

Abstract
Levodopa-induced dyskinesia (LID) is a persistent behavioral sensitization that develops after repeated levodopa (l-DOPA) exposure in Parkinson disease patients. LID is a consequence of sustained changes in the transcriptional behavior of striatal neurons following dopaminergic stimulation. In neurons, transcriptional regulation through dynamic DNA methylation has been shown pivotal to many long-term behavioral modifications; however, its role in LID has not yet been explored. Using a rodent model, we show LID development leads to the aberrant expression of DNA demethylating enzymes and locus-specific changes to DNA methylation at the promoter regions of genes aberrantly transcribed following l-DOPA treatment. Looking for dynamic DNA methylation in LID genome-wide, we used reduced representation bisulfite sequencing and found an extensive reorganization of the dorsal striatal methylome. LID development led to significant demethylation at many important regulatory areas of aberrantly transcribed genes. We used pharmacologic treatments that alter DNA methylation bidirectionally and found them able to modulate dyskinetic behaviors. Together, these findings demonstrate that l-DOPA induces widespread changes to striatal DNA methylation and that these modifications are required for the development and maintenance of LID.
SIGNIFICANCE STATEMENT Levodopa-induced dyskinesia (LID) develops after repeated levodopa (l-DOPA) exposure in Parkinson disease patients and remains one of the primary obstacles to effective treatment. LID behaviors are a consequence of striatal neuron sensitization due to sustained changes in transcriptional behavior; however, the mechanisms responsible for the long-term maintenance of this cellular priming remain uncertain. Regulation of dynamic DNA methylation has been shown pivotal to the maintenance of several long-term behavioral modifications, yet its role in LID has not yet been explored. In this work, we report a pivotal role for the reorganization of DNA methylation in the development of LID and show that modification of DNA methylation may be a novel therapeutic target for use in preventing or reversing dyskinetic behaviors.
Keywords: DNA methylation, dyskinesia, l-DOPA
Introduction
Since its discovery, levodopa (l-DOPA) has remained the most effective pharmacologic treatment for Parkinson disease (PD). Although l-DOPA can produce dramatic improvement in PD symptoms, its long-term clinical utility is often limited by irreversible abnormal involuntary choreiform movements, termed levodopa-induced dyskinesia (LID), that are seen in ∼50% of patients within 5 years of beginning l-DOPA treatment (Poewe, 2009). Although the dyskinetic behaviors are only apparent during l-DOPA treatment, it is clear that the induced state of dopaminergic hypersensitivity is long-lasting and quickly reemerges after periods of treatment interruption (Bezard et al., 2001; Fahn et al., 2004; Brotchie, 2005). Recent studies in animal models have begun to provide mechanistic insight into LID, showing evidence for significant structural and synaptic plasticity. These studies have shown that fundamental to LID is a “priming effect”, whereby initial administrations of l-DOPA trigger a sensitized biochemical and transcriptional response in the striatum upon subsequent dopaminergic stimulation, resulting in the progressive and persistent dyskinetic behaviors (Brotchie, 2005; Nadjar et al., 2009). A variety of alterations in striatal function have been described as contributing to the cellular sensitization seen during LID expression, including enhanced glutamatergic signaling, increased ERK/CREB activity, l-DOPA-dependent gene expression, and heightened translational activity (Picconi et al., 2003; Eskow et al., 2007; Jenner, 2008; Crittenden et al., 2009; Santini et al., 2009; Murer and Moratalla, 2011; Södersten et al., 2014). Although these studies have provided insight in to the molecular and circuit-level mechanisms underlying LID expression, a key unanswered question still remains: Why is the neural sensitization seen in LID persistent, when delivery of l-DOPA is transient?
The prolonged duration of neuronal sensitization central to LID suggests that there are sustained changes in cellular transcriptional behavior. Epigenetic regulation of chromatin is a fundamental mechanism for inducing long-lasting changes in transcription by controlling the 3D accessibility of DNA to regulate cell-specific transcriptional networks and maintain long-term cellular memory (Ma et al., 2009; Day and Sweatt, 2010; Zovkic et al., 2013; He and Ecker, 2015; Schultz et al., 2015). Animal models of dyskinesia have shown evidence for dysregulated transcription factor activity that correlate to changes in histone post-translational modifications (Darmopil et al., 2009; Santini et al., 2009; Johnston et al., 2013; Södersten et al., 2014). While these effects likely contribute to LID, so far they have been found to be transient in nature and seem unlikely to account for the enduring effects of dopamine-dependent priming. DNA methylation results from the direct covalent modification of cytosine and is thought to be the most stable epigenetic modification. In the nervous system, dynamic regulation of DNA methylation at CpG islands (CGIs) has been shown pivotal to sustaining alterations in neuronal function in a variety of states, including the formation of place memory in the hippocampus and the development of addictive behaviors following cocaine administration (Anier et al., 2010; LaPlant et al., 2010; Colquitt et al., 2013; Li et al., 2014; Feng et al., 2015; Halder et al., 2016). Because of its sustained nature, DNA methylation is important for the maintenance of long-term neuronal and behavioral memory in many settings and is a logical candidate for the persistent behavioral sensitizations seen in LID.
Functionally, DNA methylation is thought to provide a silencing effect on local gene expression: increases in promoter methylation lead to decreased transcription, whereas promoter demethylation enhances gene expression. Given the essential role DNA methylation plays in the long-term transcriptional regulation following other models of neuronal plasticity, we hypothesized that dysregulation of dynamic DNA methylation would be important for the development and maintenance of striatal priming by l-DOPA. Using a rodent model, our studies show that the development of dyskinetic behaviors leads to the aberrant expression of DNA demethylation enzymes and a global reconfiguration of striatal methylation near many genes with established functional importance in LID. We also found that pharmacologic modulation of global DNA methylation could bidirectionally modify dyskinetic behaviors. Together, these findings indicate an essential role for dynamic DNA methylation in the cellular memory of exposure to l-DOPA and provide further insight into the mechanisms underlying the aberrant cellular sensitization seen in LID.
Materials and Methods
Animals.
Male Sprague Dawley rats, ∼60- to 90-d-old and weighing 180–200 g, were housed in pairs in plastic cages in an Association for Assessment and Accreditation of Laboratory Animal Care-approved animal care facility on a 12 h light/dark cycle with food and water available ad libitum. All procedures were performed in accordance with the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Surgical procedures.
One week after arrival, rats were given a unilateral dopamine lesion to the left medial forebrain bundle. Before surgery, rats were given injections of desipramine (25 mg/kg, i.p.) to protect norepinephrine neurons, and buprenorphine (0.03 mg/kg, i.p.) as preemptive analgesia. Animals were then anesthetized with 1%–2% isoflurane (Baxter Healthcare) mixed with oxygen (2 L/min) and placed in a stereotaxic apparatus (Kopf Instruments). Under aseptic conditions, rats were rendered hemiparkinsonian using unilateral injections of 6-OHDA (12 μg in 4 μl, Sigma) into the medial forebrain bundle (stereotaxic coordinates: anteroposterior, 1.8 mm from bregma, ±2 mm lateral from midline, and −8.6 mm from the dura). For RG-108 experiments, at the time of lesion surgery, rats were additionally implanted with subcutaneous Alzet minipumps (model 2004, Durect) connected to a unilateral guide cannula (Plastics One) targeting the dorsal striatum (stereotaxic coordinates: anteroposterior, 0 from bregma, ±3 mm lateral from midline, and −3.5 mm from the dura). To ensure that RG-108 did not alter lesion progression, additional polyethylene tubing was added to provide a 14 d prime of vehicle before RG-108 administration as DNA methyltransferase (DNMT) inhibitors are known to be cytotoxic to dopaminergic cells. Animals were given at least 7 d of postsurgical care, during which they received buprenorphine and wound care for pain management.
Pharmacology.
Daily injections commenced 3 weeks after 6-OHDA lesion with either vehicle or l-DOPA (6 mg/kg + benserazide, 15 mg/kg, s.c.) for 7 consecutive days. This 7 day paradigm ensures that dyskinesia is displayed on the first day of treatment in most rats and that stable dyskinesia is developed by day 7.
For methionine (MP Biochemicals) experiments, animals were injected subcutaneously with 0.78 g/kg l-methionine twice a day for 7 d before and the days including l-DOPA treatment. This was timed to ensure that peak change in methylation would coincide with the beginning of l-DOPA administration based upon previous results using this paradigm to increase DNA methylation in the striatum (LaPlant et al., 2010; Massart et al., 2015; Wright et al., 2015). During training, animals were injected with methionine at least 2–3 h before behavioral experiments.
For RG-108 (Sigma) experiments, we used 14 d of continuous intrastriatal delivery of 100 μm RG-108 (0.25 μl/h) dissolved in 5% hydroxypropyl β-cyclodextrin vehicle (w/v, Sigma). This dose of RG-108 has been found to decrease DNA methylation in vivo and has shown minimal neurotoxicity in previous studies (LaPlant et al., 2010; Day et al., 2013; Massart et al., 2015). RG-108 was chosen over other DNMT inhibitors, such as 5-azacytidine, because it inhibits methylation without incorporation into DNA and has a half-life of 20 d at 37°C.
Behavioral testing.
Two weeks after lesion, the forepaw adjusting steps test was performed for use as a behavioral correlate of unilateral dopamine depletion (Chang et al., 1999). Rats were held such that they had only one free forelimb; for each trial, rats were moved laterally across a table at a steady rate of 90 cm/10 s. Each stepping test consisted of 6 trials for each forepaw, alternating between forehand and backhand. To create a “percent intact” stepping score, the total number of steps with the lesioned forelimb were divided by the total number of steps with the intact forelimb and multiplied by 100. Lower scores indicate greater parkinsonian impairment. Percent intact scores were used to rank-order the rats according to degree of impairment and assign them to 2 equivalently impaired treatment groups.
The abnormal involuntary movements (AIMs) test is a metric of dyskinesia. Rats were monitored for AIMs using a procedure as previously described (Eskow et al., 2007). Following treatment with vehicle or l-DOPA (6 mg/kg, s.c.), rats were placed in clear, plastic cages and rated by trained observers for 1 min every 10 min over a 180 min period. Individual dyskinesia severity scores ranging from 0 (not present) to 4 (severe and not interruptible) were given for axial, limb, and orolingual (ALO) dyskinesias. The three AIM subtypes were summed to create a single ALO AIMs score for data analysis. Rotational behavior was assessed by the number of rotations made contralateral to the lesioned side during the 1 min observation period.
Nucleic acid isolation.
For all experiments, animals were killed by rapid decapitation after completion of the day 7 behavioral sessions. Brains were immediately removed, the dorsal striatum dissected, flash frozen on dry ice, and stored at −80°C. Striatal tissue samples were processed for nucleic acid isolation using the AllPrep DNA/RNA Mini Kit (QIAGEN) following the manufacturer's instructions. Nucleic acid concentrations were determined spectrophotometrically.
mRNA expression.
mRNA was reverse transcribed using the iScript RT-PCR kit (Bio-Rad). Specific intron-spanning primers were used to amplify cDNA regions for transcripts of interest (primer sequences available upon request). qPCR amplifications were performed in triplicate using an iQ5 real-time PCR system (Bio-Rad) at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s, and then incubation at 72°C for 10 min followed by real-time melt analysis to verify product specificity. Actin was used as an internal control for normalization using the ΔΔCt method (Livak and Schmittgen, 2001).
Global DNA methylation analysis.
We used Epigentek's Methylamp Global DNA Methylation Quantification Ultra Kit according to the manufacturer's instructions. Raw values were colorimetrically quantified and total methylation levels estimated by generating a standard curve from Epigentek's methylated DNA standard. Values are then represented as percent of methylation relative to the vehicle/unlesioned control.
Methylated DNA immunoprecipitation (MeDIP).
Methylated DNA immunoprecipitation was performed using a 5-methylcytosine antibody (4 μl per sample, mouse monoclonal, Epigentek #A-1014) as described previously with minor modifications (Day et al., 2013). A total of 1 μg of DNA per sample was removed and sheared using the Bioruptor Pico (Diagenode) 3 times (30 s ON, 90 s off) to obtain 300 to 600 bp fragments. The quality of DNA shearing was assessed by running 600 ng of sheared DNA in a 1% agarose gel. Sonicated DNA was incubated for 1 h with 4 μl 5-methylcytosine antibody and then methylated DNA was collected with protein A-coated magnetic beads (Invitrogen), washed (Bind Wash Buffer, Epimark kit, New England BioLabs), extracted for 2 h at 60°C with proteinase K in TE buffer with 1% SDS, and purified (QIAGEN PCR purification kit). Methylation at selected DNA regions was assayed via qPCR on an iQ5 real-time PCR system (Bio-Rad) at 95°C for 5 min, followed by 45 cycles of 95°C for 15 s and 60°C for 60 s, and then incubation at 72°C for 10 min followed by real-time melt analysis to verify product specificity. Ct values for IP samples were normalized to unprocessed (input) DNA. Gapdh, which did not change across samples, was used as an internal normalization control (MeDIP primer sequences available upon request). We also validated that MeDIP enriches methylated DNA fragments with little cross-enrichment of nearly identical nonmethylated DNA fragments. Synthetic methylated and nonmethylated control DNA fragments (1 pg each, Methyl Miner kit, Invitrogen) were spiked into biological samples before MeDIP, and levels of methylated and nonmethylated DNA capture were assayed via qPCR with primers specific for each synthetic sequence. We found that MeDIP enriched the methylated DNA fragment >200-fold compared with the nonmethylated fragment.
Reduced representation bisulfite sequencing.
A total of 1 μg of gDNA sample was used for library construction. Bisulfite-converted DNA libraries were produced and adaptor ligated, and single-end reads were sequenced on an Illumina HiSeq-2500 following library QC. Bisulfite conversion efficiency was >98% for all of the samples and we obtained an average of 68 million reads per library. Quality control on raw reads was performed with FastQC (version 0.10.1, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc), and adaptor trimming and removal of trimmed reads shorter than 20 bp was performed with Trim Galore (version 0.3.7, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore). Trimmed reads were mapped to the UCSC Rattus norvegicus rn5 genome with the methylation-aware mapper bismark (version 0.13.0). Samtools (version 0.1.19–96b5f2294a) used to sort SAM files produced by bismark and deduplicate reads. SAM files were analyzed using MethylKit (version 0.9.2) in R (Akalin et al., 2012). Reads were filtered based on coverage, with a cutoff of at least three reads per site, and normalized for coverage before analysis. The genome was tiled at 250 bp, and regions were counted if they contained at least 2 identified CpGs per tile. Differential methylation was defined as a sliding linear model correct p value of <0.01 and, for highly dynamic regions, included at least a 5% change. All GO analyses were performed by WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt/) with default settings.
Statistical analysis.
Data analysis was performed in R and graphed in GraphPad Prism for Mac OS X (version 6.00; GraphPad Sofware) or MATLAB (version R2015b, The MathWorks). Normality was formally tested and verified where appropriate. Statistical significance was designated at p < 0.05 for all analyses. Statistical significance was measured using two-way ANOVAs for all biochemical results. Repeated-measures ANOVAs were used for all behavioral analysis. For ANOVAs, post hoc comparisons were performed when appropriate. For correlations between methylation change and AIM behaviors, data were analyzed using Spearman's rank correlation coefficient. For reduced representation bisulfite sequencing (RRBS) data, pairwise comparisons between differential region and gene expression groups were performed using Fisher's exact test using the whole rat genome as universe. Any grid containing p > 0.05 is considered nonsignificant, and its odds ratio is not shown. Additional data analysis was performed in R and graphed in GraphPad Prism for Mac OS X (version 6.00; GraphPad Software) or MATLAB (version R2015b, The MathWorks).
Results
Chronic l-DOPA treatment in a dopamine-depleted context leads to the aberrant expression of DNA demethylases
To examine the role of DNA methylation in LID, we rendered rats hemiparkinsonian through unilateral lesioning of nigrostriatal dopaminergic neurons by injection of 6-OHDA into the medial forebrain bundle. This lesion paradigm leads to a consistent phenotype, including the unilateral loss of >95% of striatal dopaminergic axons as well as significant motor abnormalities. After a 3 week recovery period, animals were treated with l-DOPA for 7 consecutive days, leading to the progressive development of abnormal involuntary movements (assessed by ALO score) and a sensitization in rotational behavior (Fig. 1a) (Santini et al., 2009; Heiman et al., 2014; Suárez et al., 2014). After behavioral measures on the seventh day, we isolated RNA from the striatum and confirmed the aberrant transcriptional expression of Arc and FosB selectively in the dopamine-depleted striatum, as previously reported (Fig. 1b) (Santini et al., 2009; Heiman et al., 2014; Suárez et al., 2014; Charbonnier-Beaupel et al., 2015). We then investigated the expression of enzymes involved in active DNA methylation and demethylation, targets not previously examined in this model. We found selective enhancements in the expression of DNA demethylases, Tet3 and Gadd45b, whereas the expression of all three DNMTs remained unaltered (Fig. 1c–e) (Ma et al., 2009; LaPlant et al., 2010; Guo et al., 2011; Kaas et al., 2013; Colquitt et al., 2014; Li et al., 2014; Feng et al., 2015; Park et al., 2016). Collectively, these observations point to the possibility that activation of demethylating mechanisms may contribute to the sustained changes in gene expression underlying the development of LID.
Figure 1.

Aberrant regulation of DNA methylation-modifying enzymes. a, Behavioral data demonstrating that repeated l-DOPA treatment induces a sensitized dyskinetic response, measured by ALO score (top) or rotational behavior (bottom). *p < 0.05 versus l-DOPA-treated day 1 (repeated-measures ANOVA). n = 8 per group. bi–iii, l-DOPA induces enhanced expression of immediate-early genes. ci–iii, DNA methyltransferase expression is not affected by dyskinesia development. di–iii, TET3 is aberrantly expressed in dyskinetic animals. ei–iii, Gadd45b and Gadd45 g expression is enhanced following dyskinesia development (two-way ANOVA; Tukey's post hoc tests; n = 6 or 7 per group). Error bars indicate SEM. *p < 0.05 for interaction between lesion and treatment.
LID development leads to the dynamic DNA methylation on a locus-specific level
To directly examine alterations in dorsal striatal DNA methylation, we measured total cytosine methylation but found l-DOPA had minimal effect globally (Fig. 2a). Searching for more selective modifications to DNA methylation, we used methylated DNA immunoprecipitation-qPCR (MeDIP) to target the promoter regions of LID-related genes. We focused on genes known to show aberrant expression in LID and have previously shown dynamic DNA methylation in other models of dopamine-dependent striatal plasticity (Day et al., 2013; Heiman et al., 2014; Massart et al., 2015). We found the promoter regions of Arc, FosB, and Reelin exhibited lesion-dependent methylation changes similar to the locus-specific methylation effects previously reported in PD patient samples (Fig. 2c) (Arc: Unlesioned(UL)/Sal = 1.00 ± 0.04, UL/l-DOPA = 0.95 ± 0.10, Lesion(L)/Sal = 1.31 ± 0.13, L/l-DOPA = 1.24 ± 0.08; FosB: UL/Sal = 1.00 ± 0.09, UL/l-DOPA = 0.70 ± 0.07, L/Sal = 0.32 ± 0.22, L/l-DOPA = 0.091 ± 0.24; Reelin: UL/Sal = 1.00 ± 0.08, UL/l-DOPA = 0.81 ± 0.061, L/Sal = 0.68 ± 0.26, L/l-DOPA = 0.45 ± 0.17). (Jowaed et al., 2010; Pihlstrøm et al., 2015). Interestingly, decreases in methylation at the promoter regions of Ntrk2, Dab1, and Esr1 were found selectively in the striatum ipsilateral to 6-OHDA lesion following chronic l-DOPA treatment (Fig. 2d) (Ntrk2: UL/Sal = 1.00 ± 0.03, UL/l-DOPA = 1.03 ± 0.05, L/Sal = 0.95 ± 0.06, L/l-DOPA = 0.76 ± 0.05; Dab1: UL/Sal = 1.00 ± 0.05, UL/l-DOPA = 1.03 ± 0.05, L/Sal = 0.94 ± 0.04, L/l-DOPA = 0.73 ± 0.05; Esr1: UL/Sal = 1.00 ± 0.07, UL/l-DOPA = 1.04 ± 0.07, L/Sal = 1.00 ± 0.05, L/l-DOPA = 0.54 ± 0.09). Together, these data show that locus-specific changes to striatal DNA methylation are coincident with LID development and further implicate dynamic DNA methylation as a mechanism contributing to the persistent sensitization of motor behaviors.
Figure 2.
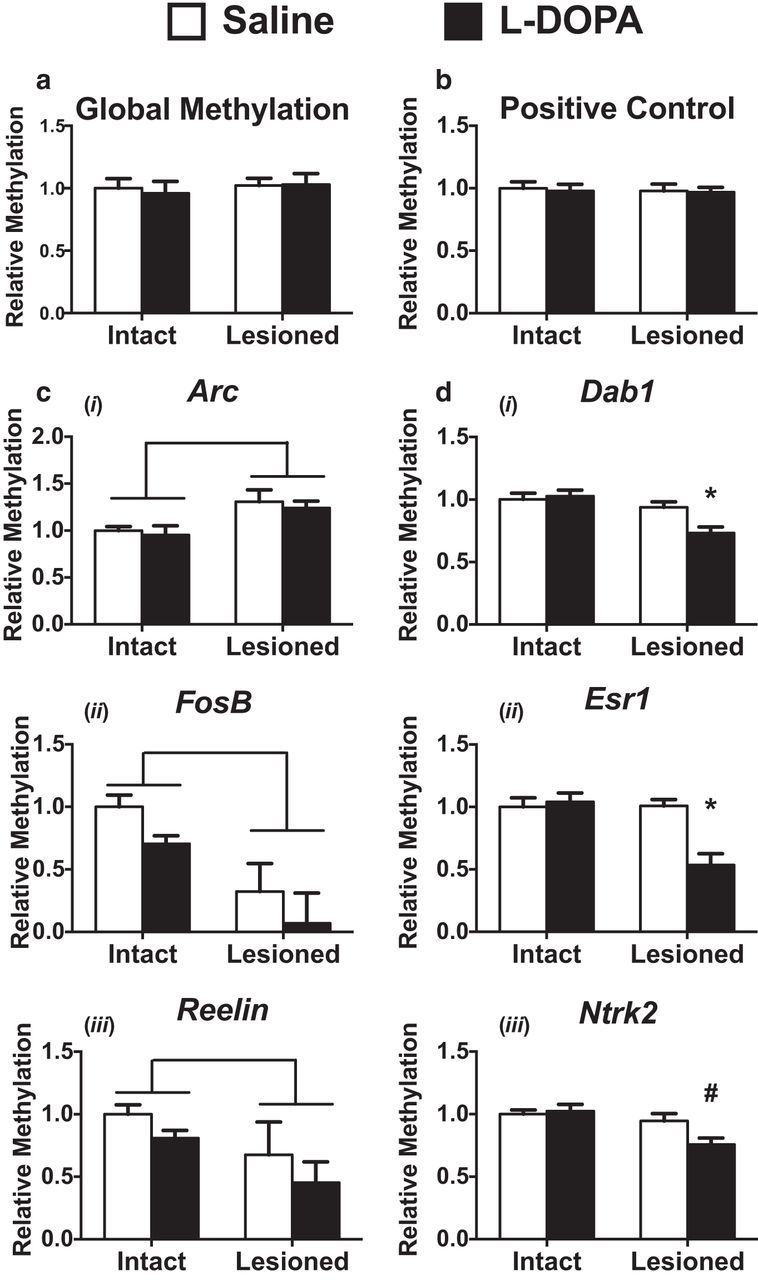
Aberrant regulation of DNA methylation following the development of LID is in a locus-specific manner. a, LID development has no effect on global methylation levels as measured by ELISA (two-way ANOVA; n = 7 per group). b, MeDIP enrichment is similar across all samples for a spiked-in positive control. ci–iii, MeDIP at the promoter regions of immediate early genes aberrantly transcribed in LID show lesion-dependent effects (two-way ANOVA). Brackets indicate p < 0.05 for a main effect of lesion (n = 3 or 4 per group). di–iii, MeDIP at regions previously shown to have dynamic methylation in other models of striatal plasticity display reductions in DNA methylation at promoter regions in a l-DOPA and lesion-dependent manner (two-way ANOVA; Tukey's post hoc tests; n = 7 per group) (Massart et al., 2015). Error bars indicate SEM. *p < 0.05 versus all other groups. #p < 0.05 versus both intact groups.
LID induces a global rearrangement of the striatal methylome
To evaluate the breadth of dynamic methylation following LID development, we investigated methylation genome-wide using RRBS. RRBS captures the majority of genomic methylation by preferentially sequencing areas rich in CpGs, the primary methyl context, and determining methylation levels at base-pair resolution (Meissner et al., 2005). Because of the difficulty of interpreting the functional effects of methylation changes at solitary cytosines, we analyzed these data with a regional approach using MethylKit, tiling the entire genome into 250 bp bins and including only those tiles that had at least 2 read CpGs (Akalin et al., 2012). Differentially methylated regions (DMRs) were identified by comparing the dopamine-depleted striata of l-DOPA- and saline-treated animals followed by filtering for a sliding linear model corrected p value of <0.01. This analysis identified ∼20,000 DMRs, including many regions near genes known to be aberrantly transcribed following LID development (Fig. 3a) (Heiman et al., 2014; Södersten et al., 2014; Charbonnier-Beaupel et al., 2015). These changes were found to be evenly distributed across the genome and unrestricted to specific chromosomal regions (Fig. 3b). As predicted by the enhanced expression of DNA demethylation enzymes (Fig. 1b), significantly more DMRs displayed decreased rather than increased methylation (Fig. 3c). Additionally, the degree of methylation change in many tiled regions correlated with dyskinesia severity, including those near genes whose expression has been previously directly associated with LID behaviors (Fos, Grin2a, Nedd4l, and Rasgrp1; Fig. 3d) (Santini et al., 2009; Heiman et al., 2014; Charbonnier-Beaupel et al., 2015).
Figure 3.

Development of LID induces a global rearrangement of the striatal methylome. a, DMRs were identified between saline (n = 8) and l-DOPA (n = 7) treated striata following 6-OHDA lesion using MethylKit and filtered for a corrected p value of <0.01. Regions near genes with established roles in LID were highlighted with red dots to support the functional relevance of the identified changes. b, DMRs were found to be evenly distributed across the genome, unbiased to chromosome. c, A direct correlation was observed between absolute methylation difference and distance from the transcriptional start site (Pearson's correlation coefficient, r = 0.138209, p = 2.2 × 10−16). d, Changes in methylation levels at DMRs nonparametrically correlated with dyskinetic behavioral changes (Spearman's correlation). Regions near genes whose expression has been directly correlated with dyskinetic behaviors were emphasized with red dots to highlight their relevance in LID. ei–iv, Using MeDIP on an independent cohort of animals, regions identified by genome-wide analysis were confirmed to exhibit dynamic demethylation in a lesion- and l-DOPA-dependent manner near genes aberrantly transcribed in LID (two-way ANOVA; Tukey's post hoc tests; n = 7 per group). *p < 0.05 versus all other groups. #p < 0.05 versus both intact groups.
To determine the functional relevance of the observed striatal methylation changes, we decided to focus on those DMRs exhibiting at least 5% change and designated these as highly dynamic regions (HDRs; ∼4000 DMRs; Fig. 4a). Although RRBS enriches for CpG dense areas of the genome, HDRs were mainly found outside of CGIs and shores, indicating a relative stability in these methyl-dense regions pivotal to cellular identity (Fig. 4b,c). Functionally, these findings fit with the cellular pathology observed in LID as basic neuronal behavior is maintained in the dopamine-depleted striatum, but repeated l-DOPA leads to a sensitization in select genes following dopaminergic stimulation. The majority of HDRs were found in intragenic and extragenic regions, areas thought to be important for the regulation of transcriptional expression levels and alternative splicing, whereas few promoters exhibited significant change (Fig. 4b,c) (Colquitt et al., 2013; Li et al., 2014; Feng et al., 2015). Correspondingly, we found a direct correlation between the effect size of methylation changes and increasing distances from transcriptional start sites (Fig. 3c), further indicating that the transcriptional priming observed in the striatum following repeated l-DOPA stimulation may be due to the dynamic regulation of epigenetic modifiers in gene regulatory regions (Santini et al., 2009; Heiman et al., 2014; Charbonnier-Beaupel et al., 2015). Based upon proximity, a list of 4005 genes adjacent to HDRs was created for further investigation. Gene ontology analysis revealed an enrichment for processes relevant to neuronal plasticity and similar to those seen in other studies of activity-dependent dynamic DNA methylation (Fig. 4d) (Colquitt et al., 2013; Li et al., 2014; Feng et al., 2015; Massart et al., 2015; Halder et al., 2016). We further compared our HDR gene list with a recently published study profiling transcriptional changes in D1- or D2-receptor-expressing medium spiny neurons following LID development (Heiman et al., 2014). We found a significant enrichment between these two datasets when comparing all 3971 genes aberrantly regulated in LID, and found our methylation dataset to be specifically enriched for genes that increased their expression in D1 neurons (571 of 2667 aberrantly transcribed genes were near HDRs) or decreased expression in D2 neurons (52 of 165 aberrantly transcribed genes were near HDRs) (Fig. 4e).
Figure 4.

Highly dynamic DMRs are involved in transcriptional regulation and synaptic plasticity. a, Heatmap of highly differentially methylated regions (at least 5% change) showing a significant enrichment of DMRs exhibiting active demethylation (Fisher's exact test, p < 0.0001). Color key represents the direction and percentage change in methylation identified. b, Highly dynamic DMRs are outside of CGIs or shores and instead enriched extragenically and intragenically. Left graphs, Distribution of all regions identified by our analysis. Right graphs, Those regions containing HDRs. c, When normalized for the genomic coverage following RRBS enrichment, the highest proportion of HDRs are in intragenic and extragenic regions, whereas few CGIs display dynamic change. d, Ontological analysis of genes associated with DMRs shows enrichment for processes shown previously relevant to neuronal plasticity and synaptic function. e, Candidate gene targets associated with the newly identified DMRs are enriched for genes previously shown to be aberrantly transcribed in LID (Fisher's exact test) (Heiman et al., 2014).
To validate our genome-wide results, we created a new group of dyskinetic animals and used MeDIP to selectively target DMRs identified by RRBS. Candidate DMRs representing extragenic, intronic, and promoter regions were selected near genes exhibiting aberrant expression following the development of LID-dependent plasticity (Arc, FosB, Nedd4l, Rasip) (Heiman et al., 2014; Charbonnier-Beaupel et al., 2015) (Arc: UL/Sal = 1.00 ± 0.04, UL/l-DOPA = 1.04 ± 0.04, L/Sal = 1.00 ± 0.04, L/l-DOPA = 0.78 ± 0.05; FosB: UL/Sal = 1.00 ± 0.07, UL/l-DOPA = 1.08 ± 0.04, L/Sal = 1.04 ± 0.04, L/l-DOPA = 0.73 ± 0.05; Nedd4l: UL/Sal = 1.00 ± 0.06, UL/l-DOPA = 1.03 ± 0.06, L/Sal = 0.94 ± 0.06, L/l-DOPA = 0.65 ± 0.08; Rasip: UL/Sal = 1.00 ± 0.04, UL/l-DOPA = 1.04 ± 0.03, L/Sal = 0.93 ± 0.03, L/l-DOPA = 0.80 ± 0.05). All targeted regions exhibited l-DOPA and lesion-dependent demethylation in accordance with sequencing results (Fig. 3e).
Experimental parkinsonism leads to striatal methylation changes
Although LID development is dependent in part to repeated l-DOPA exposure, expression of dyskinetic behaviors on the initial day of treatment indicates that dopaminergic denervation is an important contributor to the striatal priming underlying LID. To determine whether 6-OHDA lesion leads to methylation changes that may contribute to LID development, we used RRBS to compare the striatal methylation changes between the lesioned and unlesioned striatum in saline-treated animals. Analyzing the data using the same region based analysis as described previously, we were able to identify ∼27,000 DMRs in the striatum following dopaminergic denervation (Fig. 5a). These changes were again found to be evenly distributed across the genome, and significantly more DMRs displayed decreases to DNA methylation (Fisher's exact test, p < 0.0001). Similar to the l-DOPA-dependent effects, the regions displaying the most change due to lesion effect were found not in the promoter regions or CGIs, but rather within introns and extragenically where they could contribute to the aberrant transcriptional regulation following initial l-DOPA treatment (Fig. 5b) (Södersten et al., 2014; Charbonnier-Beaupel et al., 2015).
Figure 5.

Parkinsonism alters dorsal striatal methylation at genomic regions affected by LID development. a, DMRs were identified between intact (n = 8) and lesioned (n = 7) striata following 6-OHDA treatment using MethylKit and filtered for a corrected p value of <0.01. b, Highly dynamic DMRs (>5% change) are outside of CGIs or shores and instead enriched extragenically and intragenically when normalized for genomic coverage during RRBS enrichment. c, There is a significant overlap between the DMRs exhibiting dynamic DNA methylation following both dopaminergic lesion and l-DOPA treatment. d, A strong correlation was observed between l-DOPA and lesion-dependent methylation effects in the striatum (Pearson's correlation coefficient, r = 0.5590431, p = 2.2 × 10−16). However, a subset of regions (highlighted in blue) exhibited demethylation following both effects. e, Ontological analysis of genes near DMRs showing dynamic demethylation following l-DOPA treatment and 6-OHDA lesion shows enrichment for processes relevant to neuronal plasticity and function. f, Candidate gene targets associated with the DMRs demethylated by lesion- and l-DOPA-dependent effects are enriched for genes previously shown to be aberrantly transcribed in D1 neurons following LID development (Fisher's exact test) (Heiman et al., 2014).
To determine whether any of the methylation changes due to the loss of dopaminergic input were related to those found following repeated l-DOPA treatment, we compared our two datasets on lesion- and l-DOPA-dependent effects finding a significant overlap in the genomic regions undergoing dynamic DNA methylation (Fig. 5c) (Fisher's exact test, p < 0.0001). Interestingly, these overlapping regions were found to have a strong negative correlation between the lesion- and l-DOPA-dependent effect, indicating that many of lesion-dependent striatal methylation effects are actually reversed following l-DOPA treatment (Fig. 5d). However, there remained a subset of regions showing significant demethylation effects following both the loss of dopaminergic innervation and l-DOPA treatment; thus, these are regions where dopamine depletion reduces methylation, and this change is further exacerbated by l-DOPA treatment. Focusing on this subset of DMRs, we found them to be near genes heavily enriched for neuronal plasticity, and to represent the same gene ontological processes previously identified in our analysis of l-DOPA-dependent HDRs as well as other studies of experience-dependent neuronal plasticity (Fig. 5e) (Heiman et al., 2014; Feng et al., 2015; Halder et al., 2016). Specifically focusing on transcripts known to be aberrantly expressed following LID development, we found that the 869 genes near regions demethylated by lesion-dependent and l-DOPA-dependent processes were strongly enriched for those transcripts known to increase their transcription in D1 neurons following l-DOPA treatment (97 of 869 demethylated regions were near genes aberrantly transcribed) (Fig. 5f) (Heiman et al., 2014).
Modulation of global DNA methylation is able to bidirectionally modulate dyskinetic behaviors
Collectively, these data predict that modifications increasing methylation would reduce LID development, whereas interventions that decrease DNA methylation would exacerbate dyskinetic behaviors. To test these hypotheses, we administered bidaily methionine injections in a regimen previously shown to enhance DNA methylation, beginning 1 week before and continuing during treatment with l-DOPA (LaPlant et al., 2010; Massart et al., 2015; Wright et al., 2015). We found that methionine supplementation in this paradigm decreased LID development (Fig. 6a). To verify methionine's effect on DNA methylation, we then used MeDIP to study several locations that undergo LID-dependent demethylation. We found that several of these displayed increased DNA methylation following methionine supplementation (Fig. 6b) (Ntrk2: UL/l-DOPA = 1.00 ± 0.05, UL/Met = 1.02 ± 0.03, L/l-DOPA = 0.75 ± 0.03, L/Met = 0.94 ± 0.03; Dab1: UL/l-DOPA = 1.00 ± 0.06, UL/Met = 1.05 ± 0.04, L/l-DOPA = 0.78 ± 0.04, L/Met = 0.95 ± 0.02; FosB: UL/l-DOPA = 1.00 ± 0.05, UL/Met = 0.99 ± 0.03, L/l-DOPA = 0.47 ± 0.03, L/Met = 0.66 ± 0.02; Nedd4l: UL/l-DOPA = 1.00 ± 0.05, UL/Met = 1.04 ± 0.05, L/l-DOPA = 0.72 ± 0.06, L/Met = 0.98 ± 0.05). Chronic striatal infusion of RG-108, a selective DNMT inhibitor, was achieved using implanted mini osmotic pumps and timed so that drug delivery started 1 week before and continued throughout l-DOPA treatment (LaPlant et al., 2010; Massart et al., 2015). We found that RG-108 enhanced the development and expression of LID (Fig. 6c). We assessed global striatal methylation levels at the end of the experiment and found them markedly reduced by RG-108, providing evidence of on-target efficacy. We also used MeDIP to examine locus-specific methylation effects following RG-108 treatment at regions known to undergo demethylation following LID development and found that four of the regions previously identified did indeed exhibit reduced methylation after RG-108 treatment (Fig. 6d) (Ntrk2: UL/l-DOPA = 1.00 ± 0.03, UL/RG-108 = 1.01 ± 0.03, L/l-DOPA = 0.85 ± 0.01, L/RG-108 = 0.72 ± 0.02; Dab1: UL/l-DOPA = 1.00 ± 0.02, UL/RG-108 = 1.05 ± 0.02, L/l-DOPA = 0.81 ± 0.03, L/RG-108 = 0.69 ± 0.02; FosB: UL/l-DOPA = 1.00 ± 0.03, UL/RG-108 = 0.98 ± 0.03, L/l-DOPA = 0.81 ± 0.01, L/RG-108 = 0.70 ± 0.01; Arc: UL/l-DOPA = 1.00 ± 0.03, UL/RG-108 = 1.01 ± 0.03, L/l-DOPA = 0.85 ± 0.01, L/RG-108 = 0.69 ± 0.01). Together, these data provide conclusive functional evidence that a reorganization of the striatal methylome is necessary for LID development.
Figure 6.

Global modifications of DNA methylation are able to bidirectionally influence LID development and expression. a, Twice daily administration of methionine (7.2 mmol/kg, s.c.), at doses known to enhance global methylation, hinders LID ALO development and expression. *p < 0.05 versus vehicle (two-way ANOVA; Tukey's post hoc tests). n = 7 per group. bi–iv, Methionine supplementation increased DNA methylation at regions undergoing LID-dependent demethylation (two-way ANOVA; Tukey's post hoc tests). n = 7 per group. c, Chronic striatal infusion of RG-108 (100 μm), a DNMT inhibitor, enhances LID behavioral sensitization. p < 0.05 versus vehicle (two-way ANOVA; Tukey's post hoc tests). n = 10 per group. di–iv, RG-108 decreased DNA methylation at regions undergoing LID-dependent demethylation (two-way ANOVA; Tukey's post hoc tests). n = 7 per group. *p < 0.05 versus all groups. #p < 0.05 versus all groups. Error bars indicate SEM.
Discussion
Our results reveal an essential role for dynamic striatal DNA methylation in the development of LID. We observed extensive locus-specific changes in DNA methylation, including a preponderance of demethylation, in the dorsal striatum following the development of dyskinetic behaviors in our animal model system. Changes in DNA methylation were concentrated in putative regulatory regions of many genes known to be aberrantly transcribed following l-DOPA exposure and enriched for genes relevant to mechanisms of synaptic plasticity. In the areas of the genome exhibiting the highest levels of effect, the magnitudes of change to methylation were strongly correlated with dyskinetic behaviors. Further demonstrating the importance of changes in DNA methylation to LID development, we show that manipulation of methylation levels using pharmacological treatments can bidirectionally modify behavioral sensitization to l-DOPA in a PD animal model. Collectively, these data indicate that striatal dynamic DNA methylation is essential to LID development.
Previous studies of the molecular mechanisms underlying the striatal priming effect observed in LID have shown a pivotal role for aberrant transcriptional regulation (Bezard et al., 2001; Brotchie, 2005; Heiman et al., 2014; Charbonnier-Beaupel et al., 2015). During dyskinesia development, one of the key mechanisms to striatal sensitization is enhanced MAPK/ERK signaling leading to increased CREB and AP-1-dependent transcription. The alterations in AP-1 transcription factor activity are known to lead to downstream transcriptional and synaptic changes that are not well compensated for by homeostatic mechanisms and contribute to pathologic neuronal activity (Engeln et al., 2013; Suárez et al., 2014; Charbonnier-Beaupel et al., 2015). Attempts to explain the aberrant transcription in LID through alterations to histone modifications have shown a role for increased histone phosphoacetylation; however, the effects observed have been transient in nature, and pharmacologic treatments inhibiting histone acetyltransferase activity have led to counterintuitive results on dyskinetic behaviors (Nicholas et al., 2008; Darmopil et al., 2009; Santini et al., 2009; Johnston et al., 2013). Thus, although histone modifications may be part of the pathway through which LID is expressed, they are insufficient to fully explain the long-term nature of l-DOPA-induced priming and maintenance of LID. Our data indicate that dynamic regulation of DNA methylation contributes to the long-term cellular sensitization cardinal to LID and advances our understanding of the aberrant epigenetic regulation necessary for dyskinesia development. The aberrant expression of DNA demethylases TET3 and GADD45b provides further insight into the observed effects on DNA methylation (Fig. 1b) as AP-1 transcription factors are known to help localize these enzymes across the genome leading to targeted chromatin remodeling, colocalization with CREB binding protein, and increased histone acetylation (Biddie et al., 2011; Damez-Werno et al., 2012; Ostuni et al., 2013; Heiman et al., 2014). A recent study investigating GADD45's role in LID has similarly shown enhanced expression of GADD45b following dyskinesia development; interestingly, however, they also found that genetic deletion of GADD45b expression leads to an enhancement in dyskinetic behaviors and overexpression of GADD45b leads to reduced LID, suggesting that this upregulation may be compensatory (Park et al., 2016). As we also found upregulation of TET3 following dyskinesia development, it will be of particular importance to assess the role of this family of demethylases relative to other components of the systems regulating gene methylation to identify specific therapeutic targets for the dynamic DNA demethylation contributing to LID.
Several lines of clinical and experimental evidence have shown that the extent of dopaminergic degeneration is one of the primary risk factors in LID development (Jenner, 2008; Poewe, 2009). It remains well established that LID requires striatonigral degeneration, and many studies have documented significant structural and synaptic plasticity following the loss of striatal dopamine (Picconi et al., 2003; Suárez et al., 2014). Our data expand upon these findings by showing that dopaminergic denervation induces an extensive reorganization of DNA methylation across the genome (Fig. 5a) and similar to the locus-specific methylation changes previously documented in PD patient samples (Jowaed et al., 2010; Pihlstrøm et al., 2015). Interestingly, we found that l-DOPA treatment reversed many of the alterations in striatal DNA methylation caused by 6-OHDA lesion but had additive effects on dynamic demethylation at a select subset of genomic regions (Figs. 2c, 5d). These regions were in close proximity to genes aberrantly transcribed following l-DOPA treatment in dyskinetic animals, likely indicating that the loss of dopaminergic signaling leads to DNA methylation changes that are facilitatory to striatal sensitization (Fig. 5e,f). Together, these findings add to the growing consensus that the loss of striatonigral dopamine leads to a maladaptive neuronal state in the striatum that is unique in dopaminergic responsiveness and contributory to the molecular adaptations necessary for dyskinesia development.
The striatum is primarily composed of direct pathway D1-receptor expressing neurons and indirect pathway D2-receptor expressing neurons that are thought to have antagonistic effects on motor behaviors (Albin et al., 1989; Gradinaru et al., 2009). Many of the cellular and molecular adaptations following chronic l-DOPA treatment have been found in D1-receptor expressing neurons, known to be kinetogenic following dopaminergic stimulation and strongly suggesting their importance to LID development (Darmopil et al., 2009; Fieblinger et al., 2014; Heiman et al., 2014). Although our data are sampled from a heterogeneous cellular population, D1-expressing neurons are known to undergo the majority of transcriptional change, and we found an extensive enrichment of DMRs near those genes known to be aberrantly transcribed in this neuronal subset (Figs. 3e, 5f) (Heiman et al., 2014). Recent studies on experience-dependent plasticity have additionally shown that glial cell populations undergo relatively few changes in DNA methylation or histone modifications during behavioral memory formation, indicating that the effects we have observed are likely due to changes in neuronal DNA methylation (Guo et al., 2011; Feng et al., 2015; Halder et al., 2016). From these data, we hypothesize that the majority of DNA methylation differences observed in LID are specifically within D1 expressing striatal neurons. Recent advances in neuronal nuclear profiling will enable future studies to directly evaluate the contributions of different striatal cellular populations to the epigenetic changes observed following LID development and will help clarify their roles in this maladaptive striatal plasticity (Mo et al., 2015; Halder et al., 2016).
The majority of DNA methylation present in the genome is found within CpG contexts, where it is clustered into CGI thought important to transcriptional silencing and cellular identity. In this study, we used RRBS, a method enriching for CpG-containing regions, but somewhat surprisingly found that many of the changes induced by chronic l-DOPA were outside of the CpG-dense promoter regions and not within CGIs (Figs. 4b, 5b). Instead, we found that induction of LID induces targeted changes in intronic and extragenic gene-regulatory regions (Figs. 4b, 5b). These areas are thought to control transcript expression levels and alternative splicing, potentially explaining the complex transcriptional dysfunction observed in LID (Malik et al., 2014; Feng et al., 2015; Halder et al., 2016; Joo et al., 2016). These data would fit within our broader understanding of genomic function, as simple promoter accessibility is likely insufficient to explain the wide variety of alterations in transcriptional behavior following LID development. For example, LID priming is known to be dependent on both the enhanced expression and alternative splicing of FosB causing sustained protein expression and increased AP-1 transcriptional activity (Nestler et al., 2001; Santini et al., 2009; Heiman et al., 2014). The importance of FosB activity to LID development is of additional interest in light of the present results as the AP-1 family are known pioneer transcription factors capable of remodeling the enhancer landscape to cause long-term alterations in DNA methylation and gene expression levels (Biddie et al., 2011; Damez-Werno et al., 2012; Ostuni et al., 2013; Heiman et al., 2014). The identified changes in DNA methylation are likely to be dependent on pathologic pioneer transcription factor activity controlling locus-specific epigenetic remodeling near enhancer regions leading to the induction of LID. Overall, these data indicate that the striatal methylome undergoes an extensive reorganization following LID development and are consistent with the growing hypothesis that the regulation of neuronal enhancers is pivotal to the formation of long-term behavioral memory.
Clearly, better means of preventing or reversing LID are necessary in current PD treatment. The ability of pharmacologic treatment with methionine supplementation or RG-108 to bidirectionally modify dyskinetic behaviors creates a potential opportunity for methylation-based epigenetic therapeutics in PD (Fig. 6). Our data suggest that targeting mechanisms to increase striatal methylation could hinder LID development and have a substantial effect on PD clinical outcomes. Although methionine supplementation is likely to be too nonselective to be clinically useful, several new drugs targeting epigenetic modifiers have begun clinical trials as chemotherapeutics and neurotherapeutics that could potentially be used at lower doses as adjunctive therapies for PD treatment. Additionally, the identification of selective DNA methylation modifications in LID opens the door for more targeted approaches to dysregulated genomic regions using locus-specific chromatin remodeling with TALENs or the Crispr-Cas9 system (LaPlant et al., 2010; Murer and Moratalla, 2011; Engeln et al., 2013).
In conclusion, our results reveal an essential role for dynamic striatal DNA methylation in the establishment of LID and demonstrate that manipulation of DNA methylation can modify behavioral sensitization to l-DOPA in a PD animal model. These results suggest that modulation of DNA methylation, either globally or selectively, could be a useful therapeutic approach for the improvement of l-DOPA therapy in PD patients. These findings also provide further evidence that maladaptive striatal plasticity is an unintended consequence of nonphysiologic dopaminergic stimulation and that associated long-term behavioral modifications following this plasticity are dependent on changes in chromatin regulation.
Footnotes
This work was supported by the Alacare Mary Sue Beard Predoctoral Fellowship, American Parkinson Disease Association, and National Institute of Neurological Disorders and Stroke Grant F31NS090641 to D.F. We thank K. Bowling, B. Lasseinge, R. Myers, and the Hudson Alpha Genomic Services Core for sequencing assistance; and J.D. Sweatt for guidance and technical assistance.
The authors declare no competing financial interests.
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 2010;35:2450–2461. doi: 10.1038/npp.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2:577–588. doi: 10.1038/35086062. [DOI] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, Stamatoyannopoulos JA, Hager GL. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43:145–155. doi: 10.1016/j.molcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie JM. Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov Disord. 2005;20:919–931. doi: 10.1002/mds.20612. [DOI] [PubMed] [Google Scholar]
- Chang JW, Wachtel SR, Young D, Kang UJ. Biochemical and anatomical characterization of forepaw adjusting steps in rat models of Parkinson's disease: studies on medial forebrain bundle and striatal lesions. Neuroscience. 1999;88:617–628. doi: 10.1016/S0306-4522(98)00217-6. [DOI] [PubMed] [Google Scholar]
- Charbonnier-Beaupel F, Malerbi M, Alcacer C, Tahiri K, Carpentier W, Wang C, During M, Xu D, Worley PF, Girault JA, Hervé D, Corvol JC. Gene expression analyses identify Narp contribution in the development of l-DOPA-induced dyskinesia. J Neurosci. 2015;35:96–111. doi: 10.1523/JNEUROSCI.5231-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquitt BM, Allen WE, Barnea G, Lomvardas S. Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc Natl Acad Sci U S A. 2013;110:14682–14687. doi: 10.1073/pnas.1302759110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquitt BM, Markenscoff-Papadimitriou E, Duffié R, Lomvardas S. Dnmt3a regulates global gene expression in olfactory sensory neurons and enables odorant-induced transcription. Neuron. 2014;83:823–838. doi: 10.1016/j.neuron.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Cantuti-Castelvetri I, Saka E, Keller-McGandy CE, Hernandez LF, Kett LR, Young AB, Standaert DG, Graybiel AM. Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc Natl Acad Sci U S A. 2009;106:2892–2896. doi: 10.1073/pnas.0812822106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damez-Werno D, LaPlant Q, Sun H, Scobie KN, Dietz DM, Walker IM, Koo JW, Vialou VF, Mouzon E, Russo SJ, Nestler EJ. Drug experience epigenetically primes Fosb gene inducibility in rat nucleus accumbens. J Neurosci. 2012;32:10267–10272. doi: 10.1523/JNEUROSCI.1290-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits l-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;66:603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD. DNA methylation regulates associative reward learning. Nat Neurosci. 2013;16:1445–1452. doi: 10.1038/nn.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeln M, Fasano S, Ahmed SH, Cador M, Baekelandt V, Bezard E, Fernagut PO. Levodopa gains psychostimulant-like properties after nigral dopaminergic loss. Ann Neurol. 2013;74:140–144. doi: 10.1002/ana.23881. [DOI] [PubMed] [Google Scholar]
- Eskow KL, Gupta V, Alam S, Park JY, Bishop C. The partial 5-HT(1A) agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves l-DOPA efficacy. Pharmacol Biochem Behav. 2007;87:306–314. doi: 10.1016/j.pbb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, Olanow CW, Tanner C, Marek K. Levodopa and the progression of Parkinson's disease. N Engl J Med. 2004;351:2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- Feng J, Shao N, Szulwach KE, Vialou V, Huynh J, Zhong C, Le T, Ferguson D, Cahill ME, Li Y, Koo JW, Ribeiro E, Labonte B, Laitman BM, Estey D, Stockman V, Kennedy P, Couroussé T, Mensah I, Turecki G, et al. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat Neurosci. 2015;18:536–544. doi: 10.1038/nn.3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, Surmeier DJ. Cell type-specific plasticity of striatal projection neurons in parkinsonism and l-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102–110. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- He Y, Ecker JR. Non-CG methylation in the human genome. Annu Rev Genomics Hum Genet. 2015;16:55–77. doi: 10.1146/annurev-genom-090413-025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Heilbut A, Francardo V, Kulicke R, Fenster RJ, Kolaczyk ED, Mesirov JP, Surmeier DJ, Cenci MA, Greengard P. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2014;111:4578–4583. doi: 10.1073/pnas.1401819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of l-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Johnston TH, Huot P, Damude S, Fox SH, Jones SW, Rusche JR, Brotchie JM. RGFP109, a histone deacetylase inhibitor attenuates l-DOPA-induced dyskinesia in the MPTP-lesioned marmoset: a proof-of-concept study. Parkinsonism Relat Disord. 2013;19:260–264. doi: 10.1016/j.parkreldis.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, Kim TK. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci. 2016;19:75–83. doi: 10.1038/nn.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains. J Neurosci. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y, Bredy TW. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A. 2014;111:7120–7125. doi: 10.1073/pnas.1318906111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle. 2009;8:1526–1531. doi: 10.4161/cc.8.10.8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, Greenberg ME. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci. 2014;17:1330–1339. doi: 10.1038/nn.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart R, Barnea R, Dikshtein Y, Suderman M, Meir O, Hallett M, Kennedy P, Nestler EJ, Szyf M, Yadid G. Role of DNA methylation in the nucleus accumbens in incubation of cocaine craving. J Neurosci. 2015;35:8042–8058. doi: 10.1523/JNEUROSCI.3053-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, Eddy SR, Ecker JR, Nathans J. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron. 2015;86:1369–1384. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer MG, Moratalla R. Striatal signaling in l-DOPA-induced dyskinesia: common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front Neuroanat. 2011;5:51. doi: 10.3389/fnana.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson's disease: a feature inherent to the treatment or the disease? Prog Neurobiol. 2009;87:1–9. doi: 10.1016/j.pneurobio.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, Self DW. DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci U S A. 2001;98:11042–11046. doi: 10.1073/pnas.191352698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AP, Lubin FD, Hallett PJ, Vattem P, Ravenscroft P, Bezard E, Zhou S, Fox SH, Brotchie JM, Sweatt JD, Standaert DG. Striatal histone modifications in models of levodopa-induced dyskinesia. J Neurochem. 2008;106:486–494. doi: 10.1111/j.1471-4159.2008.05417.x. [DOI] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Park HY, Ryu YK, Kim YH, Park TS, Go J, Hwang JH, Choi DH, Rhee M, Lee CH, Kim KS. Gadd45beta ameliorates l-DOPA-induced dyskinesia in a Parkinson's disease mouse model. Neurobiol Dis. 2016;89:169–179. doi: 10.1016/j.nbd.2016.02.013. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in l-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Pihlstrøm L, Berge V, Rengmark A, Toft M. Parkinson's disease correlates with promoter methylation in the alpha-synuclein gene. Mov Disord. 2015;30:577–580. doi: 10.1002/mds.26073. [DOI] [PubMed] [Google Scholar]
- Poewe W. Treatments for Parkinson disease: past achievements and current clinical needs. Neurology. 2009;72:S65–S73. doi: 10.1212/WNL.0b013e31819908ce. [DOI] [PubMed] [Google Scholar]
- Santini E, Alcacer C, Cacciatore S, Heiman M, Hervé D, Greengard P, Girault JA, Valjent E, Fisone G. l-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem. 2009;108:621–633. doi: 10.1111/j.1471-4159.2008.05831.x. [DOI] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, Lin S, Lin Y, Jung I, Schmitt AD, Selvaraj S, Ren B, Sejnowski TJ, Wang W, Ecker JR. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523:212–216. doi: 10.1038/nature14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Södersten E, Feyder M, Lerdrup M, Gomes AL, Kryh H, Spigolon G, Caboche J, Fisone G, Hansen K. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet. 2014;10:e1004574. doi: 10.1371/journal.pgen.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez LM, Solís O, Caramés JM, Taravini IR, Solís JM, Murer MG, Moratalla R. l-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry. 2014;75:711–722. doi: 10.1016/j.biopsych.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Wright KN, Hollis F, Duclot F, Dossat AM, Strong CE, Francis TC, Mercer R, Feng J, Dietz DM, Lobo MK, Nestler EJ, Kabbaj M. Methyl supplementation attenuates cocaine-seeking behaviors and cocaine-induced c-Fos activation in a DNA methylation-dependent manner. J Neurosci. 2015;35:8948–8958. doi: 10.1523/JNEUROSCI.5227-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]