

Abstract
G protein–coupled receptor (GPCR) kinases (GRKs) play a critical role in cardiac function by regulating GPCR activity. GRK2 suppresses GPCR signaling by phosphorylating and desensitizing active GPCRs, and through protein-protein interactions that uncouple GPCRs from their downstream effectors. Several GRK2 interacting partners, including Gαq, promote maladaptive cardiac hypertrophy, which leads to heart failure, a leading cause of mortality worldwide. The regulator of G protein signaling (RGS) domain of GRK2 interacts with and inhibits Gαq in vitro. We generated TgβARKrgs mice with cardiac-specific expression of the RGS domain of GRK2 and subjected these mice to pressure overload to trigger adaptive changes that lead to heart failure. Unlike their nontransgenic littermate controls, the TgβARKrgs mice exhibited less hypertrophy as indicated by reduced left ventricular wall thickness, decreased expression of genes linked to cardiac hypertrophy, and less adverse structural remodeling. The βARKrgs peptide, but not endogenous GRK2, interacted with Gαq and interfered with signaling through this G protein. These data support the development of GRK2-based therapeutic approaches to prevent hypertrophy and heart failure.
INTRODUCTION
GRK2 [G protein–coupled receptor (GPCR) kinase 2, also known as β-adrenergic receptor kinase 1 (βARK1)] regulates GPCR-mediated intracellular signaling by phosphorylating agonist-occupied receptors to promote the binding of β-arrestin, thereby sterically blocking further G protein activation and attenuating downstream effector signaling (1–3). Ongoing research has demonstrated great diversity in the functional roles of GRK2, including phosphorylation of non-GPCR substrates and numerous phosphorylation-independent regulatory protein-protein interactions (3–5). GRKs have a tridomain structure with a central catalytic domain, flanked by N- and C-terminal domains containing elements involved in GRK regulation as well as key binding elements for effectors and protein-protein interactions. One such binding element is the regulator of G protein signaling (RGS)–like domain that has been identified within the N terminus (residues 51 to 173) of GRK2, which binds to and regulates Gαq activity in vitro (6–9). Classically, RGS proteins bind to Gα proteins, allosterically increasing their rate of guanosine 5′-triphosphate (GTP) hydrolysis and thereby inhibiting their activity (8, 10). Unlike canonical RGS proteins, the RGS domain of GRK2 appears to directly interact with Gαq and inhibit signaling without altering its GTPase activity (9, 11, 12). The domain of GRK2 responsible for Gαq binding appears to be distinct from the binding sites for RGS4 and RGS9, suggesting potential distinct functions of the GRK2 RGS peptide within its N terminus (8, 13).
Gαq is the final common trigger of maladaptive cardiac hypertrophy after pressure overload (14). Left ventricular hypertrophy and remodeling are often seen in hypertensive patients as an adaptive response to normalize cardiac wall stress in response to the increased pressure required to overcome systemic hypertension and achieve appropriate delivery of blood to the tissues (14–17). Left ventricular hypertrophy alters cellular signaling within cardiomyocytes and cardiac fibroblasts, increasing gene transcription, apoptosis, and fibrosis as well as altering coronary circulation (14–17). This cardiac remodeling, particularly in the presence of diabetes, obesity, high salt intake, or genetic factors, may become maladaptive and contribute to cardiac decompensation with thinning of the left ventricular wall, chamber dilation, and contractile dysfunction, resulting in heart failure (14–17). Transgenic mice with cardiac-specific overexpression of Gαq have a phenotype of decompensated cardiac hypertrophy (18–20), whereas cardiac overexpression of RGS4 blocks Gαq-mediated hypertrophy and heart failure (10). Furthermore, transgenic mice with cardiac expression of a Gαq inhibitor (GqI), which is the last 50 amino acids of murine Gαq that targets the Gαq-activated GqPCR interface, have significantly lessened ventricular hypertrophy and ventricular dysfunction after pressure overload (21–23). Therefore, a selective interaction of the GRK2 RGS domain with Gαq may alter hypertrophic responses, revealing a role for GRK2 and a potential therapeutic target to limit maladaptive cardiac hypertrophy. We have begun to address this by generating transgenic mice with cardiac-specific expression of the RGS domain of GRK2 (we have termed this peptide βARKrgs). Further studies use transgenic lines representing a Gαq-mediated antihypertrophic phenotype as well as lines that are susceptible to or protected from heart failure to further elucidate the mechanism of action of the βARKrgs peptide. Using a transaortic constriction (TAC) model of pressure overload to trigger cardiac hypertrophy followed by chronic de-compensation and heart failure in these mouse lines, we investigated whether the βARKrgs could serve as a noncanonical inhibitor of Gαq-mediated hypertrophic signaling in the heart and pave the way for novel GRK2-based therapeutic approaches to prevent maladaptive hypertrophy.
RESULTS
βARKrgs transgenic mice exhibit an antihypertrophic phenotype after pressure overload
To generate the TgβARKrgs line, the cDNA that encodes bovine GRK2 residues 50 to 185 was cloned into a vector driven by the α-myosin heavy chain (MYH6) promoter with a C-terminal Flag tag. Transgenic mice did not show gross phenotypic changes compared to nontransgenic littermate control mice. Transgenic expression was confirmed by Western blotting of cardiac lysates using a mouse monoclonal Flag antibody to detect the ~17-kD band (Fig. 1A). We analyzed the cardiovascular function and adrenergic responsiveness of the TgβARKrgs mice using terminal hemodynamics at baseline and upon challenge with increasing doses of the βAR agonist isoproterenol in transgenic and nontransgenic mice. No difference was observed in mean systemic pressure (Fig. 1B), heart rate responses to isoproterenol (Fig. 1C), or dP/dt maximum and minimum, measures of the rate of increase in left ventricular pressure during systole and decrease in pressure during diastole that indicate cardiac contractility and relaxation, respectively, at baseline and in response to isoproterenol (Fig. 1, D and E). These data demonstrate that βAR responsiveness was not altered in the TgβARKrgs animals.
Fig. 1. Cardiac-specific βARKrgs expression does not alter hemodynamic function.
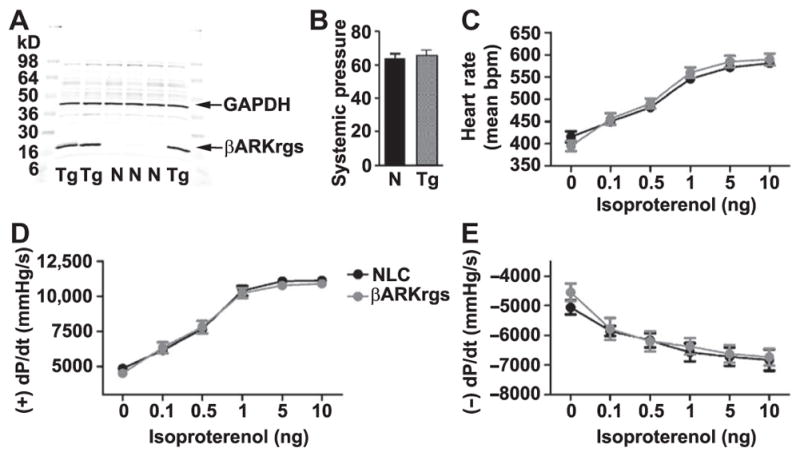
(A) Representative Western blot of TgβARKrgs (Tg) and nontransgenic littermate control (NLC or N) ventricular lysates probed for Flag-tagged βARKrgs and the loading control glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (B to E) Hemodynamics were recorded from 10- to 12-week-old TgβARKrgs and nontransgenic mice. Quantification of mean systemic pressure (B), heart rate (C), left ventricular +dP/dt average maximum (D), and left ventricular −dP/dt average minimum (E) at baseline and with increasing doses of isoproterenol. Statistics are relative to non-transgenic littermate control mice by nonparametric Student’s t test for (B) and two-way analysis of variance (ANOVA) with repeated measures and Bonferroni post hoc test for (C) to (E) as appropriate. The genotype of all animals was confirmed by Western blot before unblinding the analysis and results, meaning n > 10 independent experiments for (A); n = 12 mice per group for (B) to (E).
To determine whether the expression of the RGS peptide of GRK2 could alter Gαq-mediated signaling and hypertrophy in an animal model of pressure overload, TgβARKrgs and nontransgenic mice underwent TAC or sham surgery (24), and echocardiography was performed at baseline and at 2 and 4 weeks to monitor cardiac function and dimensions. The systolic pressure gradient as measured by pulsed wave Doppler of the aortic arch 24 hours after surgery confirmed TAC and showed no differences between nontransgenic and transgenic groups (fig. S1A). Over this time course, we typically found no effect of TAC on left ventricular ejection fraction (Fig. 2A) or fractional shortening (fig. S1B), two parameters that measure the efficiency of the heart to eject blood with each contraction. However, there was hypertrophic growth of the heart in this time period as determined by several parameters. Left ventricular posterior wall thickness during systole increased soon after TAC by about 47% in non-transgenic mice compared to an about 19% increase in the TgβARKrgs animals (Fig. 2B). Further, the left ventricular posterior wall thickness during diastole significantly increased in nontransgenic mice but not in transgenic mice, despite similar systolic pressure gradients (Fig. 2C). The pronounced post-TAC growth in the left ventricular mass in the nontransgenic mice (a 109% increase) was also significantly blunted in the βARKrgs mice (a 47% increase) (Fig. 2D). TgβARKrgs mice also had reduced heart weight normalized to tibia length after TAC, although there was no difference between transgenic and nontransgenic sham controls (Fig. 2E) or in tibia length (fig. S1C). Furthermore, left atrial weight was preserved in TgβARKrgs animals (Fig. 2F). These data support a notable antihypertrophic effect of βARKrgs peptide expression after TAC.
Fig. 2. TgβARKrgs mice exhibit a similar decrease in cardiac hypertrophy as TgGqI mice after TAC.

(A to F) TgβARKrgs and nontransgenic littermate control sham (Sh) and post-TAC animals were analyzed at baseline and at 2 and/or 4 weeks after surgery. Percent left ventricular ejection fraction (A). Serial measures for left ventricular (LV) posterior wall thickness during systole (LVPWs) (B), diastole (LVPWd) (C), and left ventricular mass (D). Measures of heart weight (HW) normalized to tibia length (TL) (HW/TL) (E) and left atrial weight (F) 4 weeks after surgery. (G and H) TgGqI and nontransgenic sham and post-TAC animals were analyzed at baseline and at 2 and/or 4 weeks after surgery. Serial measures for LVPWs (G) and LV mass (H). (I) Measures of HW/TL. *P = 0.01; **P = 0.001; ***P < 0.0001 as determined by two-way ANOVA with repeated measures and Bonferroni post hoc test for (A) to (D) and (G) to (H) and by one-way ANOVA with Tukey post hoc test for (E), (F), and (I) relative to nontransgenic sham mice. τP = 0.01; τττP < 0.0001 by two-way ANOVA with repeated measures and Bonferroni post hoc test relative to nontransgenic TAC mice. tP = 0.01 by one-way ANOVA with Tukey post hoc test relative to nontransgenic TAC mice. n = 10 to 16 mice per group for (A) to (I).
We next investigated whether the antihypertrophic phenotype of the TgβARKrgs mice was consistent with inhibition of Gαq-mediated signaling. We used mice with MYH6-driven expression of the C-terminal 50 amino acids of murine Gαq (GqI), a peptide that competes with Gαq for binding to GPCRs (21). This genetic manipulation inhibits Gαq-mediated signaling, particularly 7 days after TAC, and the mice show blunted responses to Gαq-coupled GPCR agonists, as measured by diminished Gαq-mediated activation of phospholipase C (PLC) and production of the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) and downstream activation of mitogen-activated protein kinase (MAPK), and reduced left ventricular weight (21–23). Similar to the TgβARKrgs mice, the TgGqI animals showed decreased hypertrophy 4 weeks after TAC because the significant increases in left ventricular posterior wall thickness during systole (Fig. 2G) and diastole (fig. S1D) and left ventricular mass (Fig. 2H) in nontransgenic mice were decreased in GqI mice. Furthermore, heart weight normalized to tibia length was also reduced to a similar extent in TgGqI mice after TAC as in βARKrgs mice (Fig. 2I). These data are consistent with the βARKrgs peptide inhibiting pressure overload–induced hypertrophy through altered Gαq-mediated signaling, thereby imparting a similar degree of protection as that seen with GqI.
To further elucidate whether the phenotype of the mice resulted from a selective interaction of βARKrgs with Gαq rather than a GRK2-mediated effect, we used two different mouse lines. The first line was cardiac-targeted βARKct transgenic mice (TgβARKct), which express a peptide inhibitor of GRK2 that elicits cardioprotection by binding to Gβγ in place of GRK2. Thus, Gβγ facilitates the membrane translocation of βARKct, which does not contain the catalytic domain, rather than the full-length GRK2 (25). The second line was cardiac GRK2 overexpressing mice (TgGRK2) in which GRK2 abundance is similar to that observed in failing human hearts (26). Unlike βARKrgs and GqI transgenic mice, cardiac hypertrophy was unaltered in TgβARKct or TgGRK2 mice after TAC relative to their non-transgenic controls (Fig. 3). Left ventricular posterior wall thickness during systole and left ventricular mass significantly increased to a similar extent in TgβARKct or TgGRK2 mice and their respective nontransgenic controls after TAC (Fig. 3, A to D). Heart weight normalized to tibia length and left atrial weight increased to a similar extent in TgβARKct and TgGRK2 mice as their nontransgenic controls (Fig. 3, C to H). Together, these data demonstrate that peptide inhibition or, conversely, overexpression of full-length GRK2 does not alter cardiac hypertrophy, further supporting our hypothesis that the RGS domain of GRK2 is a distinct interaction that prevents hypertrophic signaling in the heart. These data also suggest that the RGS domain within full-length GRK2 does not appear to substantially interact with Gαq in cardiomyocytes even when overexpressed.
Fig. 3. Hypertrophy is unaltered in TgβARKct and TgGRK2 mice after TAC.
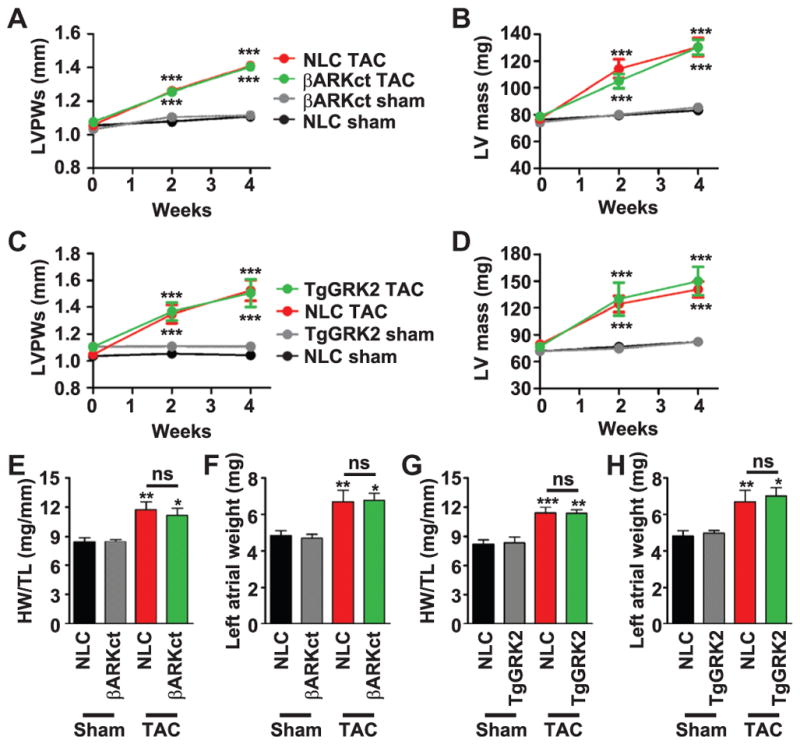
(A and B) Serial measures of TgβARKct and nontransgenic littermate control sham and post-TAC animals for LVPWs (A) and LV mass (B). (C and D) Serial measures of TgGRK2 and nontransgenic sham and post-TAC animals for LVPWs (C) and LV mass (D). (E and F) Measures of HW/TL (E) and left atrial weight (F) in TgβARKct and nontransgenic sham and post-TAC animals 4 weeks after surgery. (G and H) Measures of HW/TL (G) and left atrial weight (H) in TgGRK2 and nontransgenic sham and post-TAC animals 4 weeks after surgery. ***P < 0.0001 by two-way ANOVA with repeated measures and Bonferroni post hoc test for (A) to (D). *P = 0.01; **P = 0.001; ***P < 0.0001 by one-way ANOVA with Tukey post hoc test (E to H) relative to corresponding nontransgenic sham mice. n = 8 to 14 mice per group for (A) to (H).
βARKrgs expression protects against decompensated hypertrophy and heart failure after pressure overload
To investigate whether the antihypertrophic effect of βARKrgs expression could impart cardioprotection, we monitored cardiac function in a cohort of mice up to 14 weeks after TAC with serial echocardiography. Nontransgenic mice exhibited pronounced left ventricular decompensation from weeks 10 to 14, exhibiting significant chamber dilation, thinning of the ventricular wall, and contractile dysfunction with a reduction in ejection fraction from ~60% at baseline to 31% at 14 weeks after TAC (Fig. 4A) and a corresponding reduction in fractional shortening (fig. S2A). In contrast, ejection fraction was stable in TgβARKrgs mice throughout the chronic time course and similar to the sham controls, despite similar systolic pressure gradients (fig. S2B). Although left ventricular mass continually expanded in nontransgenic mice, this measure increased much more slowly in TgβARKrgs mice from 4 to 14 weeks after TAC (Fig. 4B). Similar trends were observed for left ventricular internal diameter during systole and left ventricular posterior wall thickness during diastole (fig. S2, C and D). The progressive reduction in left ventricular posterior wall thickness during systole that began in nontransgenic mice at 8 weeks after TAC, an index of decompensation, was absent in the TgβARKrgs mice (Fig. 4C). Furthermore, the increase in lung weight due to pulmonary congestion, which is characteristic of heart failure, was prevented in TgβARKrgs mice, and the left atrial weight was preserved (Fig. 4, D and E). Also, heart weight normalized to tibia length did not increase to the same extent in TgβARKrgs mice as in nontransgenic mice, and the tibia length did not differ between these two groups (fig. S2, E and F). These data confirm that the reduction in hypertrophy triggered by βARKrgs expression was sufficient to prevent decompensation and heart failure.
Fig. 4. Chronic pressure overload–induced heart failure is inhibited by cardiac βARKrgs expression.

(A to E) Serial measures of nontransgenic littermate control and TgβARKrgs sham and post-TAC animals for percent LV ejection fraction (A), LV mass (B), and LVPWs (C). Lung weight normalized to TL (LW/TL) (D) and left atrial weight (E) 14 weeks after surgery. *P = 0.01; **P = 0.001; ***P < 0.0001 by two-way ANOVA with repeated measures and Bonferroni post hoc test for (A) to (C). ***P < 0.0001 by one-way ANOVA with Tukey post hoc test for (D) to (E) relative to nontransgenic sham mice. τττP < 0.0001 by two-way ANOVA with repeated measures and Bonferroni post hoc test relative to nontransgenic TAC mice. n = 8 to 15 mice per group for (A) to (E).
The βARKrgs peptide limits adverse ventricular remodeling after pressure overload
Reverse transcription polymerase chain reaction (RT-PCR) revealed that hypertrophic gene expression, including members of the fetal gene program such as ANF (which encodes atrial natriuretic factor), BNP (which encodes brain natriuretic peptide), and βMHC (which encodes β-myosin heavy chain), was increased after TAC to a lesser extent in both TgβARKrgs and TgGqI mice than in their nontransgenic controls (Fig. 5, A to F, and fig. S3, A to C). These data indicate that the βARKrgs peptide prevented the molecular alterations that occur during hypertrophy and are consistent with our echocardiography results. At the protein level, the abundance and activity of GRK2 are increased after cardiac injury, when it serves as a prodeath kinase in the heart (27–29). We therefore measured GRK2 abundance by Western blot and found that the increase in GRK2 in nontransgenic mice was attenuated in TgβARKrgs mice (Fig. 5G and fig. S3D). GRK2 abundance was also lower after TAC in TgβARKct mice (Fig. 5H and fig. S3E), although hypertrophic gene induction was not altered in these mice after TAC (fig. S3, A to C). Thus, the βARKct-mediated decrease in GRK2 abundance and activity after TAC was not sufficient to prevent maladaptive hypertrophy, further suggesting that the βARKrgs peptide prevents hypertrophic signaling through a distinct mechanism.
Fig. 5. Molecular markers of left ventricular dysfunction are reduced in TgβARKrgs and TgGqI mice but not in TgβARKct mice.
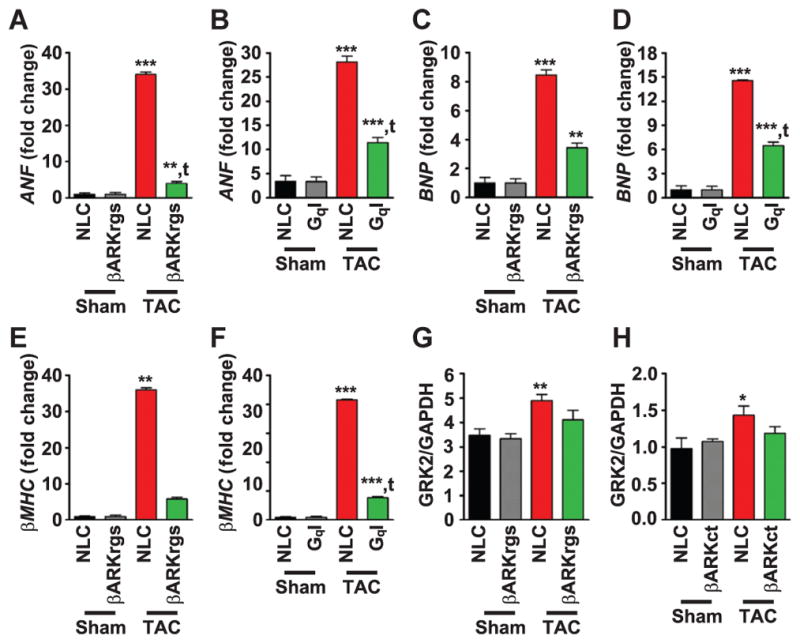
(A to F) Quantification of RT-PCR data in nontransgenic littermate control and transgenic sham and post-TAC mice 4 weeks after surgery showing fold change in mRNA expression of atrial natriuretic factor (ANF) in TgβARKrgs (A) and TgGqI (B) mice, brain natriuretic peptide (BNP) in TgβARKrgs (C) and TgGqI (D) mice, and β-myosin heavy chain (βMHC) in TgβARKrgs (E) and TgGqI (F) mice. **P = 0.001; ***P < 0.0001 by one-way ANOVA with Tukey post hoc test relative to nontransgenic sham mice. tP = 0.001; ttP < 0.0001 by one-way ANOVA with Tukey post hoc test relative to nontransgenic TAC mice. n = 8 to 10 mice per group for (A), (C), and (E), and n = 12 to 13 mice per group for (B), (D), and (F). (G and H) Quantification of GRK2 protein abundance normalized to GAPDH from Western blots in TgβARKrgs (G) and TgβARKct (H) sham and post-TAC animals 4 weeks after surgery. See fig. S3, D and E, for representative Western blots. *P = 0.01; **P = 0.001 by nonparametric one-way ANOVA with Dunn post hoc test relative to nontransgenic sham mice. n = 5 to 10 mice per group and 4 blots for (G) and 3 blots for (H).
To visualize the effect of hindering molecular responses to TAC on myocardial structure, we performed wheat germ agglutinin (WGA) staining to measure cardiomyocyte cross-sectional area (CSA) in these TgβARKrgs and nontransgenic mice 4 weeks after TAC. Although we found no difference between the sham groups, the increase greater than twofold in CSA in the nontransgenic mice was significantly blunted with βARKrgs peptide expression (Fig. 6, A and B), confirming reduced cardiomyocyte hypertrophy. Furthermore, we performed Masson trichrome staining on paraffin sections taken at the level of the aortic outflow tract where left ventricular dimensions are at their maximum. Four chamber images of the myocardium revealed no difference in sham hearts but a partial preservation of left ventricular architecture in the TgβARKrgs hearts 4 weeks after TAC compared to non-transgenic mice (Fig. 6C). To further investigate the integrity of the myocardium, we analyzed higher-magnification images of the left ventricle (Fig. 6D) and found that interstitial fibrosis was significantly reduced in TgβARKrgs mice compared to nontransgenic mice 4 weeks after TAC, with no difference in the total tissue area (Fig. 6E and fig. S4). Together, these data demonstrate that βARKrgs peptide expression reduces both adaptive and maladaptive left ventricular remodeling after TAC.
Fig. 6. Left ventricular structural remodeling is inhibited in TgβARKrgs mice.

(A) Representative images of WGA and 4′,6-diamidino-2-phenylindole (DAPI)–stained murine heart sections from nontransgenic littermate control and TgβARKrgs hearts 4 weeks after sham or TAC surgery. Scale bar, 50 μm. (B) Quantification of cardiomyocyte CSA in these animals. */tP = 0.0001 by one-way ANOVA with repeated measures and Tukey post hoc test relative to nontransgenic sham and TAC animals, respectively. n = 5 to 9 hearts per group, 40 images per heart. (C) Representative images of Masson trichrome–stained murine heart sections from nontransgenic and TgβARKrgs hearts 4 weeks after sham or TAC surgery. Scale bar, 2000 μm. (D) Representative higher magnification (×40) images of Masson trichrome–stained post-TAC murine heart sections from nontransgenic and TgβARKrgs mice demonstrating interstitial fibrosis. Scale bar, 200 μm. (E) Quantification of the percent of fibrotic tissue in the left ventricular area in nontransgenic and TgβARKrgs hearts 4 weeks after TAC. ***P < 0.0001 by Student’s t test relative to non-transgenic TAC mice. n = 108 images from nine hearts for nontransgenic mice (~11 images per heart) and 78 images from seven hearts for TgβARKrgs mice.
βARKrgs sequesters Gαq from effector binding to prevent downstream signaling
To confirm our hypothesis that the βARKrgs peptide inhibits Gαq-mediated hypertrophic signaling by directly interacting with Gαq, we performed immunoprecipitation assays on nontransgenic and TgβARKrgs mice at baseline and 4 weeks after sham or TAC surgery. Flag-tagged βARKrgs coimmunoprecipitated with Gαq (Fig. 7A), and this coimmunoprecipitation was significantly increased after pressure overload with no difference in total immunoprecipitated βARKrgs-Flag (Fig. 7, B and C, and fig. S5, A to C), indicating that Gαq-coupled GPCR activation and dissociation of Gαq facilitate binding of the βARKrgs peptide to Gαq. Further, Flag-tagged βARKrgs did not coimmunoprecipitate with Gαs or Gαi at baseline or 4 weeks after TAC (fig. S5, D to G), demonstrating the specificity of the binding of βARKrgs to Gαq. That GRK2 overexpression did not alter the cardiac hypertrophic response suggests that the RGS domain within full-length GRK2 does not appear to interact with Gαq in cardiomyocytes even when overexpressed. Therefore, it was important to determine the extent of the interaction between full-length GRK2 and Gαq and how this differed from that of βARKrgs. We performed immunoprecipitations on cardiac tissue from nontransgenic and TgβARKrgs mice at baseline and 4 weeks after TAC and found that despite efficient immunoprecipitation, Gαq did not associate with full-length GRK2 in sham or post-TAC animals (Fig. 7D and fig. S5, A and B). We next investigated whether this interaction translated to a functional effect by measuring Gαq-mediated activation of PLC by quantifying IP3. As expected, IP3 concentrations were increased in nontransgenic mice after TAC, presumably as a result of enhanced Gαq-coupled GPCR activation; however, this increase was partially attenuated by βARKrgs peptide expression (Fig. 7E). Together, these data support our hypothesis that the βARKrgs peptide inhibits Gαq-mediated hypertrophic signaling by directly interacting with Gαq.
Fig. 7. The βARKrgs peptide interacts with endogenous Gαq in the heart.
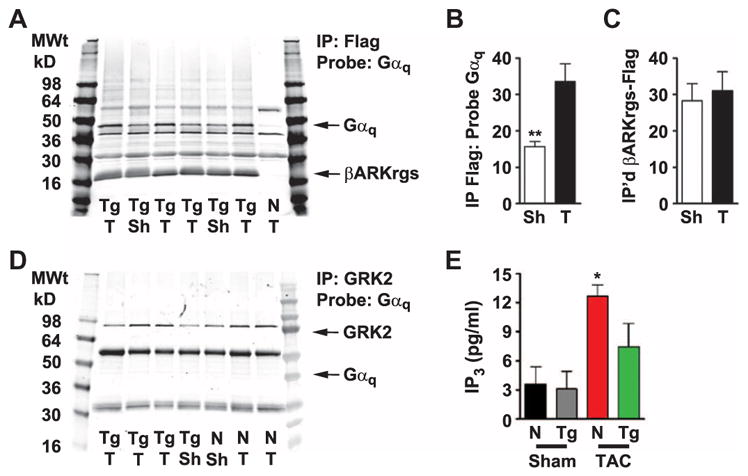
(A) Representative image of a monoclonal Flag immunoprecipitation (IP) followed by Western blotting probing for rabbit polyclonal Gαq and rabbit polyclonal Flag-tagged βARKrgs in cardiac lysates from nontransgenic littermate control and TgβARKrgs mice. (B and C) Quantification of (B) Gαq normalized to βARKrgs-Flag and (C) total βARKrgs in TgβARKrgs lysates after monoclonal Flag IP 4 weeks after sham or TAC (T) surgery. (D) Representative image of a polyclonal GRK2 IP followed by Western blotting probing with goat polyclonal Gαq/11 and mouse monoclonal GRK2 in cardiac lysates from nontransgenic and TgβARKrgs mice. **P < 0.001 by non-parametric Student’s t test relative to TgβARKrgs TAC mice. n = 6 sham hearts and 8 TAC hearts and 4 Western blots each with sham and TAC hearts run side by side. (E) Quantification of IP3 concentrations from non-transgenic and TgβARKrgs lysates 4 weeks after sham or TAC surgery. *P < 0.01 by nonparametric one-way ANOVA with Dunn post hoc test relative to nontransgenic sham mice. n = 6 to 8 mice per group.
DISCUSSION
Here, we investigated whether the RGS domain within the N terminus of GRK2 inhibited Gαq-mediated hypertrophic signaling in the heart. Mice with cardiac-specific TgβARKrgs expression were not only resistant to hypertrophic growth after TAC, but also protected from heart failure during chronic pressure overload. TgβARKrgs expression provided the same antihypertrophic effect as inhibition of Gαq receptor association (TgGqI) and reduced several markers of maladaptive remodeling after TAC. The ability to selectively inhibit Gαq-mediated signaling may offer an advancement in the treatment of human hypertrophy and heart failure because Gαq represents the common driving force for maladaptive cardiac hypertrophy (Fig. 8) (14, 21).
Fig. 8. Model for GPCR signaling in the heart.

Agonist binding to GPCRs in the heart leads to activation and dissociation of the heterotrimeric G protein complex. Gα and Gβγ subunits then bind to downstream effector proteins triggering signaling cascades that control cardiac function. Catecholamine binding to β1-adrenergic receptors (β1ARs) leads to activation of Gαs, which directly stimulates adenylyl cyclase at the membrane, altering heart rate and contractility. Concomitantly, GRK2 associates with dissociated Gβγ subunits and translocates to the membrane where it phosphorylates the agonist-bound receptor, leading to receptor desensitization and internalization. Agonist binding to Gαq-coupled receptors leads to activation of multiple Gαq effector proteins involved in cardiomyocyte hypertrophy and vascular function. This study suggests that a direct interaction of the βARKrgs peptide, but not the RGS domain within endogenous GRK2, with Gαq competes for binding to other Gαq effector proteins, inhibiting downstream signaling and cardiac hypertrophy.
Activated Gαq interacts with the N terminus of GRK2, which in vitro inhibits Gαq-mediated activation of PLCβ (6). This inhibition is independent of the kinase activity of GRK2, is enhanced in the presence of agonist, and occurs not only upon GPCR stimulation but also in the presence of constitutively actively Gαq, supporting the concept that it is due to a direct interaction with Gαq (6). Biochemical and structural studies have mapped the residues important for this physical interaction, specifically identifying a critical proline residue unique to Gαq that is absent in Gαi or Gαs (13, 30). Mutating the residues important for association with GRK2 does not alter the binding of RGS4 or RGS2 to Gαq because GRK2 does not use the RGS binding domain on Gαq (30). Rather, GRK2 binds within the effector binding domain of Gαq that facilitates the association of Gαq with effector proteins such as PLC for downstream signaling (30, 31). Further, the RGS domain of GRK2 binds Gαq through an α-helix domain similar to that in both PLCβ and Rho-GEF, using a conserved leucine structurally similar to their ALXXPI binding motif to associate with the effector binding domain of Gαq (31). These studies suggest that the direct interaction of the RGS domain of GRK2 with Gαq acts in competition with binding to other Gαq effector proteins, thereby inhibiting downstream signaling. This is consistent with our data demonstrating reduced IP3 production 4 weeks after TAC in the TgβARKrgs compared to nontransgenic mice, suggesting an inhibition of Gαq-mediated signaling through PLC. In particular, this Gαq-centric mechanism of inhibition could apply to any GPCR coupled to Gαq and various effector proteins within a given cell type. Although we did not see adverse phenotypes in our cardiac-specific TgβARKrgs animals, it will be important to determine the potential effects of such an inhibitor of Gαq signaling within other cell types to determine the optimal path for therapeutic development.
Notably, we observed no antihypertrophic effect with cardiac GRK2 over-expression up to 4 weeks after TAC. These data are in contrast to numerous in vitro studies showing that binding of GRK2 as well as the N-terminal RGS domain inhibit effector signaling to an equal extent and that increased Gαq-coupled GPCR activation acts as a driving force to enhance GRK2 association with Gαq (6). It is difficult to extrapolate whether such interactions will occur in the complex milieu in vivo, as opposed to the overexpression of a limited number of interacting partners in an in vitro system. The lack of interaction between full-length GRK2 and Gαq at baseline or during pressure overload was consistent with our data demonstrating the lack of anti-hypertrophic effect of GRK2 overexpression. It is possible that whereas GRK2 can bind to Gαq, as demonstrated in vitro, other protein-protein interactions that occur within the holoenzyme sterically or allosterically block this interaction in vivo. Two such presumed N-terminal GRK2 binding partners are caveolin (residues 63 to 71) (32) and calmodulin (residues 18 to 37) (33), but these interactions and their functional consequences have not been confirmed in vivo or characterized. Notably, the in vitro interaction between GRK2 and Gαq is enhanced by c-Src kinase–mediated phosphorylation of GRK2 that can be regulated by Gαq-coupled GPCR activation, such as the M1-muscarinic receptor (34). Of the tyrosine residues targeted by c-Src (Tyr13, Tyr86, and Tyr92), two are within the βARKrgs. Therefore, it may be interesting to elucidate any differences in the tyrosine phosphorylation of full-length GRK2 compared to the βARKrgs peptide after TAC. Investigation of the endogenous protein binding partners for βARKrgs, compared to full-length GRK2, and the regulation of these interactions during cardiovascular disease will be a major focus of future studies.
Given the mild hypertrophy observed in the TgβARKrgs mice that is preserved 14 weeks after TAC and does not decompensate to heart failure, one consideration is whether the stress response in these animals represents a “physiological hypertrophy.” Pathological hypertrophy occurs in response to cardiac stress or disease and is irreversible, whereas physiological hypertrophy occurs with chronic exercise or during pregnancy and is reversible. Numerous functional and biochemical differences have been well characterized, with pathological hypertrophy often identified by interstitial fibrosis, cell death, and dysfunction, whereas physiological hypertrophy presents with a proportional increase in wall thickness and chamber dimension (35, 36). Consistent with this idea is the fact that we observed significantly reduced interstitial fibrosis in our TgβARKrgs compared to nontransgenic mice after TAC. Although reduced fibrosis in the TgβARKrgs mice after TAC supports the notion of beneficial cardiac growth, this may simply be due to enhanced cardiomyocyte survival in the TgβARKrgs heart during pressure overload. Gene profiling may be necessary to assess the type of hypertrophy that occurs when this RGS domain of GRK2 is expressed in the heart.
Moving forward, a main focus of this research will be to determine the therapeutic potential of βARKrgs to reverse established hypertrophy or heart failure. This work will follow two lines of investigation: one into the gene therapy potential of this peptide delivered exogenously to cardiac injury models, and the other elucidating whether a small-molecule inhibitor could be developed to mimic βARKrgs activity. Our laboratory has successfully translated the study of the βARKct peptide of GRK2 from a transgenic murine model to a successful gene therapy in a porcine model of heart failure (37, 38). Furthermore, the use of gene therapy is expanding and is the subject of numerous clinical trials worldwide (39). Alternatively, the concept of a small-molecule mimic of a peptide inhibitor has been supported by work with gallein, a drug that mimics the effect of βARKct by inhibiting GRK2 coupling to, and translocation by, Gβγ (40, 41). This concept of peptidomimetics is gaining ground as a viable method for structure-based drug design in numerous fields, particularly as an approach to inhibiting highly conserved and yet selective effector sites, such as within large families of related kinases (42).
In summary, we found that transgenic expression of the RGS domain of GRK2 (βARKrgs) demonstrates a marked antihypertrophic effect in a murine model of pressure overload. The reduction in hypertrophy was similar to that induced by inhibition of Gαq activation and was not replicated by βARKct or GRK2 overexpression. Further, this inhibition of Gαq signaling was cardioprotective, preventing decompensation to heart failure and supporting a therapeutic role for noncanonical inhibition of Gαq signaling by βARKrgs. Moreover, βARKrgs inhibited induction of the fetal gene program, preserved left ventricular structure, and reduced Gαq-mediated downstream signaling through a selective functional interaction. This study emphasizes that investigation of distinct protein-interacting domains of GRK2 may enable the development of GRK2-based therapeutic approaches to prevent hypertrophy and heart failure, and highlights the importance of understanding the functional relevance of the expanding GRK2 interactome.
MATERIALS AND METHODS
Study design
Our central hypothesis was that the RGS domain of GRK2 would act as a noncanonical inhibitor of Gαq-mediated hypertrophic signaling in the heart after TAC. Statistical powering was initially performed using the nQuery Advisor 3.0 software (Statistical Solutions) for estimation of sample size. For all experiments, the calculations use α = 0.05 and β = 0.2 (power, 0.80). On the basis of these calculations, our target was a minimum of 5 animals (powered for indices of TAC) per group to attain statistical significance, with a preference for 10 or more. Each experiment was performed a minimum of three times. The number and composition of the replicates were determined on the basis of the availability of transgenic and non-transgenic animals, the surgeon, and echocardiography and hemodynamic machinery. All animals and resulting samples were monitored by mouse number only until data quantification was complete, meaning that all data analyses were blinded. Data were then decoded by gene expression and surgical group for statistical analysis. All results were substantiated by repetition. Data were only excluded if their validity was undermined by the condition of the animal or cells before or during the experiment, such as loss of the specimen.
Experimental animals
All animal procedures were carried out according to National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Temple University. To generate the TgβARKrgs line, the cDNA that encodes bovine GRK2 residues 50 to 185 was cloned into a vector driven by the MYH6 promoter with a C-terminal Flag tag, as previously described for βARKnt (43). For all in vivo experiments, nontransgenic littermate control mice were used. Mice with αMHC-targeted expression of the βARKct peptide (C-terminal 194 residues) containing the Gβγ binding domain were used as a control for GRK2 inhibition, whereas mice with MYH6-targeted expression of GRK2, causing a two- to threefold increase in protein abundance similar to the increase observed during human heart failure, were used as a control for enhanced GRK2 activity. Mice with MYH6-targeted expression of the GqI peptide, the last 50 amino acids of murine Gαq, which targets the Gαq-activated GPCR interface, were used as a control for inhibition of Gαq activation.
TAC was performed as described previously (24). To measure global cardiac function, echocardiography was performed at 8 to 10 weeks of age before TAC and at 2 and 4, or 2, 4, 8, 10, 12, and 14 weeks after TAC. In vivo cardiac hemodynamic function was measured at baseline and after administration of the βAR agonist isoproterenol (0.1, 0.5, 1, 5, and 10 ng) as described (44). Echocardiography was performed using the Vevo 2100 imaging system from VisualSonics as described (27). Briefly, two-dimensional echocardiographic views of the midventricular short axis were obtained at the level of the papillary muscle tips below the mitral valve. M-mode measurements were determined at the plane bisecting the papillary muscles according to the American Society of Echocardiography leading-edge method.
RNA isolation and semiquantitative PCR
RNA isolation and analysis was performed as previously described (45). Total RNA isolation was performed using TRIzol reagent (Life Technologies) and a VCX 130PB ultrasonic processor from Sonics and Materials Inc. for homogenization. After RNA isolation, cDNA was synthesized from 1 mg of total RNA using the iScript cDNA Synthesis Kit from Bio-Rad Laboratories. Semiquantitative PCR was carried out on cDNA using SYBR Green (Bio-Rad) and 150 nM gene-specific oligonucleotides for 18S, ANF, BNP, and βMHC on CFX96 Real-Time System with Bio-Rad CFX Manager 2.1 software. Quantitation was established by comparing 18S mRNA, which was similar between groups, for normalization and was compared using the ΔΔCt method.
Histological sectioning and staining
Subgroups of hearts were harvested at 4 and 14 weeks after TAC for analyses of structure, fibrosis, and biochemistry. Trichrome staining was performed as previously described (44). Briefly, mice were euthanized, and after cardioperfusion, hearts were fixed for 1 to 3 days in 4% paraformaldehyde at 4°C. Hearts were dehydrated and paraffinized using the Microm STP 120 from Thermo Fisher Scientific, embedded in paraffin using a HistoStar apparatus (Thermo Fisher), and sectioned (4 to 6 μm) using the Microm HM 325 (Thermo Fisher). Tissue sections were then stained with Weigert’s iron hematoxylin and Masson trichrome (Sigma-Aldrich) according to the manufacturer’s instructions. Interstitial fibrosis was quantified by color threshold measures using ImageJ. Alternatively, tissue sections were deparaffinized and rehydrated according to the trichrome staining protocol, but after the wash with deionized water, heart sections were washed three times for 5 min with 1× phosphate-buffered saline (PBS) followed by staining with Alexa Fluor 488–conjugated WGA (1:10 in 1× PBS) (Invitrogen) for 1 hour at room temperature in a humidified chamber in the dark. Sections were again washed three times for 5 min with 1× PBS followed by mounting with coverslips using Fluoromount-G mounting media containing DAPI nuclear stain (Southern Biotech).
Immunoblotting and immunoprecipitation
Immunoprecipitation was performed as described previously (6). Briefly, cardiac lysates were centrifuged at 13,000g at 4°C for 30 min, protein concentration was measured using a bicinchoninic acid assay (Pierce), and 1 mg of protein was used for each immunoprecipitation. GRK2 and βARKrgs-Flag immunoprecipitations were conducted using anti-GRK2 agarose conjugate (Santa Cruz Biotechnology) and anti-Flag M2 affinity gel (Sigma-Aldrich), respectively. Control immunoprecipitations were conducted using normal rabbit or mouse immunoglobulin G–agarose conjugates (Santa Cruz Biotechnology). Western blotting was performed as described previously (45). After SDS–polyacrylamide gel electrophoresis and transfer to nitrocellulose membranes, primary antibody incubations were performed overnight at 4°C. Primary antibodies used were as follows: rabbit polyclonal GRK2 (Santa Cruz Biotechnology), monoclonal GRK2 (Millipore), monoclonal GAPDH (Santa Cruz), monoclonal and polyclonal Flag (Sigma-Aldrich), goat polyclonal Gαq/11 (Santa Cruz Biotechnology), rabbit polyclonal Gαq (Santa Cruz Biotechnology), mouse monoclonal Gαs (Santa Cruz Biotechnology), and rabbit polyclonal Gαi (Santa Cruz Biotechnology). Visualization of Western blot signals was performed using secondary antibodies coupled to Alexa Fluor 680 (Invitrogen Molecular Probes) or IRDye 800 (LI-COR Biosciences) and imaged using the Odyssey CLx infrared imager (LI-COR Biosciences). Odyssey version 1.2 imaging software was used to process all images.
IP3 quantification
IP3 concentration was quantified using a mouse IP3 enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (MyBioSource).
Statistical analysis
All values in the text and figures are presented as means ± SEM of independent experiments for given n sizes. Statistical significance was determined by two-way ANOVA with Bonferroni post hoc, one-way ANOVA with Tukey post hoc, or Student’s t test, as appropriate. Probabilities of 0.05 or less were considered to be statistically significant.
Supplementary Material
Fig. S1. TgβARKrgs mice exhibit a similar decrease in cardiac hypertrophy as TgGqI mice after TAC.
Fig. S2. Chronic pressure overload–induced heart failure is inhibited by cardiac βARKrgs expression.
Fig. S3. Molecular markers of left ventricular dysfunction are reduced in TgβARKrgs and TgGqI mice but not in TgβARKct mice.
Fig. S4. Left ventricular structural remodeling is inhibited in TgβARKrgs mice.
Fig. S5. The βARKrgs peptide interacts with endogenous Gαq in the heart.
Acknowledgments
We thank D. Yu for guidance on appropriate statistical analysis and Z. Qu for technical assistance.
Funding: This work was supported by a Brody Family Medical Trust Fund Fellowship to S.M.S. and NIH grants R37 HL061690, P01 HL108806, and P01 HL075443 (W.J.K.).
Footnotes
Author contributions: S.M.S. and W.J.K. wrote the manuscript and designed the experiments. S.M.S. conducted most of the experiments and analyzed the data. M.C. and M.L. were a medical student and a graduate student, respectively, who trained under S.M.S. and assisted in performing some experiments. M.L. performed blinded imaging for the CSA and fibrosis quantification. E.G. and S.M.S. performed the sham and TAC surgeries, and E.G. performed the in vivo hemodynamics. S.M.S. and E.G. performed statistical analysis. J.K.C. and W.J.K. provided intellectual guidance and manuscript revision.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Penn RB, Pronin AN, Benovic JL. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc Med. 2000;10:81–89. doi: 10.1016/s1050-1738(00)00053-0. [DOI] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Sato PY, Chuprun JK, Schwartz M, Koch WJ. The evolving impact of G protein-coupled receptor kinases in cardiac health and disease. Physiol Rev. 2015;95:377–404. doi: 10.1152/physrev.00015.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evron T, Daigle TL, Caron MG. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci. 2012;33:154–164. doi: 10.1016/j.tips.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penela P, Murga C, Ribas C, Lafarga V, Mayor F., Jr The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T. Selective regulation of Gαq/11 by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem. 1999;274:34483–34492. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- 7.Sallese M, Mariggio S, D’Urbano E, Iacovelli L, De Blasi A. Selective regulation of Gq signaling by G protein-coupled receptor kinase 2: Direct interaction of kinase N terminus with activated Gαq. Mol Pharmacol. 2000;57:826–831. [PubMed] [Google Scholar]
- 8.Sterne-Marr R, Tesmer JJG, Day PW, Stracquatanio RP, Cilente JAE, O’Connor KE, Pronin AN, Benovic JL, Wedegaertner PB. G protein-coupled receptor Kinase 2/Gαq/11 interaction. A novel surface on a regulator of G protein signaling homology domain for binding G alpha subunits. J Biol Chem. 2003;278:6050–6058. doi: 10.1074/jbc.M208787200. [DOI] [PubMed] [Google Scholar]
- 9.Usui H, Nishiyama M, Moroi K, Shibasaki T, Zhou J, Ishida J, Fukamizu A, Haga T, Sekiya S, Kimura S. RGS domain in the aminoterminus of G protein-coupled receptor kinase 2 inhibits Gq-mediated signaling. Int J Mol Med. 2000;5:335–340. doi: 10.3892/ijmm.5.4.335. [DOI] [PubMed] [Google Scholar]
- 10.Rogers JH, Tsirka A, Kovacs A, Blumer KJ, Dorn GW, II, Muslin AJ. RGS4 reduces contractile dysfunction and hypertrophic gene induction in Gαq overexpressing mice. J Mol Cell Cardiol. 2001;33:209–218. doi: 10.1006/jmcc.2000.1307. [DOI] [PubMed] [Google Scholar]
- 11.Sterne-Marr R, Dhami GK, Tesmer JJG, Ferguson SSG. Characterization of GRK2 RH domain-dependent regulation of GPCR coupling to heterotrimeric G proteins. Methods Enzymol. 2004;390:310–336. doi: 10.1016/S0076-6879(04)90020-1. [DOI] [PubMed] [Google Scholar]
- 12.Dhami GK, Anborgh PH, Dale LB, Sterne-Marr R, Ferguson SSG. Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J Biol Chem. 2002;277:25266–25272. doi: 10.1074/jbc.M203593200. [DOI] [PubMed] [Google Scholar]
- 13.Day PW, Tesmer JJG, Sterne-Marr R, Freeman LC, Benovic JL, Wedegaertner PB. Characterization of the GRK2 binding site of Gαq. J Biol Chem. 2004;279:53643–53652. doi: 10.1074/jbc.M401438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorn GW, II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadruz W. Myocardial remodeling in hypertension. J Hum Hypertens. 2015;29:1–6. doi: 10.1038/jhh.2014.36. [DOI] [PubMed] [Google Scholar]
- 16.Lorell BH, Carabello BA. Left ventricular hypertrophy: Pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. doi: 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- 17.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- 18.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorn GW, II, Tepe NM, Lorenz JN, Koch WJ, Liggett SB. Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gαq-overexpressing mice. Proc Natl Acad Sci USA. 1999;96:6400–6405. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., II Enhanced Gαq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 22.Esposito G, Naga Prasad SV, Rapacciuolo A, Mao L, Koch WJ, Rockman HA. Cardiac overexpression of a Gq inhibitor blocks induction of extracellular signal–regulated kinase and c-Jun NH2-terminal kinase activity in in vivo pressure overload. Circulation. 2001;103:1453–1458. doi: 10.1161/01.cir.103.10.1453. [DOI] [PubMed] [Google Scholar]
- 23.Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 24.Martini JS, Raake P, Vinge LE, DeGeorge BR, Jr, Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc Natl Acad Sci USA. 2008;105:12457–12462. doi: 10.1073/pnas.0803153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 26.Rockman HA, Chien KR, Choi D-J, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Wang X, Qiu G, Gumpert A, Harris DM, Eckhart AD, Most P, Koch WJ. Level of G protein–coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res. 2010;107:1140–1149. doi: 10.1161/CIRCRESAHA.110.221010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen M, Sato PY, Chuprun JK, Peroutka RJ, Otis NJ, Ibetti J, Pan S, Sheu SS, Gao E, Koch WJ. Prodeath signaling of G protein–coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal–regulated kinase-dependent heat shock protein 90–mediated mitochondrial targeting. Circ Res. 2013;112:1121–1134. doi: 10.1161/CIRCRESAHA.112.300754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan Q, Chen M, Zuo L, Shang X, Huang MZ, Ciccarelli M, Raake P, Brinks H, Chuprun KJ, Dorn GW, II, Koch WJ, Gao E. Myocardial ablation of G protein–coupled receptor kinase 2 (GRK2) decreases ischemia/reperfusion injury through an anti-intrinsic apoptotic pathway. PLOS One. 2013;8:e66234. doi: 10.1371/journal.pone.0066234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJG. Snapshot of activated G proteins at the membrane: The Gαq-GRK2-Gβγ complex. Science. 2005;310:1686–1690. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 31.Lyon AM, Taylor VG, Tesmer JJG. Strike a pose: Gαq complexes at the membrane. Trends Pharmacol Sci. 2014;35:23–30. doi: 10.1016/j.tips.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carman CV, Lisanti MP, Benovic JL. Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem. 1999;274:8858–8864. doi: 10.1074/jbc.274.13.8858. [DOI] [PubMed] [Google Scholar]
- 33.Pronin AN, Satpaev DK, Slepak VZ, Benovic JL. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J Biol Chem. 1997;272:18273–18280. doi: 10.1074/jbc.272.29.18273. [DOI] [PubMed] [Google Scholar]
- 34.Mariggiò S, García-Hoz C, Sarnago S, De Blasi A, Mayor F, Jr, Ribas C. Tyrosine phosphorylation of G-protein-coupled-receptor kinase 2 (GRK2) by c-Src modulates its interaction with Gαq. Cell Signal. 2006;18:2004–2012. doi: 10.1016/j.cellsig.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 36.McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 37.Raake PWJ, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, Pleger S, Mier W, Haberkorn U, Koch WJ, Katus HA, Most P, Müller OJ. AAV6.βARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J. 2013;34:1437–1447. doi: 10.1093/eurheartj/ehr447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno-associated virus serotype 6–βARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012—An update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 40.Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CMB, Smrcka AV, Blaxall BC. Small molecule disruption of Gβγ signaling inhibits the progression of heart failure. Circ Res. 2010;107:532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamal FA, Mickelsen DM, Wegman KM, Travers JG, Moalem J, Hammes SR, Smrcka AV, Blaxall BC. Simultaneous adrenal and cardiac G-protein–coupled receptor-Gβγ inhibition halts heart failure progression. J Am Coll Cardiol. 2014;63:2549–2557. doi: 10.1016/j.jacc.2014.02.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rettenmaier TJ, Sadowsky JD, Thomsen ND, Chen SC, Doak AK, Arkin MR, Wells JA. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc Natl Acad Sci USA. 2014;111:18590–18595. doi: 10.1073/pnas.1415365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keys JR, Greene EA, Cooper CJ, Naga Prasad SV, Rockman HA, Koch WJ. Cardiac hypertrophy and altered β-adrenergic signaling in transgenic mice that express the amino terminus of β-ARK1. Am J Physiol Heart Circ Physiol. 2003;285:H2201–H2211. doi: 10.1152/ajpheart.00112.2003. [DOI] [PubMed] [Google Scholar]
- 44.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445–1453. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang ZM, Gao E, Fonseca FV, Hayashi H, Shang X, Hoffman NE, Chuprun JK, Tian X, Tilley DG, Madesh M, Lefer DJ, Stamler JS, Koch WJ. Convergence of G protein–coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci Signal. 2013;6:ra95. doi: 10.1126/scisignal.2004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TgβARKrgs mice exhibit a similar decrease in cardiac hypertrophy as TgGqI mice after TAC.
Fig. S2. Chronic pressure overload–induced heart failure is inhibited by cardiac βARKrgs expression.
Fig. S3. Molecular markers of left ventricular dysfunction are reduced in TgβARKrgs and TgGqI mice but not in TgβARKct mice.
Fig. S4. Left ventricular structural remodeling is inhibited in TgβARKrgs mice.
Fig. S5. The βARKrgs peptide interacts with endogenous Gαq in the heart.