule cell to Purkinje cell synapse (Beierlein et al., 2007; Fioravante et al., 2012), and at the calyx of Held synapse (Korogod et al., 2007; Xue and Wu, 2010; Genc et al., 2014). Furthermore, the PKC activator PDBu enhances synaptic transmission at many synapses and occludes PTP (Malenka et al., 1986; Gustafsson et al., 1988; Searl and Silinsky, 1998; Brager et al., 2002, 2003; Rhee et al., 2002; Korogod et al., 2007; Wierda et al., 2007). However, the specificity of PKC inhibitors and activators have been called into question, because widely used PKC inhibitors block several other kinases with varying potency (Toullec et al., 1991; Beltman et al., 1996; Alessi, 1997; Hers et al., 1999; Roberts et al., 2005; Lee et al., 2008), and the PKC activator PDBu binds to the DAG-binding domain of PKC (Fig. 1A), Munc13 (Newton, 1995; Betz et al., 1998), chimaerins (Ahmed et al., 1990; Caloca et al., 2001), PKD (Valverde et al., 1994), RasGRPs (Ebinu et al., 1998; Lorenzo et al., 2000), and DAG kinase γ (Kazanietz, 2000; Shindo et al., 2001; Brose and Rosenmund, 2002). Moreover, inactive PKC inhibitor analogues suppressed PTP at the calyx of Held synapse (Lee et al., 2008) and PKC inhibitors cannot prevent the synaptic enhancement-induced by PDBu (Searl and Silinsky, 1998; Rhee et al., 2002; but see Wierda et al., 2007).
Figure 1.

Effects of PKC inhibitor on post-tetanic potentiation (PTP) at Hippocampal CA3→CA1 synapses. A, Domain structures of Ca2+-dependent PKC isoforms (classical PKCs). B, CA3 to CA1 synapses were stimulated at 0.4 Hz and synaptic responses were measured with an extracellular electrode. As indicated, different protocols were used to induce PTP with tetanic stimulation time t = 0. Left, Average normalized field EPSPs (fEPSPs). Right, representative traces of the averages of baseline responses (black) and the first three responses after tetanic stimulation (gray). These five protocols were used in the same slice, and three to five trials per protocol were recorded for the average (n = 12, 4; 12 slices from 4 animals, denoted similarly in other figures). Scale bar: 0.2 mV, 10 ms. C, Similar experiments were conducted as in A using the tetanic protocol 50 stim at 50 Hz to induce PTP, but with paired stimulation (Δt = 50 ms) to monitor the paired-pulse ratio (PPR). Inset, Scaled representative traces of the averages of baseline responses (black) and the first three responses after tetanic stimulation (gray; n = 47, 16). D, Similar experiments as in B, but with whole-cell voltage-clamp recordings from CA1 neurons (n = 29, 10). E, PTP induced at CA3→CA1 synapses was monitored with or without the presence of broad spectrum PKC inhibitor GF (2 or 10 μm; 1 h preincubation). Left, Average normalized fEPSPs. Right, representative traces of the averages of baseline responses (black) and the first three responses after tetanic stimulation (gray). (Control: n = 42, 15; 2 μm GF: n = 10, 2; 10 μm GF: n = 8, 2). Scale bar: 0.2 mV, 10 ms. F, Similar recordings as in E, but PTP was induced with 400 stimuli at 100 Hz (Control: n = 17, 7; 2 μm GF: n = 10, 2; 10 μm GF: n = 8, 2). Scale bar: 0.2 mV, 10 ms.
Limitations of pharmacological studies have been overcome by using genetic approaches to assess the involvement of calcium-sensitive PKCs in PTP. The Ca2+-binding PKC isoforms (also termed classical PKC isoforms) composed of PKCα, PKCβ, and PKCγ are widely expressed with differential expression patterns (Brandt et al., 1987; McGinty et al., 1991; Steinberg, 2008). PKCα and PKCβ both mediate PTP at the granule cell to Purkinje cell synapse (Fioravante et al., 2012). Calcium-sensitive PKC isoforms also mediate a component of PTP at the calyx of Held synapse (Fioravante et al., 2011) and this is thought to involve the phosphorylation of Munc18-1 (Genc et al., 2014).
The findings that PKC mediates PTP at the calyx of Held and cerebellar synapses, pharmacological studies implicate PKC in PTP at many other synapses, and the widespread expression of calcium-dependent PKC isoforms, suggested that PKC might be the calcium sensor for PTP at most synapses. Here we use PKC knock-out mice to test the hypothesis that calcium-dependent PKC isoforms mediate PTP at the hippocampal CA3 to CA1 synapse. This is an important model synapse where pharmacological studies have implicated PKC in PTP (Brager et al., 2003; Wierda et al., 2007). We find that PTP is unaffected in PKCαβγ triple knock-out (TKO) animals. Moreover, PKC activators occlude PTP and high concentrations of PKC inhibitors suppress PTP equivalently in wild-type and TKO animals. These findings indicate that PKC isoforms are not the calcium sensors that mediate PTP at the CA3 to CA1 synapse.
Materials and Methods
Animals.
All animal experiments were completed in accordance with guidelines set by the Harvard Medical Area Standing Committee on Animals. PKCαβγ TKO mice were obtained through breeding of PKCα, PKCβ, and PKCγ knock-out animals, whereas wild-type mice were derived from the same lines with the same genetic background (mixed BL6 and 129S2). PKCα and PKCβ KO mice were generated by Leitges et al. (1996, 2002) and were obtained from PKC Research Consult (contact M. Leitges, michael.leitges@biotek.uio.no). PKCγ KO mice (Abeliovich et al., 1993) were obtained from The Jackson Laboratory.
Preparation of brain slices.
Mice of either sex aged 18–25 d were anesthetized with isoflurane and decapitated. Acute transverse slices (320–350 μm thick) containing the hippocampus were cut in ice-cold solution consisting of the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.1 CaCl2, 6 MgCl2, 25 glucose or choline-based solution: 110 choline-Cl, 7 MgSO4, 2.5 KCl, 1.2 NaH2PO4, 0.5 CaCl2, 25 glucose, 11.6 Na-ascorbate, 2.4 Na-pyruvate, and 25 NaHCO3. Slices were then incubated at 32°C for 20 min in a bicarbonate-buffered solution composed of the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, and 25 glucose. The slices were kept in the chamber at room temperature until recording or protein extraction.
Electrophysiology.
Recordings were conducted at 30°–32°C. The hippocampal CA3 region was cut from the CA1 region with a scalpel blade to prevent recurrent excitation. The external solution was the same solution used for incubating slices but supplemented with the following drugs (in μm): 20 bicuculline, 2 CGP, 2 CPP, and 1 AM-251. A stimulation electrode filled with ACSF was placed at least 500 μm away from the recording site to stimulate the Schaffer collateral fibers. The recording pipettes (0.3–2 MΩ) were filled with ACSF and placed in the stratum radiatum in the CA1 dendritic area. For all recordings an input–output curve with the stimulation range 10–80 μA was first analyzed, and the stimulation intensity that elicited approximately one-half of the maximum response without evoking population spikes was chosen. Whole-cell voltage-clamp recordings (holding potential −65 mV) from CA1 neurons were obtained using 3–5 MΩ pipettes. The internal solution contained the following (in mm): 115 Cs-methanesulfonate, 25 TEA, 10 HEPES, 0.5 EGTA, 5 QX-314-Cl, 4 NaCl, 4 MgATP, 0.4 Na3GTP, and 10 Na2phosphocreatine, 315 mOsm, pH 7.3. Custom-written programs in IgorPro (WaveMetrics) were used to analyze data. Pairwise comparisons were performed using Student's t tests. Data are expressed as mean ± SEM. Blind experiments were difficult because TKO mice are smaller than WT littermates. Consequently, most experiments were not done blind. However, in a subset of experiments one investigator cut slices and a second investigator did experiments blind to phenotype. For these studies, as for the rest of our experiments, PTP evoked by 50 stimuli at 50 Hz was similar in WT (1.55 ± 0.04-fold increase, n = 7) and TKO animals (1.51 ± 0.04, n = 8) and phorbol-ester-mediated enhancement was similar in WT (2.39 ± 0.37, n = 4) and TKO animals (1.82 ± 0.45, n = 4).
Western blotting.
Brain slices were prepared as for the electrophysiological recordings. Transverse slices excluding the cerebellum were incubated in ACSF in the presence or absence of 1 μm PDBu for 20 min at room temperature. To test the blocking efficacy of the PKC inhibitor GF, slices were preincubated in the presence of GF for 1 h before being transferred to solutions containing 1 μm PDBu concurrent with the inhibitors. Slices were then harvested in ice-cold lysis buffer containing 150 mm NaCl, 25 mm HEPES, 4 mm EGTA, phosphatase inhibitor (Roche, 04906845001), and protease inhibitor (Sigma-Aldrich, P8340). Total protein concentration was determined by a BCA assay (Pierce), and 30 μg of protein in Laemmli sample buffer was loaded onto 10% polyacrylamide gels (Bio-Rad). After SDS-PAGE, gels were transferred onto nitrocellulose membranes and blocked in 5% nonfat milk (Cell Signaling Technology) in TBS-Tween 20. Membranes were incubated overnight at 4°C with primary antibodies, followed by incubation with HRP-conjugated secondary antibodies (Cell Signaling Technology) for 2 h at room temperature. Blots were then washed, incubated with chemiluminescence substrate (Bio-Rad), and imaged with Gel Doc (Bio-Rad). Non-saturated images were used and analyzed with the Gel-Doc built-in software Image Lab. To quantify Western blot experiments (see Fig. 4), fluorescent secondary antibodies were used and membranes were imaged in Odyssey Classic (LI-COR). Non-saturated images were quantified in ImageJ. The fluorescent intensity of entire lane (range between ∼60 and 250 kDa) was first normalized to β-actin fluorescent intensity and then normalized to the averaged wild-type control values. Data are expressed as mean ± SEM, and statistical analysis were conducted using paired t tests. The following antibodies were used: rabbit anti-PKCαβγ (1:3000; Abcam, ab179522; immunogens for generating antibodies were composed of PKCα + PKCβ + PKCγ), mouse β-Actin (1:5000; Sigma-Aldrich, A1978), rabbit phospho-PKC substrates (1:1000; Cell Signaling Technology, 6967), HRP-conjugated anti-rabbit IgG (1:3000; Cell Signaling Technology, 7074S), HRP-conjugated anti-mouse IgG (1:3000; Cell Signaling Technology, 7076S), and fluorescent antibodies (1:10,000; IRDye 800CW Donkey anti-Rabbit IgG, LI-COR 926-32213 and IRDye 680RD Donkey anti-Mouse IgG, LI-COR 926-68072).
Figure 4.
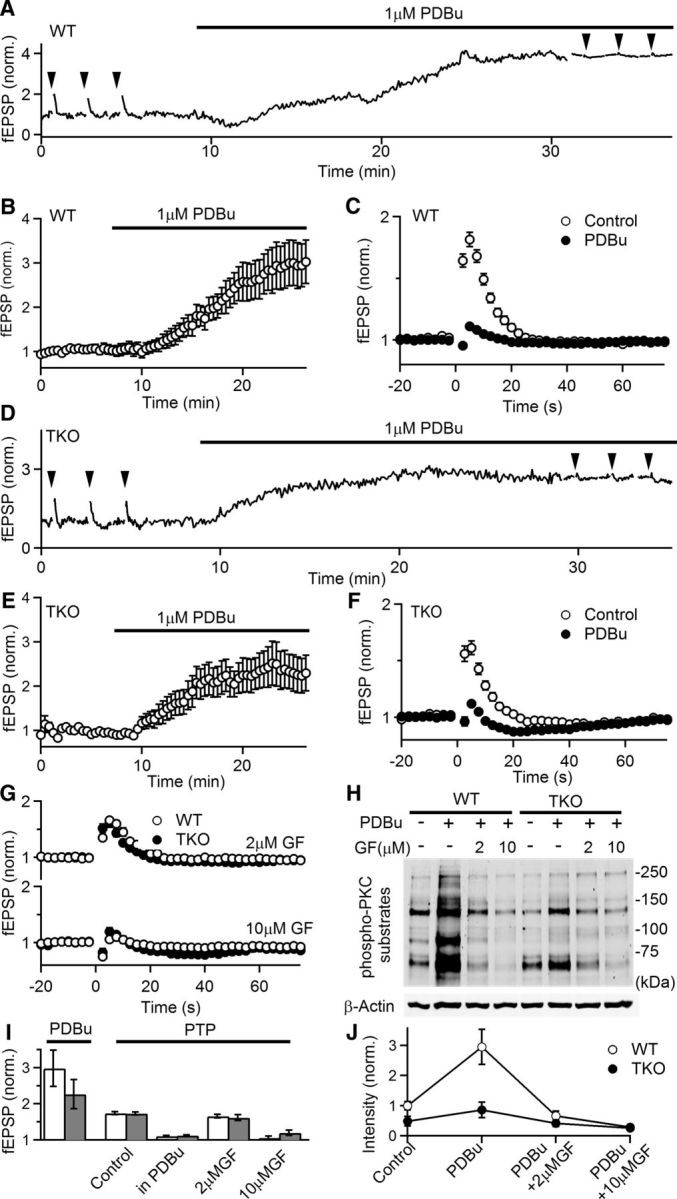
The phorbol ester PDBu enhances synaptic responses and occludes PTP in both WT and TKO animals. A, Representative experiment showing normalized fEPSPs as a function of time in a WT slice. fEPSPs were recorded in 1.5 mm external Ca2+ and PTP (50 stimuli at 50 Hz induction) was monitored before and after the application of the PKC activator PDBu (1 μm). Induction of PTP is indicated by arrowheads. B, Average normalized fEPSPs during PDBu application in WT animals (n = 17, 7). fEPSPs were recorded every 5 s to monitor the effects of PDBu but displayed in the plot every 25 s for clarity. C, Comparison of PTP before and after the application of PDBu in WT slices. D–F, Similar to A–C, but in TKO animals (n = 13, 4). G, PTP (50 stimuli at 50 Hz induction) in the presence of the indicated concentrations of the PKC inhibitor GF in WT (open symbols) and TKO (filled symbols) animals. (WT control: n = 42, 15; WT + 2 μm GF: n = 10, 2; WT + 10 μm GF: n = 8, 2; TKO control: n = 29, 11; TKO + 2 μm GF: n = 7, 2; TKO + 10 μm GF: n = 8, 2). H, Protein analysis of phosphorylated levels of PKC substrates in the presence of PKC activator PDBu (1 μm) with and without the preincubation in the indicated concentrations of GF for 1 h. I, Summary of physiology experiments. Empty bars: WT; filled bars: TKO. J, Quantification of phosphorylated levels of PKC substrates in H. Intensity is normalized to WT control conditions (n = 5 for both genotypes).
Immunohistochemistry.
Age-matched animals were perfused and fixed with cold paraformaldehyde and stored at 4°C overnight. Brains were transferred to phosphate buffer (Sigma-Aldrich) and stored at 4°C until further processing. Transverse hippocampal slices (50 μm thick) were cut with the vibratome (LEICA VT1000S) and then incubated in blocking solution (0.25% Triton X-100 and 10% normal goat serum in PBS; PBST) for 1 h at room temperature. Slices were then incubated with primary antibodies in PBST overnight at 4°C, followed by incubation with secondary antibodies in PBST for 2 h at room temperature. Slices were mounted with anti-fade medium (Invitrogen) and allowed to dry for at least 24 h before imaging. The following antibodies were used: anti-VGluT1 guinea pig polyclonal (Synaptic Systems, 135304), anti-PKCα rabbit monoclonal (Abcam, 32376), anti-PKCβI rabbit polyclonal (Santa Cruz Biotechnology, sc-209), anti-PKCγ rabbit polyclonal (Santa Cruz Biotechnology, sc-211), goat anti-guinea pig rhodamine-conjugated (Life Technologies, A-11074) and goat anti-rabbit FITC-conjugated secondaries (Abcam, 150085). All antibodies were used at a 1:500 dilution.
For parallel comparisons, slices were taken blind to the genotype and immunostained with the same solutions at the same time. Images from different genotypes were acquired on the same day with an Olympus FV1000 confocal microscope using a 60 × 1.42 N.A oil lens along with similar parameter settings. Excitation was set at 543 nm for rhodamine (VGluT1) and 488 nm for FITC (PKCs). Images were processed and analyzed with ImageJ using similar brightness and contrast settings.
Results
To determine a consistent and reliable protocol for inducing PTP at hippocampal CA3→CA1 synapses, we varied the number of stimuli in a 50 Hz induction train and measured the resulting field EPSPs (fEPSPs; Fig. 1B). PTP decayed exponentially with a time constant τ and the magnitude of PTP increased with stimulation number (Fig. 1B; 5 stim: 1.15 ± 0.02-fold, τ = 5.7 ± 0.7 s; 10 stim: 1.33 ± 0.02-fold, τ = 6.2 ± 0.5 s; 25 stim: 1.60 ± 0.04-fold, τ = 8.0 ± 0.8 s; 50 stim: 1.73 ± 0.06-fold, τ = 8.7 ± 0.5 s). We also assessed the efficacy of a 400 stimuli 100 Hz stimulus train, which has been used previously to induce PTP at hippocampal slice cultures (Brager et al., 2003), and found that it produced similar PTP than that induced by 50 stimuli at 50 Hz (Fig. 1B; 1.55 ± 0.15-fold τ = 8.7 ± 0.5 s). We found that prolonged stimulation at 100 Hz did not reliably activate CA3 axons, and that may have limited the magnitude of PTP. We also found that following prolonged tetanic stimulation (4 s at 100 Hz) an initial period of enhancement was followed by a somewhat longer lasting depression. It is often the case that multiple forms of short-term plasticity coexist at synapses (Zucker and Regehr, 2002), and these findings suggest that a slow form of depression complicates the interpretation of PTP following 400 stimuli at 100 Hz. Based on these findings we used an induction protocol of 50 stimuli at 50 Hz for most studies of PTP.
Previous studies have shown that increases in release probability (p) contribute to PTP at many synapses and that such increases in p are accompanied by a decrease in the magnitude of facilitation as measured by the paired-pulse ratio (PPR) of two closely spaced stimuli (Zucker and Regehr, 2002; Brager et al., 2003; Habets and Borst, 2005; Korogod et al., 2005). We therefore used test pulses of two stimuli (Δt = 50 ms) to determine whether the induction of PTP was accompanied by a decrease in PPR at CA3→CA1 synapses. We found that when PTP was induced by 50 stimuli at 50 Hz, PPR measured with fEPSPs decayed from 1.38 ± 0.02 to 1.11 ± 0.02 (n = 31, p < 0.001, paired t test). A plot of the PPR normalized to the pre-tetanus value revealed that PPR decreased to 77 ± 1% and recovered exponentially with a time constant similar to the time constant of synaptic enhancement (Fig. 1C; τ for PPR: 10.7 ± 2.3 s, n = 39 vs τ for fEPSP: 8.6 ± 0.4 s, n = 46, p = 0.3). The extent of the reduction of the PPR ratio and the duration of this reduction were dependent on the number of stimuli in a 50 Hz induction train (5 stim: 96%, τ = 3.7 s; 10 stim: 93%, τ = 4.3 s; 25 stim: 81%, τ = 9.4 s; 50 stim: 77%, τ = 9.8 s). We also conducted whole-cell recordings to measure EPSCs in voltage-clamp and used 50 stimuli at 50 Hz to induce PTP. We found that the magnitude and decay time constant of PTP were similar to those measured with fEPSPs (Fig. 1D; 1.65 ± 0.05-fold, τ = 8.3 ± 1.7 s, n = 29). For the remainder of the paper we used extracellular methods and fEPSPs to quantify PTP because such measurements are more stable than whole-cell methods and yield PTP with the same amplitude and time course.
It was previously found that PKC inhibitors suppress PTP at CA3 to CA1 synapses in slice cultures (Brager et al., 2003). To determine whether or not PKC also regulates PTP at CA3 to CA1 synapses in acute slices, we measured PTP in the presence of the PKC inhibitor, GF 109203X (GF; also known as bisindolylmaleimide I or Gö 6850). We found that as in slice cultures, 10 μm GF eliminated PTP induced by 50 stimuli at 50 Hz and depression became apparent (Fig. 1E; control: 1.75 ± 0.05-fold vs 10 μm GF: 1.09 ± 0.05-fold; p < 0.001). We also found that 10 μm GF eliminated PTP induced by 400 stimuli at 100 Hz (Fig. 1F; control: 1.51 ± 0.07-fold vs 10 μm GF: 0.79 ± 0.13-fold; p < 0.001). However, the high concentration of GF used in these studies was a cause for concern, because the concentration of 10 μm GF is much higher than the IC50 of 0.02 μm required to block Ca2+-dependent PKC isoforms (GF IC50 for PKCα, PKCβ, and PKCγ: 0.008, 0.018, and 0.016 μm, respectively; Toullec et al., 1991; Alessi, 1997). Moreover, previous studies showed that PTP is abolished by lower concentrations of GF at several synapses. Two micromoles of GF can block PTP at the granule cell to Purkinje cell synapses (Fioravante et al., 2012) and at the calyx of Held synapses (although PTP was also blocked by 2 μm of GF's inactive analog; Lee et al., 2008), and 1 μm GF reduced PTP at granule cell to interneurons synapses in the hippocampus (Alle et al., 2001; Galván et al., 2010). We therefore tested a lower concentration of GF (2 μm) in PTP at CA3 to CA1 synapses. We found that 2 μm GF did not significantly reduce PTP induced by 50 stimuli at 50 Hz (Fig. 1E; 2 μm GF: 1.67 ± 0.05-fold; p = 0.9) and did not significantly reduce the magnitude of PTP induced by 100 stimuli at 400 Hz (Fig. 1F; 2 μm GF: 1.71 ± 0.13-fold; p = 0.45). These findings suggest that the blockade of PTP by 10 μm GF could be due to off-target effects (Hers et al., 1999; Barry and Kazanietz, 2001; Roberts et al., 2005). This adds to the concern that at CA3 to CA1 synapses, the need for very high concentrations of GF to block PTP may be because PKC is not involved in the induction of PTP at this synapse.
As a result of the described limitations of pharmacological methods, we sought to use PKC knock-out animals to test whether or not PKC mediates PTP at CA3 to CA1 synapses. The hypothesis that a tetanic stimulus train increases the Ca2+ concentration in presynaptic boutons, which in turn activates the downstream signaling to regulate short-term synaptic plasticity, is well established (Zucker and Regehr, 2002; Fioravante and Regehr, 2011; Regehr, 2012). Therefore, we used knock-out animals, in which all three Ca2+-dependent PKC isoforms were removed (PKCαβγ TKO animals; Fig. 2). Protein analysis of brain lysate confirmed that these Ca2+-dependent PKC isoforms were strongly expressed in WT but absent in TKO mice (Fig. 2A). It was previously shown that CA3 pyramidal cells express PKCα, PKCβ, and PKCγ (Brandt et al., 1987; McGinty et al., 1991). To determine the expression of different PKC isoforms in the hippocampal CA1 regions, we used immunohistochemistry and co-stained for both the PKC isoforms and the presynaptic marker vesicular glutamate transporter 1 (VGluT1; Fig. 2B). Antibodies to all three isoforms resulted in strong labeling in the CA1 region of wild-type animals that was absent in PKCαβγ TKO animals. To rule out the possibility that the antibodies have cross-reactivity against different isoforms that could confound results, we also used PKC double knock-out (DKO) animals to determine the specificity of each antibody (Fig. 2B). For all three isoforms, we observed a similar pattern in DKO and WT animals. The pattern of labeling suggests that all isoforms are present in the CA1 region, with labeling apparent both in the postsynaptic CA1 pyramidal cells and in stratum radiatum where CA3 synaptic boutons can be visualized with VGluT1 labeling.
Figure 2.

Expression of PKC isoforms in the hippocampal CA1 region. A, Western blot of classical PKC isoforms from WT and PKCαβγ TKO brain lysates. B, Confocal images of Ca2+-dependent PKC isoforms α, β, and γ in the hippocampal CA1 region from the indicated genotypes. SP, Stratum pyramidale; SR, stratum radiatum. Scale bar, 25 μm.
Because PTP often reflects an increase in release probability (Zucker and Regehr, 2002), we used two approaches to determine whether the initial probability of release was altered in PKCαβγ TKO animals. We first evoked synaptic responses with a range of stimulus intensities and compared the amplitudes of the presynaptic volley and the fEPSP. The presynaptic volley amplitude is proportional to the number of activated CA3 axons, so the ratio of the fEPSP amplitude and the presynaptic volley provides a sensitive measure of the initial probability of release. We found that the slope of the fEPSP versus volley amplitude was unchanged in PKCαβγ TKO animals (Fig. 3A,B; WT: 1.05 ± 0.15, n = 17 s; TKO: 1.21 ± 0.35, n = 19; p = 0.7). We also compared the amplitude of paired-pulse facilitation because increases in the initial probability of release are accompanied by a decrease in paired pulse plasticity. There was no significant difference in the PPR in WT and TKO animals (Fig. 3B; PPR: WT: 1.68 ± 0.04, n = 22; TKO: 1.66 ± 0.03, n = 33; p = 0.7). These findings indicate that the initial probability of release was not altered in the absence of Ca2+-dependent PKC isoforms.
Figure 3.
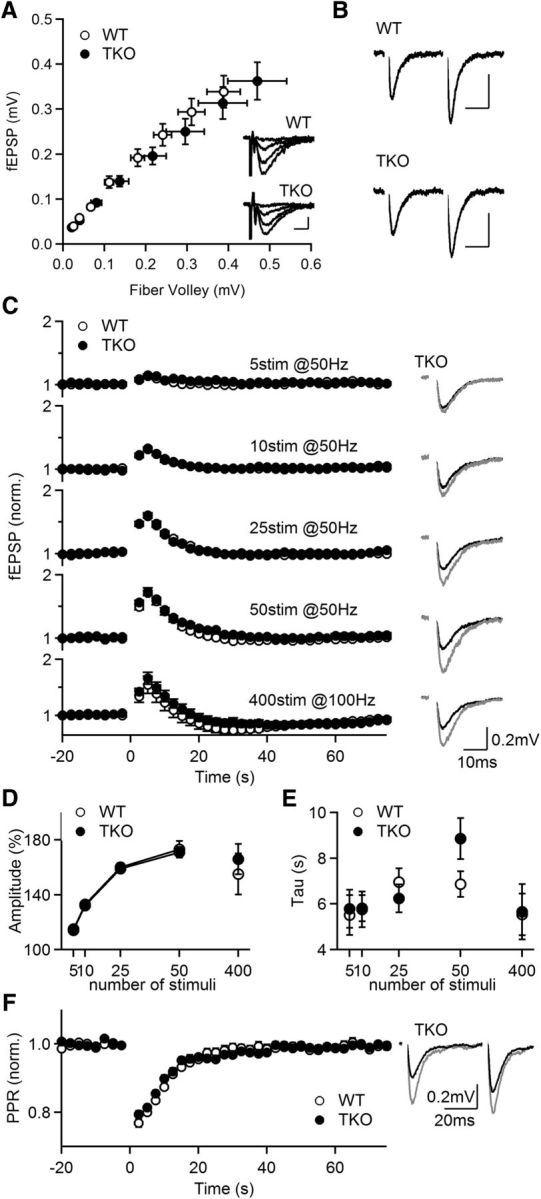
PTP at CA3→CA1 synapses is unaltered in PKCαβγ TKO animals. A, Averaged fEPSP amplitude plotted against fiber volley amplitude, for stimuli between 10–80 μA. Inset, Example extracellular recordings evoked by a range of stimulus intensities in WT and PKC TKO slices. Stimulus intensities from top: 10, 30, 50, and 70 μA, respectively. Scale bar: 0.2 mV, 5 ms. B, Representative traces of paired stimulation (Δt = 50 ms) evoked in WT and TKO slices. WT PPR: 1.68 ± 0.04, n = 22; TKO PPR: 1.66 ± 0.03, n = 34. p = 0.7. Scale bar: 0.2 mV, 20 ms. C, Normalized fEPSPs as a function of time for the indicated induction protocols in WT (open symbols; n = 12, 4) and TKO (filled symbols; n = 9, 3). Right, representative traces of the averages of baseline responses (black) and the first three responses after tetanic stimulation (gray). D, Amplitude of PTP induced by a train consisting of 5–50 stimuli at 50 Hz and for 400 stimuli at 100 Hz. E, Comparison of PTP time course induced using different protocols in WT and TKO slices. F, Similar experiments as in A using the tetanic protocol 50 stimuli at 50 Hz to induce PTP, but with paired stimulation (Δt = 50 ms) to allow for the monitoring of the PPR. Left, Average normalized fEPSP. Right, representative traces of the averages of baseline responses (black) and the first three responses after tetanic stimulation (gray; n = 36, 14).
We found that both the amplitudes and time courses of PTP induced by a 50 Hz train consisting of 5–50 stimuli, or by a 400 stimuli at 100 Hz train were similar in WT and TKO mice (Fig. 3C–E). We also found that in PKCαβγ TKO animals PTP is accompanied by a decrease in PPR to 79% of initial values with recovery time constants comparable to those observed in WT animals (τ: fEPSP: 7.8 ± 0.7 s vs PPR: 8.7 ± 0.7; n = 36, p = 0.2; Fig. 3E). Thus, PTP is indistinguishable in WT and PKCαβγ TKO mice for a range of induction protocols.
The observation that PKC activators such as phorbol esters enhance neurotransmitter release (Malenka et al., 1986; Gustafsson et al., 1988) and occlude the induction of PTP has implicated the involvement of PKC in PTP (Brager et al., 2002, 2003; Korogod et al., 2007). However, this is controversial because phorbol esters activate other proteins in addition to PKC (Ahmed et al., 1990; Valverde et al., 1994; Newton, 1995; Goda et al., 1996; Betz et al., 1998; Ebinu et al., 1998; Hori et al., 1999; Honda et al., 2000; Lorenzo et al., 2000; Caloca et al., 2001; Shindo et al., 2001; Rhee et al., 2002; Rosenmund et al., 2002; Wierda et al., 2007; for review, see Kazanietz, 2000; Brose and Rosenmund, 2002). We therefore compared the ability of the phorbol ester PDBu to increase neurotransmitter release and occlude PTP in WT and PKCαβγ TKO mice. These experiments were in lower external Ca2+ (1.5 mm) to reduce the initial release probability, because PDBu might increase the probability of release to maximal levels making the subsequent reduction of PTP in the presence of PDBu difficult to interpret. In these experiments PTP was examined in control conditions with a 50 Hz 50 stimuli induction train, PDBu was washed in, and then the effects of tetanic stimulation were reexamined in the presence of PDBu (Fig. 4A). Consistent with previous studies, in WT animals PDBu significantly increased synaptic strength (2.9 ± 0.4-fold, n = 17, p < 0.001; Fig. 4A,B) and reduced PPR (1.51 ± 0.03–1.03 ± 0.02; n = 17, p < 0.001), which is consistent with a presynaptic increase in the probability of release. PDBu also decreased the magnitude of PTP and the reduced PPR following PTP induction was occluded (Fig. 4A,C; PTP magnitude: control: 1.8 ± 0.06-fold vs PDBu: 1.1 ± 0.02; n = 17, p < 0.001; normalized PPR after PTP induction: control: 0.79 ± 0.02 vs PDBu: 0.93 ± 0.02).
We conducted similar experiments in PKCαβγ TKO animals and comparable results were observed. In TKO animals PDBu significantly enhanced synaptic strength 2.3 ± 0.4-fold (p < 0.01) and occluded PTP (Fig. 4D–F; PTP magnitude: control: 1.63 ± 0.07-fold vs PDBu: 1.1 ± 0.02-fold; n = 13, p < 0.001). Concurrently, PPR in TKO slices decreased in the presence of PDBu (control: 1.62 ± 0.05 vs PDBu 1.15 ± 0.05, n = 13, p < 0.001), and the decrease in PPR followed by PTP induction was occluded by PDBu (normalized PPR after PTP induction: reduced to 80 ± 1% in control conditions vs a reduction to 94 ± 1% in the presence of PDBu).
The observations that in PKCαβγ TKO mice PTP is unaltered and is occluded by PDBu indicate that at the CA3 to CA1 synapse PTP is mediated by mechanisms that do not involve calcium-dependent PKC isoforms. This does not necessarily mean that PTP in WT animals does not require these calcium-dependent PKC isoforms, as PTP in TKO animals could be mediated by a compensatory mechanism. We previously found that at the granule cell parallel fiber to Purkinje cell (PF to PC) synapse, PKCα and PKCβ mediate PTP in WT animals, but in PKCαβ DKO mice PTP is mediated by a compensatory PKC-independent mechanism (Fioravante et al., 2012). These experiments demonstrate a strategy to determine whether PTP is mediated by a PKC-independent compensatory mechanism. If this is the case, we expected the PKC antagonist GF to only block PTP in WT but not in TKO animals. However, we found that similar to the effects of GF in WT slices, in TKO mice 2 μm GF had only minor effects on PTP and only 10 μm GF eliminated PTP (Fig. 4G; 2 μm GF: 1.62 ± 0.08-fold; 10 μm GF: 1.2 ± 0.1-fold). These findings indicate clear differences in the properties of PTP at the CA3 to CA1 synapse versus the PF to PC synapse, where a compensatory mechanism mediates PTP that cannot be suppressed by GF in the absence of classical PKCs.
The differential effects of 2 and 10 μm GF on PTP prompted us to examine the ability of different GF concentrations to inhibit PKC activity in brain slices. In these experiments we probed the phosphorylation levels of PKC substrates with anti-phospho PKC substrate antibodies (Fig. 4H,J). In wild-type animals PDBu increased the phosphorylation of PKC substrates 2.95 ± 0.58-fold (Fig. 4H,J; lane 1 vs lane 2; n = 5, p = 0.01). GF (2 and 10 μm) inhibited the phosphorylation of PKC substrates induced by PDBu. In TKO animals the phosphorylation of PKC substrates was reduced compared with wild-type animals, indicating that classic PKC isoforms help to maintain basal phosphorylation levels (Fig. 4H,J, lane 1 vs lane 5; TKO control: 0.48 ± 0.16-fold; n = 5, p = 0.04). In slices from TKO animals, PDBu also increased the extent of phosphorylation of PKC substrates (Fig. 4H,J, lane 5 vs lane 6), although the phosphorylation levels were only elevated to levels that were comparable to those observed in wild-type animals under control conditions. Preincubation with 2 μm GF also blocked PDBu-enhanced phosphorylation of PKC substrates in TKO tissues (Fig. 4H,J, lane 6 vs lane 7; PDBu: 0.86 ± 0.25-fold vs GF+PDBu: 0.42 ± 0.11-fold; n = 5, p = 0.046). Thus, in both WT and TKO animals, PDBu increased the phosphorylation of PKC substrates and this enhancement was largely prevented by 2 μm GF. These findings indicate that even though 2 μm GF does not inhibit PTP at CA3 to CA1 synapses, it penetrates slices sufficiently to strongly attenuate the phosphorylation of PKC substrates. This suggests that GF likely inhibits PTP through off-target effects (see Discussion).
Discussion
Our main finding is that at the hippocampal CA3 to CA1 synapse PTP does not require the classical PKC isoforms PKCα, PKCβ, and PKCγ. Moreover, we find that activating or inhibiting PKC has the same effects in wild-type and PKCαβγ triple knock-out mice. We conclude that a leading hypothesis for the mechanism of PTP, namely that calcium activation of classical PKCs leads to PTP, does not account for PTP at CA3 to CA1 synapse.
Genetic removal of PKCα, PKCβ, and PKCγ did not affect PTP at the hippocampal CA3 to CA1 synapses. We tested several tetanic protocols to induce PTP, ranging from 5 stimuli to 50 stimuli at 50 Hz and 400 stimuli at 100 Hz. We observed that the magnitude of PTP increased with the number of stimuli delivered at 50 Hz but did not become larger when using 400 stimuli at 100 Hz. For this range of induction protocols the magnitude and kinetics of PTP were similar in WT and PKCαβγ TKO animals (Fig. 3).
Our study also provides insight into the roles of non-classical PKC isoforms in PTP at CA3 to CA1 synapses. We confirmed that a high concentration of GF (10 μm) blocked PTP at CA3 to CA1 synapses (Brager et al., 2003; Fig. 1E). This leaves open the possibility that calcium-independent PKC isoforms might be required for PTP. However, the concentration of GF that did not block PTP (2 μm) is still much higher than the IC50s of GF for PKC isoforms- α, β, γ, δ, and ε. (0.008, 0.018, 0.016, 0.21, and 0.13 μm, respectively; Toullec et al., 1991; Alessi, 1997). Moreover, 2 μm GF strongly reduced PDBu-mediated phosphorylation of PKC substrates (Fig. 4H,J). This raises the possibility that disruption of PTP by high concentrations of GF may not involve any PKC isoform and that 10 μm GF eliminates PTP by affecting a mechanism that is independent of PKC. GF is also known to inhibit many kinases such as P70-S6 kinase and MAPKAP kinase with an IC50 <0.1 μm; myosin light chain kinase, phosphorylase kinase, and glycogen synthase kinase with an IC50 of <1 μm; and PKA with an IC50 of 2 μm (Toullec et al., 1991; Alessi, 1997; Hers et al., 1999; Roberts et al., 2005).
The sensitivity of PTP to GF also provides insight into whether there is a compensatory mechanism that mediates PTP in the absence of classical PKCs. At the parallel fiber to Purkinje cell synapse, the observation that 2 μm GF blocks PTP in wild-type animals but not in PKCαβ knock-out animals suggested that a PKC-independent compensatory mechanism mediates PTP in the absence of Ca2+-binding PKC isoforms (Fioravante et al., 2012). If that is the case for CA3 to CA1 synapses, PTP in TKO animals should not be affected by 10 μm GF. However, our finding demonstrated that 10 μm GF completely abolished PTP in PKCαβγ TKO animals (Fig. 4G), as in WT animals. This indicates that PTP is not mediated by a compensatory PKC-independent mechanism at CA3 to CA1 synapses in TKO animals.
The occlusion of PTP by phorbol esters must also be interpreted with caution. We found that in WT animals PDBu increased evoked synaptic responses 2.9-fold and occluded PTP, but it also enhanced synaptic responses 2.3-fold and occluded PTP in PKCαβγ TKO animals (Fig. 4I). This indicates that although calcium-dependent PKCs might contribute partially to PDBu-dependent enhancement, PDBu enhanced transmission at the CA3 to CA1 synapses primarily by acting on targets other than calcium-dependent PKC isoforms. Indeed, PKC inhibitors did not block PDBu-induced augmentation of neurotransmitter release in hippocampal cultures (Rhee et al., 2002; Wierda et al., 2007) or at the rat neuromuscular junction (Searl and Silinsky, 1998). It seems likely that PDBu enhances synaptic transmission by activating one of the many proteins containing the DAG-binding domain that include chimaerin, RasGRPs, PKD1, and Munc13 (Hori et al., 1999; Honda et al., 2000; Rhee et al., 2002; Rosenmund et al., 2002; Wierda et al., 2007). Munc13 is the most promising candidate to mediate enhancement by phorbol esters because of the importance of the Munc13 DAG binding domain in phorbol-mediated synaptic enhancement for cultured hippocampal cells (Betz et al., 1998; Rhee et al., 2002; Rosenmund et al., 2002).
Thus, we have established that the leading mechanism for PTP, that calcium activates calcium-sensitive PKCs to enhance transmission, does not account for PTP at the CA3 to CA1 synapse. This establishes that PTP is mediated by different mechanisms at different synapses. Our findings highlight the importance of combining pharmacological and molecular genetic approaches to clarify the mechanism of PTP at the CA3 to CA1 synapse and elsewhere.
Footnotes
This work was supported by NIH Grants NS032045 to W.G.R, a William Randolph Hearst Fellowship to C.C.W, and a Boehringer Ingelheim Fonds PhD fellowship to C.W. We thank P. Kaeser, S. Jackman, S. Rudolph, J. Turecek, L. Witter, and C. Guo for comments on the paper; the Neurobiology Department and the Neurobiology Imaging Facility for consultation and instrument availability that supported this work, in part by the Neural Imaging Center as part of an NINDS P30 Core Center Grant NS072030; K. McDaniels and E. Ellis for helping with genotyping; and the Kaeser Laboratory in the Neurobiology Department at Harvard Medical School for sharing reagents and technical advice.
The authors declare no competing financial interests.
References
- Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Kozma R, Monfries C, Hall C, Lim HH, Smith P, Lim L. Human brain n-chimaerin cDNA encodes a novel phorbol ester receptor. Biochem J. 1990;272:767–773. doi: 10.1042/bj2720767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR. The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1β (Rsk-2) and p70 S6 kinase. FEBS Lett. 1997;402:121–123. doi: 10.1016/S0014-5793(96)01510-4. [DOI] [PubMed] [Google Scholar]
- Alle H, Jonas P, Geiger JR. PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc Natl Acad Sci U S A. 2001;98:14708–14713. doi: 10.1073/pnas.251610898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry OP, Kazanietz MG. Protein kinase C isozymes, novel phorbol ester receptors and cancer chemotherapy. Curr Pharm Des. 2001;7:1725–1744. doi: 10.2174/1381612013397041. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fioravante D, Regehr WG. Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron. 2007;54:949–959. doi: 10.1016/j.neuron.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltman J, McCormick F, Cook SJ. The selective protein kinase C inhibitor, Ro-31-8220, inhibits mitogen-activated protein kinase phosphatase-1 (MKP-1) expression, induces c-Jun expression, and activates Jun N-terminal kinase. J Biol Chem. 1996;271:27018–27024. doi: 10.1074/jbc.271.43.27018. [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/S0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Brager DH, Capogna M, Thompson SM. Short-term synaptic plasticity, simulation of nerve terminal dynamics, and the effects of protein kinase C activation in rat hippocampus. J Physiol. 2002;541:545–559. doi: 10.1113/jphysiol.2001.015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Cai X, Thompson SM. Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat Neurosci. 2003;6:551–552. doi: 10.1038/nn1067. [DOI] [PubMed] [Google Scholar]
- Brandt SJ, Niedel JE, Bell RM, Young WS., 3rd Distinct patterns of expression of different protein kinase C mRNAs in rat tissues. Cell. 1987;49:57–63. doi: 10.1016/0092-8674(87)90755-0. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- Caloca MJ, Wang H, Delemos A, Wang S, Kazanietz MG. Phorbol esters and related analogs regulate the subcellular localization of beta 2-chimaerin, a non-protein kinase C phorbol ester receptor. J Biol Chem. 2001;276:18303–18312. doi: 10.1074/jbc.M011368200. [DOI] [PubMed] [Google Scholar]
- Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ. The α-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron. 1995;14:591–597. doi: 10.1016/0896-6273(95)90315-1. [DOI] [PubMed] [Google Scholar]
- Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Myoga MH, Leitges M, Regehr WG. Adaptive regulation maintains posttetanic potentiation at cerebellar granule cell synapses in the absence of calcium-dependent PKC. J Neurosci. 2012;32:13004–13009. doi: 10.1523/JNEUROSCI.0683-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumara F, Milanese C, Corradi A, Giovedì S, Leitinger G, Menegon A, Montarolo PG, Benfenati F, Ghirardi M. Phosphorylation of synapsin domain A is required for post-tetanic potentiation. J Cell Sci. 2007;120:3228–3237. doi: 10.1242/jcs.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván EJ, Cosgrove KE, Mauna JC, Card JP, Thiels E, Meriney SD, Barrionuevo G. Critical involvement of postsynaptic protein kinase activation in long-term potentiation at hippocampal mossy fiber synapses on CA3 interneurons. J Neurosci. 2010;30:2844–2855. doi: 10.1523/JNEUROSCI.5269-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc O, Kochubey O, Toonen RF, Verhage M, Schneggenburger R. Munc18-1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. Elife. 2014;3:e01715. doi: 10.7554/eLife.01715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF, Tonegawa S. Phorbol ester effects at hippocampal synapses act independently of the gamma isoform of PKC. Learn Mem. 1996;3:182–187. doi: 10.1101/lm.3.2-3.182. [DOI] [PubMed] [Google Scholar]
- Gustafsson B, Huang YY, Wigström H. Phorbol ester-induced synaptic potentiation differs from long-term potentiation in the guinea pig hippocampus in vitro. Neurosci Lett. 1988;85:77–81. doi: 10.1016/0304-3940(88)90432-6. [DOI] [PubMed] [Google Scholar]
- Habets RLP, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hers I, Tavaré JM, Denton RM. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999;460:433–436. doi: 10.1016/S0014-5793(99)01389-7. [DOI] [PubMed] [Google Scholar]
- Honda I, Kamiya H, Yawo H. Re-evaluation of phorbol ester-induced potentiation of transmitter release from mossy fibre terminals of the mouse hippocampus. J Physiol. 2000;529:763–776. doi: 10.1111/j.1469-7793.2000.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Takai Y, Takahashi T. Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci. 1999;19:7262–7267. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanietz MG. Eyes wide shut: protein kinase C isozymes are not the only receptors for the phorbol ester tumor promoters. Mol Carcinog. 2000;28:5–11. doi: 10.1002/(SICI)1098-2744(200005)28:1. 3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca(2+) sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007;104:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Lee KH, Ho WK, Lee SH. Target cell-specific involvement of presynaptic mitochondria in post-tetanic potentiation at hippocampal mossy fiber synapses. J Neurosci. 2007;27:13603–13613. doi: 10.1523/JNEUROSCI.3985-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci. 2008;28:7945–7953. doi: 10.1523/JNEUROSCI.2165-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase cbeta-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV, Letiges M. Knockout of PKC alpha enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16:847–858. doi: 10.1210/mend.16.4.0809. [DOI] [PubMed] [Google Scholar]
- Lorenzo PS, Beheshti M, Pettit GR, Stone JC, Blumberg PM. The guanine nucleotide exchange factor RasGRP is a high-affinity target for diacylglycerol and phorbol esters. Mol Pharmacol. 2000;57:840–846. [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Couce ME, Bohler WT, Ways DK. Protein kinase C subspecies distinguish major cell types in rat hippocampus: an immunocytochemical and in situ hybridization histochemical study. Hippocampus. 1991;1:293–301. doi: 10.1002/hipo.450010320. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Regehr WG. Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol. 2012;4:a005702. doi: 10.1101/cshperspect.a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C, Brose N. β Phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/S0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Roberts NA, Haworth RS, Avkiran M. Effects of bisindolylmaleimide PKC inhibitors on p90RSK activity in vitro and in adult ventricular myocytes. Br J Pharmacol. 2005;145:477–489. doi: 10.1038/sj.bjp.0706210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/S0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Searl TJ, Silinsky EM. Increases in acetylcholine release produced by phorbol esters are not mediated by protein kinase C at motor nerve endings. J Pharmacol Exp Ther. 1998;285:247–251. [PubMed] [Google Scholar]
- Shindo M, Irie K, Ohigashi H, Kuriyama M, Saito N. Diacylglycerol kinase gamma is one of the specific receptors of tumor-promoting phorbol esters. Biochem Biophys Res Commun. 2001;289:451–456. doi: 10.1006/bbrc.2001.5935. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Maler L. Differential roles of Ca2+/calmodulin-dependent kinases in posttetanic potentiation at input selective glutamatergic pathways. Proc Natl Acad Sci U S A. 1998;95:7133–7138. doi: 10.1073/pnas.95.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Xu J, He L, Wu LG. Role of Ca(2+) channels in short-term synaptic plasticity. Curr Opin Neurobiol. 2007;17:352–359. doi: 10.1016/j.conb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Xue L, Wu LG. Post-tetanic potentiation is caused by two signalling mechanisms affecting quantal size and quantal content. J Physiol. 2010;588:4987–4994. doi: 10.1113/jphysiol.2010.196964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]