

Abstract
Postsynaptic density (PSD)-95, the most abundant postsynaptic scaffolding protein, plays a pivotal role in synapse development and function. Continuous palmitoylation cycles on PSD-95 are essential for its synaptic clustering and regulation of AMPA receptor function. However, molecular mechanisms for palmitate cycling on PSD-95 remain incompletely understood, as PSD-95 depalmitoylating enzymes remain unknown. Here, we isolated 38 mouse or rat serine hydrolases and found that a subset specifically depalmitoylated PSD-95 in heterologous cells. These enzymes showed distinct substrate specificity. α/β-Hydrolase domain-containing protein 17 members (ABHD17A, 17B, and 17C), showing the strongest depalmitoylating activity to PSD-95, showed different localization from other candidates in rat hippocampal neurons, and were distributed to recycling endosomes, the dendritic plasma membrane, and the synaptic fraction. Expression of ABHD17 in neurons selectively reduced PSD-95 palmitoylation and synaptic clustering of PSD-95 and AMPA receptors. Furthermore, taking advantage of the acyl-PEGyl exchange gel shift (APEGS) method, we quantitatively monitored the palmitoylation stoichiometry and the depalmitoylation kinetics of representative synaptic proteins, PSD-95, GluA1, GluN2A, mGluR5, Gαq, and HRas. Unexpectedly, palmitate on all of them did not turn over in neurons. Uniquely, most of the PSD-95 population underwent rapid palmitoylation cycles, and palmitate cycling on PSD-95 decelerated accompanied by its increased stoichiometry as synapses developed, probably contributing to postsynaptic receptor consolidation. Finally, inhibition of ABHD17 expression dramatically delayed the kinetics of PSD-95 depalmitoylation. This study suggests that local palmitoylation machinery composed of synaptic DHHC palmitoylating enzymes and ABHD17 finely controls the amount of synaptic PSD-95 and synaptic function.
SIGNIFICANCE STATEMENT Protein palmitoylation, the most common lipid modification, dynamically regulates neuronal protein localization and function. Its unique reversibility is conferred by DHHC-type palmitoyl acyl transferases (palmitoylating enzymes) and still controversial palmitoyl-protein thioesterases (depalmitoylating enzymes). Here, we identified the membrane-anchored serine hydrolases, ABHD17A, 17B, and 17C, as the physiological PSD-95 depalmitoylating enzymes that regulate PSD-95 palmitoylation cycles in neurons. This study describes the first direct evidence for the neuronal depalmitoylating enzyme and provides a new aspect of the dynamic regulatory mechanisms of synaptic development and synaptic plasticity. In addition, our established APEGS assay, which provides unbiased and quantitative information about the palmitoylation state and dynamics, revealed the distinct regulatory mechanisms for synaptic palmitoylation.
Keywords: ABHD17, ABHD protein, AMPA receptor, palmitoylation, PSD-95, synapse
Introduction
Precise localization of proteins to specialized subcellular regions is essential for neuronal development and function. The postsynaptic density (PSD) of excitatory synapses represents such a specialized membrane region. During synapse development, neurotransmitter receptors, signaling molecules, and scaffolding proteins accumulate at the PSD and are poised to respond to synaptic stimuli. In addition, neurotransmitter receptors, such as AMPA receptors, are rapidly recruited to the PSD in response to synaptic activity and this dynamic process underlies aspects of synaptic plasticity. Thus, protein trafficking to the PSD plays critical roles in synapse development and synaptic plasticity (Kim and Sheng, 2004; Choquet and Triller, 2013; Huganir and Nicoll, 2013).
Protein palmitoylation, a common lipid modification, is an important mechanism for the postsynaptic targeting of proteins (Linder and Deschenes, 2007; Fukata and Fukata, 2010; Chamberlain and Shipston, 2015). By adding the lipid palmitate, palmitoylation increases the hydrophobicity of proteins and regulates diverse aspects of protein trafficking and function. Examples of palmitoylated proteins include H- and NRas small GTPases, heterotrimeric G-protein α subunits, Fyn tyrosine kinase, and various transmembrane proteins, such as AMPA and NMDA receptors. The unique reversible nature of palmitoylation is considered to allow proteins to dynamically relocalize between the cytoplasm and the intracellular and plasma membranes.
PSD-95, a member of the membrane-associated guanylate kinase (MAGUK) family (Zhu et al., 2016), is the most representative palmitoylated protein in neurons (El-Husseini et al., 2002). Palmitoylation of PSD-95 at cysteines 3 and 5 is essential for its postsynaptic targeting (Craven et al., 1999). Synaptic PSD-95 binds to AMPA receptor/TARPs complex and determines the number of synaptic AMPA receptors (Elias and Nicoll, 2007). Interestingly, synaptic PSD-95 undergoes continuous intraspine palmitoylation cycles to maintain the PSD-95 clusters and to flexibly control the number of membrane-bound PSD-95 in response to synaptic activity (Fukata et al., 2013). Although the DHHC protein family has emerged as palmitoylating enzymes [palmitoyl-acyl transferases (PATs; Linder and Deschenes, 2007; Fukata and Fukata, 2010; Chamberlain and Shipston, 2015] and synaptic DHHC2 mediates PSD-95 palmitoylation (Fukata et al., 2004, 2013; Noritake et al., 2009), depalmitoylating enzymes remain controversial. So far, Lypla1 (also known as Apt1) and lysosomal Ppt1 were biochemically purified as candidates for several limited substrates, such as HRas and Gα (Camp and Hofmann, 1993; Duncan and Gilman, 1998; Yeh et al., 1999). However, it remains unknown whether Lypla1 and Ppt1 depalmitoylate PSD-95.
Depalmitoylating enzymes (ie, palmitoyl-protein thioesterases) catalyze hydrolysis of S-acylated cysteine residues and enzymologically belong to the metabolic serine hydrolase superfamily (∼115 genes in the human genome), which also includes proteases, lipases, esterases, and amidases (Bachovchin et al., 2010). A recent study showed that hexadecylfluorophosphonate (HDFP), an inhibitor of a subset of serine hydrolases including Lypla1 and Ppt1, blocks the depalmitoylation of the Ras GTPases, Gα proteins, and MAGUK proteins (MPP1 and MPP6; Martin et al., 2012). Because 21 kinds of HDFP-sensitive serine hydrolases mostly have an α/β-hydrolase fold domain as a catalytic domain, here we asked whether certain HDFP-sensitive serine hydrolases and related α/β-hydrolase domain-containing (ABHD) proteins (Lord et al., 2013) function as depalmitoylating enzymes for PSD-95.
Materials and Methods
All animal studies were reviewed and approved by the ethic committees at NINS and were performed according to the institutional guidelines concerning the care and handling of experimental animals. The sex of the species studied is described in the following subsections.
Materials.
The following reagents were used: [3H]palmitic acid (PerkinElmer), maleimide-conjugated PEGs (mPEG-2k and mPEG-5k; NOF), N-ethyl maleimide (NEM; Wako), hydroxylamine hydrochloride (NH2OH; Sigma-Aldrich), tris-(2-carboxyethyl) phosphine (TCEP; Thermo Fisher Scientific), and 2-bromopalmitate (2-BP; Sigma-Aldrich).
Antibodies.
The antibodies used here included: rabbit polyclonal antibodies to ABHD17A (Catalog #15854-1-AP; Proteintech), AMPA receptor GluA1 (Catalog #AB1504; EMD Millipore), GABAA receptor γ2 (Catalog #224003; Synaptic Systems), NMDA receptor GluN2A (Catalog #GluRe1C-Rb-Af542-1; Frontier Science), mGluR5 (Catalog #AB5675; EMD Millipore), transferrin receptor (TfR; Catalog #ab84036; Abcam), and vGluT1 (Catalog #135303; Synaptic Systems); rabbit monoclonal antibodies to ABHD16A/BAT5 (Catalog #ab185549; Abcam), Gαq (Catalog #ab199533; Abcam), and HRas (Catalog #ab32417; Abcam); mouse monoclonal antibodies to bassoon (Catalog #ab82958; Abcam), FLAG (Catalog #F3165; Sigma-Aldrich), GFP (Catalog #M048-3; MBL), GluA2 (Catalog #75-002; NeuroMab), HA (Catalog #11583816001; Sigma-Aldrich), PSD-95 (Catalog #MA1-046; Thermo Scientific), pan-Shank (Catalog #75-089; NeuroMab), and synaptophysin (Catalog #S5768; Sigma-Aldrich); a guinea pig polyclonal antibody to vGluT1 (Catalog #AB5905; EMD Millipore); and rat monoclonal antibodies to FLAG (Catalog #NBP1-06712; Novus Biologicals) and HA (Catalog #11867423001; Roche). Rabbit polyclonal antibodies to ABHD17B and ABHD17C were raised against ABHD17B (aa 20–91) and ABHD17C (aa 310–320), respectively, and affinity-purified. A recombinant antibody against palmitoylated PSD-95 with human-Fc (hPF11; Fukata et al., 2013) and rabbit polyclonal antibodies to PSD-95 and GFP (Noritake et al., 2009) were described previously.
Plasmid constructions.
The cDNAs of rat ABHD1 (NM_001008520), ABHD9 (NM_001108988), ABHD14A (NM_001009670), ABHD14B (NM_001007664), ABHD15 (NM_001107025), ABHD16B (NM_001014052), ABHD17A (NM_001006983), Ppt2 (NM_019367), Acot7 (NM_013214), and Tex30 (NM_001106909) were cloned from female rat brain total RNA by RT-PCR. The cDNAs of mouse ABHD2 (NM_018811), ABHD3 (NM_134130), ABHD4 (NM_134076), ABHD5 (NM_026179), ABHD6 (NM_025341), ABHD7 (NM_001001804), ABHD8 (NM_022419), ABHD10 (NM_172511), ABHD11 (NM_145215), ABHD12 (NM_024465), ABHD13 (NM_001081119), ABHD16A (NM_178592), ABHD17B (NM_146096), ABHD17C (NM_133722), Lypla1 (NM_008866), Lypla2 (NM_011942), Lyplal1 (NM_146106), Ppt1 (NM_008917), Aadacl1 (NM_178772), Dpp8 (NM_028906), Faah (NM_010173), thioesterases domain (TE, aa 2201–2504) of Fasn (NM_007988), Lipe (NM_010719.5), Pafah2 (NM_133880), Pgap1 (NM_001163314), Pnpla6 (NM_015801), and Pnpla8 (NM_026164) were cloned from female mouse brain total RNA. These cDNAs were subcloned into pCAGGS-FLAG (C-terminal tag) vector. cDNAs of Pafah2 and Ppt2 were subcloned into pcDNA3.1-FLAG-Hisx6 (C-terminal tag). pCAGGS-mouse ABHD17B mutants, pCAGGS-rat PSD-95, pCAGGS-rat PSD-95-FLAG, pCAGGS-FLAG-mouse HRas, and their palmitoyl-cysteine mutants were generated by standard PCR methods. To construct pEGFP-N-rat ABHD17A, -mouse Lypla2, and pCAGGS-mCherry-N-human TfR, corresponding cDNAs were subcloned into pEGFP-N or pCAGGS-mCherry-N vectors. pmCherry-Galactosyl transferase (GalT) was described previously (Tsutsumi et al., 2009). For AAV production of mouse ABHD17B, wild-type ABHD17B-HA and inactive ABHD17B-D235A-HA were subcloned into pAAV vector with CAG promoter.
Rat ABHD17A, 17B, and 17C were knocked down by the miR-RNAi system (Thermo Fisher Scientific), and the following targeting sequences were used: miR-ABHD17A, 5′-TGAGAAGAACCTCTATGCTG-3′; miR-ABHD17B, 5′-ATACCAAGAAGACCTACTGT-3′; miR-ABHD17C, 5′-CGCGCATCAACTGCAACATC-3′; and miR-LacZ (β-galactosidase), 5′-GACTACACAAATCAGCGATTT-3′ as a negative control. Targeting sequences were subcloned into pcDNA6.2-EmGFP-miR. For triple knockdown of ABHD17A, 17B, and 17C, three chained miRNAs were subcloned into the 3′UTR of GFP. The resulting GFP-miRNA cassette was subcloned into pAAV vector with CAG promoter. All PCR products were analyzed by DNA sequencing (Functional Genomics Facility, National Institute for Basic Biology).
Metabolic labeling with [3H]palmitate.
Transfected HEK293T cells or COS7 cells were labeled with 0.4 mCi/ml [3H]palmitic acid for 4 h. Then, the whole-cell lysate was separated by SDS-PAGE for fluorography and Western blotting or Coomassie brilliant blue (CBB) staining as described previously (Fukata et al., 2004, 2006). In the fluorography, we confirmed that no signal was detected at the same size as substrates in the mock sample (ie, palmitoylation levels of tested substrates were not affected by other endogenous palmitoylated proteins). To quantify the relative PSD-95 palmitoylation levels in Figure 1C, we normalized the levels of [3H]palmitate-labeled PSD-95-GFP to the expression levels of PSD-95-GFP (quantified using ImageJ).
Figure 1.

Screening for PSD-95 depalmitoylating enzymes. A, B, Isolated serine hydrolase (SH) clones were individually cotransfected with PSD-95-GFP into HEK293T cells (A) and COS7 cells (B). After metabolic labeling with [3H]palmitate, proteins were separated by SDS-PAGE, followed by fluorography (top) and Western blotting (WB) with GFP antibody (bottom). Arrowheads indicate the position of PSD-95-GFP. WT, Wild-type; C3,5S, mutated at cysteines 3 and 5 to serines. Normalized palmitoylation levels of PSD-95 relative to control (%) in COS7 cells are indicated (B). C, Effects of SH clones on palmitoylation levels of PSD-95 in HEK293T cells. PSD-95-GFP palmitoylation level was normalized by the expression level of PSD-95-GFP. Note that ABHD1, 3, and 4 reduced the PSD-95-GFP expression. n = 3 independent experiments. D, Expression of individual FLAG-tagged SH clones was confirmed. Asterisks indicate bands detected by overexposing the film.
AAV production and infection.
DJ-type AAV vectors were prepared using AAV-DJ Helper Free Packaging System (Cell Biolabs). For overexpression experiments (see Figs. 5, 6A–C), cultured hippocampal neurons from embryonic day 18 rats of both sexes [12–13 d in vitro (DIV)] were infected for 7 d. For knock-down experiments (see Fig. 8), neurons (1 DIV) were infected for 13 d, followed by the acyl-PEGyl exchange gel shift (APEGS) assay or immunofluorescence. The knock-down efficiency was validated by real-time PCR using the StepOnePlus system (Applied Biosystems). Following primer sets were used: ABHD17A, 5′-CATCATCCACGGCACAGAAG-3′ and 5′-CGTAGGCGCTCCAGGTATTG-3′; ABHD17B, 5′-GGACCGTACCGTCTGTGGAC-3′ and 5′-CCGCATTCCTGAGGTCAAAG-3′; ABHD17C, 5′-ATTGGCACTGTCCCCACTGT-3′ and 5′-GGAAAAGCAACACGCAATCC-3′; and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-GACATGCCGCCTGGAGAAAC-3′ and 5′-AGCCCAGGATGCCCTTTAGT-3′. The expression of ABHD17s was normalized to that of GAPDH.
Figure 5.

Expression of ABHD17 selectively depalmitoylates PSD-95 in neurons. Neurons infected with AAV vectors, mock (−), wild-type (WT) ABHD17B, or inactive ABHD17B-D235A mutant (D235A), were processed for the APEGS assay. The obtained PEGylated samples were subjected to WB with indicated antibodies. Note that ABHD17B robustly depalmitoylated PSD-95 in neurons. Closed and open arrowheads indicate the positions of palmitoylated and non-palmitoylated proteins, respectively. Palm*, the number of palmitoylation sites is not determined. We found that mGluR5 was palmitoylated at one specific site (data not shown). GABAARγ2, GABAA receptor γ2.
Figure 6.

Expression of ABHD17 reduces synaptic clustering of PSD-95 and AMPA receptor in neurons. A–C, ABHD17B expression significantly reduced the intensity of synaptic PSD-95 (A, B) and AMPA receptor clusters (A, C). Bassoon is shown in blue. Scale bar, 5 μm. ***p < 0.001 by one-way ANOVA with post hoc Tukey's test; n = 3 independent experiments (9 neurons). D–F, Effects of ABHD17B expression on the dendritic spine density (E) and the spine morphology (F). M, Mushroom-; S, stubby-; F, filopodia-like spine. Scale bar, 5 μm. ***p < 0.001 one-way ANOVA with post hoc Tukey's test. n = 30 neurons from two independent experiments. n.s., Not significant.
Figure 8.
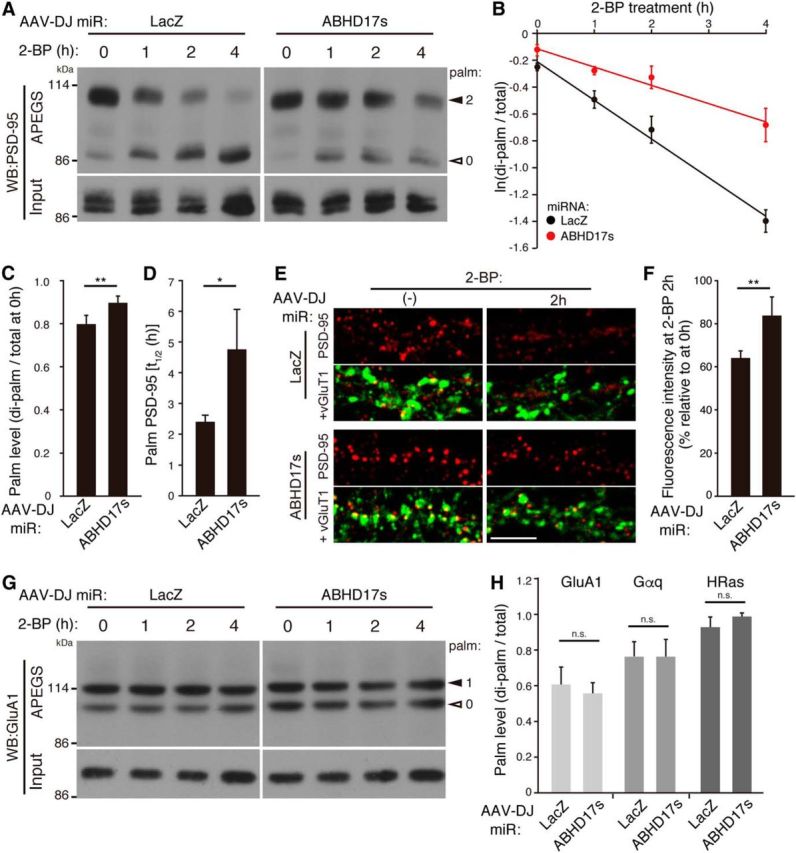
ABHD17 specifically mediates PSD-95 depalmitoylation in neurons. A–D, Triple knockdown of ABHD17 members (miR-ABHD17s) in rat hippocampal neurons significantly delayed PSD-95 depalmitoylation process (A, B, D). A graph showing that ABHD17 knockdown caused the increase in the basal level of palmitoylation stoichiometry of PSD-95 (C; n = 5 independent cultures) and prolonged the half-life (t1/2) of palmitoylated PSD-95 (D; n = 4 independent cultures). E, F, ABHD17 knockdown inhibited 2-BP-induced PSD-95 declustering. Scale bar, 5 μm. n = 3 independent experiments (9 neurons). G, H, ABHD17 knockdown did not affect GluA1 depalmitoylation process (G), and the basal levels of the palmitoylation stoichiometry of GluA1 (mono-palmitoylated; n = 5), Gαq (n = 4), and HRas (n = 3) (H). *p < 0.05, **p < 0.01 by the Student's t test. n.s., Not significant. Closed and open arrowheads indicate the positions of palmitoylated and non-palmitoylated proteins, respectively (A, G).
Subcellular fractionation.
The method was followed as described previously (Noritake et al., 2009). Five adult female rat brains were homogenized in buffer containing 320 mm sucrose, 10 mm HEPES, pH 7.4, and 0.2 mm PMSF. Homogenate was centrifuged for 10 min at 1000 × g to remove crude nuclear fraction (P1). The supernatant (S1) was centrifuged at 9000 × g for 15 min to produce a pellet (P2) and supernatant (S2). The S2 was centrifuged at 100,000 × g for 1 h to produce a pellet (P3) and supernatant (S3). The P2 fraction was resuspended in the homogenization buffer. Discontinuous sucrose gradients containing 3 ml of the resuspended P2 material and 3 ml each of 0.8, 1.0, and 1.2 m sucrose solutions were run for 2 h at 58,000 × g. The band between 1.0 and 1.2 m sucrose was obtained as a synaptosome fraction (Syn). This synaptosome fraction was extracted with 0.5% Triton X-100, and then centrifuged at 32,800 × g for 20 min to divide into soluble (Triton-Sol) and insoluble fractions (PSD1). The PSD1 fraction was resuspended in 0.5% Triton X-100, and centrifuged at 200,000 × g for 1 h to produce a pellet (PSD2). Fifty microgram proteins of each fraction were analyzed by Western blotting.
Immunoprecipitation.
Hippocampal neurons infected with AAV vectors were lysed by buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Igepal CA-630, 0.5% deoxycholate, 0.1% SDS, and 50 μg/ml PMSF. After extraction for 20 min on ice and centrifugation at 11,000 × g for 10 min, the samples were incubated with anti-HA antibody for 1 h at 4°C. Protein A sepharose (GE Healthcare) was added and incubated for 1 h at 4°C. Immunoprecipitates were washed three times with buffer containing 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 100 mm NaCl, and 1% Triton X-100. Immunoprecipitates were resolved by SDS-PAGE, followed by Western blotting.
APEGS assay.
Cultured cells and one adult female rat cerebrum were homogenized with Buffer A (4% SDS, 5 mm EDTA, 50 μg/ml PMSF, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A in PBS) and Buffer A containing 8 m urea, respectively. After sonication and centrifugation at 100,000 × g for 15 min, supernatant proteins (0.3 mg/ml, 150 μg) were reduced with 25 mm TCEP for 1 h at 55°C (without urea) or at room temperature (RT; with urea), and free cysteine residues were alkylated with 50 mm NEM for 3 h at RT to be blocked. After chloroform/methanol precipitation (CM ppt), resuspended proteins in PBS with 4% SDS and 5 mm EDTA were incubated in buffer (1% SDS, 5 mm EDTA, 1 m NH2OH, pH 7.0) for 1 h at 37°C to cleave palmitoylation thioester bonds. As a negative control, 1 m Tris-HCl, pH7.0, was used (NH2OH−). After CM ppt, resuspended proteins (0.5 mg/ml, 50 μg) in PBS with 4% SDS were PEGylated with 20 mm mPEGs for 1 h at RT to label newly exposed cysteinyl thiols. We selected mPEG-2k and mPEG-5k for <60 and >60 kDa proteins, respectively. As a negative control, 20 mm NEM was used instead of mPEG (mPEG−). After CM ppt, protein precipitates were resuspended with SDS-sample buffer and boiled at 100°C for 3 min. Protein concentration was measured by BCA protein assay at individual steps. In addition, we optimized the doses and reaction time for the TCEP, NEM, NH2OH, and mPEG treatment to completely accomplish individual reactions. For quantitative Western blotting, the EzFastBlot HMW transfer buffer (ATTO) was used to get higher transfer efficiency. Then, blotted membranes were visualized with a cooled CCD camera (Light-Capture II; ATTO) and the optimal specific bands were analyzed with CS Analyzer 3.0 software (ATTO).
To purify the PEGylated PSD-95 for Figure 4D, we first performed the APEGS assay on HEK293T cell lysates in which PSD-95-WT-FLAG or PSD-95-C3,5S-FLAG was cotransfected with DHHC3. Then, proteins were extracted from the final CM ppt with ultrasonication in PBS containing 1% SDS and 50 μg/ml PMSF. Buffer containing 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 100 mm NaCl, and 1% Triton X-100 was then added to reduce the SDS concentration to 0.1%. After centrifugal clarification, PSD-95-FLAG was immunoaffinity-purified using anti-FLAG M2 affinity agarose (Sigma-Aldrich). The bands of di-PEGylated PSD-95-WT-FLAG and non-PEGylated PSD-95-C3,5S-FLAG were quantified by CBB gel staining using bovine serum albumin as the standard. Mock-transfected cell sample was used to subtract the background signal. Purified proteins were then analyzed by CBB staining (0.2 μg) and Western blotting (50 fmol) with anti-PSD-95 antibody.
Figure 4.

The APEGS assay for quantifying in vivo palmitoylation stoichiometries. A, The principle of the APEGS assay. Disulfide bonds are broken by TCEP (1) and free thiols are blocked by NEM (2). Palmitoyl-thioester linkages are cleaved by hydroxylamine (NH2OH; 3) and newly formed thiols are labeled by mPEG (4). WB with antibodies to proteins of interest visualizes the palmitoylation states. B, C, The APEGS assay precisely detects palmitoylation-dependent mobility shifts of PSD-95 (B) and FLAG-HRas (C) in transfected HEK293T cells. DHHC3 was coexpressed with PSD-95. mPEG-5k and mPEG-2k were used for labeling PSD-95 and HRas, respectively. NH2OH (−) and mPEG (−) lanes indicate the negative controls. D, PEGylation does not affect the sensitivity of WB. An equal amount of purified di-PEGylated PSD-95-WT-FLAG and non-PEGylated PSD-95-C3,5S-FLAG was subjected to SDS-PAGE, followed by CBB staining (0.2 μg each) and WB with PSD-95 antibody (50 fmol each). Fold intensity of individual bands is shown. Asterisks indicate the bands used for quantification of PSD-95-C3,5S-FLAG. E, Adult rat cerebrum homogenate and cultured rat hippocampal (hipp) neuron lysate (21 DIV) were processed for the APEGS assay and samples were analyzed by Western blotting with anti-PSD-95 antibody. F, The APEGS assay detects a wide range of palmitoylation stoichiometries with linearity. PEGylated rat cerebrum lysate was serially diluted with unlabeled lysate to generate the standards and analyzed by WB with anti-PSD-95 antibody. Actual stoichiometries of PSD-95 palmitoylation calculated by ratio of band intensities were plotted. Closed and open arrowheads indicate the positions of palmitoylated and non-palmitoylated proteins, respectively.
For the analysis of the depalmitoylation process, cultured neurons were treated with 100 μm of 2-BP for 1–6 h. We confirmed that treatment with anisomycin (25 μm; EMD Millipore), an inhibitor of protein synthesis, did not affect depalmitoylation kinetics of substrates tested in the presence of 2-BP under our experimental conditions. So, we pooled these data (with and without anisomycin) for statistic analysis (see Fig. 7).
Figure 7.

Unique dynamic palmitate turnover on PSD-95. A–C, To monitor the depalmitoylation process of PSD-95, hippocampal neurons were treated with 2-bromopalmitate (2-BP) for the indicated periods and subjected to the APEGS assay, followed by WB with anti-PSD-95 antibody. B, A graph showing that the depalmitoylation kinetics of PSD-95 decelerated as neurons developed. C, The half-life of palmitate on PSD-95 was prolonged as neurons developed from 7 to 21 DIV. *p < 0.05, **p < 0.01 by one-way ANOVA with post hoc Dunnet's test; n = 3 independent cultures. D, Rapid palmitate turnover was observed only in PSD-95, but not in GluA1, GluN2A, mGluR5, Gαq, and HRas in 7 and 21 DIV neurons. Closed and open arrowheads indicate the positions of palmitoylated and non-palmitoylated proteins, respectively. ND, Not determined due to the low expression of GluN2A. n = 2–4 independent cultures.
Immunofluorescence analysis.
Dissociated hippocampal neuron cultures were prepared from embryonic day 18 rats of both sexes as previously described (Fukata et al., 2013). Neurons were fixed with methanol for 10 min at −30°C (see Figs. 3A, 6A, 8E) or with 4% paraformaldehyde for 10 min at RT and permeabilized with methanol for 10 min at −30°C [see Fig. 3D; or permeabilized with 0.1% Triton X-100 for 10 min on ice; see Fig. 6D)]. Then, neurons were blocked with PBS containing 10 mg/ml BSA for 10 min on ice and incubated with indicated primary antibodies, visualized with AlexaFluor 488, Cy-3 or/and Alexa Fluor 647-conjugated secondary antibodies. For Figures 3A–D and 6D–F, neurons were transfected by Lipofectamine 2000, followed by immunofluorescence or live imaging (Fig. 3B,C). Live imaging was done within 24 h after transfection.
Figure 3.

Subcellular distribution of ABHD17. A, Differential subcellular distribution of FLAG-tagged ABHD proteins and Lypla2. Note that some populations of ABHD17A localized at the postsynaptic membranes labeled by pan-Shank antibody (red) in 14 DIV rat hippocampal neurons. Scale bars: top, 10 μm; bottom, 5 μm. B, Endosomal localization of ABHD17A. TfR, transferrin receptor. Scale bars: left, 10 μm; right, 2 μm. C, Localization of Lypla2 at the Golgi apparatus in the cell body. GalT, galactosyl transferase. Scale bar, 10 μm. D, Localization of FLAG-tagged ABHD17B mutants (green) was examined. PSD-95 and TfR are shown in red and blue, respectively. An arrow indicates a residual spine with ABHD17B and PSD-95. A spine indicated by the arrowhead is magnified. Scale bars: left, 5 μm; right, 1 μm. E, CAG promoter drives EGFP with synthetic miRNA sequences targeting rat ABHD17A, 17B, and 17C (miR-ABHD17s). F, G, Infection of rat hippocampal neurons with AAV-expressing miR-ABHD17s caused large reduction in the amount of ABHD17A, 17B, and 17C mRNAs (F), and proteins (G). miR-LacZ indicates a control miRNA. H, ABHD17 members were enriched in the P3 microsomal and synaptosome fractions, and some populations localized in the PSD fraction. Syph, synaptophysin. Asterisks (G, H) indicate the confirmed cross-reactivity to ABHD17B. I, Enzyme-substrate interaction of ABHD17B and PSD-95 in neurons. Neurons were infected with the indicated AAV vectors, and ABHD17B-HA was immunoprecipitated (IP) with HA antibody.
Fluorescent images were acquired with a confocal laser scanning microscopy system (TCS SP5II; Leica) equipped with a HCX PL APO 63×/1.40–0.60 oil-immersion lens. Microscope control and all image analyses were performed with Leica LAS AF software, and images were acquired with the same laser power and detector settings in the datasets. To quantify the synaptic PSD-95, the mean intensity of all the detectable PSD-95 clusters apposed to vGluT1 was measured along the stretch of primary dendrites in the individual image (∼100 clusters/neuron). The quantification of synaptic GluA1 clusters, which were adjacent to both bassoon and PSD-95 was measured by the same criteria. To quantify the spine density and the spine morphology, each image was reconstructed by a z-series maximal projection of five to eight images, taken at 0.5 μm depth intervals (see Fig. 6D). The morphology of dendritic spines was classified into filopodia (long and thin protrusions without a bulbous head), stubby (spines without clear neck), and mushroom (spines with necks and large heads). The number of three types of spines on the dendritic length (along the stretch of first branches from primary dendrites) was counted.
Statistical analysis.
For paired sample comparisons, Student's t test was used. For multiple test subjects, one-way ANOVA with post hoc Tukey's or Dunnett's test was used. Error bars denote the SD. Box-and-whisker plots are shown to identify the median, 25th, and 75th percentiles.
Results
Screening for PSD-95 depalmitoylating enzymes
We isolated mouse or rat cDNA clones of 21 HDFP-sensitive serine hydrolases (Martin et al., 2012) and additional 17 related clones, which mainly contain ABHD proteins. We individually transfected these serine hydrolase cDNAs together with PSD-95 into HEK293T cells and assessed palmitoylation levels of PSD-95 by metabolic labeling with [3H]palmitate. We found that PSD-95 palmitoylation was reduced with the expression of ABHD12, 13, 17A (also known as FAM108A), 17B (FAM108B), 17C (FAM108C), Lypla1, and Lypla2 (<80% of control levels; Fig. 1A,C, D). The greatest effect was seen with ABHD17A and 17B. Expression of ABHD1, 3, and 4 reduced the signal of palmitoylated PSD-95 by reducing the expression of PSD-95 protein in HEK293T cells. We performed a similar screen in COS7 cells, and similar strong effects of ABHD17A, 17B, and 17C and moderate effects of Lypla1 and Lypla2 were obtained (Fig. 1B).
Substrate specificity for ABHD proteins
We next asked whether ABHD proteins show substrate specificity. We cotransfected selected serine hydrolases (ABHD13, 16A, 17B, and Lypla2) with neuronal palmitoyl proteins in HEK293T cells. PSD-95 and GAP43 have two palmitoyl cysteines at their N-terminal region whereas palmitoyl cysteines of Fyn are located near the N-terminal myristoylated glycine. HRas has two palmitoyl cysteines at its C-terminal region. We found that ABHD17B robustly depalmitoylated GAP43 and HRas, as well as PSD-95, but did not show any activity toward Fyn (Fig. 2A). In contrast, Lypla2 greatly reduced the Fyn palmitoylation, but showed less activity to PSD-95. ABHD13 showed moderate activity to PSD-95 but no activity to GAP43, Fyn, and HRas. ABHD16A heavily reduced the protein level of Fyn. Thus, ABHD17B, Lypla2, and ABHD13 show exquisite substrate specificity. The phylogenetic tree of ABHD proteins suggests that we could classify ABHD proteins into several subfamilies (Fig. 2B).
Figure 2.

Enzymatic characterization of ABHD17. A, Substrate specificity of ABHD proteins and Lypla2 was tested in HEK293T cells as in Figure 1. B, Phylogenetic tree of mouse ABHD genes. The tree is based on alignment of the α/β hydrolase fold domains. Putative subgroups for PSD-95 depalmitoylating enzyme candidates are shown by ovals in different colors. C, Increasing amounts of cotransfected ABHD17B plasmid caused PSD-95 depalmitoylation (0.5 μg plasmid) in a dose-dependent manner. D, ABHD17B mutant constructs. ABHD17B–5CS (C10,11,14,15,18S), palmitoylation-deficient ABHD17B; N-myr, myristoylated ABHD17B in which the N-terminal palmitoylation motif (aa 1–20) was replaced with myristoylation motif of Src (aa 1–20). The catalytic triad (S170/D235/H264) is indicated. E, F, Mutagenesis of ABHD17B showed that the catalytic triad (S170/D235/H264; E) and the N-terminal palmitoylation motif (F) were essential for the depalmitoylating activity of ABHD17B to PSD-95. Note that wild-type ABHD17B, but not ABHD17B–5CS, was highly palmitoylated. The weak [3H]palmitate labeling of N-myr ABHD17B might reflect N-palmitoylation, as N-myristoyl transferase can use palmitic acid as a substrate in vitro, albeit at a reduced efficiency compared with myristate (Kishore et al., 1993; Martin et al., 2011). SD-AA, ABHD17B-S170A/D235A; SDH-AAA, ABHD17B-S170A/D235A/H264A.
Enzymatic characterization of ABHD17
Next, we wondered whether the effect of ABHD17 on PSD-95 palmitoylation is mediated by the thioesterase enzyme activity. We first examined the dose-dependent effect of ABHD17B on PSD-95 palmitoylation. When the amount of cotransfected ABHD17B plasmid was decreased, ABHD17B depalmitoylated PSD-95 even at a 1:100 transfection ratio (Fig. 2C), suggesting that the depalmitoylating activity of ABHD17B is potent and catalytic. We then identified critical amino acids for the ABHD17B catalytic activity. The catalytic triad of α/β hydrolase fold is generally composed of serine, aspartate, and histidine residues. Based on the consensus motif of active serine residue (Gly-X-Ser-X-Gly; X is any amino acid), we identified Ser170 in ABHD17B (Fig. 2D). By three-dimensional structural modeling (http://toolkit.tuebingen.mpg.de/hhpred), we predicted critical Asp235 and His264 residues, which are located close to Ser170. Consistently, mutating postulated catalytic triad to alanine blocked its effect on PSD-95 depalmitoylation (Fig. 2E), and this catalytic triad is all conserved among ABHD17 members.
It was reported that ABHD17 members are palmitoylated at their N-terminal cysteine cluster and this palmitoylation is essential for their plasma membrane targeting in HeLa cells (Martin et al., 2012). To investigate the role of this palmitoylation, we constructed a palmitoylation-deficient mutant [ABHD17B-5CS (C10,11,14,15,18S)] and a myristoylated ABHD17B mutant (N-myr-ABHD17B), in which an N-terminal palmitoylation motif was replaced with a myristoylation motif of Src (Fig. 2D). Interestingly, both mutants lost the depalmitoylating activity toward PSD-95 (Fig. 2F). These results raise the possibility that the enzymatic activity of ABHD17 depends on its own palmitoylation or, alternatively, that palmitoylation of ABHD17 is critical (but myristoylation is not sufficient) for its targeting to the specific membrane domain where palmitoylated PSD-95 is localized (Fig. 3D).
Subcellular localization of ABHD17 in neurons
To explore the physiological PSD-95 depalmitoylating enzymes, we first examined the subcellular locus for candidate clones (ABHD17A, Lypla2, ABHD12, and ABHD13) in hippocampal neurons. FLAG-tagged ABHD17A localized at the plasma membrane, and some populations were distributed in the dendrites as vesicular-like structures and partly colocalized with postsynaptic scaffolding protein Shank in dendritic spines (Fig. 3A). In contrast, Lypla2 localized to the GalT-positive Golgi apparatus and in the cytoplasm (Fig. 3A,C). ABHD12 and ABHD13 were neither present at the plasma membrane nor in the dendritic spines. We then examined the ABHD17A localization using various marker proteins and found that ABHD17A localized mainly on TfR-positive recycling endosomes in the dendritic shafts and spines (Fig. 3B).
We further examined the localization of ABHD17B mutants (Fig. 2D) together with endogenous PSD-95 in neurons (Fig. 3D). Although overexpression of wild-type ABHD17B robustly reduced the PSD-95 cluster intensity and spine density (analyzed in detail in Fig. 6), ABHD17B was overlapped with or apposed to endogenous PSD-95 in the surviving dendritic spine. Enzymatically inactive ABHD17B-D235A, which did not affect PSD-95 clusters, was distributed to the plasma membrane and some vesicular-like structure in dendritic shafts and spines. ABHD17B-D235A clearly showed the colocalization with or apposition to PSD-95 and TfR in the dendritic spines. Palmitoylation-deficient ABHD17B-5CS showed diffuse cytoplasmic localization in the dendritic shaft and spines, and myristoylated N-myr-ABHD17B was distributed to the plasma membrane of dendritic shafts/spines more diffusely without making clear clusters than ABHD17B WT and D235A. Interestingly, ABHD17B-5CS and N-myr-ABHD17B, which were partially overlapped with endogenous PSD-95 in the dendritic spine, did not affect the PSD-95 clusters, possibly correlated with their loss of enzymatic activity (Fig. 2F).
We then biochemically examined the subcellular localization of ABHD17 members in the brain. Antibodies against ABHD17A, 17B, and 17C detected the corresponding endogenous proteins in Western blotting, as the band intensity recognized by these antibodies was greatly reduced in the sample of neurons infected with the AAV vector carrying a chained validated miRNA construct (Fig. 3E–G). All the ABHD17 members were distributed to the microsomal P3 (containing ER, Golgi apparatus, and parts of endosomes) and synaptosome fractions, and some populations localized in the PSD fraction (Fig. 3H). The presence of ABHD17 in the PSD is not just contamination, because: (1) synaptophysin, a synaptic vesicle protein, which was also enriched in P3 and synaptosome fractions was completely excluded from the PSD fractions; and (2) ABHD17 proteins show a very similar distribution pattern to TfR, which was colocalized with ABHD17 on the recycling endosomes (Fig. 3B,D) and is recycled between the cytoplasm and plasma membrane in the dendritic spine (Kennedy et al., 2010). These results suggest that the membrane-anchored ABHD17 members localize in dendritic spines and have opportunity to interact with PSD-95 in the PSD. We therefore investigated whether ABHD17 interacts with PSD-95 in neurons. Although the enzyme-substrate interaction is too transient to be detected, the catalytically inactive mutant sometimes serves as a substrate-trapping mutant. In fact, endogenous PSD-95 was coimmunoprecipitated with catalytically inactive ABHD17B, but not with wild-type ABHD17B (Fig. 3I). This strongly suggests that ABHD17 directly acts on PSD-95 in neurons. Together with the results that: (1) the enzymatic activity of ABHD12, 13, and Lypla2 was much less than that of ABHD17s in heterologous cells; and (2) ABHD12, 13, and Lypla2 did not localize at the plasma/spine membrane in neurons where depalmitoylation of PSD-95 should occur (Fukata et al., 2013), we hereafter focused on ABHD17s' function in neurons.
The APEGS assay for monitoring the palmitoylation states of synaptic proteins in neurons
Next, to examine whether ABHD17 could catalyze PSD-95 depalmitoylation in neurons, we attempted to quantitatively monitor the PSD-95 palmitoylation level in neurons. Here, we combined the acyl-biotinyl exchange method (Drisdel and Green, 2004) and the polyethylene glycol switch method (Rexach et al., 2010), and named the APEGS assay (Fig. 4A). The APEGS assay consists of four chemical steps: (1) break of disulfide bonds with TCEP, (2) blockade of free cysteine thiols with NEM, (3) specific cleavage of palmitoylation thioester linkages with hydroxylamine (NH2OH), and (4) labeling of newly exposed cysteinyl thiols with mPEG-5k or -2k, which causes the mobility shift of palmitoylated proteins by SDS-PAGE. Western blotting with antibodies against proteins of interest would separate palmitoylated bands from non-palmitoylated. Quantifying the relative intensities of bands would provide information about: (1) the palmitoylation stoichiometry (ie, the palmitoylation levels in cells, tissues, or any samples), and (2) palmitoylation states (ie, mono-, di-, or multiple palmitoylated).
We first validated whether mobility shifts observed in the APEGS assay reflect the number of palmitoylation sites. We selected PSD-95 and HRas as model substrates for mPEG-5k and mPEG-2k labeling, respectively, and prepared a series of palmitoylation-site mutants (El-Husseini et al., 2000b; Roy et al., 2005). HEK293T cells were transfected with wild-type or palmitoyl-cysteine mutants, and cell lysates were subjected to the APEGS assay. As expected, wild-type PSD-95 showed quantitative two-step mobility shifts; mono-palmitoylated C3L and C5L mutants of PSD-95 showed the one-step shift; and a palmitoylation-deficient C3,5S mutant did not show any shift (Fig. 4B). We confirmed that these mobility shifts of PSD-95 depend on the treatments with NH2OH and mPEG. HRas and its palmitoylation-site mutants also showed the expected mobility shifts by the APEGS assay (Fig. 4C).
To precisely quantify palmitoylation stoichiometries, we carefully optimized the individual chemical reactions to completely and saturably proceed (see Materials and Methods). In addition, we asked whether PEGylation could affect the sensitivity of Western blotting because of an attachment of the high molecular weight polymer to proteins. Di- and non-PEGylated FLAG-tagged PSD-95 was immunoaffinity-purified from the APEGS samples of HEK293T cells, which were transfected with PSD-95-WT or PSD-95-C3,5S plus DHHC3 (one of PSD-95 PATs). Western blotting with PSD-95 antibody showed that the same amount of di- and non-PEGylated PSD-95 was detected with the similar band intensity (Fig. 4D), indicating that PEGylation of PSD-95 does not influence the detection of PSD-95.
Next, we performed the APEGS assay using the adult rat brain homogenate and 21 DIV hippocampal neuron lysates. We confirmed the specific PEGylation of endogenous PSD-95 and found that ∼90% and 80% of PSD-95 were di-palmitoylated in the brain and cultured neurons, respectively, and mono-palmitoylated PSD-95 was hardly detected in either sample (Fig. 4E). Thus, the APEGS assay is applicable to endogenous proteins even in the crude tissue lysates and for the first time provides the quantitative information that most of PSD-95 is di-palmitoylated in the brain and mature cultured neurons.
We also tested the sensitivity and linearity of the APEGS assay using the standard samples with different stoichiometries of palmitoylated PSD-95, which were generated by serial dilution of PEG-labeled brain homogenate with unlabeled homogenate. We found that the APEGS assay could detect as little as 9% of palmitoylated PSD-95 and show a linearity over the wide range of stoichiometries (Fig. 4F). Thus, the APEGS assay was established as a simple, specific, and sensitive assay for quantifying palmitoylation stoichiometries of any biological samples.
Overexpression of ABHD17 depalmitoylates PSD-95 in neurons
Using the APEGS assay, we examined the effect of ABHD17 overexpression on the PSD-95 palmitoylation state in neurons. When wild-type ABHD17B was overexpressed by an AAV vector, the amount of di-palmitoylated PSD-95 was greatly reduced and that of non-palmitoylated PSD-95 was inversely increased (Fig. 5). In contrast, the expression of inactive ABHD17B-D235A did not show any effect. The strong effect of ABHD17B is specific to PSD-95, as the palmitoylation levels of GluA1 AMPA receptor subunit and GluN2A NMDA receptor subunit were slightly reduced and those of GluA2, Gαq, HRas, and GABAA receptor γ2 subunit were hardly changed. In addition, we found that mGluR5 and vGluT1 are novel palmitoyl-protein candidates and their mobility shift was not affected by ABHD17B overexpression.
Consistent with biochemical data, expression of ABHD17B dramatically reduced the intensity of synaptic PSD-95 clusters (Fig. 6A,B). The reason why PSD-95 immunoreactivity seems absent from the dendrites by ABHD17B expression is probably because: (1) depalmitoylated PSD-95 is diffused into the cytoplasm of the dendritic shafts, and the intensity of diffused endogenous PSD-95 may be less than the detection threshold and/or; (2) depalmitoylated cytosolic PSD-95 is not efficiently fixed by methanol and thereby leaks out from neurons. This effect of ABHD17B on PSD-95 clusters depends on its enzymatic activity, as expression of inactive ABHD17B-D235A did not show any effects. Furthermore, we found that the synaptic clustering of GluA1 AMPA receptor subunit, which is anchored by PSD-95, was significantly reduced by ABHD17B overexpression (Fig. 6A,C). Because palmitoylation-deficient GluA1 mutant shows the normal cell-surface distribution (Hayashi et al., 2005), it is suggested that the reduction of synaptic GluA1 is mediated by decreased PSD-95 scaffolds rather than slight GluA1 depalmitoylation. We also found that expression of ABHD17B largely reduced the spine density (Fig. 6D,E), and reduced the proportion of mushroom-like and filopodia-like spines (Fig. 6D,F). This is consistent with the previous observations that PSD-95 expression increases the number of dendritic spines (El-Husseini et al., 2000a). Together, overexpressed ABHD17B in neurons specifically depalmitoylates endogenous PSD-95 and reduces the synaptic clusters of PSD-95 and AMPA receptors.
Rapid and robust palmitate cycling on PSD-95 in neurons
The APEGS assay enables us not only to monitor stoichiometries of palmitoylated proteins in neurons, but also to examine the dynamic palmitate turnover on proteins. To monitor the depalmitoylation process of proteins, cultured hippocampal neurons were treated with 2-BP to block the newly occurring palmitoylation. The APEGS assay showed that ∼60% (60.2 ± 6.1%) of PSD-95 was di-palmitoylated and ∼0% was mono-palmitoylated in young (7 DIV) neurons (Fig. 7A,B). Upon 2-BP treatment, the amount of di-palmitoylated PSD-95 was rapidly reduced and that of non-palmitoylated PSD-95 was reciprocally increased. This is the first evidence that the almost entire population of palmitoylated PSD-95 (90% at 7 DIV) participates in dynamic palmitoylation cycles. We noted that the stoichiometry of PSD-95 palmitoylation in young neurons is lower compared with those in aged (21 DIV) neurons and in adult rat brain (Fig. 4E). We therefore asked whether the stoichiometry and the rate of palmitate cycling are changed as neurons develop from 7 to 21 DIV, during when synapses mature. We found that the basal palmitoylation stoichiometry of PSD-95 was increased to >70% after 10 DIV (74.2 ± 2.6%, p < 0.05; compared with 7 DIV; Fig. 7A,B). The half-life of palmitate on PSD-95 was prolonged from ∼1 h (7 DIV) to 3 h (21 DIV; Fig. 7B,C). Thus, the depalmitoylation kinetics of PSD-95 is more dynamic (plastic) in the early stage of synaptic maturation and decelerates as synapses develop. In addition, we found that stably palmitoylated population of PSD-95, which remained di-palmitoylated even at 6 h after 2-BP treatment, was significantly increased in mature aged neurons (Fig. 7B; at 6 h).
We next asked whether such a dynamic palmitate cycling could be observed among other palmitoyl-proteins. Unexpectedly, palmitate on GluA1, GluN2A, mGluR5, Gαq, and HRas hardly turned over in 7 and 21 DIV neurons within 6 h after 2-BP treatment (Fig. 7D). These results highlight the distinct and tight regulatory control of synaptic palmitoylation in neurons.
ABHD17 mediates PSD-95 depalmitoylation in neurons
Finally, to investigate whether ABHD17 mediates PSD-95 depalmitoylation in neurons, we examined ABHD17 loss of function using a knock-down approach. When we simultaneously knocked-down ABHD17A, 17B, and 17C by the AAV-miRNA vector (Fig. 3E), the basal palmitoylation level of PSD-95 was significantly increased (Fig. 8A,C; di-palmitoylated/total ratio 0.80 ± 0.04 for miR-LacZ and 0.89 ± 0.03 for miR-ABHD17s, 14 DIV). Then, we treated neurons with 2-BP and performed the APEGS assay. Upon 2-BP treatment, the amount of di-palmitoylated PSD-95 was reduced over time and the amount of non-palmitoylated PSD-95 was increased as observed in Figure 7A. Consistent with the data in Figure 7C and previous pulse chase data with [125I]palmitate (El-Husseini et al., 2002), the half-life of palmitate on PSD-95 was 2.4 ± 0.2 h at 14 DIV. Importantly, triple-knockdown of ABHD17 members apparently delayed PSD-95 depalmitoylation process (Fig. 8A, B) and significantly prolonged the half-life of palmitate on PSD-95 to 4.8 ± 1.3 h (Fig. 8D). When neurons were treated with 2-BP for 2 h, the intensity of synaptic PSD-95 clusters was significantly reduced (Fig. 8E,F), indicating rapid depalmitoylation of PSD-95 at local synapses. Consistent with biochemical data, this reduction of PSD-95 clusters was significantly suppressed in neurons in which ABHD17 members were knocked-down. The effect of ABHD17 was specific to PSD-95, as knockdown of ABHD17 members affected neither the palmitate cycling on GluA1 (Fig. 8G) nor the basal palmitoylation levels of GluA1, Gαq, and HRas (Fig. 8H).
Discussion
By screening a subset of serine hydrolases, this study identified three ABHD17 isoforms as PSD-95 depalmitoylating enzymes in neurons. However, we noted that PSD-95 depalmitoylation still occurred with slow kinetics in neurons, in which ABHD17 members were knocked-down (Fig. 8A, B, D). Reasons for this partial effect are probably due to: (1) incomplete knockdown (6∼17% mRNAs of ABHD17s remain; Fig. 3F), and (2) the molecular redundancy or compensation by other serine hydrolases, such as ABHD12, ABHD13, Lypla1, and Lypla2. Although the enzymatic activity of ABHD12 and 13 was much less than that of ABHD17 (Fig. 1A–C), it is noteworthy that ABHD12, 13, and 17 are originated from a common yeast ortholog, YNL320W. In addition, ABHD13 shows high homology with parasitic Toxoplasma protein TgPpt1 that regulates the Toxoplasma host cell invasion and depalmitoylates Toxoplasma proteins (Child et al., 2013), suggesting that ABHD13 compensates ABHD17's functions. Another genetic study in Caenorhabditis elegans has identified AHO-3 that is essential for starvation-induced thermotactic plasticity. Importantly, this abnormal behavior observed in AHO-3 mutant worms is rescued by human ABHD17B (Nishio et al., 2012). These genetic analyses suggest an important role of ABHD17 and ABHD13 in mammals. Future studies of their multiple knock-out mice will clarify their molecular redundancy, physiological substrates, and physiological significance.
Recent studies indicate enormous potential of the metabolic serine hydrolase superfamily for new enzymatic activity. For example, ABHD16A and ABHD12 were reported to function as a phosphatidylserine lipase and a lysophosphatidylserine lipase, respectively (Kamat et al., 2015). MAGL, ABHD6, and ABHD12 hydrolyze endocannabinoid 2-arachidonylglycerol (Blankman et al., 2007). Notum, a secreted protein, deacylates Wnt (ie, cleaves a carboxylester linkage between a serine residue and palmitoleic acid; Kakugawa et al., 2015). Because the α/β-hydrolase domain of Notum has homology with that of ABHD17, the future structural analysis of ABHD17 will clarify the different modes of action between thioesterases and carboxylesterases. We speculate that future characterization of metabolic serine hydrolases would not only discover the new enzymatic activity, but also contribute to providing new drug targets for various disorders.
While preparing this paper, Lin and Conibear (2015) reported that ABHD17 enhances the depalmitoylation of overexpressed NRas and PSD-95 in non-neuronal cells, although its role for endogenous NRas and PSD-95 was not tested. Our group also found that overexpression of ABHD17 reduces the palmitoylation level of overexpressed NRas and HRas, as well as PSD-95 in HEK293T cells (Fig. 2A; data not shown). However, the dynamic palmitate cycling on endogenous HRas was not readily detected in neurons (Fig. 7D), and ABHD17 did not depalmitoylate HRas in neurons (Fig. 5). One possibility for this discrepancy is due to different cellular contexts examined. Another is that overexpression of enzymes or substrates can result in artificial enzyme–substrate reactions that do not endogenously occur in native cells. In fact, fast-cycling palmitoylation events (ie, depalmitoylation-involved events) are rather exceptional in T-cell hybridoma cells (Martin et al., 2012). Therefore, identification of physiological depalmitoylating enzymes requires the analysis against endogenous substrate protein.
The APEGS assay developed here provides information about the palmitoylation stoichiometries and the number of palmitoylation sites, which cannot be obtained by current ABE and metabolic labeling methods. Advantageous features of the APEGS assay include: (1) versatile application to any biological samples (even tissue samples); (2) no need for protein purification (ie, high-yield and unbiased sample preparation); (3) reliably examining whether proteins of interest are palmitoylated or not if antibodies are available, in fact we identified novel palmitoyl-protein candidates, mGluR5 and vGluT1 in Figure 5; (4) studying the interplay between palmitoylation and other modifications, such as phosphorylation if modification specific antibodies are available; and (5) investigating the dynamic changes between palmitoylation states (non- to multiple palmitoylated). On the other hand, the present APEGS assay cannot be applied to high-throughput proteomic analysis of palmitoylated proteins. Therefore, both methods would work complementarily to clarify the whole picture of protein palmitoylation. In addition to palmitoylation, the principle of the PEG-switch assay has been used for the quantification of O-GlcNAcylation (Rexach et al., 2010) and cysteine oxidation (Makmura et al., 2001; Burgoyne et al., 2013). Given that cysteine residues are subjected to various modifications, such as nitrosylation and sulfhydration, the similar method could be useful for these modifications. In fact, a piece of data were recently published, reporting the palmitoylation states of phospholemman in ventricular myocytes (Howie et al., 2014). In addition, during the revision of this paper, another related paper was published in which the palmitoylation states of Ras, calnexin, and IFITM3 were examined in cultured cells (Percher et al., 2016).
The reversible nature of palmitoylation represents one of the most important aspects of protein palmitoylation. However, it remains unknown how many proteins actually undergo palmitate cycling in cells. Unexpectedly, only PSD-95 showed apparent palmitate cycling in neurons among proteins we tested (Fig. 7D). We cannot discount the possibility that our 2-BP treatment did not completely inhibit newly occurring palmitoylation of some proteins. However, as described above, a proteomic pulse-chase analysis reported that fast-cycling palmitoylation events are, as a whole, exceptional in T-cell hybridoma cells (Martin et al., 2012). These results suggest that palmitoylation is not always dynamic in vivo, but rather only a specific protein, such as PSD-95, undergoes dynamic palmitoylation cycles under the basal conditions. It is speculated that synaptic palmitoylation is distinctively regulated by various combination of DHHC-PATs and depalmitoylating enzymes in response to extracellular stimuli. Therefore, it is worthwhile to profile changes in the palmitoylation states of various neuronal proteins following synaptic activity, and to examine whether certain ABHD and Lypla proteins are involved in this process using the APEGS assay.
Mechanisms that control the activity of ABHD17 remain incompletely understood. Our data that the enzymatic activity and the postsynaptic targeting of ABHD17B require its N-terminal palmitoylation (Figs. 2F, 3D) suggest the interdependent regulation between DHHC-PATs and ABHD17. It is conceivable that the increased ABHD17 palmitoylation by DHHC-PATs could target ABHD17 to postsynaptic membranes and enhance the depalmitoylating activity. In turn, increased depalmitoylating activity of the membrane-anchored ABHD17 may reduce the autopalmitoylation of DHHC proteins, which is essential for their PAT activity (Fukata et al., 2004; Mitchell et al., 2010). Such a negative feedback loop between synaptic palmitoylating and depalmitoylating enzymes may keep the palmitoylation level of substrates within a certain range, contributing to the synaptic homeostasis. On the other hand, palmitate turnover on PSD-95 is augmented by enhanced synaptic activity (El-Husseini et al., 2002; Fukata et al., 2013). Therefore, whether and how ABHD17 activity is regulated by synaptic activity and whether ABHD17 is involved in the AMPA receptor-mediated synaptic transmission and plasticity represent an important future goal and require further studies. Furthermore, it might be worthwhile to examine whether the developmental changes in PSD-95 palmitoylation cycles might play a role in developmental synaptic plasticity. Our study opens the new avenue for understanding the whole picture of palmitate cycling in synaptic development and plasticity.
Footnotes
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (Grant 15H04279 and 15H01299 to Y.F. and Grant 26291045, 15H01570, 16H01371, and 16K14560 to M.F.); the Ministry of Health, Labour, and Welfare (Intramural Research Grant [H27-7] for Neurological and Psychiatric Disorders of NCNP to Y.F.); Takeda Science Foundation to Y.F.; and The Uehara Memorial, Ono Medical Research, The Naito, and Daiichi Sankyo Foundations to M.F. We thank Dr. B. Martin (University of Michigan) for helpful discussion; R. Kageyama for technical assistance; Drs. Y. Kubo and M. Tateyama (NIPS) for helpful discussion; Dr. R. Zechner (University of Graz, Austria) for cDNAs of mouse Pnpla6 and Pnpla7; Dr. V. Verkhusha (Albert Einstein College of Medicine) for cDNA of human transferrin receptor; Center for Radioisotope Facilities, Okazaki Research Facilities, NINS for technical support, and the members of the Fukata laboratory for their kind supports.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne JR, Oviosu O, Eaton P. The PEG-switch assay: a fast semi-quantitative method to determine protein reversible cysteine oxidation. J Pharmacol Toxicol Methods. 2013;68:297–301. doi: 10.1016/j.vascn.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Camp LA, Hofmann SL. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J Biol Chem. 1993;268:22566–22574. [PubMed] [Google Scholar]
- Chamberlain LH, Shipston MJ. The physiology of protein S-acylation. Physiol Rev. 2015;95:341–376. doi: 10.1152/physrev.00032.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child MA, Hall CI, Beck JR, Ofori LO, Albrow VE, Garland M, Bowyer PW, Bradley PJ, Powers JC, Boothroyd JC, Weerapana E, Bogyo M. Small-molecule inhibition of a depalmitoylase enhances Toxoplasma host-cell invasion. Nat Chem Biol. 2013;9:651–656. doi: 10.1038/nchembio.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D, Triller A. The dynamic synapse. Neuron. 2013;80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron. 1999;22:497–509. doi: 10.1016/S0896-6273(00)80705-9. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Green WN. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- Duncan JA, Gilman AG. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS) J Biol Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000a;290:1364–1368. doi: 10.1126/science.290.5495.1364. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Craven SE, Chetkovich DM, Firestein BL, Schnell E, Aoki C, Bredt DS. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol. 2000b;148:159–172. doi: 10.1083/jcb.148.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/S0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Iwanaga T, Fukata M. Systematic screening for palmitoyl transferase activity of the DHHC protein family in mammalian cells. Methods. 2006;40:177–182. doi: 10.1016/j.ymeth.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rumbaugh G, Huganir RL. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron. 2005;47:709–723. doi: 10.1016/j.neuron.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Howie J, Reilly L, Fraser NJ, Vlachaki Walker JM, Wypijewski KJ, Ashford ML, Calaghan SC, McClafferty H, Tian L, Shipston MJ, Boguslavskyi A, Shattock MJ, Fuller W. Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proc Natl Acad Sci U S A. 2014;111:17534–17539. doi: 10.1073/pnas.1413627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakugawa S, Langton PF, Zebisch M, Howell SA, Chang TH, Liu Y, Feizi T, Bineva G, O'Reilly N, Snijders AP, Jones EY, Vincent JP. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519:187–192. doi: 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat SS, Camara K, Parsons WH, Chen DH, Dix MM, Bird TD, Howell AR, Cravatt BF. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat Chem Biol. 2015;11:164–171. doi: 10.1038/nchembio.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kishore NS, Wood DC, Mehta PP, Wade AC, Lu T, Gokel GW, Gordon JI. Comparison of the acyl chain specificities of human myristoyl-CoA synthetase and human myristoyl-CoA:protein N-myristoyltransferase. J Biol Chem. 1993;268:4889–4902. [PubMed] [Google Scholar]
- Lin DT, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife. 2015;4:e11306. doi: 10.7554/eLife.11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Lord CC, Thomas G, Brown JM. Mammalian alpha beta hydrolase domain (ABHD) proteins: lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim Biophys Acta. 2013;1831:792–802. doi: 10.1016/j.bbalip.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makmura L, Hamann M, Areopagita A, Furuta S, Muñoz A, Momand J. Development of a sensitive assay to detect reversibly oxidized protein cysteine sulfhydryl groups. Antioxid Redox Signal. 2001;3:1105–1118. doi: 10.1089/152308601317203611. [DOI] [PubMed] [Google Scholar]
- Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods. 2012;9:84–89. doi: 10.1038/nchembio.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DD, Beauchamp E, Berthiaume LG. Post-translational myristoylation: Fat matters in cellular life and death. Biochimie. 2011;93:18–31. doi: 10.1016/j.biochi.2010.10.018. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Mitchell G, Ling Y, Budde C, Deschenes RJ. Mutational analysis of Saccharomyces cerevisiae Erf2 reveals a two-step reaction mechanism for protein palmitoylation by DHHC enzymes. J Biol Chem. 2010;285:38104–38114. doi: 10.1074/jbc.M110.169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio N, Mohri-Shiomi A, Nishida Y, Hiramatsu N, Kodama-Namba E, Kimura KD, Kuhara A, Mori I. A novel and conserved protein AHO-3 is required for thermotactic plasticity associated with feeding states in Caenorhabditis elegans. Genes Cells. 2012;17:365–386. doi: 10.1111/j.1365-2443.2012.01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percher A, Ramakrishnan S, Thinon E, Yuan X, Yount JS, Hang HC. Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proc Natl Acad Sci U S A. 2016;113:4302–4307. doi: 10.1073/pnas.1602244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol. 2010;6:645–651. doi: 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Plowman S, Rotblat B, Prior IA, Muncke C, Grainger S, Parton RG, Henis YI, Kloog Y, Hancock JF. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol. 2005;25:6722–6733. doi: 10.1128/MCB.25.15.6722-6733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R, Fukata Y, Noritake J, Iwanaga T, Perez F, Fukata M. Identification of G protein alpha subunit-palmitoylating enzyme. Mol Cell Biol. 2009;29:435–447. doi: 10.1128/MCB.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh DC, Duncan JA, Yamashita S, Michel T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca2+-calmodulin. J Biol Chem. 1999;274:33148–33154. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- Zhu J, Shang Y, Zhang M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nat Rev Neurosci. 2016;17:209–223. doi: 10.1038/nrn.2016.18. [DOI] [PubMed] [Google Scholar]