

Abstract
Purpose
Acid sphingomyelinase (ASMase) catalyzes the hydrolysis of sphingomyelin to ceramide and mediates multiple responses involved in inflammatory and apoptotic signaling. However, the role ASMase plays in ischemic retinal injury has not been investigated. The purpose of this study was to investigate how reduced ASMase expression impacts retinal ischemic injury.
Methods
Changes in ceramide levels and ASMase activity were determined by high performance liquid chromatography-tandem mass spectrometry analysis and ASMase activity. Retinal function and morphology were assessed by electroretinography (ERG) and morphometric analyses. Levels of TNF-α were determined by ELISA. Activation of p38 MAP kinase was assessed by Western blot analysis.
Results
In wild-type mice, ischemia produced a significant increase in retinal ASMase activity and ceramide levels. These increases were associated with functional deficits as measured by ERG analysis and significant structural degeneration in most retinal layers. In ASMase+/− mice, retinal ischemia did not significantly alter ASMase activity, and the rise in ceramide levels were significantly reduced compared to levels in retinas from wild-type mice. In ASMase+/− mice, functional and morphometric analyses of ischemic eyes revealed significantly less retinal degeneration than in injured retinas from wild-type mice. The ischemia-induced increase in retinal TNF-α levels was suppressed by the administration of the ASMase inhibitor desipramine, or by reducing ASMase expression.
Conclusions
Our results demonstrate that reducing ASMase expression provides partial protection from ischemic injury. Hence, the production of ceramide and subsequent mediators plays a role in the development of ischemic retinal injury. Modulating ASMase may present new opportunities for adjunctive therapies when treating retinal ischemic disorders.
Keywords: acid sphingomyelinase, ischemia, neuroprotection, retinal degeneration, sphingolipids
Retinal ischemia is commonly associated with multiple retinal diseases, such as glaucoma, anterior ischemic optic neuropathy, retinal and choroidal vessel occlusion, diabetic retinopathy, and traumatic optic neuropathy.1 However, due to the absence of effective therapies for retinal ischemia, these diseases remain common causes of blindness.1 Therefore, it is important to understand the underlying events that contribute to ischemic retinal degeneration and the developing drugs that target these processes.
Sphingolipids are a class of lipids containing 18-carbon amino-alcohol backbones called sphingosines. They were initially identified as important structural components of cell membranes; however, they are now also recognized as vital regulators of diverse biological processes, including vascular angiogenesis, cell growth arrest, cell differentiation, apoptosis, chemotaxis, inflammation, and neuronal degeneration.2,3 In the eye, studies have shown that sphingolipid signaling plays important roles in the homeostasis of photoreceptors and retinal pigment epithelium and the modulation of conventional outflow resistance.4–11 However, the roles of these molecules in retinal ischemic injury are unknown.
Sphingomyelinases are enzymes that catalyze the hydrolysis of sphingomyelin to ceramide, a central modulator of inflammatory cytokines and apoptotic signaling.12 To date, the following multiple sphingomyelinases have been identified: acidic, secretory, Mg2+-dependent neutral, and Mg2+-independent neutral, and alkaline sphingomyelinase.13,14 Both the acid sphingomyelinase (ASMase) and Mg2+-dependent neutral sphingomyelinase (NSMase) are considered two major candidates for mediating stress-induced production of ceramide.15 This study focused on ASMase, as the activity of this enzyme is optimized and accounts for most of the sphingomyelinase activity under acidic conditions, as occurs in the retina following ischemia.16–18 Upon cell stimulation, ASMase is translocated from intracellular compartments to the plasma membrane outer leaflet, which is enriched in sphingomyelin and represents the substrate pool for ceramide production.19–21 Hydrolysis of sphingomyelin into ceramide by sphingomyelinase facilitates receptor clustering, inflammatory signaling, and apoptosis.22,23
The activation of ASMase is an early response to various cellular stresses15 and precedes ceramide production, which accelerates the signaling pathways involved in cell death. Previous publications have shown that elevation of ceramide levels occurs in various in vivo ischemia models, including heart,24–29 brain,30–35 liver,36,37and kidney.38 In the brain, the upregulation of ASMase activity is responsible for the ischemia-induced rise in astroglia ceramide levels.39 The extent of tissue damage in the ischemic brain, heart, and liver can be attenuated by the genetic deletion of ASMase, or in vivo administration of an ASMase inhibitor such as desipramine or short interfering RNA.28,35,40 In the eye, Opreanu et al.41,42 provided evidence that cytokine-induced inflammatory signaling can be suppressed by down-regulating ASMase in human retinal endothelial cells. However, the role of ASMase in ischemia-induced retinal neuronal degeneration has not been investigated. The purpose of this study was to investigate whether sphingolipid signaling is involved in retinal ischemic injury and whether suppression of ASMase expression suppresses the ischemic injury.
Materials and Methods
Animals
The ASMase knockout mice on a C57/BL6 background were the generous gift of Edward H. Schuman (Icahn Medical Institute, New York, NY). Heterozygous (ASMase+/−) mice were bred and genotyped as previously described.43 Animals were reared under 12L:12D conditions with ambient light intensity (150 ± 20 lux). Mice 10 to 12 weeks of age were used for the experiments. Because previous studies have demonstrated that ASMase knockout mice exhibit outer retinal dysfunction and degeneration,44 ASMase+/− mice were used for most of the experiments in this study. In select experiments, wild-type control mice were treated with the ASMase inhibitor desipramine (Sigma-Aldrich Corp., St. Louis, MO, USA). For those experiments, desipramine was dissolved in 1× phosphate-buffered saline (PBS; 0.8 mg/mL) and injected intraperitoneally (10 mg/kg) 1 hour before the retinal ischemia injury. Control animals were injected with an equal volume of sterile 1× PBS.
All experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research; and the study protocol was approved by the MUSC Animal Care and Use Committee.
Retinal Ischemia
Retinal ischemia was induced using techniques described previously45 with minor modification. Briefly, mice were anesthetized with 300 mg/kg 1.25% Avertin solution (1.25 g 2,2,2-tribromoethanol; Sigma-Aldrich Corp.), 2.5 mL tertiary-amyl alcohol (Sigma-Aldrich Corp.) in 100 mL PBS. Proparacaine (5 μL, 0.5%; Akorn, Inc., Buffalo Grove, IL, USA) was applied for cornea analgesia. Body temperature was maintained on a temperature-controlled heat pad (Bioanalytical System, Inc., West Lafayette, IN, USA) at 37°C during the experiment. The anterior chamber was cannulated with a 33-gauge needle (World Precision Instruments, Inc., Sarasota, FL, USA), which was connected to a reservoir of sterile PBS, pH 7.4. The container was elevated to raise the IOP to 120 mm Hg for 50 minutes. The IOP was monitored by a calibrated pressure transducer (Argon Medical Devices, Athens, TX, USA), and the absence of retinal blood flow was confirmed by direct ophthalmoscopy. The contralateral eye was untreated and served as a control. Following ischemic injury and recovery from anesthesia, the pupil light reflex was verified as present and not different from the contralateral control eye. Then the eyes were allowed to reperfuse for a different period of time depending on the experimental endpoint being evaluated. To evaluate structural and functional injury, retinas were allowed to reperfuse for 7 days. To evaluate changes in ASMase activity and TNF-α, tissues were collected at 90 minutes and 4 hours, respectively. Changes in sphingolipids were evaluated following 2 and 24 hours of reperfusion.
Sphingolipid Analyses
Retinas were collected in lysis buffer (50 mM Tris-base, 1.0 mM EDTA, 0.2% Triton X-100, 1× protease inhibitor, pH 7.4). These retinas were lysed by brief sonication. Tissue debris and unbroken cells were pelleted and removed by centrifugation at 300g for 5 minutes at 4°C. Cellular homogenates (0.5 mg) were used for the sphingolipid analyses, which were performed by Lipidomics Core Facility (Medical University of South Carolina, Charleston, SC, USA), using high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) as previously described, using electrospray ionization/tandem mass spectrometry (ESI-MS/MS), using a triple-stage quadrupole mass spectrometer (TSQ 7000; Thermo Finnigan, San Jose, CA, USA), operating in a multiple reaction monitoring positive ionization mode as described previously.46
Electroretinograms
Mice were dark-adapted overnight and anesthetized using xylazine (20 mg/kg, intraperitoneally) and ketamine (80 mg/kg, intraperitoneally). Pupils were dilated with phenylephrine hydrochloride (2.5%; Akorn) and atropine sulfate (1%; Bausch & Lomb, Tampa, FL, USA). Contact lens electrodes were placed on both eyes accompanied by 2.5% hypromellose ophthalmic demulcent solution (Goniovisc; HUB Pharmaceuticals, LLC, Rancho Cucamonga, CA, USA). Full-field scotopic electroretinograms (ERGs) were recorded as described previously,47 using universal testing and electrophysiologic system 2000 (UTAS-2000; LKC Technologies, Gaithersburg, MD, USA). Single white flashes (10 ms) with intensity of 2.48 cd/s/m2 were used for stimulation. No frequency filtering was used. A-wave amplitudes were measured from baseline to the a-wave trough. The b-wave amplitudes were measured from the a-wave trough to the peak of the b-wave. ERGs were recorded 24 hours before the retinal ischemia and 7 days post ischemia.
Histology
For morphometric analyses, mouse eyes were enucleated and fixed in freshly made 4% paraformaldehyde in 0.1 M PBS for 2 hours at 4°C. After fixation, the tissues were dehydrated and embedded in paraffin. Retinal cross-sections (5 μm thick) were then cut and stained with hematoxylin and eosin (Sigma-Aldrich Corp.). Only sections through the optic nerve were used. Retinal sections were photographed and measured approximately 2 to 3 disc diameters from the optic nerve, using an Axioplan II microscope (Carl Zeiss, Inc., Germany) and a 20× objective lens. The number of cells in the retinal ganglion cell layer was determined by cell counts over a distance scale of 200 μm. Morphologic values for each retina were determined from three sections for each eye, and n ≥6 eyes were included for each group.
ASMase Activity Assay
Retinas were isolated 90 minutes after the ischemic injury, and lysed in lysis buffer (50 mM sodium acetate, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail, pH 5.0) by brief sonication. The lysates were centrifuged at 12,000g for 4 minutes, and the supernatants were used for measuring ASMase activity by using commercial Amplex Red sphingomyelinase assay kit following the manufacture's instruction (Molecular Probes, Eugene, OR, USA). The samples or negative controls were incubated with 10 μL of the 5 mM sphingomyelin solution for 1 hour at 37°C, protected from light; then working solution of 100 μM Amplex Red reagent containing 2 U/mL horseradish-peroxidase, 0.2 U/mL choline oxidase, and 8 U/mL alkaline phosphatase, was added and incubated for 2 hours. Fluorescence was measured using a fluorescence microplate reader at excitation of 530 nm and emission detection at 590 nm. The actual readings were corrected for background fluorescence by subtracting the values derived from the negative control and normalized to protein levels.
TNF-α Analysis
Retinas were collected at 4 hours after ischemia, and retina extracts were used to measure TNF-α by commercial ELISA kits following the manufacture's instruction (eBioscience, San Diego, CA, USA). Briefly, retina extracts were incubated in 96-well plates coated with monoclonal antibody specific for mouse TNF-α. After washing, biotin-conjugated anti-mouse TNF-α (polyclonal) and avidin-horseradish peroxidase antibodies were added and incubated in accordance with the manufacturer's instructions. After washing, a substrate solution was added to each well. The enzyme reaction was ended by adding stop solution. Absorbance was measured at 450 nm. Using a standard curve prepared from different dilutions of recombinant TNF-α standard, we calculated each concentration of TNF-α for the retina extracts.
Cell Culture
The primary human optic nerve head astrocytes were isolated and cultured as previously described.48 Prior to the study, confluent astrocyte cultures were serum-starved overnight and pretreated with desipramine (20 μM; Sigma-Aldrich Corp.) or vehicle for 1 hour. Cells were then treated with TNF-α (10 ng/mL; R&D System, Inc., Minneapolis, MN, USA) for 15 minutes.
Western Blotting
Following the treatment period, cells were harvested and homogenized in lysis buffer (50 mM Tris-base; 10 mM EDTA; 0.5 mM sodium orthovanadate; 0.5% sodium deoxycholic acid; 1% Triton X-100 and protease inhibitor cocktail (Roche Diagnostics Corp., Indianapolis, IN, USA); pH 7.4). Equal amounts of total protein from each lysate were loaded onto NuPAGE Novex 4-12% bis-Tris protein gels (Life Technologies Corp., Carlsbad, CA, USA) and subjected to Western blot analysis using primary antibodies, followed by horseradish-peroxidase-labeled secondary antibody (1:5000 dilution;, Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The signals were detected using enhanced chemoluminescence reagents (Thermo Scientific, Waltham, MA, USA). The primary antibodies against p-p38 (1:1000 dilution) and p38 antibody (1:1000 dilution) were purchased from Cell Signaling Technology (Danvers, MA, USA). β-Actin antibody (1:2000 dilution) was purchased from Sigma-Aldrich Corp.
Statistics
For all experiments, data are mean ± SEM. Data were analyzed using a 2-tailed Student's t-test or ANOVA. A P value < 0.05 was considered significant.
Results
Effect of Retinal Ischemia on Sphingolipid Profile
To investigate whether acute retinal ischemic injury could alter the sphingolipid profile, retinas were isolated 2 or 24 hours after ischemia, and homogenates were analyzed by mass spectrophotometry. As shown in Figure 1A, 2 hours after ischemic injury, the ceramide levels were elevated in ischemic retinas (Fig. 1, black bars) compared with the contralateral retinas (Fig. 1, white bars). Ceramide lipids (C14-Cer, C16-Cer, C18-Cer, C18:1-Cer, C26-Cer, dehydro C16-Cer) and sphingosine displayed significant increases (P < 0.05). Among these ceramides, C16-Cer, C18-Cer, and C20-Cer were the dominant retinal forms and exhibited robust elevations (110.5 ± 19.3%, 107.1 ± 38.0%, and 145.8 ± 19.9%, respectively) following ischemic injury. No significant changes were detected in the sphingomyelin profile between contralateral and ischemic retinas (Fig. 1B). The elevation of ceramide was maintained up to 24 hours postischemic injury, and no significant differences in total ceramide levels were measured between 2 and 24 hours (Fig. 2). A comparison of retinal ceramide levels from contralateral eyes of wild-type mice with those of ASMase+/− mice demonstrated that total retinal ceramide in ASMase+/− retinas was significantly lower than that in wild-type mice. This reduction was primarily due to the reduction in isoforms C16-Cer and C18-Cer (Fig. 3). Analysis of retinas from ASMase+/− mice 2 hours following ischemic injury demonstrated that the ischemia-induced rise in total ceramide and individual isoforms (C16-Cer, C18-Cer, C18:1-Cer, and C20-Cer) were significantly reduced compared to the ischemic retinas from wild-type mice (Fig. 3).
Figure 1.

Ceramide and SM profiles of contralateral and ischemic retinas in wild-type mice 2 hours after the retinal ischemic injury. (A) Ceramide profile and (B) sphingomyelin profile in contralateral (white) and ischemic (black) retinas. Data are mean ± SEM; n = 4. *P < 0.05.
Figure 2.

Comparison of ceramide profiles in wild-type retinas at 2 and 24 hours after the retinal ischemic injury. (Inset) Total ceramide level at 2 and 24 hours after the retinal ischemic injury. Data are mean ± SEM; n = 4.
Figure 3.

Ceramide profile in ASMase+/+ and ASMase+/− retinas in control and ischemic retinas 2 hours after the ischemic injury. Inset presents the total ceramide level in ASMase+/+ and ASMase+/− retinas. Data are mean ± SEM; n = 4. *Significant differences from ASMase+/− contralateral to wild-type contralateral eyes (P < 0.05); **Significant differences from ASMase+/− ischemic eyes to wild-type ischemic eyes.
To determine whether ischemia alters retinal ASMase activity, retinas from both the wild-type and the ASMase+/− mice were isolated 90 minutes after ischemia. In wild-type mice, ischemia induced a 29.8% increase in ASMase activity compared with retinas from contralateral eyes. In control retinas from ASMase+/− mice, ASMase activity was reduced by 35 ± 7.8% compared to that in the corresponding retinas from wild-type mice (Fig. 4). No increase in retinal ASMase activity was measured in the ASMase+/− mice following ischemia.
Figure 4.

Effect of retinal ischemia on ASMase activity changes in ASMase+/+ (black) and ASMase+/− (white) retinas before and 90 minutes after the retinal ischemic injury. Data are mean ± SEM; n = 4. *P < 0.05.
Effect of Reduced ASMase Expression on Retinal Ischemic Injury
In wild-type mice (n = 5), baseline ERG a- and b-wave amplitudes were 309.9 ± 17.9 and 584.54 ± 28.8 μV, respectively. In ASMase+/− mice (n = 8), baseline ERG amplitudes (a-wave: 321.0 ± 10.5; and b-wave: 583.8 ± 18.7 μV) were not significant from amplitudes measured in wild-type mice. Seven days postischemic injury, wild-type mice exhibited a significant decrease in mean a- and b-wave amplitudes from baseline levels of 61 ± 0.9% and 61 ± 12.3%, respectively (Fig. 5). In ASMase+/− mice, 7 days postischemic injury, ERG a-and b-waves were reduced by 38 ± 9.1% and 25 ± 4.6% of baseline levels, respectively. These reductions in amplitude in ASMase+/− mice were significant (P ≤ 0.05) compared to baseline measurements. However, comparing post ischemic a- and b-wave amplitudes in ASMase+/− mice to that in wild-type mice demonstrated that post ischemic reductions of a- and b-wave amplitudes were significantly less (P ≤ 0.05) in ASMase+/− mice. No significant differences in contralateral a- and b-wave amplitudes between wild-type and ASMase+/− mice were measured at study day 7 (Fig. 5).
Figure 5.

Effect of ASM inhibition on ERG response. Representative ERGs in (A) ASMase+/+ and ASMase+/− mice 24 hours before ischemia and 7 days postischemic injury. (B) Analyses of ERG a-wave amplitudes and (C) b-wave amplitudes before and 7 days postischemic injury in ASMase+/+ (white) and ASM+/− (black) mice. ERGs were obtained by averaging two responses to 2.48 cd/s/m2 flashes with an interstimulus interval of 2 minutes. Data were normalized to the percentage of pre-ischemia baseline level and are mean ± SEM; n = 4. *P < 0.05.
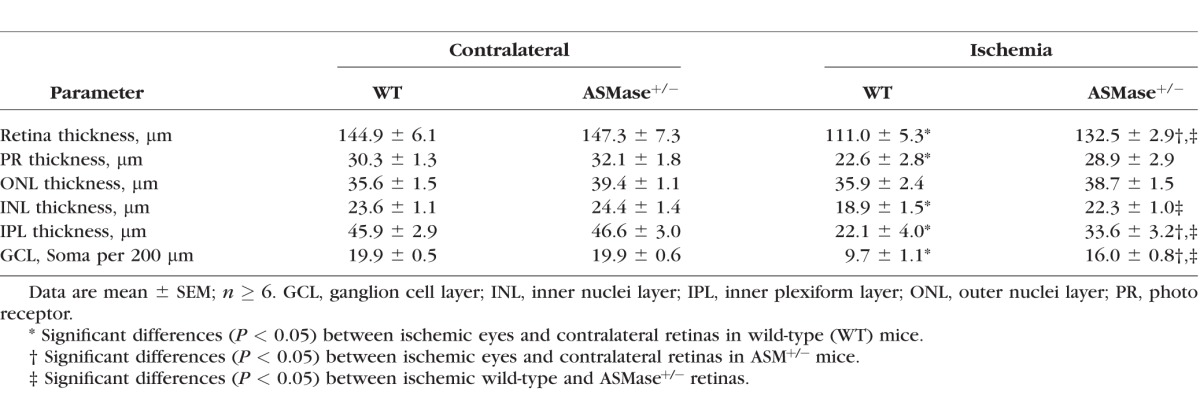
Morphometric analysis was used to assess retina structural changes 7 days following unilateral retinal ischemic injury. In wild-type mice, the ischemic injury produced 23 ± 3.7% decrease in overall retina thickness compared to that in contralateral eyes (Fig. 6; Table). This reduction in thickness was mainly due to the thinning of the inner plexiform layer, inner nuclei layer, and photoreceptor layers. Following ischemic injury, wild-type mice also exhibited a significant loss (51 ± 5.5%) in cell bodies from the ganglion cell layer (Table). In ASMase+/− mice, a significant thinning in the inner plexiform layer and total retina thickness was measured, as well as a reduction in cell body numbers in the ganglion cell layer. However, a comparison between retinal morphology of wild-type and that of ASMase+/− mice revealed that at 7 days postischemic injury retinal layer thickness and ganglion cell body counts in retinas from ASMase+/− were significantly greater than corresponding measurements from wild-type mice. No significant morphologic differences were detected in contralateral retinas between wild-type and ASMase+/− mice at study day 7 (Fig. 6; Table).
Figure 6.

Effect of ASM inhibition on retinal morphology change after retinal ischemic injury. Images of representative retinal cross-sections of contralateral eyes and ischemic eyes from ASMase+/+ and ASMase+/− mice. All images were taken 2 to 3 disc diameters from the optic nerve.
Table.
Analyses of Contralateral and Ischemic Retinas in Wild-Type and ASMase+/− Mice
Effect of Reduced ASMase Expression on Inflammatory Signaling
Retinas were collected at 4 hours after ischemia, and retinal TNF-α levels were determined by ELISA. In retinas from wild-type mice, ischemia induced a 3.2-fold increase in TNF-α compared to the levels measured in the contralateral retinas (Fig. 7). Although TNF-α levels also increased in ischemic ASMase+/− retinas, those levels were significantly lower than the levels from ischemic wild-type retinas. In the wild-type mice, administration of the ASMase inhibitor desipramine (10 mg/kg) significantly suppressed the ischemia-induced rise in TNF-α levels by 52 ± 12.4% compared to the ischemic retinas from control mice.
Figure 7.

Measurement of TNF-α levels in contralateral and ischemic retinas from ASMase+/+ and ASMase+/− mice and desipramine-treated ASMase+/+ mice by ELISA. Data are mean ± SEM; n = 4. *P < 0.05.
Astrocytes play a major role in modulating ischemic retinal injury. Hence, cultured primary human astrocytes, derived from the optic nerve head, were utilized to assess the involvement of ASMase on activation (phosphorylation) on the stress-activated MAP kinase, p38. Astrocyte cultures treated with TNF-α (10 ng/mL) for 15 minutes exhibited a significant increase in p38 phosphorylation over the control level (Fig. 8). This increase in p38 phosphorylation was significantly reduced in cultures pretreated (15 minutes) with the ASMase inhibitor, desipramine (20 μM). Treatment of astrocytes with desipramine alone did not significantly alter the levels of p38 phosphorylation.
Figure 8.

Effect of the ASMase inhibitor desipramine on TNF-α-induced activation of p38. Representative Western blot of phospho-p38 MAP kinase in human astrocytes cultured from optic nerve head with and without the pretreatment of desipramine (15 minutes). Densitometric analysis of changes in phospho-p38 compared to levels in controls. Data are mean ± SEM; n = 4. *P < 0.05.
Discussion
Ceramides are a family of bioactive lipid molecules, which have been shown to modulate many physiological functions. In addition, these lipids have been shown to contribute to the pathogenesis of various cancers, diabetes, obesity, inflammatory conditions, microbial infections, and neurodegenerative disorders.3,49,50 One of the most studied roles of ceramides is their function as proapoptotic molecules. Several in vivo ischemia-reperfusion models have provided evidence that ceramides can contribute to the apoptotic process.24,26–29,31,33–38
The endogenous levels and turnover of the sphingolipid molecules are regulated by multiple enzymes and pathways. Ceramides can be produced through multiple mechanisms including hydrolysis of sphingomyelin by sphingomyelinase, de novo synthetic and salvage pathways.3,51,52 In the normal nonischemic retina, the three dominant ceramides are C16, C18, and C20 (Fig. 1). This pattern provides evidence that ceramide synthase 1, 4, and 6 (CerS1, CerS4, and CerS6) are the predominant isoforms contributing to de novo ceramide synthesis in retinas.53–55
In the current study, we found that acute retinal ischemic injury significantly increased the ceramide levels as early as 2 hours after injury and that these levels remained elevated up to 24 hours post ischemia. As shown in Figure 3, the increase in ceramides was dependent on the normal expression of ASMase. However, we did not measure any significant changes in the level of sphingomyelins following ischemic injury. Stress-induced elevations in ceramides, in the absence of measureable reductions in sphingomyelin levels, can indicate an increase in de novo pathway activity.56 However, in tissues where the sphingomyelin-to-ceramide ratio is high (e.g., 10:1), SMase-dependent increases in ceramide levels have been measured without corresponding significant reductions in sphingomyelin.3,57 In the retina, sphingomyelins-to-corresponding ceramide ratios ranged from 12:1 to 122:1 based on our sphingolipid profiles (Fig. 1). Hence, significant increases in the ceramide production via ASMase hydrolysis can likely occur without significantly reducing the levels of sphingomyelin. Overall, our results provide evidence that the ischemia-induced increase in ceramides is dependent upon ASMase hydrolysis; however, the involvement of the de novo and salvage pathways in this increase cannot be completely discounted. It should also be noted that activation of NSMase has been linked to ischemic brain injury.25,32–34,58,59 Hence, understanding all the pathways that contribute to the rise in ceramides observed in ischemic retinas from ASMase+/− mice will require additional study.
Previous studies in the brain have demonstrated that ASMase plays a critical role in ischemia-induced cytokine production and neuronal apoptosis and that ASMase inhibition can reduce both inflammation and apoptosis.35 Mechanistic studies have shown that ASMase can regulate both cytokine production and p38 MAPK activation.60,61 However, the ability of ASMase to influence p38 signaling is not observed in all cells.62,63 Our results demonstrated that ischemic retinal injury in wild-type mice increased the levels of ceramides and TNF-α and that by comparison both responses were suppressed in retinas from ASMase+/− mice. Using cultured astrocytes, we demonstrated that the ASMase inhibitor desipramine also blocked the TNF-α-induced activation of p38 MAP kinase, revealing a critical role for ASMase in TNF-α signaling in astrocytes. Although the underlying mechanisms regarding how ASMase modulates these signals in astrocytes will require additional investigations, these mechanisms may include the transactivation of cytokines (e.g., IL-1β or IL-6) potentiates the activation of ASMase-dependent sphingolipid signaling and ultimately results in autocrine activation by sphingosine-1-phosphate.60,63 Despite the complexities of retinal sphingolipid metabolism, our results support the idea that the increased ceramide levels contribute to inflammatory signaling in the ischemic retina and the resulting neurodegenerative response to ischemia is due to the increased ASMase activity and/or expression.
To investigate whether reduced expression of ASMase provides functional and structural protection of the retina, wild-type and ASMase+/− mice received unilateral ischemic retinal injury and allowed to recover for seven days. As shown in Figure 5, ERG responses from wild-type mice 7 days post injury exhibited mean deficits in a- and b-wave amplitudes of 61 ± 0.9% and 61 ± 12.3%, respectively. In ASMase+/− mice that received unilateral ischemic retinal injury, ERG a- and b- wave amplitudes exhibited significant declines compared to contralateral (nonischemic) eyes; however, these reductions were significantly less than those measured in wild-type mice. However, it should be noted that using contralateral eyes as controls may obscure contralateral changes resulting from ipsilateral retinal and optic nerve degeneration. Morphologic measurements demonstrated that ischemic injury produced significant degeneration in most retinal layers; however, this degenerative response was also significantly reduced in ASMase+/− mice. These results are consistent with the idea that suppressing ASMase activity can provide structural and functional protection from ischemic injury.
Finally, it should also be noted that wild-type mice used in these studies exhibited greater a-wave deficits than those observed in previous studies without corresponding increases in photoreceptor degeneration.64 These results provide evidence that maintaining photoreceptor architecture is not sufficient to preserve photoreceptor function and that the C57/BL6 is functionally more sensitive to ischemic injury than previous mouse strains evaluated by this laboratory.64
In summary, studies in brain, heart, liver, and kidney have shown that the activation of ASMase is an early event in apoptotic and necrotic processes.15 The current study revealed that acute retinal ischemia results in the rapid increase in ASMase activity leading to a rise in the levels of multiple ceramides, the secretion of TNF-α and eventual retinal degeneration. In addition, we found that reducing ASMase expression suppressed this sequelae of events and partially protected the retina from retinal ischemic injury. These results provided evidence that sphingolipid metabolism plays an important role in the development of ischemic retinal injury. Hence, the identifying strategies to inhibit ASMase may provide new opportunities to treat ischemic retinal disorders in adults.
Acknowledgments
The authors thank Luanna Bartholomew, PhD, for critical review of the manuscript.
Supported by U.S. National Institutes of Health grant EY021368 (CEC), an unrestricted grant to Department of Ophthalmology, Medical University of South Carolina (MUSC), from Research to Prevent Blindness; Lipidomics Shared Resource, Hollings Cancer Center, MUSC grant P30 CA138313; Lipidomics Core, SC Lipidomics and Pathobiology Center of Biomedical Research Excellence, Department of Biochemistry, MUSC grant P20 RR017677; and National Center for Research Resources and Office of the Director of the NIH grant C06 RR018823.
Disclosure: J. Fan, None; B.X. Wu, None; C.E. Crosson, None
References
- 1. Osborne NN,, Casson RJ,, Wood JP,, Chidlow G,, Graham M,, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004; 23: 91–147. [DOI] [PubMed] [Google Scholar]
- 2. Hannun YA,, Luberto C. Lipid metabolism: ceramide transfer protein adds a new dimension. Curr Biol. 2004; 14: R163–165. [PubMed] [Google Scholar]
- 3. Hannun YA,, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008; 9: 139–150. [DOI] [PubMed] [Google Scholar]
- 4. Acharya U,, Patel S,, Koundakjian E,, Nagashima K,, Han X,, Acharya JK. Modulating sphingolipid biosynthetic pathway rescues photoreceptor degeneration. Science. 2003; 299: 1740–1743. [DOI] [PubMed] [Google Scholar]
- 5. Ali M,, Ramprasad VL,, Soumittra N,, et al. A missense mutation in the nuclear localization signal sequence of CERKL (p.R106S) causes autosomal recessive retinal degeneration. Mol Vis. 2008; 14: 1960–1964. [PMC free article] [PubMed] [Google Scholar]
- 6. Auslender N,, Sharon D,, Abbasi AH,, Garzozi HJ,, Banin E,, Ben-Yosef T. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Invest Ophthalmol Vis Sci. 2007; 48: 5431–5438. [DOI] [PubMed] [Google Scholar]
- 7. German OL,, Miranda GE,, Abrahan CE,, Rotstein NP. Ceramide is a mediator of apoptosis in retina photoreceptors. Invest Ophthalmol Vis Sci. 2006; 47: 1658–1668. [DOI] [PubMed] [Google Scholar]
- 8. Skoura A,, Sanchez T,, Claffey K,, Mandala SM,, Proia RL,, Hla T. Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J Clin Invest. 2007; 117: 2506–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tuson M,, Marfany G,, Gonzalez-Duarte R. Mutation of CERKL a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am J Hum Genet. 2004; 74: 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie B,, Shen J,, Dong A,, Rashid A,, Stoller G,, Campochiaro PA. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J Cell Physiol. 2009; 218: 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boussommier-Calleja A,, Bertrand J,, Woodward DF,, Ethier CR,, Stamer WD,, Overby DR. Pharmacologic manipulation of conventional outflow facility in ex vivo mouse eyes. Invest Ophthalmol Vis Sci. 2012; 53: 5838–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanfer JN,, Young OM,, Shapiro D,, Brady RO. The metabolism of sphingomyelin. I. Purification and properties of a sphingomyelin-cleaving enzyme from rat liver tissue. J Biol Chem. 1966; 241: 1081–1084. [PubMed] [Google Scholar]
- 13. Goni FM,, Alonso A. Sphingomyelinases: enzymology and membrane activity. FEBS Lett. 2002; 531: 38–46. [DOI] [PubMed] [Google Scholar]
- 14. Samet D,, Barenholz Y. Characterization of acidic and neutral sphingomyelinase activities in crude extracts of HL-60 cells. Chem Phys Lipids. 1999; 102: 65–77. [DOI] [PubMed] [Google Scholar]
- 15. Marchesini N,, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004; 82: 27–44. [DOI] [PubMed] [Google Scholar]
- 16. Jenkins RW,, Canals D,, Hannun YA. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009; 21: 836–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyake T,, Nishiwaki A,, Yasukawa T,, Ugawa S,, Shimada S,, Ogura Y. Possible implications of acid-sensing ion channels in ischemia-induced retinal injury in rats. Jpn J Ophthalmol. 2013; 57: 120–125. [DOI] [PubMed] [Google Scholar]
- 18. Tan J,, Ye X,, Xu Y,, Wang H,, Sheng M,, Wang F. Acid-sensing ion channel 1a is involved in retinal ganglion cell death induced by hypoxia. Mol Vis. 2011; 17: 3300–3308. [PMC free article] [PubMed] [Google Scholar]
- 19. Grassme H,, Schwarz H,, Gulbins E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun. 2001; 284: 1016–1030. [DOI] [PubMed] [Google Scholar]
- 20. Schissel SL,, Jiang X,, Tweedie-Hardman J,, et al. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J Biol Chem. 1998; 273: 2738–2746. [DOI] [PubMed] [Google Scholar]
- 21. Smith EL,, Schuchman EH. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 2008; 22: 3419–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ko YG,, Lee JS,, Kang YS,, Ahn JH,, Seo JS. TNF-alpha-mediated apoptosis is initiated in caveolae-like domains. J Immunol. 1999; 162: 7217–7223. [PubMed] [Google Scholar]
- 23. Martin MU,, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002; 1592: 265–280. [DOI] [PubMed] [Google Scholar]
- 24. Argaud L,, Prigent AF,, Chalabreysse L,, Loufouat J,, Lagarde M,, Ovize M. Ceramide in the antiapoptotic effect of ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2004; 286: H246–H251. [DOI] [PubMed] [Google Scholar]
- 25. Bielawska AE,, Shapiro JP,, Jiang L,, et al. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol. 1997; 151: 1257–1263. [PMC free article] [PubMed] [Google Scholar]
- 26. Cordis GA,, Yoshida T,, Das DK. HPTLC analysis of sphingomyelin, ceramide and sphingosine in ischemic/reperfused rat heart. J Pharm Biomed Anal. 1998; 16: 1189–1193. [DOI] [PubMed] [Google Scholar]
- 27. Cui J,, Engelman RM,, Maulik N,, Das DK. Role of ceramide in ischemic preconditioning. J Am Coll Surg. 2004; 198: 770–777. [DOI] [PubMed] [Google Scholar]
- 28. Der P,, Cui J,, Das DK. Role of lipid rafts in ceramide and nitric oxide signaling in the ischemic and preconditioned hearts. J Mol Cell Cardiol. 2006; 40: 313–320. [DOI] [PubMed] [Google Scholar]
- 29. Zhang DX,, Fryer RM,, Hsu AK,, et al. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res Cardiol. 2001; 96: 267–274. [DOI] [PubMed] [Google Scholar]
- 30. Herr I,, Martin-Villalba A,, Kurz E,, et al. FK506 prevents stroke-induced generation of ceramide and apoptosis signaling. Brain Res. 1999; 826: 210–219. [DOI] [PubMed] [Google Scholar]
- 31. Kubota M,, Kitahara S,, Shimasaki H,, Ueta N. Accumulation of ceramide in ischemic human brain of an acute case of cerebral occlusion. Jpn J Exp Med. 1989; 59: 59–64. [PubMed] [Google Scholar]
- 32. Kubota M,, Narita K,, Nakagomi T,, et al. Sphingomyelin changes in rat cerebral cortex during focal ischemia. Neurol Res. 1996; 18: 337–341. [DOI] [PubMed] [Google Scholar]
- 33. Nakane M,, Kubota M,, Nakagomi T,, et al. Lethal forebrain ischemia stimulates sphingomyelin hydrolysis and ceramide generation in the gerbil hippocampus. Neurosci Lett. 2000; 296: 89–92. [DOI] [PubMed] [Google Scholar]
- 34. Takahashi K,, Ginis I,, Nishioka R,, et al. Glucosylceramide synthase activity and ceramide levels are modulated during cerebral ischemia after ischemic preconditioning. J Cereb Blood Flow Metab. 2004; 24: 623–627. [DOI] [PubMed] [Google Scholar]
- 35. Yu ZF,, Nikolova-Karakashian M,, Zhou D,, Cheng G,, Schuchman EH,, Mattson MP. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production and neuronal apoptosis. J Mol Neurosci. 2000; 15: 85–97. [DOI] [PubMed] [Google Scholar]
- 36. Bradham CA,, Stachlewitz RF,, Gao W,, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 1997; 25: 1128–1135. [DOI] [PubMed] [Google Scholar]
- 37. Kukimoto M,, Nishiyama M,, Ohnuki T,, et al. Identification of interaction site of pseudoazurin with its redox partner, copper-containing nitrite reductase from Alcaligenes faecalis S-6. Protein Eng. 1995; 8: 153–158. [DOI] [PubMed] [Google Scholar]
- 38. Zager RA,, Conrad S,, Lochhead K,, Sweeney EA,, Igarashi Y,, Burkhart KM. Altered sphingomyelinase and ceramide expression in the setting of ischemic and nephrotoxic acute renal failure. Kidney Int. 1998; 53: 573–582. [DOI] [PubMed] [Google Scholar]
- 39. Ohtani R,, Tomimoto H,, Kondo T,, et al. Upregulation of ceramide and its regulating mechanism in a rat model of chronic cerebral ischemia. Brain Res. 2004; 1023: 31–40. [DOI] [PubMed] [Google Scholar]
- 40. Llacuna L,, Mari M,, Garcia-Ruiz C,, Fernandez-Checa JC,, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006; 44: 561–572. [DOI] [PubMed] [Google Scholar]
- 41. Opreanu M,, Lydic TA,, Reid GE,, McSorley KM,, Esselman WJ,, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2010; 51: 3253–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Opreanu M,, Tikhonenko M,, Bozack S,, et al. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes. 2011; 60: 2370–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horinouchi K,, Erlich S,, Perl DP,, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. 1995; 10: 288–293. [DOI] [PubMed] [Google Scholar]
- 44. Wu BX,, Fan J,, Boyer NP,, et al. Lack of acid sphingomyelinase induces age-related retinal degeneration. PLoS One. 2015; 10: e0133032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Husain S,, Potter DE,, Crosson CE. Opioid receptor-activation: retina protected from ischemic injury. Invest Ophthalmol Vis Sci. 2009; 50: 3853–3859. [DOI] [PubMed] [Google Scholar]
- 46. Bielawski J,, Pierce JS,, Snider J,, Rembiesa B,, Szulc ZM,, Bielawska A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv Exp Med Biol. 2010; 688: 46–59. [DOI] [PubMed] [Google Scholar]
- 47. Fan J,, Rohrer B,, Moiseyev G,, Ma JX,, Crouch RK. Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc Natl Acad Sci U S A. 2003; 100: 13662–13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang P,, Hernandez MR. Purification of astrocytes from adult human optic nerve heads by immunopanning. Brain Res Brain Res Protoc. 2003; 12: 67–76. [DOI] [PubMed] [Google Scholar]
- 49. Wu D,, Ren Z,, Pae M,, et al. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J Immunol. 2007; 179: 4829–4839. [DOI] [PubMed] [Google Scholar]
- 50. Zeidan YH,, Hannun YA. Translational aspects of sphingolipid metabolism. Trends Mol Med. 2007; 13: 327–336. [DOI] [PubMed] [Google Scholar]
- 51. Haimovitz-Friedman A,, Kan CC,, Ehleiter D,, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994; 180: 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kitatani K,, Idkowiak-Baldys J,, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008; 20: 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ebel P. VomDorp K,, Petrasch-Parwez E,, et al. Inactivation of ceramide synthase 6 in mice results in an altered sphingolipid metabolism and behavioral abnormalities. J Biol Chem. 2013; 288: 21433–21447. [DOI] [PMC free article] [PubMed]
- 54. Ginkel C,, Hartmann D,, vom Dorp K,, et al. Ablation of neuronal ceramide synthase 1 in mice decreases ganglioside levels and expression of myelin-associated glycoprotein in oligodendrocytes. J Biol Chem. 2012; 287: 41888–41902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Riebeling C,, Allegood JC,, Wang E,, Merrill AH, Jr,, Futerman AH. Two mammalian longevity assurance gene (LAG1) family members trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem. 2003; 278: 43452–43459. [DOI] [PubMed] [Google Scholar]
- 56. Yu J,, Novgorodov SA,, Chudakova D,, et al. JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J Biol Chem. 2007; 282: 25940–25949. [DOI] [PubMed] [Google Scholar]
- 57. Bielawski J,, Szulc ZM,, Hannun YA,, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006; 39: 82–91. [DOI] [PubMed] [Google Scholar]
- 58. Hernandez OM,, Discher DJ,, Bishopric NH,, Webster KA. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ Res. 2000; 86: 198–204. [DOI] [PubMed] [Google Scholar]
- 59. Zhai ST,, Liu GY,, Xue F,, et al. Changes of sphingolipids profiles after ischemia-reperfusion injury in the rat liver. Chin Med J (Engl). 2009; 122: 3025–3031. [PubMed] [Google Scholar]
- 60. Perry DM,, Newcomb B,, Adada M,, et al. Defining a role for acid sphingomyelinase in the p38/interleukin-6 pathway. J Biol Chem. 2014; 289: 22401–22412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sakata A,, Ochiai T,, Shimeno H,, et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology. 2007; 122: 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Manthey CL,, Schuchman EH. Acid sphingomyelinase-derived ceramide is not required for inflammatory cytokine signalling in murine macrophages. Cytokine. 1998; 10: 654–661. [DOI] [PubMed] [Google Scholar]
- 63. Saldeen J,, Jaffrezou JP,, Welsh N. The acid sphingomyelinase inhibitor SR33557 counteracts TNF-alpha-mediated potentiation of IL-1beta-induced NF-kappaB activation in the insulin-producing cell line Rinm5F. Autoimmunity. 2000; 32: 241–254. [DOI] [PubMed] [Google Scholar]
- 64. Fan J,, Alsarraf O,, Dahrouj M,, et al. Inhibition of HDAC2 protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2013; 54: 4072–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]