

Abstract
Purpose
Mutations in the gene ARL13B cause the classical form of Joubert syndrome, an autosomal recessive ciliopathy with variable degrees of retinal degeneration. As second-site modifier alleles can contribute to retinal pathology in ciliopathies, animal models provide a unique platform to test how genetic interactions modulate specific phenotypes. In this study, we analyzed the zebrafish arl13b mutant for retinal degeneration and for epistatic relationships with the planar cell polarity protein (PCP) component vangl2.
Methods
Photoreceptor and cilia structure was examined by light and electron microscopy. Immunohistochemistry was performed to examine ciliary markers. Genetic interactions were tested by pairwise crosses of heterozygous animals. Genetic mosaic animals were generated by blastula transplantation and analyzed by fluorescence microscopy.
Results
At 5 days after fertilization, photoreceptor outer segments were shorter in zebrafish arl13b−/− mutants compared to wild-type larvae, no overt signs of retinal degeneration were observed by light or electron microscopy. Starting at 14 days after fertilization (dpf) and continuing through 30 dpf, cells lacking Arl13b died following transplantation into wild-type host animals. Photoreceptors of arl13b−/−;vangl2−/− mutants were more compromised than the photoreceptors of single mutants. Finally, when grown within a wild-type retina, the vangl2−/− mutant cone photoreceptors displayed normal basal body positioning.
Conclusions
We show that arl13b−/− mutants have shortened cilia and photoreceptor outer segments and exhibit a slow, progressive photoreceptor degeneration that occurs over weeks. The data suggest that loss of Arl13b leads to slow photoreceptor degeneration, but can be exacerbated by the loss of vangl2. Importantly, the data show that Arl13b can genetically and physically interact with Vangl2 and this association is important for normal photoreceptor structure. The loss of vangl2, however, does not affect basal body positioning.
Keywords: cilia, photoreceptor, Arl13b, Vangl2, planar polarity, retina, zebrafish
Ciliopathies refer to the collection of human disorders caused by defects in cilia.1,2 The primary cilium is a microtubule-based “cellular antenna” protruding from the surface of nearly all vertebrate cells.3 These disorders vary across a wide clinical spectrum and involve organ systems, such as the eye, kidney, nervous system, bones, liver, heart, and gonads.1 Syndromic disorders, such as Joubert syndrome (MIM 213300), Bardet-Biedl syndrome (MIM 209900), Jeune syndrome (MIM 208500), Meckel-Grüber syndrome (MIM 249000), or Senior-Løken syndrome (MIM 266900), are defined by the specific organs affected and the relative severity of the symptoms.
Joubert syndrome is a rare, recessive ciliopathy characterized by congenital ataxia and a distinctive midbrain–hindbrain malformation known as the “molar tooth sign.” Hypotonia, hepatic fibrosis, kidney cysts, polydactyly, and varying degrees of ocular disorders also can be observed.4–6 Approximately 30% of individuals with Joubert syndrome exhibit retinal dystrophy, making this the most common secondary diagnostic feature.7 To date, 22 causative genes for Joubert syndrome have been identified and the encoded proteins localize at or near the primary cilium. A genotype–phenotype correlation analysis of 440 individuals with Joubert syndrome revealed that retinal dystrophy was strongly correlated with mutations in the genes for CEP290 and AHI1, although the limitations inherent to analyses of rare disorders precluded associating phenotypes with specific mutant alleles.7
Mutations in ARL13B (accession number NM_182896) are responsible for approximately 1% to 2% of Joubert syndrome cases7 and patients with mutations in ARL13B exhibit abnormal eye movements, ptosis, and signs of coloboma.5 The electroretinogram responses (ERGs), however, can range from undetectable to normal,5,8 leaving the role of Arl13b in retinal function unresolved. Arl13b is a member of the ADP-ribosylation factor-like (ARL) family of small GTPases9,10 and localizes to cilia, but its function(s) are poorly understood, particularly in photoreceptor cell biology. As genetic interactions between mutant alleles of ciliopathy loci can modulate pathologic expression,11–13 it is possible that mutant alleles of ARL13B also may contribute to other diseases and that alleles at other loci modulate the penetrance of retinal phenotypes in Joubert syndrome patients harboring ARL13B mutations.
The role of Arl13b in cilia formation was first documented in mouse14 and zebrafish.15 The mouse hennin (hnn/Arl13b) exhibited embryonic lethality and altered cilia architecture,14 while the zebrafish scorpion/arl13b mutant had kidney cysts and cilia defects.10,15 In both cases, the retinal phenotype was not explored. Although Arl13b itself does not undergo intraflagellar transport (IFT) movement, axonemal defects have been observed in arl13 mutant worms16 and Arl13b mutant mice,14 which result in mislocalization of ciliary receptor proteins and destabilized movement of the IFT particle. These results suggest that phenotypes result from altered ciliary structure rather than ciliogenesis defects.16 Such structural abnormalities could perturb transport and/or localization of ciliary proteins, resulting in slow degeneration of cilia. We examined the role of Arl13b in photoreceptors and tested the hypothesis that genetic interactions between Arl13b and Vangl2, a component of the planar cell polarity (PCP) pathway, exacerbate the ciliary defects in photoreceptors. Mutation of arl13b with vangl2 (accession number NM_153674) resulted in shortened cilia and photoreceptor outer segments and Arl13b biochemically interacted with Vangl2.
Materials and Methods
Zebrafish Care and Maintenance
All animals were raised and maintained at 28.5°C on a 14/10-hour light/dark cycle in integrated AHAB housing systems (Aquatic Habitats, Apopka, FL, USA) and performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH; Bethesda, MD, USA). The protocols were approved by the Institutional Animal Care and Use Committees at Texas A&M University and the Cleveland Clinic, and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Wild-type zebrafish were of a mixed AB*-Ekkwill strain. The arl13b (sco)hi459 and vangl2m209 mutant lines were obtained from the Zebrafish International Resource Center (ZIRC) at the University of Oregon. The Tg(−3.2gnat2:eGFP) transgenic line, referred to as the TαC:GFPucd1, was used to label cone photoreceptors and was obtained from Sue Brockerhoff (University of Washington, Seattle, WA, USA). In this line, sequences from the cone transducin α promoter drive expression of GFP.17
Histologic Analysis
Larvae were euthanized in ice water and fixed in a primary fixation solution of 2.5% glutaraldehyde, 1.0% paraformaldehyde, and 2% tannic acid in PBS18 at 28.5°C for 20 minutes, followed by an overnight incubation at 4°C with rotation. Specimens subsequently were processed using cold microwave technology in a BioWave microwave (Ted Pella, Inc., Redding, CA, USA). Primary fixation was completed at 20°C by a 6-minute microwave cycle (2 minutes on, 2 minutes off, 2 minutes on) at 200 watts (W), with intermittent vacuum cycles (30 seconds on, 30 seconds off). Specimens were washed two times in 0.06 M phosphate rinse buffer (0.046 M NaH2PO4, 0.302 M Na2HPO4, pH 7.35–7.40) containing 0.002% CaCl and 3% sucrose for 15 minutes at room temperature with rotation. Specimens were postfixed in 1% OsO4 (prepared in 0.06 phosphate rinse buffer) for 1.5 hours at 4°C before completing fixation at 20°C by a 6-minute microwave cycle as described above. Specimens were washed twice in 0.06 M phosphate rinse buffer for 15 minutes at room temperature. Specimens were dehydrated through a series of ethanol–water steps (10%–100%) in the cold microwave at 20°C and 200 W. Ethanol was replaced with propylene oxide (PO) for 15 minutes at room temperature with rotation (×2). Specimens then were infiltrated with epoxy resin at room temperature. Transverse semithin sections (1 μm) and thin sections (100 nM) were processed as described previously.19,20 Grids then were imaged in a JEOL 1200EX transmission electron microscope and whole images were adjusted for brightness and contrast using Adobe Photoshop (Adobe Systems, San Jose, CA, USA).
Immunohistochemistry and In Situ Hybridization
Larvae were processed for immunohistochemistry, whole-mount antibody staining, and in situ hybridization as described previously.21,22 Antibodies used included the 1D1 monoclonal antibody (a gift from Jim Fadool, Florida State University, Tallahassee, FL, USA) at 1:200 dilution, the mouse anti-acetylated α tubulin (T7451, clone 6-11B-1; Sigma-Aldrich Corp., St. Louis, MO, USA) at 1:1000 dilution, the mouse monoclonal antibody Zpr-1 (Fret-43; Zebrafish International Resource Center, Eugene, OR, USA) at 1:200 dilution, and the rabbit polyclonal anti-Ift88 antibody23 at 1:5000 dilution. Alexa Fluor secondary antibodies (Life Technologies, Frederick, MD, USA) were used at 1:500 dilutions. We used 4′,6-diamidino-2-phenylendole (DAPI; 1:10,000) as a nuclear stain. Images were generated as z-stacks of optical sections collected using a Zeiss ImagerZ1 fluorescence microscope fitted with an ApoTome and AxioCam (Carl Zeiss, Thornhill, NY, USA) and processed with ImageJ and Adobe Photoshop. At least two independent experiments were performed for all immunohistochemistry studies and at least six larvae were sectioned and stained in each experiment. A minimum of 12 retinas were evaluated in each study. Statistics were calculated and graphed using GraphPad Prism6 (GraphPad Software, La Jolla, CA, USA). Immunohistochemistry and imaging of adult retinas for basal body analysis was performed as described previously.21
Mosaic Analysis
Mosaic retinas were created through blastomere transplantation.24 Embryos were collected from pairwise crosses of arl13b+/−;Tg(−3.2gnat:eGFP)+/+ adults or vangl2+/−;Tg(−3.2gnat:eGFP)+/+ adults and injected with 5% solution of lysine-fixable rhodamine-dextran (Life Technologies) at the 1- to 8-cell stage. Between 3 and 4 hours after fertilization (hpf), 20 to 40 donor cells were transplanted into the region fated for eye and forebrain of wild-type hosts.25 After 24 hpf, embryos were allowed to develop in 200 μM PTU solution to prevent melanosome pigmentation. The phenotype of donor embryos was determined at 3 to 4 dpf. Host embryos were assessed for the presence of GFP+ donor cells in the retina by fluorescence microscopy at 4 days after fertilization (dpf). Mosaic fish were raised to the indicated time points and assessed by immunohistochemistry. Between 10 and 12 animals were evaluated for each donor–host combination at each time point.
Plasmids and Cell Culture
Human ARL13b cDNA was obtained from OriGene (Rockville, MD, USA) in the pCMV6-AC-GFP shuttle vector, which expresses a turbo green fluorescent protein (tGFP). The human VANGL2 cDNA was obtained from Origene, amplified by PCR and cloned into pCS2+8 (Addgene, Cambridge, MA, USA). Clones were validated by Sanger sequencing. HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) in 5% fetal bovine serum (FBS) in 6-well plates according to standard procedures. DNA was transfected with Lipofectamine2000 (Life Technologies) when cells were approximately 70% confluent and cultured for 24 hours in serum-free media to induce ciliogenesis.
Immunoprecipitation and Western Blotting
Cultured cells were washed with PBS and lysed with m-PER buffer (Thermo Fisher Scientific, Inc., Rockford, IL, USA) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Immunoprecipitation was performed using the Pierce Crosslink Magnetic IP and Co-IP Kit (Thermo Fisher Scientific) per manufacturer's instructions. Eluates were resuspended in 5× Laemmli-DTT buffer and resolved on 4% to 12% Bis-Tris Plus SDS polyacrylamide gels before being transferred to polyvinylidine fluoride (PVDF) membranes. Blots were blocked for 45 minutes with Tris-buffered saline with 0.1% Tween-20 (TBST) buffer in 5% milk and subsequently incubated for 45 minutes at room temperature with primary antibodies. Blots were washed in TBST and incubated for 45 minutes with secondary antibodies diluted in TBST in 0.5% milk. Blots were washed 3 times with TBST buffer and incubated with Pierce Clean-blot IP Detection Reagent for 45 minutes at room temperature. The rabbit anti-Vangl2 (EMD-Millipore; Temecula, CA, USA) and rabbit anti-ARL13b (17711-1-AP; Protein Tech, Chicago, IL, USA) were used as primary antibodies for immunoblotting. The goat anti-VANGL2 (sc-46561; Santa Cruz Biotechnology, Dallas, TX, USA) was used for immunoprecipitations.
Results
Zebrafish arl13b−/− Mutants Have Normal Retinal Anatomy
We analyzed the zebrafish arl13bhi459 mutant allele, which resulted from a proviral insertion within the 5′-UTR region of the arl13b gene and is referred to herein as the arl13b−/− mutant.15 Previous studies have shown that the arl13b transcript is supplied maternally and arl13bhi459 represents a zygotic null allele.10 Zebrafish carrying homozygous mutations in the arl13b gene exhibit defects in body axis curvature and kidney cysts (Figs. 1A, 1B), traits commonly observed in zebrafish with ciliary defects.15 Phenotypically wild-type siblings (arl13b+/+ and arl13b+/–) were grouped together (arl13b+/?). At 5 dpf the rod and cone photoreceptors have differentiated and outer segments can be identified. The arl13b−/− mutant larvae had normally laminated retinas with all nuclear layers present (Figs. 1C–F). The photoreceptor outer segments appeared intact. We analyzed photoreceptor ultrastructure by transmission electron microscopy and again saw no qualitative differences in photoreceptor disc membrane organization (Figs. 1G, 1H). These results were in contrast to several other zebrafish mutants and morphants of cilia and Joubert-related genes, such as components of the intraflagellar transport particle,19,23,26 cep290,27 and cc2d2a,28 which all exhibited severe photoreceptor degeneration and disorganization in the outer segments.
Figure 1.
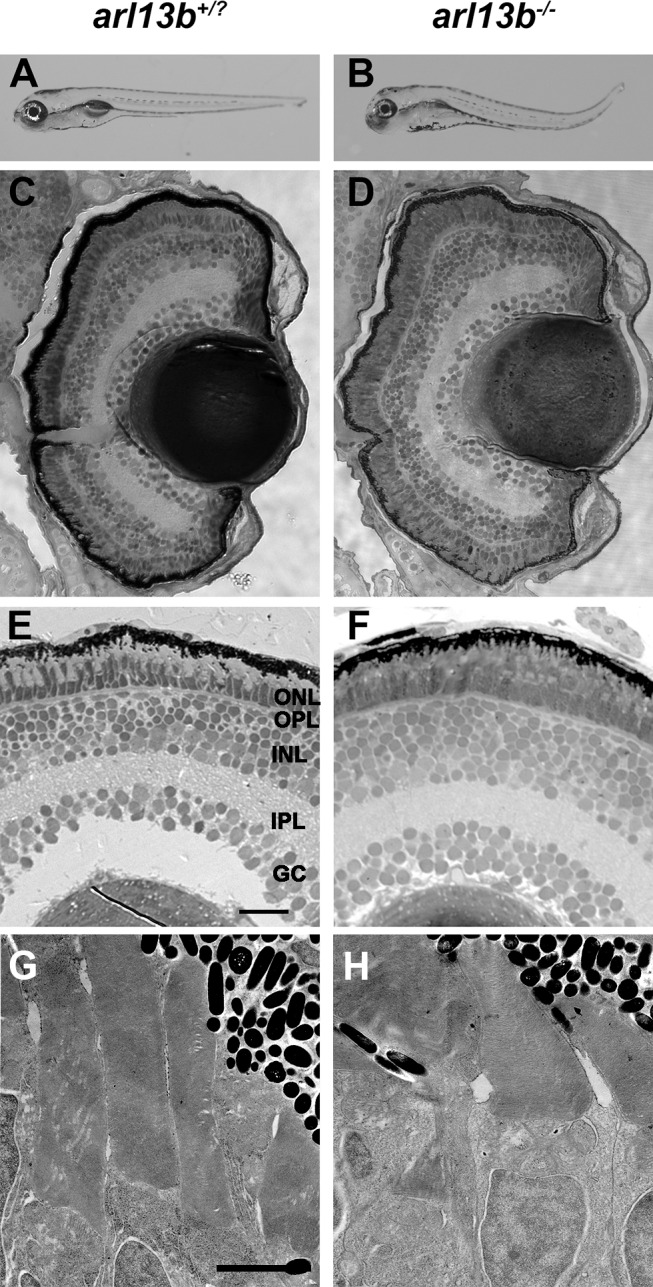
arl13b is not required for zebrafish retinal development. (A, B) Lateral view of larvae at 5 dpf. (C–F) Methylene blue-stained plastic sections of 5 dpf wild-type siblings (arl13b+/?) and arl13b−/− mutant retinas. The outer nuclear layer (ONL), outer plexiform layer (OPL), inner nuclear layer (INL), inner plexiform layer (IPL), and ganglion cell layer (GC) were present in wild-type and mutant retinas. (G, H) Transmission electron micrographs of 5 dpf retinas do not show any evidence of photoreceptor outer segment disorganization or ciliary defects. Scale bars: 200 μm (A, B), 20 μm (C, D), 10 μm (E, F), and 2 μm (G, H).
Unlike motile cilia, which consist of nine outer microtubule doublets and two inner microtubule doublets (9 + 2 configuration), the photoreceptor outer segment is a modified primary cilium that adopts a 9 + 0 axonemal architecture that lacks the central pair. Defects in the outer microtubules of the axoneme were previously reported in motile cilia of the node in mouse Arl13b−/− mutants,14 but microtubule organization was normal in motile kidney cilia of zebrafish arl13b−/− mutants.10 We examined the ultrastructure of photoreceptor connecting cilia in cross-section and could not detect differences between wild-type and mutants (Fig. 2). In cross-section, photoreceptor cilia appeared normal and were surrounded by numerous electron-lucent regions that are likely the calyceal processes (Fig. 2A, arrows). The lack of structural defects in kidney and photoreceptor cilia may reflect species-specific differences between zebrafish and mouse or that the architectural abnormalities only manifest in some ciliated tissues.
Figure 2.

Ultrastructural analysis of photoreceptor connecting cilia by transmission electron microscopy. (A, B). Horizontal sections through photoreceptors of wild-type and arl13b−/− larvae at 5 dpf show the connecting cilia in the vicinity of the mitochondria (M). Calyceal processes surround the cilium (arrows). (C, D) Higher magnification images revealed no obvious defects in microtubule organization in arl13b−/− mutants. Scale bars: 0.5 μm (A, B) and 0.2 μm (C, D).
Loss of arl13b Leads to Shorter Outer Segments and Cilia
We next used immunohistochemistry to determine if the localization and trafficking of photoreceptor ciliary proteins occurred normally in arl13b−/− mutants. At 4 dpf, rhodopsin localized normally to the rod outer segments of wild-type and arl13b−/− mutants and no inner segment mislocalization was seen (Figs. 3A, 3B). We noticed, however, that the size of the rhodopsin-positive outer segment was considerably smaller in arl13b−/− mutants. We used rhodopsin immunoreactivity as a surrogate for outer segment length and quantified the length of the outer segment as extent of rhodopsin staining along the proximal–distal axis of the outer segment (Fig. 3C). Wild-type outer segments were 8.3 μm in length (n = 16 embryos, 112 outer segments), while arl13b−/− mutant outer segments were 5.1 μm in length (38.3% shorter; n = 9 embryos, 116 outer segments). To determine if this correlated with smaller cilia, we then quantified cilia length by measuring the extent of labeling with antibodies against acetylated α-tubulin (Figs. 3D–F). Immunoreactivity against Ift88, a component of the IFT particle, was used to label the connecting cilium.23 We found that arl13b−/− mutant cilia were significantly shorter than wild-type (3.9 vs. 1.6 μm; n ≥ 8 embryos, 170 cilia per genotype). While this is consistent with previous studies that reported smaller cilia in Arl13b-null mice and zebrafish,10,14 we cannot rule out the possibility that our results reflect reduced posttranslational acetylation of tubulin, which was shown to occur in Arl13b−/− null mouse embryonic fibroblasts.29 It should be noted that unlike many other zebrafish cilia mutants and morphants,19,26,30–32 we did not observe any other signs of photoreceptor degeneration or loss (e.g., pyknotic nuclei, acellular holes, mislocalized opsin) in plastic sections or by immunohistochemistry of arl13b−/− mutants by 4 to 5 dpf. Taken together, these results indicated that arl13b is not required to form cilia or photoreceptor outer segments but may be necessary for elongation of ciliary structures.33 Because mutant larvae die by 8 to 9 dpf, it is not clear if long-term survival of photoreceptors requires Arl13b.
Figure 3.

arl13b−/− mutants have shorter photoreceptor outer segments and cilia at 4 dpf. (A, B) Immunohistochemistry on transverse cryosections from 4 dpf wild-type and arl13b−/− mutant retinas stained for rhodopsin (red). (C) Quantification of the length of rhodopsin staining along the long axis of the photoreceptor. (D, E) Immunohistochemistry for acetylated α-tubulin (red) and Ift88 (green). (F) Quantification of average cilia length. All sections were counterstained with DAPI to label nuclei. Scale bars: 10 μm. Error bars: ± SEM. *P < 0.0001 using Student's t-test with Welch's correction.
Arl13b-Deficient Photoreceptors Undergo Progressive Degeneration
To determine if loss of Arl13b would eventually lead to photoreceptor death, we generated genetically mosaic animals to place arl13b-deficient cells into wild-type animals that would live to ages beyond the lifespan of the arl13b−/− mutants. We hypothesized that arl13b− deficient cells would eventually degenerate and die. We bred the arl13b mutation to the TαC:GFPucd1 transgenic line (see Methods) to genetically label cone photoreceptors with GFP. Embryos arising from a cross of heterozygous arl13b+/−; TαC:GFPucd1 adults were grown to blastula stage (∼ 4 hpf) and donor cells then were transplanted into nontransgenic wild-type blastula hosts (Fig. 4A). Cell transplantation did not affect viability of hosts or donors. The mutant and wild-type donors then were distinguished by phenotype at 4 dpf. Hosts containing GFP-positive cones at 4 dpf then were selected and subsequently raised to 14 to 30 dpf. These hosts were analyzed by immunohistochemistry at the stated time points. Green fluorescent protein-labeled photoreceptors would populate eyes unilaterally or bilaterally and would vary in clone size and location within the retina (Figs. 4B, 4C). To determine if cells showed signs of degeneration, sections were stained with 1D1 to label rhodopsin. Clones of wild-type cells were identified by GFP-positive cones. Rods within these clones showed opsin localized exclusively to the outer segments (Fig. 4D). In clones of arl13b-deficient cells, we observed mislocalization of opsin (Fig. 4E, arrow), disorganization of the outer segment layer, and gaps where GFP-positive cones were missing. To estimate the extent of photoreceptor loss, both eyes from at least 10 hosts at 14, 21, 25, and 30 dpf of age were cryosectioned and analyzed for the presence of GFP-positive cones. If a single GFP-expressing cone was found in any section from a given retina, that host was considered to be “positive” for GFP donor cells. At 14, 21, and 25 dpf, 12/12 (100%) hosts still contained GFP-expressing cones from wild-type donors, while 11/12 (92%) hosts had GFP-expressing cones from wild-type donors at 30 dpf (Fig. 4E). As early at 14 dpf, only 7/10 (70%) hosts still contained arl13b-deficient GFP-expressing cones. By 30 dpf, only 4/10 (40%) of the donors still had any GFP-expressing cones from arl13b−/− mutant donors (Fig. 4E). These results strongly suggest that photoreceptors lacking Arl13b progressively degenerate and die while wild-type cells incorporate into the neural retina and survive at least a month after transplantation. Unfortunately, attempts to directly identify dying cells by TUNEL labeling or by immunoreactivity to caspase-9 were unsuccessful. At least two explanations could account for these results. First, cell death was only sampled every 4 to 7 days. TUNEL-positive cells typically are cleared within 24 hours and dying cells may have escaped detection during the larger window. Second, smaller clones undergoing progressive degeneration may lose cells in small numbers over a broad window of time, which also may be difficult to detect.
Figure 4.

Progressive degeneration of arl13b−/− photoreceptors. (A) Overview of the blastula transplant strategy. Heterozygous arl13+/− fish were crossed to the TαC:GFPucd1 transgenic line to produce arl13b+/−; TαC:GFPucd1 animals. Progeny from heterozygous crosses were injected with rhodamine-dextran and donor cells transplanted into the animal pole of unlabeled wild-type hosts. At 4 dpf, arl13b−/−;TαC:GFPucd1 mutants were identified phenotypically (top). Wild-type hosts containing photoreceptors from mutant donors were identified by mosaic GFP fluorescence within the eye (bottom). (B, C) Cryosections of 14 dpf larvae showing GFP+ clones populating retinas bilaterally and unilaterally. (D, E) Immunofluorescence image of 16 dpf larval retinas immunolabeled for rhodopsin (red). Cone photoreceptors from donor embryos express GFP (green). Transplanted cone photoreceptors from an arl13b−/−; TαC:GFPucd1 mutant donors exhibited partial rhodopsin mislocalization within the donor clone ([E], arrows). Sections were counterstained with DAPI to show nuclei. (E) Graph showing the percentage of hosts (n > 11 retinas per genotype) still containing GFP-positive cells from wild-type or arl13b−/− mutant donors at specified time points. Scale bar: 50 μm.
Genetic Interactions Between arl13b and vangl2
Because genetic interactions (i.e., epistasis) between cilia-related genes can have additive or synergistic effects on ciliary phenotypes,12,34–36 we hypothesized that phenotypes associated with loss of arl13b could be exacerbated by partial or complete loss of function in other genes regulating cilia function. We previously demonstrated that the basal bodies of zebrafish cone photoreceptors localize at one side of the apical domain of the inner segment, in a phenomenon known as translational polarity.21 This kind of polarity is regulated by the PCP pathway.37 The transmembrane protein Vangl2 is a core member of the PCP pathway and is essential for PCP signaling. We found that vangl2 was expressed throughout the central nervous system in 5 dpf larvae (Fig. 5A). Within the retina, vangl2 expression was strongest in the RPE and photoreceptor layer in zebrafish larvae at 5 dpf (Fig. 5B). While the loss of Vangl2 does not impinge on cilia formation, asymmetric cilia orientation often is a critical factor in proper ciliary and cellular function. Therefore, we reasoned that arl13b may genetically interact with PCP genes. Using immunohistochemistry, connecting cilia lengths and photoreceptor outer segment area were analyzed in retinas of arl13b−/− and vangl2−/− single mutants and arl13b−/−;vangl2−/− double mutant larvae at 5 dpf (Fig. 6). Cilia in the dorsocentral region of the retina were stained with antibodies against acetylated α-tubulin and quantified (Figs. 6A–D, 6I). Compared to cilia in wild-type larvae (4.5 μm) loss of arl13b reduced cilia length (3.6 μm; P < 0.0001), whereas loss of vangl2 had no effect on cilia length (Fig. 6I). This is consistent with previous maternal-zygotic vangl2 mutants that have normal cilia length.38 The arl13b−/−;vangl2−/− double mutants had a significant reduction in cilia length (1.87 μm; P < 0.0001). Similar results were observed when rhodopsin staining was used to quantify outer segment area (Figs. 6E–H, 6J). Wild-type outer segments averaged 29.5 ± 1.3 μm2 (mean ± SEM) in area. Loss of arl13b reduced outer segment area by 31% (20.2 μm2; P < 0.0001), whereas no change in outer segment length was observed in vangl2 mutants (27.9 μm2; P > 0.05). Loss of arl13b and vangl2 reduced outer segment area by 46% (15.8 μm2; P < 0.0001).
Figure 5.

Expression of vangl2 in 5 dpf zebrafish larvae. (A) Whole-mount in situ hybridization of vangl2 in 5 dpf larvae shows expression in the central nervous system. (B) Retinal expression was highest in the RPE and photoreceptor layers. Scale bar: 40 μm.
Figure 6.

Suppression of arl13b and PCP components result in shortened photoreceptor cilia and outer segments. (A–D) Immunohistochemistry using antibodies against acetylated α-tubulin (red) to label cilia in retinal cryosections of 5 dpf zebrafish larvae. (E–H) Immunohistochemistry using rhodopsin antibodies to label photoreceptor outer segments in retinal cryosections of 5 dpf larvae. (I) Quantification of cilia length. (J) Quantification of outer segment area. All sections were counterstained with DAPI. Results are significant (****P < 0.0001) using a 1-way ANOVA with Tukey's multiple comparisons test correction. Error bars: ± SEM. Scale bar: 10 μm.
Arl13b Forms a Complex With Vangl2
We next tested if a biochemical link exists between these two proteins. HEK293-T cells were transiently transfected with constructs expressing human Vangl2 and/or a human Arl13b-GFP fusion protein. Protein extracts were used for immunoprecipitation using an anti-Vangl2 antibody and subsequently immunoblotted with antibodies against Arl13b. A band at approximately 85 kDa (Fig. 7A, black arrow), which corresponded to Arl13b-GFP, was detected in protein extracts containing Vangl2 (Fig. 7A, lane 1), but not in the absence of Vangl2 (Fig. 7A, lanes 2, 3). The specificity of the Arl13b antibody was confirmed by immunoprecipitating extracts with the Arl13b antibody and immunoblotting with the same antibody (Fig. 7A, lanes 4, 5). This antibody also detected two nonspecific bands of lower molecular weight (Fig. 7A, white arrows). In reciprocal experiments, protein extracts were immunoprecipitated with antibodies against Arl13b and immunoblotted with anti-Vangl2 antibodies. Vangl2 was detected as a single band at approximately 60 kDa in extracts containing Arl13b (Fig. 7B, lane 1), but not in extracts lacking Arl13b (Fig. 7B, lane 3). These results suggested that Arl13b and Vangl2 may exist in a specific protein complex important for outer segment development or stability.
Figure 7.

Arl13b biochemically interacts with Vangl2. (A) Protein extracts from HEK293-T cells transiently transfected with Vangl2 and/or Arl13b and immunoprecipitated (IP) with antibodies against Vangl2 (lanes 1–3) or Arl13b (4–5) and subsequently immunoblotted (IB) with antibodies against Arl13b. Immunoblots of input samples show the presence of Arl13b from different extracts used for IP (lanes 6–8). Black arrows indicate bands corresponding to Arl13b, while open arrows indicate nonspecific bands. (B) Protein extracts from HEK293-T cells transiently transfected with Vangl2 and/or Arl13b and immunoprecipitated with antibodies against Arl13b (lanes 1–3) or Vangl2 (lanes 4–5) and subsequently subjected to IB with antibodies against Vangl2. Immunoblot of input samples to show the presence of Vangl2 in different protein extracts (lanes 6–8, black arrow). The high molecular weight band (white arrow) likely represents Vangl2 homodimers. Molecular mass of protein markers is denoted in kDa.
Vangl2 Is Not Essential for Basal Body Positioning or Photoreceptor Survival
We next wanted to know if Vangl2 could have an important role in establishing the translational polarity of basal bodies in adult cone photoreceptors, or in long-term photoreceptor survival. Zebrafish vangl2−/− mutants are lethal and do not survive beyond larval stages. To further investigate the requirements of Vangl2 in adult photoreceptors, blastula transplantations were conducted to place vangl2−/−;Tg(TαC:GFP) cells into wild-type host embryos and the hosts were grown to adulthood. We previously documented that in wild-type zebrafish red, green, and blue cones, the basal bodies align on the cell periphery, on the edge leading toward the optic nerve.21 To assess basal body positioning in vangl2−/− mutant cells, cryosections of flat-mounted retinas were stained with antibodies against γ-tubulin. The vangl2−/− mutant cones could be identified by GFP fluorescence in the cell body, while wild-type cells lacked GFP fluorescence. The identity of the specific cone subtypes was identified by the position within the row mosaic and the vertical tiering within the outer nuclear layer. We quantified the angular positions of basal bodies within multiple fields of wild-type and vangl2−/− mutant cells relative to the optic nerve and graphed on circular plot (a detailed description of the methodology was published previously21). As noted previously, the basal bodies of wild-type red-green double cones localized on the apical edge toward the optic nerve (Figs. 8A, 8C). The basal bodies of vangl2−/− mutant red- and green-sensitive cones also exhibited strong polarization toward the optic nerve (Fig. 8C), suggesting that Vangl2 is not essential for translational polarity. In wild-type and vangl2−/− mutant clones, the UV-sensitive cones failed to exhibit the same translational polarity (Figs. 8B, 8D), as we reported previously.21 In transverse cryosections of wild-type hosts containing vangl2−/− mutant clones, we found no difference in peanut agglutinin (PNA) staining of cone outer segments within the clone compared to cones outside the clone (Fig. 8E), suggesting vangl2−/− is not required for cone outer segment formation or cone survival. Similarly, we found no evidence of rhodopsin mislocalization or shortening of outer segments in vangl2−/− mutant rods, that would have indicated rod degeneration (Fig. 8F).
Figure 8.

Vangl2 is not essential for photoreceptor outer segments. (A, B) Representative fields of cells showing basal bodies of (A) red-/green-sensitive cones and (B) UV-sensitive cones. Basal bodies were stained by γ-tubulin (red) and GFP (green) marked vangl2−/−;Tg(−3.2gnat2:GFP) donor cells. Clones of donor cells are marked with white dotted lines. Nuclei were counterstained with DAPI (blue). (C, D) Angular dimensions of wild-type (red points) and vangl2−/− mutant basal bodies (green points) were plotted around a unit circle. The mean vectors are indicated as black arrows. The optic nerve is up in all Figures. (E, F) Transverse cryosections of mosaic animals containing vangl2−/−;Tg(−3.2gnat2:GFP) donor cells and stained for PNA or rhodopsin (red). No differences were seen in cells inside the clone versus those outside the clone. Scale bars: 10 μm (A, B) or 50 μm (E, F).
Discussion
We showed that: (1) mutation of arl13b in zebrafish results in shortened cilia and photoreceptor outer segments, (2) arl13b−/− photoreceptors degenerate over weeks, (3) arl13b genetically interacts with vangl2 to regulate the length of photoreceptor cilia, (4) Arl13b and Vangl2 biochemically interact in cell culture, but (5) Vangl2 is not essential for photoreceptor survival or for basal body positioning in zebrafish adult cones. To our knowledge, our work is the first to examine the consequences of arl13b loss on retinal anatomy in any species, including humans, and provides insight to the epistatic relationships that may influence disease variability in ciliopathies.
Retinal degeneration is considered to be a common feature of many ciliopathies,2 but the degree of visual dysfunction varies greatly within Joubert syndrome patients.39 The mutations causing Joubert syndrome occur in at least 22 genes coding for proteins that localize to the primary cilium or basal body. While some estimates suggest that 70% to 100% of Joubert syndrome patients have ocular abnormalities,40 this number often reflects reports of ocular nystagmus and little is known about retinal histopathology. Fundus examinations in a small study of 8 unrelated patients with unknown genetic lesions found that 3 individuals (38%) showed signs of retinal dystrophy, but all 8 patients had normal ERGs.39 A separate study of 13 Joubert syndrome patients, however, found that 6 had undetectable ERGs and pallor of the optic disc, indicating that retinal involvement in Joubert syndrome varies. Importantly, the clinical variability in retinal degeneration cannot be attributed primarily to the causative mutation, as siblings with identical mutations can have different clinical features.5,41 The presence of additional modifier alleles may explain this variability and reinforces the need for studies that investigate potential modifier alleles.12
Arl13b Function in Cilia
In many cell types, Arl13b localizes to proximal ciliary compartments and positively regulates cilia length.14,29,33,42 Arl13b is thought to function as the GEF for Arl3,43 an Arl family member that is important for trafficking of lipidated proteins. In mouse, zebrafish, and worms, shortened cilia are the only common phenotype observed to date in the absence of Arl13b. Defects in axonemal microtubules can occur in Caenorhabditis elegans amphid cilia and the mouse node,14,16 but microtubule structure was normal in zebrafish kidney and photoreceptor cilia (Duldulao et al.10 and this study). Furthermore, loss of Arl13b reduces the posttranslational acetylation and glutamylation of tubulin.29 It is unclear, however, if Arl13b directly regulates these posttranslational modifications or if these phenotypes are secondary to the architectural changes in the axonemes of Arl13b-deficient cells. Missense mutations in the GTPase domain cause Joubert syndrome in humans, and expression of GTPase mutant forms of Arl13b do not rescue the mutant phenotypes in zebrafish and cannot increase ciliary length when overexpressed.10,33 These mutant proteins do, however, localize normally to cilia, suggesting GTP hydrolysis is required for Arl13b function but not ciliary targeting.5,10,33
Slow Degeneration of arl13b−/− Photoreceptors
The time course of photoreceptor degeneration observed in zebrafish arl13b−/− mutants differs from that seen in other zebrafish models of Joubert syndrome and other ciliopathies. For example, mutation of cc2d2a or morpholino knockdown of cep290, two other causative genes for Joubert syndrome, resulted in abnormal outer segment assembly, trafficking defects during larval stages, and cell death.27,28 Our previous work demonstrated that mutation of IFT genes ift88 and ift172 completely blocked outer segment formation in zebrafish19 and that loss of bbs4 led to photoreceptor degeneration within 5 dpf.44 Although immunoreactivities for markers of the outer segments and cilia were shorter in arl13b−/− mutants, we did not observe phenotypes consistent with early rapid degeneration, such as reduced numbers of photoreceptors, mislocalization of opsin, or disorganized outer segment disc membranes at 5 dpf, which is seen typically in other mutants. Rather, arl13b-deficient photoreceptors were lost over the course of weeks and this was determined only through blastula-transplantation.
Interactions Between Arl13b and Components of the PCP Pathway
The genetic and physical interactions between Arl13b and Vangl2 provide a new link between cilia and the PCP pathway. It is well documented that genetic interactions between ciliopathy genes can modulate phenotypic severity in species ranging from worms to humans.11,12,16,45 In C. elegans, epistatic relationships exist between arl13 and the genes bbs-8, nphp-2, and nphp-4,16,46 which are the orthologs to the ciliopathy genes BBS8, NPHP2, and NPHP4. Epistatic interactions between BBS genes lead to more severe phenotypes in humans and zebrafish.11,13 The links between ciliopathy genes and PCP components also have been described. Genetic interactions between Bbs genes and Vangl2 caused early embryonic lethality and hair cell defects in mice.47 The absence of Bbs8 or Ift20 altered the membrane localization of Vangl2 in the cochlea, possibly by disrupting physical interactions within a complex.48 In zebrafish, genetic interactions between bbs genes and vangl2 regulated CE phenotypes during gastrulation.47,49 Our knowledge of genetic interactions modulating photoreceptor phenotypes has thus far been limited to interactions between known ciliopathy genes12,45 and almost nothing is known about the function of PCP genes in photoreceptor biology. We previously showed that basal bodies in zebrafish cones show exquisite translational polarity and align asymmetrically on the cell edge nearest the optic nerve, consistent with a role for planar polarity.21 Somewhat surprisingly, loss of vangl2 did not affect basal body positioning. At least two possibilities exist to explain these results. It is possible that Vangl2 may be functionally redundant with Vangl1 and loss of both proteins may be required to disrupt basal body positioning. Alternatively, Vangl2 may not be required at all and other signaling pathways function to establish translational polarity. Either way, the translational polarity of basal bodies is unlikely to occur through chance and active mechanisms would be required to maintain such asymmetries.
Implications for Human Disease
Although mutations in ARL13B account for a small percentage of Joubert syndrome cases, the data from zebrafish may provide insight into the time course of retinal degeneration in these patients. To date, two families have been identified with mutations in ARL13B and retinal degeneration is variable in children under the age of 10.5 In one family, one sibling presented with obesity, pigmentary retinopathy, and no recordable electroretinogram, while his affected siblings had normal ERGs and no obesity.50 One prediction is that the child with early degeneration carries pathogenic alleles in second-site modifier genes, while the siblings possess mutations only in ARL13B and will undergo a slower retinal degeneration. Thus far, none of the core PCP genes has been implicated as a causative or modifying allele in human ciliopathies. Given the growing evidence from animal studies that PCP components regulate cilia function, we hypothesized that PCP genes represent likely candidates for such modifier alleles.
Acknowledgments
The authors thank Kristen Denton and Rhonda Patterson (Texas A&M University) for their care of the Texas A&M zebrafish facility and Don Zeisloft for his care of the zebrafish facility at the Cleveland Clinic. The authors also thank the other members of the Perkins lab, Bela Anand-Apte, and Ivy Samuels for comments on earlier drafts of the manuscript.
Supported by NIH Grant EY017037, a Doris and Jules Stein Professorship Award from Research to Prevent Blindness (BDP), and by an NIH Ruth L. Kirschstein NRSA (F32) EY02514 (JF). The authors alone are responsible for the content and writing of this paper.
Disclosure: P. Song, None; L. Dudinsky, None; J. Fogerty, None; R. Gaivin, None; B.D. Perkins, None
References
- 1. Hildebrandt F,, Benzing T,, Katsanis N. Ciliopathies. N Engl J Med. 2011; 364: 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Novarino G,, Akizu N,, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011; 147: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Satir P,, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007; 69: 377–400. [DOI] [PubMed] [Google Scholar]
- 4. Doherty D. Joubert syndrome: insights into brain development cilium biology, and complex disease. Sem Ped Neurol. 2009; 16: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantagrel V,, Silhavy JL,, Bielas SL,, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008; 83: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sattar S,, Gleeson JG. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol. 2011; 53: 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bachmann-Gagescu R,, Dempsey JC,, Phelps IG,, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015; 52: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Romano S,, Boddaert N,, Desguerre I,, et al. Molar tooth sign and superior vermian dysplasia: a radiological, clinical, and genetic study. Neuropediatrics. 2006; 37: 42–45. [DOI] [PubMed] [Google Scholar]
- 9. Verpy E,, Weil D,, Leibovici M,, et al. Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature. 2008; 456: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duldulao NA,, Lee S,, Sun Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development. 2009; 136: 4033–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badano JL,, Leitch CC,, Ansley SJ,, et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006; 439: 326–330. [DOI] [PubMed] [Google Scholar]
- 12. Khanna H,, Davis EE,, Murga-Zamalloa CA,, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009; 41: 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zaghloul NA,, Liu Y,, Gerdes JM,, et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2010; 107: 10602–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caspary T,, Larkins CE,, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007; 12: 767–778. [DOI] [PubMed] [Google Scholar]
- 15. Sun Z,, Amsterdam A,, Pazour GJ,, Cole DG,, Miller MS,, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004; 131: 4085–4093. [DOI] [PubMed] [Google Scholar]
- 16. Cevik S,, Hori Y,, Kaplan OI,, et al. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol. 2010; 188: 953–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kennedy BN,, Alvarez Y,, Brockerhoff SE,, et al. Identification of a zebrafish cone photoreceptor-specific promoter and genetic rescue of achromatopsia in the nof mutant. Invest Ophthalmol Vis Sci. 2007; 48: 522–529. [DOI] [PubMed] [Google Scholar]
- 18. Sambrook J,, Fritsch EF,, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 19. Sukumaran S,, Perkins BD. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57 ift88 and ift172 intraflagellar transport mutants. Vision Res. 2009; 49: 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krock BL,, Bilotta J,, Perkins BD. Noncell-autonomous photoreceptor degeneration in a zebrafish model of choroideremia. Proc Natl Acad Sci U S A. 2007; 104: 4600–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramsey M,, Perkins BD. Basal bodies exhibit polarized positioning in zebrafish cone photoreceptors. J Comp Neurol. 2013; 521: 1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lunt S,, Haynes T,, Perkins BD. Zebrafish ift57 ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect Hedgehog signaling. Dev Dyn. 2009; 238: 1744–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krock BL,, Perkins BD. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci. 2008; 121: 1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ho RK,, Kane DA. Cell-autonomous action of zebrafish spt-1 mutation in specific mesodermal precursors. Nature. 1990; 348: 728–730. [DOI] [PubMed] [Google Scholar]
- 25. Kimmel CB,, Warga RM,, Schilling TF. Origin and organization of the zebrafish fate map. Development. 1990; 108: 581–594. [DOI] [PubMed] [Google Scholar]
- 26. Tsujikawa M,, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004; 42: 703–716. [DOI] [PubMed] [Google Scholar]
- 27. Murga-Zamalloa CA,, Ghosh AK,, Patil SB,, et al. Accumulation of the Raf-1 kinase inhibitory protein (Rkip) is associated with Cep290-mediated photoreceptor degeneration in ciliopathies. J Biol Chem. 2011; 286: 28276–28286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bachmann-Gagescu R,, Phelps IG,, Stearns G,, et al. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet. 2011; 20: 4041–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Larkins CE,, Aviles GD,, East MP,, Kahn RA,, Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell. 2011; 22: 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Insinna C,, Baye LM,, Amsterdam A,, Besharse JC,, Link BA. Analysis of a zebrafish dync1h1 mutant reveals multiple functions for cytoplasmic dynein 1 during retinal photoreceptor development. Neural Dev. 2010; 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krock BL,, Mills-Henry I,, Perkins BD. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest Ophthalmol Vis Sci. 2009; 50: 5463–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsujikawa M,, Omori Y,, Biyanwila J,, Malicki J. Mechanism of positioning the cell nucleus in vertebrate photoreceptors. Proc Natl Acad Sci. U S A 2007; 104: 14819–14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu H,, Toh MT,, Narasimhan V,, Thamilselvam SK,, Choksi SP,, Roy S. A function for the Joubert syndrome protein Arl13b in ciliary membrane extension and ciliary length regulation. Dev Biol. 2015; 397: 225–236. [DOI] [PubMed] [Google Scholar]
- 34. Chang B,, Khanna H,, Hawes N,, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006; 15: 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. den Hollander AI,, Koenekoop RK,, Yzer S,, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006; 79: 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fahim AT,, Bowne SJ,, Sullivan LS,, et al. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS One. 2011; 6: e23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boutin C,, Labedan P,, Dimidschstein J,, et al. A dual role for planar cell polarity genes in ciliated cells. Proc Natl Acad Sci U S A. 2014; 111: E3129–E3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borovina A,, Superina S,, Voskas D,, Ciruna B. Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat Cell Biol. 2010; 12: 407–412. [DOI] [PubMed] [Google Scholar]
- 39. Khan AO,, Oystreck DT,, Seidahmed MZ,, et al. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008; 115: 2286–2289. [DOI] [PubMed] [Google Scholar]
- 40. Maria BL,, Boltshauser E,, Palmer SC,, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999; 14: 583–590; discussion 590–581. [DOI] [PubMed] [Google Scholar]
- 41. Makino S,, Tampo H. Ocular findings in two siblings with Joubert syndrome. Clin Ophthalmol. 2014; 8: 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cevik S,, Sanders AA,, Van Wijk E,, et al. Active transport and diffusion barriers restrict Joubert Syndrome-associated ARL13B/ARL-13 to an Inv-like ciliary membrane subdomain. PLoS Genet. 2013; 9: e1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gotthardt K,, Lokaj M,, Koerner C,, Falk N,, Giessl A,, Wittinghofer A. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. Elife. 2015; 4: e11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang H,, Chen X,, Dudinsky L,, et al. Exome capture sequencing identifies a novel mutation in BBS4. Mol Vis. 2011; 17: 3529–3540. [PMC free article] [PubMed] [Google Scholar]
- 45. Rachel RA,, May-Simera HL,, Veleri S,, et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J Clin Invest. 2012; 122: 1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warburton-Pitt SR,, Silva M,, Nguyen KC,, Hall DH,, Barr MM. The nphp-2 and arl-13 genetic modules interact to regulate ciliogenesis and ciliary microtubule patterning in C. elegans. PLoS Genet. 2014; 10: e1004866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ross AJ,, May-Simera H,, Eichers ER,, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005; 37: 1135–1140. [DOI] [PubMed] [Google Scholar]
- 48. May-Simera HL,, Petralia RS,, Montcouquiol M,, et al. Ciliary proteins Bbs8 and Ift20 promote planar cell polarity in the cochlea. Development. 2015; 142: 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. May-Simera HL,, Kai M,, Hernandez V,, Osborn DP,, Tada M,, Beales PL. Bbs8, together with the planar cell polarity protein Vangl2, is required to establish left-right asymmetry in zebrafish. Dev Biol. 2010; 345: 215–225. [DOI] [PubMed] [Google Scholar]
- 50. Thomas S,, Cantagrel V,, Mariani L,, et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur J Hum Genet. 2015; 23: 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]