

Abstract
Clinical risk assessment for cancer predisposition includes a three-generation pedigree and physical examination to identify inherited syndromes. Additionally genetic and genomic biomarkers may identify individuals with a constitutional basis for their disease that may not be evident clinically. Genomic biomarker testing may detect molecular variations in single genes, panels of genes, or entire genomes. The strength of evidence for the association of a genomic biomarker with disease risk may be weak or strong. The factors contributing to clinical validity and utility of genomic biomarkers include functional laboratory analyses and genetic epidemiologic evidence. Genomic biomarkers may be further classified as low, moderate or highly penetrant based on the likelihood of disease. Genomic biomarkers for breast cancer are comprised of rare highly penetrant mutations of genes such as BRCA1 or BRCA2, moderately penetrant mutations of genes such as CHEK2, as well as more common genomic variants, including single nucleotide polymorphisms, associated with modest effect sizes. When applied in the context of appropriate counseling and interpretation, identification of genomic biomarkers of inherited risk for breast cancer may decrease morbidity and mortality, allow for definitive prevention through assisted reproduction, and serve as a guide to targeted therapy.
Keywords: Genetics, Genomics, Breast oncology, Biomarkers, Prophylactic, Chemoprevention, Genetic counseling, Genetic testing, BRCA
As inherited variation in DNA sequence has been shown to correlate with future disease risk, genomic tests constitute objective “biomarkers” of an individual’s susceptibility to cancer [1]. A family history of breast cancer has long been thought to indicate the presence of inherited genetic events that predispose to this disease. Although familial breast cancer has been recognized since the nineteenth century, the detailed medical description of inherited breast (and ovarian cancer) in families took place in the 1970s [2, 3]. Subsequently, up to 15 % of patients diagnosed with invasive breast cancer were shown to have at least one first-degree female relative (mother, sister, or daughter) with the disease. Testing for genetic biomarkers of risk has evolved over the past two decades to complement family history and physical findings. The most notable of these genetic biomarkers emerged from genetic analysis of families affected by multiple cases of early-onset (50 years of age) breast cancer, leading to the discovery of the breast cancer susceptibility genes, BRCA1 and BRCA2 [4–6]. The genetic mapping of BRCA1 strongly suggested an inherited risk of breast cancer resulting from genetic alterations located on chromosome 17q21 [7]. The subsequent discovery of BRCA1, and later BRCA2 [8, 9], initiated widespread interest in hereditary breast cancer. These discoveries also galvanized resource allocation to investigators exploring translation of this information to improve clinical care for those with breast cancer susceptibility. In the late 1990s, mutations in BRCA1/2 were established as the main contributors to familial breast cancer, and population specific frequencies of mutations in these genes were compiled [10–14]. In the 10 years following, the clinical utility and the benefits of clinical genetic biomarkers became evident, as genetic testing led to individualized risk reduction strategies including preventive surgeries, chemoprophylaxis and targeted therapies [15, 16].
Although genetic tests for cancer risk constitute “biomarkers” in a general sense, these genomic markers are distinct from non-genetic biomarkers in that they reflect the impact of modifiers of penetrance, population-specific differences in allele frequencies, and influence of gene-environment interactions. As genomic testing continues to evolve, biomarkers of various strength and significance are being routinely detected and gene-gene and gene-environment interactions are beginning to emerge [17–22]. Understanding the functional significance of genomic alterations is conceptually critical in assessing the potential utility of genetic variants as biomarkers. The type of alteration and the location of an aberration in a gene, i.e., a synonymous missense variant, a nonsense missense variant, a deletion/duplication, a translocation, or an inversion, all bear on the assessment of a gene test as a “biomarker” of inherited cancer risk. Thus, understanding the type of genetic change is as important as the fact that the gene is altered.
Novel biomarkers are being revealed by next generation sequencing and tend to be associated with low and moderate penetrance genomic loci [23]. As more is known, algorithms will be required to weigh multiple biomarkers simultaneously and hence allow clinicians to most informatively provide recommendations pertaining to risk reduction surgeries, surveillance guidelines, family planning, apply novel therapies, and modify and dose-adjust existing therapies.
Genetics in Breast Cancer Predisposition
Although the ease of testing for different genetic biomarkers is appealing in the “information age,” the ability to contextualize this information remains a challenge. Statements from the American Society of Clinical Oncology (ASCO) have stressed the process of offering predictive genetic testing and the elements pertaining to medical, social, and psychological consequences of positive, negative and yet to be determined results. Provided here is an updated algorithm of the contents of informed consent for genomic testing for inherited genetic changes (Table 1).
Table 1.
HUGO Gene ID, inheritance pattern, clinical manifestations and context dependent guidelines for highly penetrant breast cancer predisposition syndromes
| Characterization of breast cancer predisposition syndromes | ||||||
|---|---|---|---|---|---|---|
| Gene | Syndrome | Inheritance | Overt stigmata | General surveillance | Context specific risk reduction considerations | Penetrance |
| BRCA1/BRCA2 | Hereditary breast and ovarian | AD | No | Exam, Imaging (MRI, mammography) | Chemoprevention with tamoxifen, Mastectomy, TAH/BSO | High |
| TP53 | Li Fraumeni | AD | No | Biochemical and endocrine testing, MRI whole body | +/− Mastectomy | High |
| PTEN | Cowden | AD | Yes | Clinical exam, MRI, colonoscopy, skin exams | +/− Mastectomy | High |
| STK11 | Peutz-Jeghers | AD | Yes | Clinical exam, mammography, colonoscopy, skin exam | Polypectomy, Mastectomy, TAH/BSO | High |
| CDH1 | Familial gastric cancer | AD | No | Colonoscopy, esophogealduodenaloscopy, mammography, MRI | Gastrectomy | High |
AD autosomal dominant, MRI magnetic resonance imaging, TAH/BSO total abdominal hysterectomy bilateral salpingo-oophorectomy
Genetic testing for mutations in BRCA1, BRCA2, and other breast cancer susceptibility genes has served as a model for the integration of genomics into the practice of personalized medicine, with proven efficacy required for enhanced screening and prevention strategies, and as markers for targeted therapy. The rapid pace of molecular sequencing still requires due diligence to assure that the basic tenets of genetic counseling are fulfilled. Historically, a clinical genetics visit entails rapport building, a detailed account of the family history in the form of a pedigree, documentation of medical history, a physical exam with specific focus on the presence or absence of syndrome stigmata (e.g. macrocephaly or skin findings which may be manifestations of alterations in specific breast cancer genes), review of genetic concepts, discussion of options for screening and early detection, an opportunity for questions, a link to supporting services and a plan for follow up. In cases whereby a genetic visit indicates testing, the basic elements of informed counseling remain the standard of care [24], although these may increasingly be conveyed and communicated in on-line via video conferencing as well as in-person contexts. In an era of increasing somatic genetic analysis of breast and other tumors for the purposes of “targeting” therapies, it will be important to distinguish whether the primary purpose of genomic analysis is to determine inherited susceptibilities, or whether this information may emerge as a secondary byproduct of tumor genomic analysis (Fig. 1).
Fig. 1.

Elements of informed consent
The current number of individuals having been tested for mutations in BRCA1/2 exceeds one million. Pathogenic mutations appear to account for ~ 30 % of high-risk breast cancer families and explain ~ 15 % of the breast cancer familial relative risk (the ratio of the risk of disease for a relative of an affected individual to that for the general population) (Fig. 1) [4–6, 25]. Contextualizing disease risk of inherited mutations and sequence variants in BRCA1/2 can be complex, since the pathogenicity of sequence variants is uncertain, and requires annotation and curation using existing databases (e.g. the Breast Cancer Information Core; www.research.nhgri.nih.gov/bic).
Syndromes of Breast Cancer Predisposition
Hereditary Breast and Ovarian Syndrome
BRCA1 and BRCA2 are the predominant breast cancer susceptibility genes. Pre-test probability for BRCA2 testing is higher for families with male and female breast cancer and for BRCA1 testing in families with both breast and ovarian cancer [26]. 18,000 cases of breast cancer annually are associated with an obvious hereditary predisposition. Detection of breast cancer leads to a cure rate of more than 90 % if detected at an early stage. All told more than 200,000 breast cancer survivors in the United States developed their primary cancers as a result of a constitutional (inherited) predisposition, highlighting the importance and rationale for genetic testing [27]. Estimates range from one in 150 to one in 800 individuals in the population who are genetically predisposed to developing breast cancer and in certain ethnic groups these estimates are as high as 1 in 40 [28, 29]. A woman carrying a mutation in BRCA1 has a lifetime breast cancer risk as high as 70 % by age 70 by epidemiologic analysis [29, 30–32]. In select families with a high frequency of early onset of breast or ovarian cancer risk, estimates further increase to as high as 90 % lifetime breast cancer risk [33].
Highly Penetrant Breast Cancer Genes
BRCA1 and BRCA2
The BRCA1 and BRCA2 genes function in DNA damage response and homologous recombination [34]. BRCA1 is a large gene located on chromosome 17 and is made up of 24 exons, 22 of which are coding and two of which are non-coding. BRCA2 spans greater than 70,000 bases and the gene is comprised of 27 exons (genenames.org).
Premature truncations of the BRCA1 and BRCA2 proteins by nonsense or frame-shift alterations are the predominant genomic aberrations underlying susceptibility. Variants of uncertain significance were initially observed in up to a quarter of patients, however the frequency of these predominantly missense variants of unknown significance (VUS) dropped to between 2 and 5 % as large databases of genetic variants and “high-risk” kindreds were created [35, 36]. With the uptake of commercial testing by new laboratories, and the expansion of testing criteria beyond “high-risk” kindreds, this percentage of VUS may again increase [37].
Over 2000 distinct rare variants, in the form of intronic changes, missense mutations, and small in-frame insertions and deletions, have been reported in BRCA1 and BRCA2 (Breast Cancer Information Core; www.research.nhgri.nih.gov/bic). The main domains of BRCA1, which are critical for DNA repair activity, are located in the RING finger and BRCT domains. In BRCA2, highly penetrant, pathogenic missense mutations reside mainly in the DNA binding domain [38, 39]. Large genomic rearrangements or structural variations occur in BRCA1 (14 % of mutations) and BRCA2 (2.6 % of mutations). A reason for the relative increase in structural variations in BRCA1 compared to BRCA2 results from the large number of Alu repeats in the genomic region containing the BRCA1 gene [40].
Population specific or “founder” mutations in BRCA1/2 have been described. Some of the most common founder mutations occur in individuals of Ashkenazi (eastern European) Jewish ancestry, including two mutations in BRCA1 (185delG and 5382insC) and one mutation in BRCA2 (6174delT) [41–43]. A small number of patients in the Ashkenazi population with breast cancer have non-founder mutations in BRCA1/2 (5 % of all mutations) and thus reflex full gene sequencing may be required if founder mutations are non-revealing [42, 43]. The Ashkenazi Jewish founder mutations are the best studied and described; 3 % of individuals in this population carry a founder mutation. Other examples of BRCA1 founder mutations are reported in the Dutch and Hispanic populations. Again for these populations, targeted sequencing for specific BRCA1/2 mutations is advised before reflex to full gene testing in cases of a negative result. Carriers ascertained from population studies demonstrate a lower penetrance of disease in comparison to those identified through kindred based studies, which is not surprising as a striking overt phenotype in the families prompted initial study.
Including follow up recommendations for screening and prevention for BRCA1 mutation carriers remains as a standard of care given a ~ 57 % probability of developing breast cancer and a 40 % chance of developing ovarian cancer by age 70. BRCA2 mutation carriers are estimated to have a 49 % chance of breast cancer and an 18 % chance of ovarian cancer [44]. Contributing factors to the development of cancer include environment, modifying genomic alterations and the specific type of constitutional aberration in BRCA1/2. Statistical evidence has emerged suggesting genotype-phenotype correlations with regard to ovarian cancer risk. The early literature correlated the location of mutations in BRCA1/2 with specific phenotypes and gleaned that nonsense and frameshift mutations located in the central regions of either coding sequence, termed ovarian cancer cluster regions (OCCR), were associated with a greater risk of ovarian cancer than similar mutations in the proximal and distal regions of each gene [45, 46]. Among the greater than 22,000 BRCA1/2 mutation carriers enrolled in Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) group, the relative increases in ovarian cancer and decreases in breast cancer risk for mutations in the central region of each gene and higher risk of breast cancer for mutations in the 5′ and 3′ regions of each gene have been observed. Further variability in risk is also partly explained by common genetic modifiers of breast and ovarian cancer risk in BRCA1/2 mutation carriers that have been identified through genome-wide association studies [19, 47–51]. [55, 117] (Fig. 2).
Fig. 2.
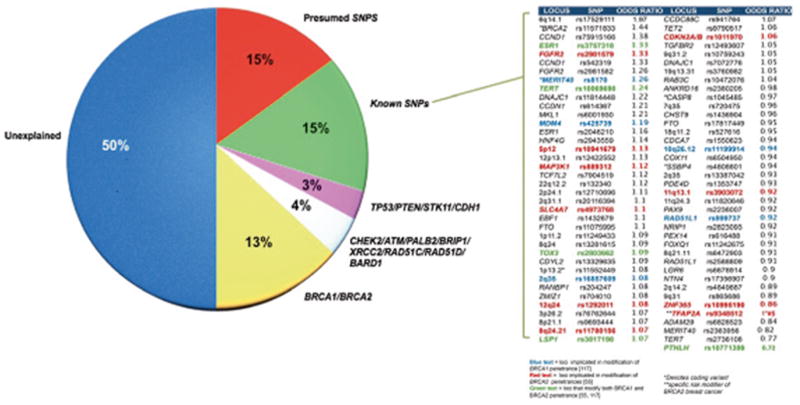
Breast cancer biomarkers
The genomic location of a patient’s BRCA1/2 mutation and the risk from modifier genes suggests that the BRCA1 mutation carriers in the highest risk category may have an 81 % or greater chance of breast cancer and a 63 % or greater chance of ovarian cancer by age 80, whereas BRCA2 mutation carriers at greatest risk may have more than an 83 % chance of breast cancer by age 80 [19, 52]. In conjunction with other variables modifying risk in BRCA1/2 mutation carriers, these emerging biomarker data on mutation location and modifier genes offer the potential for more precise risk estimates. It is also possible that such biomarkers may correlate with disease behavior. As breast cancer patients with BRCA1 mutations tend to have tumors that display features of more aggressive disease [53–56], genomic biomarkers of risk may also impact on the phenotype (e.g. estrogen receptor status) of hereditary disease.
As alluded to previously, VUS, including missense, intronic, and small in-frame insertion/deletion variants, continue to pose clinical challenges in terms of interpreting test results. Although one large testing company has classified many BRCA1/2 variants as neutral or pathogenic using data collected over years, that data have thus far not been placed into public access. Thus, laboratories now entering the clinical sequencing space have had challenges classifying variants encountered during testing. In an effort to improve the classification process for variants in all genes now offered as part of clinic genetic testing, the Clinvar (www.ncbi.nlm.nih.gov/clinvar) database has been curating variants and attempting to capture clinical information, efforts pioneered for BRCA1/2 by the international Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Consortium (see below). In 2014, the Global Alliance announced a demonstration project to create an international database of BRCA1/2 variants. These steps are crucial to allow the most accurate interpretation of these genetic biomarkers for inherited risk. In the absence of these definitive databases, evaluation of VUS has often relied on in-silico models or animal models that predict the functional impact of variants on the basis of amino acid conservation and/or structure or try equate the human disease to a different species that is not a direct homologue to humans.
To provide algorithms to the interpretation of variants of uncertain significance, expert and evidence-based committees focused on the development of quantitative risk prediction methods. One such effort is ENIGMA, which has substantially improved assessment of the pathogenicity of VUS [57]. The following elements are assessed for each variant: conservation, family history, tumor pathology, and the effects of RNA splicing [39, 57–59]. This effort also estimates the probability of pathogenicity for each variant using combined evolutionary sequence conservation (Align-GVGD) [39, 59–61], and has resulted in classification of many BRCA1/2 VUS as pathogenic or of neutral/low effect [59]. Due to the lack of statistical power for rare variants or individual VUS, high throughput quantitative cell-based in vitro assays have been developed to evaluate the effect of variants on established functions of the BRCA1 and BRCA2 proteins, with known controls of normal and pathogenic mutations as controls to asses sensitivity and specificity for VUS [39] or variant specific biomarker.
A special challenge in interpreting gene variants is the example of hypomorphic mutations, which retain some protein activity. Insights are being gained for some specific variants, i.e. the p. Arg1699Gln (R1699Q) missense mutation in the BRCT domain of BRCA1 that abrogates the repression of microRNA-155 [62] and is associated with a cumulative risk of breast cancer of 24 % by age 70 [57], and the well-known polymorphic stop codon in BRCA2, p.Lys3326X, which is associated with only a modest increase in breast cancer risk [odds ratio (OR) = 1.26] [63] and appears to have little clinical relevance. As more moderate risk variants or biomarkers in breast cancer predisposition genes are detected and clinically validated, personalized surveillance and prophylaxis measures may be developed.
Impact on Clinical Management for BRCA1 and BRCA2 Mutation Carriers
Genetic testing informs both medical decisions and family planning. While evidence-based medicine continues to evolve, BRCA mutation carriers should undergo a triple assessment for breast surveillance, including self-examination, clinician examination and mammography/Magnetic resonance imaging (MRI) [64–66].
Mammography is of limited sensitivity in BRCA mutation carriers; in one study 29 % of new tumors were missed by mammography [16]. This limitation may be due to higher breast density in younger women and as hereditary breast cancers are often more rapidly growing “triple negative” tumors (negative for estrogen and progesterone receptors and lacking HER2/neu overexpression or amplification) [67]. It is strongly recommended that women at hereditary risk begin annual mammography/MRI screening at age 25 (http://www.nccn.org/professionals/physician_gls/pdf/f_guidlines.asp#breast_risk) [68]. MRI detects twice as many breast cancers in BRCA1/2 mutation carriers as mammography or sonography [16], and is considered the standard of care. Alternatively, risk reducing mastectomy (RRM) decreases the risk of breast cancer by at least 90 % in BRCA1/2 mutation carriers [69, 70], but only 36 % of women in the United States and 22 % in Canada choose to undergo this surgery [71]. In contrast, risk-reducing salpingo-oophorectomy (RRSO) has become the standard of care for all women with BRCA1/2 mutations because ovarian cancer screening methods using serum markers and imaging are ineffective [72, 73]. RRSO has been shown to reduce the risk of BRCA-associated gynecologic cancer by 80–96 % [15, 69, 74] and to reduce the risk of breast cancer by ~ 50 %, most likely through the induction of premature menopause [15, 69, 75]. Most significantly, RRSO reduces overall mortality of women with BRCA1/2 mutations by 60 % [76]. This reduction in mortality occurs despite the 0.2 % annual risk of cancer of the peritoneal lining around the ovaries and fallopian tubes, which remains as these tissues cannot be surgically removed by RRSO [74]. Genetic testing for BRCA1/2 mutations and RRSO provided an early example of the deployment of ‘personalized’ prevention through genetics [16, 77].
Data pertaining to chemoprevention based on inherited biomarkers such as BRCA1/2 are limited. Efficacy of tamoxifen for BRCA1/2 mutations carriers was conducted as a sub-analysis as part of the 13,388 women enrolled in the National Surgical Adjuvant Breast and Bowel Project Prevention Trial (NSABP-P1). In this study, 19 BRCA1/2 mutation carriers were identified among 288 that developed breast cancer, with risk ratios for developing breast cancer with tamoxifen estimated to be 1.67 (95 % confidence interval (CI): 0.32–10.7) for BRCA1 mutation carriers and 0.38 (95 % CI: 0.06–1.56) for BRCA2 mutation carriers [78]. In a larger study of 2464 mutation carriers, tamoxifen use after a first breast cancer was associated with a reduced risk of contralateral breast cancer [79]. More refined chemoprevention options for women with mutations in BRCA1/2 may evolve. In patients with no mutations in BRCA1/2, other selective estrogen receptor modulators and aromatase inhibitors have been shown to prevent breast cancer,(http://www.nccn.org/professionals/physician_gls/pdf/f_guidlines.asp#breast_risk). Some data have also begun to emerge suggesting that modulators of RANKL signaling may be a target for chemoprevention [80].
Ovarian cancer chemoprevention studies have produced somewhat conflicting results bearing on benefits for BRCA mutation carriers [81–83], although most believe that oral contraception does decrease risk of hereditary as well as sporadic ovarian cancer. In that regard, treatment and standard of care for BRCA1/2 mutation carriers must address ovarian cancer detection and prevention. Given the unproven methods of screening and the high mortality at time of diagnosis associated with ovarian cancer, definitive counseling and recommendations for prophylactic removal of ovaries after childbearing are standards of care for BRCA1/2 mutation carriers or for women with two or more first degree relatives with ovarian cancers in the family (http://www.nccn.org/professionals/physician_gls/pdf/f_guidlines.asp#breast_risk). [15, 69, 84–88].
Finally, the identification of mutated genes as biomarkers has led to therapeutic applications. In vitro and in vivo experiments and clinical trials have shown that platinum chemotherapy is effective against BRCA1 (and, by analogy, BRCA2) mutant tumors, in part because platinum generates interstrand cross-links that can only be adequately repaired by BRCA1- and BRCA2-dependent homologous recombination DNA repair [89]. A new class of drugs that inhibit poly(ADP-ribose) polymerase (PARP), an enzyme involved in base excision repair [90, 91] shows antitumor activity in the background of BRCA-associated defects in homologous recombination-mediated DNA repair [92]. Clinical trials have explored the efficacy of PARP inhibitors in the treatment of BRCA1/2 mutant breast, ovarian, pancreatic, prostate, and other cancers, and one such compound was recently licensed for use in the U.S. for patients with previously treated BRCA mutant ovarian tumors [93]. Not all BRCA mutation carriers respond to these agents; mutations in the N-terminal BARD1 binding domain of BRCA1, such as the relatively common p.Cys61Gly (C61G), may not confer hypersensitivity to PARP ihibitors [94, 95]. Acquired resistance to PARP inhibitors has been associated with multiple mechanisms, including drug metabolism and efflux, post-transcriptional alterations of BRCA1/2, secondary mutations that restore the homologous recombination activity of BRCA1/2, and accumulation of somatic genetic alterations that counteract the sensitivity associated with BRCA1/2 mutations [95–97]. Whether combination therapies can overcome these complications remains to be determined.
Other Highly Penetrant Breast Cancer Predisposing Genes
TP53 and CDH1
Compared to BRCA1/2 mutations, TP53 mutations are rare. However when testing for BRCA1/2 is non-revealing or determined not causative, testing of TP53 may be warranted in cases with a strong family history of cancer and negative BRCA1/2 testing. Li-Fraumeni syndrome (LFS) is a multi-cancer predisposing syndrome driven by genomic alterations in the TP53 gene. TP53 encodes the tumor suppressor protein p.TP53. Patients with TP53 mutations have an increased risk of breast cancer [98]. In determining the importance to variants detected by next generation sequencing similar steps taken by ENIGMA’s efforts in assessing the BRCA genes are required. The International Association Cancer Research (IARC) hosts the TP53 locus specific database. The database curates frequency of variants, if the variant has been detected in the germline, been found in the tumor, seen in a cell line, segregation information of the variants and functional prediction of the genomic variant on protein function. National guidelines for patients with Li-Fraumeni Syndrome support TP53 testing concurrently for women ≤35 years of age or as a follow-up test after negative BRCA1/2 testing (http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf).
For carriers of TP53 mutations, it seems reasonable to consider adding annual MRI starting at age 20–25 years of age or based on earliest age of onset in the family. When patients are found to harbor a TP53 mutation, there is some laboratory based evidence that radiation exposure may be deleterious, although this remains incompletely documented. Ongoing trials are testing other approaches such as whole body MRI, PET, and other focused screening; patients should discuss approaches to novel screening and technology with their providers [99].
Reports of germline CDH1 mutations emerged in patients with hereditary diffuse gastric cancer in the late 1990s [100–104] and it was soon observed that these families also included individuals with lobular breast cancer. In screening of over 400 cases of breast cancer, three patients were found to harbor germline mutations in CDH1. Families with multi-generations affected with gastric cancer have a 30 % chance of harboring a mutation in E-Cadherin (CDH1), and 70 % of carriers of mutations in this gene develop gastric cancer. In addition to the diffuse gastric cancer risk individuals with CDH1 mutations also have approximately a 40–50 % risk of lobular cancer of the breast (http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf).
While no formal testing recommendations are established for patients with CDH1 mutations and breast cancer, Petridis et al. recently proposed CDH1 mutation screening should be considered in patients with bilateral lobular carcinoma in situ with or without invasive lobular breast cancer and with or without a family history. Gastrectomy for patients with CDH1 mutations is routinely advised. However, the identification of families with CDH1 mutations through multi-gene panel testing and no family history of gastric cancer are proving difficult to counsel, as the risk of gastric cancer in those patients is unknown. Patients are also presented options regarding mastectomy given the frequency of breast cancer in these patients or cumulative risk for breast cancer for females by age 75 years is 52 % [104].
PTEN/STK11
The majority of patients that undergo inherited genetic testing do not have overt physical manifestations of a syndrome. However, a few constitutional syndromes with overt phenotypes and genetic testing or “biomarkers” do have an increased risk of breast cancer such as Cowden syndrome/Bannayan-Riley-Ruvalcaba syndrome/PTEN hamartoma tumor syndrome (PHTS), and Peutz-Jeghers syndrome [105–107]. Major criteria to assess in diagnosing female patients suspected of having Cowden syndrome include breast cancer, endometrial cancer, follicular thyroid cancer, multiple gastrointestinal hamartomas or ganglioneuromas, macrocephaly ( > 97 %), and mucocutaneous lesions (trichilemmoma, palmoplantar keratosis, extensive mucosal papillomatosis or verrucous facial papules). Minor criteria include autism spectrum disorder, colon cancer, ≥three esophageal glycogenic acanthosis, lipomas, intellectual disability, papillary or follicular variant of thyroid cancer, thyroid structural lesions, renal cell carcinoma, single gastrointestinal hamartoma or ganglioneuroma, testicular lipomatosis, and vascular anomalies. Individuals with a family member with a known mutation, patients with autism and macrocephaly, two or more biopsy proven trichilemmomas, two or more major criteria where one has to equal macrocephaly, three major criteria without macrocephaly or one major and three minor criteria and four minor criteria [108]. Screening for patients with Cowden is as per National Comprehensive Cancer Network (NCCN) guidelines; breast MRI is part of this strategy and preventive surgeries can also be considered (http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf) (Table 1).
STK11
Peutz-Jeghers syndrome (PJS) is an autosomal dominant cancer predisposition syndrome with clinical characteristics of mucocutaneous pigmentation and gastrointestinal polyps. Patients with PJS are at increased risk of colon cancer, breast cancer, ovarian (mucinous tumors and sex cord tumors with annular tubules) [109–112]. Most mutations are small deletions/insertions or single base substitutions resulting in aberrant protein function with loss of kinase activity. In the analysis of greater than 400 patients, and close to 300 of these individuals with known STK11 mutations, the cancer risk for the development of breast cancer was 50 % by age 60 [113, 114]. However, in the largest study to date of PJS patients no differences in breast cancer risk have been found [113, 114] but the absolute numbers of kindreds with this syndrome collected for study is still small.
The major phenotype of PJS is gastrointestinal polyps. Patients require frequent endoscopic surveillance with polypectomy, which decreases the rate of intussusception and potential bowel loss. Patients with PJS should be counseled required the high rate of breast cancer and the benefits of prophylactic mastectomy and bilateral salpingo-oophorectomy after the age of 35 to prevent malignancy. In addition to monitoring of the gastrointestinal tract, routine screening of the breast (e.g. mammography and possibly MRI) should be standard of care for individuals with PJS. In addition, patients should be offered investigational pancreatic cancer screening (e.g. magnetic resonance cholangiopancreatography (MRCP) or endoscopic ultrasound) starting at an early age, as well as small bowel visualization, and pelvic exam with consideration of transvaginal ultrasound (although unproven, to address ovarian cancer risk) and annual physical exam [113, 114].
Moderate Penetrance Breast Cancer Genes or Biomarkers: CHEK2, ATM, PALB2, BRIP1, RAD51C, RAD51D, BARD1
There are no standardized guidelines for the management of other cancer risks or for the relatives of carriers with moderate penetrance gene mutations; screening recommendation should be established based on the patient’s personal and family histories.
CHEK2
CHEK2 normally functions by preventing cellular entry into mitosis when DNA is damaged. In 2000, Lee et al. reported that CHEK2 function in DNA damage by phosphorylating BRCA1 [115]. Further experiments revealed CHEK2 and BRCA1 interaction is necessary for BRCA1 to restore the survival after DNA damage. Heterozygous mutations were initially reported in a LFS-like family, suggesting CHEK2 serves as a tumor suppressor and mutations predispose individuals to cancer [116]. Subsequently mutations were shown to be associated with a moderate risk of breast cancer, rather having any association with LFS. Population studies have aimed to determine the role of CHEK2 in patients without an identifiable mutation in BRCA1/2 but a suggestive family history [117]. The truncating mutation CHEK2*1100delC affecting kinase activity was revealed in 1.1 % of healthy individuals compared to 5.1 % coming from over 700 families with breast cancer (male and female breast cancers both included) and negative BRCA1/2 testing. These data suggest a greater than a two-fold increase of breast cancer risk in females and 10-fold increase in men with the CHEK2*1100delC. As a means to assess for additional mutations in BRCA negative families with breast cancer, Shutte et al. assessed 89 kindreds with three or more individuals with breast cancer and did not find other appreciable site specific variation in CHEK2 [118]. Although studies are still in progress, it appears that the detection of a CHEK2 deleterious mutation in the setting of a strong family history of breast cancer may warrant clinical use of this biomarker in the pre-symptomatic assessment for screening. Whether the absolute level of CHEK2-associated risk meets threshold for MRI screening can be determined on an individualized basis, taking into account population derived as well as family history data.
ATM
ATM is a gene encoding a protein that allows for the efficient repair of DNA. ATM when altered manifests phenotypes from bi-allelic and arguably mono-allelic genomic alterations. Individuals with two mutations or bi-allelic or homozygous mutations develop severe disease of the immune system and are predisposed to developing leukemia and lymphoma, called Ataxia-Telangiectasia (A-T). Various degrees of evidence support or refute individuals harboring a single mutation in the ATM gene as having an increased risk of developing breast cancer, stomach, ovarian, pancreatic, or lung cancer [119–122, 123]. Approximately 1 % of the population is heterozygous for mutations in the ATM gene.
Mutant specific evidence for ATM p.S49C and p.F858L in association with increased breast cancer susceptibility show an odds ratio of 1.44 combining data from an American and Polish study [124]. When mutations that have been identified specifically in patients with ATM have been studied in mono-allelic carriers the estimated relative risk for familial breast cancer was = 2.37. The data are based on the evaluation of individuals from 443 familial breast cancer kindreds [120, 125–127]. Breast cancer-associated ATM mutations tend to be missense mutations whereas missense mutations are uncommon in individuals with A-T, even in the same host population [121].
Individuals who are carriers for ATM gene mutations should be aware that they might be sensitive to radiation, although the magnitude of this radiation sensitivity requires further study. There are no ATM mutation specific sets of recommendations for therapy, treatment, or tailored management options [128–131]. No definitive evidence has emerged regarding increased risk of mammograms in ATM mutation carriers, however, MRIs and ultrasound remain an important screening strategy. Annual breast MRI screening is recommended for women with a lifetime risk for breast cancer of 20–25 % or greater and it is generally recommended that MRI be used in conjunction with mammogram.
Regarding prevention, prophylactic mastectomy has not been evaluated extensively in individuals who are carriers for ATM gene mutations. There is no evidence concerning the effectiveness of chemoprevention in the ATM gene carrier population, although there is also no evidence that it will not be as effective as in the general population.
PALB2
PALB2 is a gene encoding a necessary protein of the Fanconi complex and is also known as the partner and localizer of BRCA2 and FANCN. PALB2 interacts with the BRCA2 protein and work together to correct and fix DNA breaks. PALB2, as it helps control the rate of cell growth and division, is a tumor suppressor (http://ghr.nlm.nih.gov/gene/PALB2). Moreover, by limiting mistakes in DNA repair, PALB2 aids in maintaining the stability of genetic information.
Literature is emerging in regards to the contribution of germline mutations of PALB2 and hereditary breast cancer. Approximately a dozen mutations have been identified in PALB2 and familial breast cancer. Mutations in PALB2 are estimated to lead to a two-fold increase in breast cancer risk. In 2007, investigators sequenced the PALB2 gene in close to 1000 individuals with breast cancer who were negative for BRCA1/2 mutations [132]. Ten out of 923 harbored PALB2 mutations conferring a 2.3-fold higher risk of breast cancer. The Q775X variant was identified in 1/50 high-risk women or 2/356 breast cancer cases and not present in any of > 6000 controls [133]. Assessing 559 women with contralateral disease and 565 women with unilateral disease as controls, fine truncating pathogenic mutations were identified. A study of Australian and New Zealand women who were negative for BRCA1/2 mutations underwent PALB2 testing and 26 out of 747 women were detected having PALB2 genomic alterations. Two women harbored nonsense mutations and two frameshift mutations. Investigators concluded that ~ 1.5 % of Australasian women in families with multiple members affected with breast cancer segregate PALB2 mutations in their families.
Recent studies analyzing the risk of breast cancer in > 150 families assessing truncating, splicing or deletions in PALB2 and family history estimated the risk of breast cancer for female carriers compared to the general population was eight to nine times as high among women younger than 40, six to eight times as high among those 40–60 years of age and five times as high for those females older than 60 years of age [134]. The estimated cumulative risk of breast cancer among female mutations carriers was 35 % by age 70 and the absolute risk ranged from 33 to 58 % depending on the extent of family history [134]. The investigators of this study concluded the breast cancer risk from PALB2 potentially overlap with that for BRCA2 mutation carriers and that loss of function mutations account for roughly 2.4 % of familial aggregation of breast cancer [134]. These data would support the role for MRI breast screening in this genetically defined population.
BRIP1
BRIP1, or alternatively named FANCJ, similar to PALB2 manifests disease in both the heterozygous and homozygous state. BRIP1 is also known as the BRCA interacting helicase. Patients with constitutional bi-allelic mutations in these two genes are notable for a Fanconi anemia phenotype. One study suggests that constitutional heterozygous carriers have a relative risk of breast cancer of 2.0 [135], however further validation studies need to be done.
RAD51C and RAD51D
Nonsense, frameshift, splice and non-functional missense mutations have been described in RAD51C, however the evidence that they are a driver of familial breast cancer is limited [136, 137]. Evidence of RAD51C mutations in familial ovarian cancer is greater than familial breast cancer [137, 138]. In a cohort of familial breast and ovarian cancer cases a distinct difference was noted between ovarian and breast cancer i.e., data revealed a relative risk of 5.88 in mutation carriers for ovarian cancer and 0.91 for breast cancer [139].
Loveday et al. also demonstrated a similar risk ratio for patients harboring mutations in RAD51D. Regarding therapeutics, the group showed that cells deficient in RAD51D are sensitive to treatment with a PARP inhibitor, suggesting a possible therapeutic approach for RAD51D mutant patients with a family history of breast and predominantly ovarian cancer.
BARD1
In Finnish families with breast and/or ovarian cancer, 5.6 % of individuals were detected to have a cys557-to-ser substitution (C557S) in the BARD1 gene compared to healthy controls (5.6 vs. 1.4 %, p = 0.005) [140]. The highest prevalence of C557S was detected in a subgroup of 94 patients with breast cancer whose family history did not include ovarian cancer (7.4 vs 1.4 %, p = 0.001). The C557S mutation is located in a region of BARD1 needed for induction of apoptosis and possibly also transcriptional regulation. The investigators concluded that C557S may be a breast cancer-predisposing allele.
Low Penetrant Polygenes
Other single nucleotide variations or single nucleotide polymorphisms have been detected conferring a moderate to low penetrant breast cancer genes or biomarkers [23] (Fig. 2). Genome-wide association studies (GWAS) have identified common genetic variants in 76 loci associated with small increases in the risk of breast cancer (Fig. 1) [63, 141]. However, most of these variants have weak effects on risk (OR < 1.10) [63]. Little is known about the relevance of these risk factors to the different molecular subtypes of breast cancer, although three of these loci (MDM4, 19p13.1, and TERT-CLPTM1L rs10069690) are exclusive to triple-negative breast cancer [142–145] and BRCA1-associated breast cancer [19]. Although the identification of causal variants and mechanism of action for most remain unclear, some variants are near known genes such as BRCA2, TGFBR2, MYC, and TET2 [63]. One mechanism of action of common variants is on gene transcription, as evidenced by the 11q31.1 locus and Cyclin D1 expression via a transcriptional enhancer and a silencer of the CCND1 gene [146], and FGFR2 expression via induction of FOXA1, ERa, and E2F1 binding to enhancers [142].
The clinical utility of these common variants as a paradigm of polygenic risk assessment for human cancer remains a work in progress [144–146]; breast cancer–associated common variants combined with traditional breast cancer risk markers had minimal impact on risk prediction models [147] or discriminatory accuracy [148]. A polygenic risk score calculated as the sum of the ORs for each allele, correlated with risk of early onset breast cancer (OR = 3.37, P = 0.03) [148] and other such studies are now under way [145], with the goal of leading to better identification of women who will benefit from enhanced screening and intervention [22].
New Paradigms for Genomic Biomarkers of Risk
Two decades of molecular biologic and genetic epidemiologic research have resulted in tests for inherited genomic variants as useful biomarkers for breast cancer risk. Tests for highly penetrant (high-risk) genetic mutations have been incorporated into clinical practice. Currently, “panel” tests for large numbers of genes, including some of unclear clinical utility, are commercially available. A pressing challenge posed by these developments is the interpretation and actionability of the large number of variants and “low penetrance” mutations discovered. To address this challenge, in 2014, the Prospective Registry of Multiplex Testing (PROMPT) began as an academic-commercial-and patient-centered initiative, and readers are encouraged to access it at https://connect.patientcrossroads.org. Such longitudinal studies would also add to the evidence base for targeted screening and prevention in genetically defined high-risk cohorts. Other federal initiatives are underway to catalogue and interpret the emerging array of genomic biomarkers of inherited cancer risk, which will only increase as screening of entire exomes and genomes becomes more feasible.
Acknowledgments
We thank S. M. Domchek, M. Robson, L. Norton, and C. Hudis for informative discussions on this subject. F.J.C. is supported by the Breast Cancer Research Foundation, NIH grants CA192393, CA176785 and CA116167, and a National Cancer Institute Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201). K.L.N. is supported by the Basser Center for BRCA Research, The Breast Cancer Research Foundation, the Rooney Family Foundation, Melanoma Research Alliance, NIH grants U01CA164947 and CA135509, the Commonwealth of Pennsylvania, and the Abramson Cancer Center Core grant (CA016520). M.W. is supported by the Robert and Kate Niehaus Clinical Genetics Initiative. K.O. is supported by the Sandra Taub Memorial Award of the Breast Cancer Research Foundation, the Sharon Levine Corzine Fund for Cancer Research, NIH grant 3P30CA008748-47, the Filomen M. D’Agostino Foundation, and the Andrew Sabin Family Fund. F.J.C. is a coholder of U.S. patents 5,837,492 and 6,033,857 held by Myriad Genetics Inc., Trustees of the University of Pennsylvania, HSC Research and Development Limited Partnership, and Endo Recherche Inc., which cover testing for mutations in the BRCA2 gene.
Contributor Information
Michael F. Walsh, Clinical Genetics Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, USA
Katherine L. Nathanson, Division of Translational Medicine and Human Genetics, Department of Medicine, Abramson Cancer Center, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA
Fergus J. Couch, Division of Experimental Pathology and Laboratory Medicine, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN 55905, USA
Kenneth Offit, Email: offitk@mskcc.org, Clinical Genetics Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA. Program in Cancer Biology and Genetics, Sloan Kettering Institute; Departments of Medicine and Public Health, Weill Cornell Medical College, New York, NY 10065, USA.
References
- 1.Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. 2010;5(6):463–466. doi: 10.1097/COH.0b013e32833ed177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch HT, Krush AJ. Carcinoma of the breast and ovary in three families. Surg Gynecol Obstet. 1971;133(4):644–648. [PubMed] [Google Scholar]
- 3.Lynch HT, Krush AJ. Genetic predictability in breast cancer risk. Surgical implications. Arch Surg. 1971;103(1):84–88. doi: 10.1001/archsurg.1971.01350070110027. [DOI] [PubMed] [Google Scholar]
- 4.Bahcall OG. iCOGS collection provides a collaborative model. Foreword. Nat Genet. 2013;45(4):343. doi: 10.1038/ng.2592. [DOI] [PubMed] [Google Scholar]
- 5.Antoniou AC, Cunningham AP, Peto J, Evans DG, Lalloo F, Narod SA, Risch HA, Eyfjord JE, Hopper JL, Southey MC, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tryggvadottir L, Syrjakoski K, Kallioniemi OP, Eerola H, Nevanlinna H, Pharoah PD, Easton DF. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer. 2008;98(8):1457–1466. doi: 10.1038/sj.bjc.6604305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010;4(3):174–191. doi: 10.1016/j.molonc.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250(4988):1684–1689. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 8.Hall JM, Friedman L, Guenther C, Lee MK, Weber JL, Black DM, King MC. Closing in on a breast cancer gene on chromosome 17q. Am J Hum Genet. 1992;50(6):1235–1242. [PMC free article] [PubMed] [Google Scholar]
- 9.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265(5181):2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 10.Couch FJ, Rommens JM, Neuhausen SL, Belanger C, Dumont M, Abel K, Bell R, Berry S, Bogden R, Cannon-Albright L, Farid L, Frye C, Hattier T, Janecki T, Jiang P, Kehrer R, Leblanc JF, McArthur-Morrison J, Meney D, Miki Y, Peng Y, Samson C, Schroeder M, Snyder SC, Simard J, et al. Generation of an integrated transcription map of the BRCA2 region on chromosome 13q12-q13. Genomics. 1996;36(1):86–99. doi: 10.1006/geno.1996.0428. [DOI] [PubMed] [Google Scholar]
- 11.Neuhausen S, Gilewski T, Norton L, Tran T, McGuire P, Swensen J, Hampel H, Borgen P, Brown K, Skolnick M, Shattuck-Eidens D, Jhanwar S, Goldgar D, Offit K. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat Genet. 1996;13(1):126–128. doi: 10.1038/ng0596-126. [DOI] [PubMed] [Google Scholar]
- 12.Oddoux C, Struewing JP, Clayton CM, Neuhausen S, Brody LC, Kaback M, Haas B, Norton L, Borgen P, Jhanwar S, Goldgar D, Ostrer H, Offit K. The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1 % Nat Genet. 1996;14(2):188–190. doi: 10.1038/ng1096-188. [DOI] [PubMed] [Google Scholar]
- 13.Peelen T, Cornelis RS, van Vliet M, Petrij-Bosch A, Cleton-Jansen AM, Meijers-Heijboer H, Klijn JG, Vasen HF, Cornelisse CJ, Devilee P. The majority of 22 Dutch high-risk breast cancer families are due to either BRCA1 or BRCA2. Eur J Hum Genet. 1996;4(4):225–230. doi: 10.1159/000472203. [DOI] [PubMed] [Google Scholar]
- 14.Tonin P, Weber B, Offit K, Couch F, Rebbeck TR, Neuhausen S, Godwin AK, Daly M, Wagner-Costalos J, Berman D, Grana G, Fox E, Kane MF, Kolodner RD, Krainer M, Haber DA, Struewing JP, Warner E, Rosen B, Lerman C, Peshkin B, Norton L, Serova O, Foulkes WD, Garber JE, et al. Frequency of recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer families. Nat Med. 1996;2(11):1179–1183. doi: 10.1038/nm1196-1179. [DOI] [PubMed] [Google Scholar]
- 15.Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, Hudis CA, Ellis NA, Boyd J, Borgen PI, Barakat RR, Norton L, Castiel M, Nafa K, Offit K. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2002;346(21):1609–1615. doi: 10.1056/NEJMoa020119. [DOI] [PubMed] [Google Scholar]
- 16.Robson M, Offit K. Clinical practice. Management of an inherited predisposition to breast cancer. N Engl J Med. 2007;357(2):154–162. doi: 10.1056/NEJMcp071286. [DOI] [PubMed] [Google Scholar]
- 17.Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P, Kosarin K, Olsh A, Bergeron J, Ellis NA, Klein RJ, Clark AG, Norton L, Dean M, Boyd J, Offit K. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA. 2008;105(11):4340–4345. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salgotra RK, Gupta BB, Stewart CN., Jr From genomics to functional markers in the era of next-generation sequencing. Biotechnol Lett. 2014;36(3):417–426. doi: 10.1007/s10529-013-1377-1. [DOI] [PubMed] [Google Scholar]
- 19.Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, Barrowdale D, Dennis J, Gaudet MM, Dicks E, Kosel M, Healey S, Sinilnikova OM, Lee A, Bacot F, Vincent D, Hogervorst FB, Peock S, Stoppa-Lyonnet D, Jakubowska A, Radice P, Schmutzler RK, Swe B, Domchek SM, Piedmonte M, Singer CF, Friedman E, Thomassen M, Hansen TV, Neuhausen SL, Szabo CI, Blanco I, Greene MH, Karlan BY, Garber J, Phelan CM, Weitzel JN, Montagna M, Olah E, Andrulis IL, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Nevanlinna H, Osorio A, Terry MB, Daly MB, van Rensburg EJ, Hamann U, Ramus SJ, Toland AE, Caligo MA, Olopade OI, Tung N, Claes K, Beattie MS, Southey MC, Imyanitov EN, Tischkowitz M, Janavicius R, John EM, Kwong A, Diez O, Balmana J, Barkardottir RB, Arun BK, Rennert G, Teo SH, Ganz PA, Campbell I, van der Hout AH, van Deurzen CH, Seynaeve C, Gomez Garcia EB, van Leeuwen FE, Meijers-Heijboer HE, Gille JJ, Ausems MG, Blok MJ, Ligtenberg MJ, Rookus MA, Devilee P, Verhoef S, van Os TA, Wijnen JT, Hebon, Embrace, Frost D, Ellis S, Fineberg E, Platte R, Evans DG, Izatt L, Eeles RA, Adlard J, Eccles DM, Cook J, Brewer C, Douglas F, Hodgson S, Morrison PJ, Side LE, Donaldson A, Houghton C, Rogers MT, Dorkins H, Eason J, Gregory H, McCann E, Murray A, Calender A, Hardouin A, Berthet P, Delnatte C, Nogues C, Lasset C, Houdayer C, Leroux D, Rouleau E, Prieur F, Damiola F, Sobol H, Coupier I, Venat-Bouvet L, Castera L, Gauthier-Villars M, Leone M, Pujol P, Mazoyer S, Bignon YJ, Collaborators GS, Zlowocka-Perlowska E, Gronwald J, Lubinski J, Durda K, Jaworska K, Huzarski T, Spurdle AB, Viel A, Peissel B, Bonanni B, Melloni G, Ottini L, Papi L, Varesco L, Tibiletti MG, Peterlongo P, Volorio S, Manoukian S, Pensotti V, Arnold N, Engel C, Deissler H, Gadzicki D, Gehrig A, Kast K, Rhiem K, Meindl A, Niederacher D, Ditsch N, Plendl H, Preisler-Adams S, Engert S, Sutter C, Varon-Mateeva R, Wappenschmidt B, Weber BH, Arver B, Stenmark-Askmalm M, Loman N, Rosenquist R, Einbeigi Z, Nathanson KL, Rebbeck TR, Blank SV, Cohn DE, Rodriguez GC, Small L, Friedlander M, Bae-Jump VL, Fink-Retter A, Rappaport C, Gschwantler-Kaulich D, Pfeiler G, Tea MK, Lindor NM, Kaufman B, Shimon Paluch S, Laitman Y, Skytte AB, Gerdes AM, Pedersen IS, Moeller ST, Kruse TA, Jensen UB, Vijai J, Sarrel K, Robson M, Kauff N, Mulligan AM, Glendon G, Ozcelik H, Ejlertsen B, Nielsen FC, Jonson L, Andersen MK, Ding YC, Steele L, Foretova L, Teule A, Lazaro C, Brunet J, Pujana MA, Mai PL, Loud JT, Walsh C, Lester J, Orsulic S, Narod SA, Herzog J, Sand SR, Tognazzo S, Agata S, Vaszko T, Weaver J, Stavropoulou AV, Buys SS, Romero A, de la Hoya M, Aittomaki K, Muranen TA, Duran M, Chung WK, Lasa A, Dorfling CM, Miron A, Bcfr, Benitez J, Senter L, Huo D, Chan SB, Sokolenko AP, Chiquette J, Tihomirova L, Friebel TM, Agnarsson BA, Lu KH, Lejbkowicz F, James PA, Hall P, Dunning AM, Tessier D, Cunningham J, Slager SL, Wang C, Hart S, Stevens K, Simard J, Pastinen T, Pankratz VS, Offit K, Easton DF, Chenevix-Trench G, Antoniou AC kConFab I, Ontario Cancer Genetics N, Cimba. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013;9(3):e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kauff ND, Offit K. Modeling genetic risk of breast cancer. JAMA. 2007;297(23):2637–2639. doi: 10.1001/jama.297.23.2637. [DOI] [PubMed] [Google Scholar]
- 21.Stadler ZK, Thom P, Robson ME, Weitzel JN, Kauff ND, Hurley KE, Devlin V, Gold B, Klein RJ, Offit K. Genome-wide association studies of cancer. J Clin Oncol. 2010;28(27):4255–4267. doi: 10.1200/JCO.2009.25.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadler ZK, Vijai J, Thom P, Kirchhoff T, Hansen NA, Kauff ND, Robson M, Offit K. Genome-wide association studies of cancer predisposition. Hematol Oncol Clin North Am. 2010;24(5):973–996. doi: 10.1016/j.hoc.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343(6178):1466–1470. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Offit K. Clinical cancer genetics: risk counseling and management. Wiley-Liss; New York: 1998. [Google Scholar]
- 25.Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999;91(11):943–949. doi: 10.1093/jnci/91.11.943. [DOI] [PubMed] [Google Scholar]
- 26.Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20(6):1480–1490. doi: 10.1200/JCO.2002.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 27.Niederhuber JE, Armitage JO, Doroshow JH, Kastan MB, Tepper JE, Abeloff MD. Abeloff’s clinical oncology. 5. Saunders Elsevier; Philadelphia: 2013. [Google Scholar]
- 28.Ford D, Easton DF. The genetics of breast and ovarian cancer. Br J Cancer. 1995;72(4):805–812. doi: 10.1038/bjc.1995.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whittemore AS, Gong G, Itnyre J. Prevalence and contribution of BRCA1 mutations in breast cancer and ovarian cancer: results from three U.S. population-based case-control studies of ovarian cancer. Am J Hum Genet. 1997;60(3):496–504. [PMC free article] [PubMed] [Google Scholar]
- 30.Claus EB, Risch N, Thompson WD. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet. 1991;48(2):232–242. [PMC free article] [PubMed] [Google Scholar]
- 31.Claus EB, Risch N, Thompson WD. The calculation of breast cancer risk for women with a first degree family history of ovarian cancer. Breast Cancer Res Treat. 1993;28(2):115–120. doi: 10.1007/BF00666424. [DOI] [PubMed] [Google Scholar]
- 32.Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer. 1996;77(11):2318–2324. doi: 10.1002/(SICI)1097-0142(19960601)77:112318::AID-CNCR213.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 33.Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343(8899):692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 34.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 35.Peshkin BN, DeMarco TA, Brogan BM, Lerman C, Isaacs C. BRCA1/2 testing: complex themes in result interpretation. J Clin Oncol. 2001;19(9):2555–2565. doi: 10.1200/JCO.2001.19.9.2555. [DOI] [PubMed] [Google Scholar]
- 36.Stoppa-Lyonnet D, Laurent-Puig P, Essioux L, Pages S, Ithier G, Ligot L, Fourquet A, Salmon RJ, Clough KB, Pouillart P, Bonaiti-Pellie C, Thomas G. BRCA1 sequence variations in 160 individuals referred to a breast/ovarian family cancer clinic. Institut Curie Breast Cancer Group. Am J Hum Genet. 1997;60(5):1021–1030. [PMC free article] [PubMed] [Google Scholar]
- 37.LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, Chen E, Gau CL, Palmaer E, Shoaepour K, Shah D, Speare V, Gandomi S, Chao E. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2000 patients. Genet Med. 2014;16(11):830–837. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro AN, Neuhausen SL, Hansen TV, Couch FJ, Vreeswijk MP consortium E. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat. 2014;35(2):151–164. doi: 10.1002/humu.22478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guidugli L, Pankratz VS, Singh N, Thompson J, Erding CA, Engel C, Schmutzler R, Domchek S, Nathanson K, Radice P, Singer C, Tonin PN, Lindor NM, Goldgar DE, Couch FJ. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013;73(1):265–275. doi: 10.1158/0008-5472.CAN-12-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judkins T, Rosenthal E, Arnell C, Burbidge LA, Geary W, Barrus T, Schoenberger J, Trost J, Wenstrup RJ, Roa BB. Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer. 2012;118(21):5210–5216. doi: 10.1002/cncr.27556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Offit K, Gilewski T, McGuire P, Schluger A, Hampel H, Brown K, Swensen J, Neuhausen S, Skolnick M, Norton L, Goldgar D. Germline BRCA1 185delAG mutations in Jewish women with breast cancer. Lancet. 1996;347(9016):1643–1645. doi: 10.1016/s0140-6736(96)91484-1. [DOI] [PubMed] [Google Scholar]
- 42.Szabo CI, King MC. Population genetics of BRCA1 and BRCA2. Am J Hum Genet. 1997;60(5):1013–1020. [PMC free article] [PubMed] [Google Scholar]
- 43.Thorlacius S, Sigurdsson S, Bjarnadottir H, Olafsdottir G, Jonasson JG, Tryggvadottir L, Tulinius H, Eyfjord JE. Study of a single BRCA2 mutation with high carrier frequency in a small population. Am J Hum Genet. 1997;60(5):1079–1084. [PMC free article] [PubMed] [Google Scholar]
- 44.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gayther SA, Harrington P, Russell P, Kharkevich G, Garkavtseva RF, Ponder BA. Rapid detection of regionally clustered germ-line BRCA1 mutations by multiplex heteroduplex analysis. UKCCCR Familial Ovarian Cancer Study Group. Am J Hum Genet. 1996;58(3):451–456. [PMC free article] [PubMed] [Google Scholar]
- 46.Gayther SA, Harrington P, Russell P, Kharkevich G, Garkavtseva RF, Ponder BA. Frequently occurring germ-line mutations of the BRCA1 gene in ovarian cancer families from Russia. Am J Hum Genet. 1997;60(5):1239–1242. [PMC free article] [PubMed] [Google Scholar]
- 47.Antoniou AC, Beesley J, McGuffog L, Sinilnikova OM, Healey S, Neuhausen SL, Ding YC, Rebbeck TR, Weitzel JN, Lynch HT, Isaacs C, Ganz PA, Tomlinson G, Olopade OI, Couch FJ, Wang X, Lindor NM, Pankratz VS, Radice P, Manoukian S, Peissel B, Zaffaroni D, Barile M, Viel A, Allavena A, Dall’Olio V, Peterlongo P, Szabo CI, Zikan M, Claes K, Poppe B, Foretova L, Mai PL, Greene MH, Rennert G, Lejbkowicz F, Glendon G, Ozcelik H, Andrulis IL, Thomassen M, Gerdes AM, Sunde L, Cruger D, Birk Jensen U, Caligo M, Friedman E, Kaufman B, Laitman Y, Milgrom R, Dubrovsky M, Cohen S, Borg A, Jernstrom H, Lindblom A, Rantala J, Stenmark-Askmalm M, Melin B, Swe B, Nathanson K, Domchek S, Jakubowska A, Lubinski J, Huzarski T, Osorio A, Lasa A, Duran M, Tejada MI, Godino J, Benitez J, Hamann U, Kriege M, Hoogerbrugge N, van der Luijt RB, van Asperen CJ, Devilee P, Meijers-Heijboer EJ, Blok MJ, Aalfs CM, Hogervorst F, Rookus M, Hebon, Cook M, Oliver C, Frost D, Conroy D, Evans DG, Lalloo F, Pichert G, Davidson R, Cole T, Cook J, Paterson J, Hodgson S, Morrison PJ, Porteous ME, Walker L, Kennedy MJ, Dorkins H, Peock S, Embrace, Godwin AK, Stoppa-Lyonnet D, de Pauw A, Mazoyer S, Bonadona V, Lasset C, Dreyfus H, Leroux D, Hardouin A, Berthet P, Faivre L, Gemo, Loustalot C, Noguchi T, Sobol H, Rouleau E, Nogues C, Frenay M, Venat-Bouvet L, Gemo, Hopper JL, Daly MB, Terry MB, John EM, Buys SS, Yassin Y, Miron A, Goldgar D, Singer CF, Dressler AC, Gschwantler-Kaulich D, Pfeiler G, Hansen TV, Jonson L, Agnarsson BA, Kirchhoff T, Offit K, Devlin V, Dutra-Clarke A, Piedmonte M, Rodriguez GC, Wakeley K, Boggess JF, Basil J, Schwartz PE, Blank SV, Toland AE, Montagna M, Casella C, Imyanitov E, Tihomirova L, Blanco I, Lazaro C, Ramus SJ, Sucheston L, Karlan BY, Gross J, Schmutzler R, Wappenschmidt B, Engel C, Meindl A, Lochmann M, Arnold N, Heidemann S, Varon-Mateeva R, Niederacher D, Sutter C, Deissler H, Gadzicki D, Preisler-Adams S, Kast K, Schonbuchner I, Caldes T, de la Hoya M, Aittomaki K, Nevanlinna H, Simard J, Spurdle AB, Holland H, Chen X, Platte R, Chenevix-Trench G, Easton DF kConFab, Ontario Cancer Genetics N, Breast Cancer Family R, Cimba. Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: implications for risk prediction. Cancer Res. 2010;70(23):9742–9754. doi: 10.1158/0008-5472.CAN-10-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoniou AC, Kartsonaki C, Sinilnikova OM, Soucy P, McGuffog L, Healey S, Lee A, Peterlongo P, Manoukian S, Peissel B, Zaffaroni D, Cattaneo E, Barile M, Pensotti V, Pasini B, Dolcetti R, Giannini G, Putignano AL, Varesco L, Radice P, Mai PL, Greene MH, Andrulis IL, Glendon G, Ozcelik H, Thomassen M, Gerdes AM, Kruse TA, Birk Jensen U, Cruger DG, Caligo MA, Laitman Y, Milgrom R, Kaufman B, Paluch-Shimon S, Friedman E, Loman N, Harbst K, Lindblom A, Arver B, Ehrencrona H, Melin B, Swe B, Nathanson KL, Domchek SM, Rebbeck T, Jakubowska A, Lubinski J, Gronwald J, Huzarski T, Byrski T, Cybulski C, Gorski B, Osorio A, Ramon y Cajal T, Fostira F, Andres R, Benitez J, Hamann U, Hogervorst FB, Rookus MA, Hooning MJ, Nelen MR, van der Luijt RB, van Os TA, van Asperen CJ, Devilee P, Meijers-Heijboer HE, Gomez Garcia EB, Hebon, Peock S, Cook M, Frost D, Platte R, Leyland J, Evans DG, Lalloo F, Eeles R, Izatt L, Adlard J, Davidson R, Eccles D, Ong KR, Cook J, Douglas F, Paterson J, Kennedy MJ, Miedzybrodzka Z, Embrace, Godwin A, Stoppa-Lyonnet D, Buecher B, Belotti M, Tirapo C, Mazoyer S, Barjhoux L, Lasset C, Leroux D, Faivre L, Bronner M, Prieur F, Nogues C, Rouleau E, Pujol P, Coupier I, Frenay M, Collaborators CS, Hopper JL, Daly MB, Terry MB, John EM, Buys SS, Yassin Y, Miron A, Goldgar D, Singer CF, Tea MK, Pfeiler G, Dressler AC, Hansen T, Jonson L, Ejlertsen B, Barkardottir RB, Kirchhoff T, Offit K, Piedmonte M, Rodriguez G, Small L, Boggess J, Blank S, Basil J, Azodi M, Toland AE, Montagna M, Tognazzo S, Agata S, Imyanitov E, Janavicius R, Lazaro C, Blanco I, Pharoah PD, Sucheston L, Karlan BY, Walsh CS, Olah E, Bozsik A, Teo SH, Seldon JL, Beattie MS, van Rensburg EJ, Sluiter MD, Diez O, Schmutzler RK, Wappenschmidt B, Engel C, Meindl A, Ruehl I, Varon-Mateeva R, Kast K, Deissler H, Niederacher D, Arnold N, Gadzicki D, Schonbuchner I, Caldes T, de la Hoya M, Nevanlinna H, Aittomaki K, Dumont M, Chiquette J, Tischkowitz M, Chen X, Beesley J, Spurdle AB, Neuhausen SL, Chun Ding Y, Fredericksen Z, Wang X, Pankratz VS, Couch F, Simard J, Easton DF, Chenevix-Trench G kConFab i, Breast Cancer Family R, Cimba. Common alleles at 6q25.1 and 1p11.2 are associated with breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2011;20(16):3304–3321. doi: 10.1093/hmg/ddr226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaudet MM, Kirchhoff T, Green T, Vijai J, Korn JM, Guiducci C, Segre AV, McGee K, Mc-Guffog L, Kartsonaki C, Morrison J, Healey S, Sinilnikova OM, Stoppa-Lyonnet D, Mazoyer S, Gauthier-Villars M, Sobol H, Longy M, Frenay M, Collaborators GS, Hogervorst FB, Rookus MA, Collee JM, Hoogerbrugge N, van Roozendaal KE, Collaborators HS, Piedmonte M, Rubinstein W, Nerenstone S, Van Le L, Blank SV, Caldes T, de la Hoya M, Nevanlinna H, Aittomaki K, Lazaro C, Blanco I, Arason A, Johannsson OT, Barkardottir RB, Devilee P, Olopade OI, Neuhausen SL, Wang X, Fredericksen ZS, Peterlongo P, Manoukian S, Barile M, Viel A, Radice P, Phelan CM, Narod S, Rennert G, Lejbkowicz F, Flugelman A, Andrulis IL, Glendon G, Ozcelik H, Ocgn, Toland AE, Montagna M, D’Andrea E, Friedman E, Laitman Y, Borg A, Beattie M, Ramus SJ, Domchek SM, Nathanson KL, Rebbeck T, Spurdle AB, Chen X, Holland H, John EM, Hopper JL, Buys SS, Daly MB, Southey MC, Terry MB, Tung N, Overeem Hansen TV, Nielsen FC, Greene MH, Mai PL, Osorio A, Duran M, Andres R, Benitez J, Weitzel JN, Garber J, Hamann U, Embrace, Peock S, Cook M, Oliver C, Frost D, Platte R, Evans DG, Lalloo F, Eeles R, Izatt L, Walker L, Eason J, Barwell J, Godwin AK, Schmutzler RK, Wappenschmidt B, Engert S, Arnold N, Gadzicki D, Dean M, Gold B, Klein RJ, Couch FJ, Chenevix-Trench G, Easton DF, Daly MJ, Antoniou AC, Altshuler DM, Offit K. Common genetic variants and modification of penetrance of BRCA2-associated breast cancer. PLoS Genet. 2010;6(10):e1001183. doi: 10.1371/journal.pgen.1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rebbeck TR, Mitra N, Domchek SM, Wan F, Friebel TM, Tran TV, Singer CF, Tea MK, Blum JL, Tung N, Olopade OI, Weitzel JN, Lynch HT, Snyder CL, Garber JE, Antoniou AC, Peock S, Evans DG, Paterson J, Kennedy MJ, Donaldson A, Dorkins H, Easton DF, Carriers BM, Rubinstein WS, Daly MB, Isaacs C, Nevanlinna H, Couch FJ, Andrulis IL, Freidman E, Laitman Y, Ganz PA, Tomlinson GE, Neuhausen SL, Narod SA, Phelan CM, Greenberg R, Nathanson KL kConFab, Epidemiological Study of B. Modification of BRCA1-Associated breast and ovarian cancer risk by BRCA1-Interacting Genes. Cancer Res. 2011;71(17):5792–5805. doi: 10.1158/0008-5472.CAN-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Pankratz VS, Fredericksen Z, Tarrell R, Karaus M, McGuffog L, Pharaoh PD, Ponder BA, Dunning AM, Peock S, Cook M, Oliver C, Frost D, Embrace, Sinilnikova OM, Stoppa-Lyonnet D, Mazoyer S, Houdayer C, Gemo, Hogervorst FB, Hooning MJ, Ligtenberg MJ, Hebon, Spurdle A, Chenevix-Trench G, Schmutzler RK, Wappenschmidt B, Engel C, Meindl A, Domchek SM, Nathanson KL, Rebbeck TR, Singer CF, Gschwantler-Kaulich D, Dressler C, Fink A, Szabo CI, Zikan M, Foretova L, Claes K, Thomas G, Hoover RN, Hunter DJ, Chanock SJ, Easton DF, Antoniou AC, Couch FJ kConFab. Common variants associated with breast cancer in genome-wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2010;19(14):2886–2897. doi: 10.1093/hmg/ddq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaudet MM, Kuchenbaecker KB, Vijai J, Klein RJ, Kirchhoff T, McGuffog L, Barrowdale D, Dunning AM, Lee A, Dennis J, Healey S, Dicks E, Soucy P, Sinilnikova OM, Pankratz VS, Wang X, Eldridge RC, Tessier DC, Vincent D, Bacot F, Hogervorst FB, Peock S, Stoppa-Lyonnet D, Peterlongo P, Schmutzler RK, Nathanson KL, Piedmonte M, Singer CF, Thomassen M, Hansen T, Neuhausen SL, Blanco I, Greene MH, Garber J, Weitzel JN, Andrulis IL, Goldgar DE, D’Andrea E, Caldes T, Nevanlinna H, Osorio A, van Rensburg EJ, Arason A, Rennert G, van den Ouweland AM, van der Hout AH, Kets CM, Aalfs CM, Wijnen JT, Ausems MG, Hebon, Embrace, Frost D, Ellis S, Fineberg E, Platte R, Evans DG, Jacobs C, Adlard J, Tischkowitz M, Porteous ME, Damiola F, Collaborators GS, Golmard L, Barjhoux L, Longy M, Belotti M, Ferrer SF, Mazoyer S, Spurdle AB, Manoukian S, Barile M, Genuardi M, Arnold N, Meindl A, Sutter C, Wappenschmidt B, Domchek SM, Pfeiler G, Friedman E, Jensen UB, Robson M, Shah S, Lazaro C, Mai PL, Benitez J, Southey MC, Schmidt MK, Fasching PA, Peto J, Humphreys MK, Wang Q, Michailidou K, Sawyer EJ, Burwinkel B, Guenel P, Bojesen SE, Milne RL, Brenner H, Lochmann M, Network G, Aittomaki K, Dork T, Margolin S, Mannermaa A, Lambrechts D, Chang-Claude J, Radice P, Giles GG, Haiman CA, Winqvist R, Devillee P, Garcia-Closas M, Schoof N, Hooning MJ, Cox A, Pharoah PD, Jakubowska A, Orr N, Gonzalez-Neira A, Pita G, Alonso MR, Hall P, Couch FJ, Simard J, Altshuler D, Easton DF, Chenevix-Trench G, Antoniou AC, Offit K Investigators KC, Ontario Cancer Genetics N. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet. 2013;9(3):e1003173. doi: 10.1371/journal.pgen.1003173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bignon YJ, Fonck Y, Chassagne MC. Histoprognostic grade in tumours from families with hereditary predisposition to breast cancer. Lancet. 1995;346(8969):258. doi: 10.1016/s0140-6736(95)91310-6. [DOI] [PubMed] [Google Scholar]
- 54.Jacquemier J, Eisinger F, Birnbaum D, Sobol H. Histoprognostic grade in BRCA1-associated breast cancer. Lancet. 1995;345(8963):1503. doi: 10.1016/s0140-6736(95)91060-3. [DOI] [PubMed] [Google Scholar]
- 55.Johannsson OT, Idvall I, Anderson C, Borg A, Barkardottir RB, Egilsson V, Olsson H. Tumour biological features of BRCA1-induced breast and ovarian cancer. Eur J Cancer. 1997;33(3):362–371. doi: 10.1016/s0959-8049(97)89007-7. [DOI] [PubMed] [Google Scholar]
- 56.Robson M, Rajan P, Rosen PP, Gilewski T, Hirschaut Y, Pressman P, Haas B, Norton L, Of-fit K. BRCA-associated breast cancer: absence of a characteristic immunophenotype. Cancer Res. 1998;58(9):1839–1842. [PubMed] [Google Scholar]
- 57.Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, Pettigrew C, Van Asperen CJ, Ausems MG, Kattentidt-Mouravieva AA, van den Ouweland AM, Dutch Belgium UVC, Lindblom A, Pigg MH, Schmutzler RK, Engel C, Meindl A, Ovarian C, Caputo S, Sinilnikova OM, Lidereau R, French Cgc, Couch FJ, Guidugli L, Hansen T, Thomassen M, Eccles DM, Tucker K, Benitez J, Domchek SM, Toland AE, Van Rensburg EJ, Wappenschmidt B, Borg A, Vreeswijk MP, Goldgar DE kConFab, Consortium E, German Consortium of Hereditary B. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J Med Genet. 2012;49(8):525–532. doi: 10.1136/jmedgenet-2012-101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iversen ES, Jr, Couch FJ, Goldgar DE, Tavtigian SV, Monteiro AN. A computational method to classify variants of uncertain significance using functional assay data with application to BRCA1. Cancer Epidemiol Biomarkers Prev (a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive) 2011;20(6):1078–1088. doi: 10.1158/1055-9965.EPI-10-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindor NM, Guidugli L, Wang X, Vallee MP, Monteiro AN, Tavtigian S, Goldgar DE, Couch FJ. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS) Hum Mutat. 2012;33(1):8–21. doi: 10.1002/humu.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29(11):1342–1354. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ Breast Cancer Information Core Steering C. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75(4):535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Haines DC, Basik M, Mai P, Poggi E, Isaacs C, Looi LM, Mun KS, Greene MH, Byers SW, Teo SH, Deng CX, Sharan SK Kathleen Cuningham Foundation Consortium for Research into Familial Breast C. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17(10):1275–1282. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, Hereditary B, van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Muller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LF, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens MR, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJ, van der Schoot CE, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JI, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MW, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE, Schumacher F, Le Marchand L, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Lindblom A, Margolin S, Hooning MJ, Hollestelle A, van den Ouweland AM, Jager A, Bui QM, Stone J, Dite GS, Apicella C, Tsimiklis H, Giles GG, Severi G, Baglietto L, Fasching PA, Haeberle L, Ekici AB, Beckmann MW, Brenner H, Muller H, Arndt V, Stegmaier C, Swerdlow A, Ashworth A, Orr N, Jones M, Figueroa J, Lissowska J, Brinton L, Goldberg MS, Labreche F, Dumont M, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Bruning T, Network G, Radice P, Peterlongo P, Manoukian S, Bonanni B, Devilee P, Tollenaar RA, Seynaeve C, van Asperen CJ, Jakubowska A, Lubinski J, Jaworska K, Durda K, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Bogdanova NV, Antonenkova NN, Dork T, Kristensen VN, Anton-Culver H, Slager S, Toland AE, Edge S, Fostira F, Kang D, Yoo KY, Noh DY, Matsuo K, Ito H, Iwata H, Sueta A, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Shu XO, Lu W, Gao YT, Cai H, Teo SH, Yip CH, Phuah SY, Cornes BK, Hartman M, Miao H, Lim WY, Sng JH, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Shen CY, Hsiung CN, Wu PE, Ding SL, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Blot WJ, Signorello LB, Cai Q, Zheng W, Deming-Halverson S, Shrubsole M, Long J, Simard J, Garcia-Closas M, Pharoah PD, Chenevix-Trench G, Dunning AM, Benitez J, Easton DF kConFab I, Breast, Ovarian Cancer Susceptibility C, Ovarian Cancer Research Group N, Australian Ovarian Cancer Study G. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353–361. 361e351–352. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burke W, Petersen G, Lynch P, Botkin J, Daly M, Garber J, Kahn MJ, McTiernan A, Offit K, Thomson E, Varricchio C. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA. 1997;277(11):915–919. [PubMed] [Google Scholar]
- 65.Kriege M, Brekelmans CT, Boetes C, Besnard PE, Zonderland HM, Obdeijn IM, Manoliu RA, Kok T, Peterse H, Tilanus-Linthorst MM, Muller SH, Meijer S, Oosterwijk JC, Beex LV, Tollenaar RA, de Koning HJ, Rutgers EJ, Klijn JG Magnetic Resonance Imaging Screening Study G. Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med. 2004;351(5):427–437. doi: 10.1056/NEJ-Moa031759. [DOI] [PubMed] [Google Scholar]
- 66.Morris EA, Liberman L, Ballon DJ, Robson M, Abramson AF, Heerdt A, Dershaw DD. MRI of occult breast carcinoma in a high-risk population. AJR Am J Roentgenol. 2003;181(3):619–626. doi: 10.2214/ajr.181.3.1810619. [DOI] [PubMed] [Google Scholar]
- 67.Collett K, Stefansson IM, Eide J, Braaten A, Wang H, Eide GE, Thoresen SO, Foulkes WD, Akslen LA. A basal epithelial phenotype is more frequent in interval breast cancers compared with screen detected tumors. Cancer Epidemiol Biomarkers Prev (a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive) 2005;14(5):1108–1112. doi: 10.1158/1055-9965.EPI-04-0394. [DOI] [PubMed] [Google Scholar]
- 68.Mettler FA, Upton AC, Kelsey CA, Ashby RN, Rosenberg RD, Linver MN. Benefits versus risks from mammography: a critical reassessment. Cancer. 1996;77(5):903–909. [PubMed] [Google Scholar]
- 69.Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’t Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E, Olopade OI, Weber BL Prevention, Observation of Surgical End Points Study G. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med. 2002;346(21):1616–1622. doi: 10.1056/NEJMoa012158. [DOI] [PubMed] [Google Scholar]
- 70.Domchek SM, Rebbeck TR. Prophylactic oophorectomy in women at increased cancer risk. Curr Opin Obstet Gynecol. 2007;19(1):27–30. doi: 10.1097/GCO.0b013e32801195da. [DOI] [PubMed] [Google Scholar]
- 71.Metcalfe KA, Birenbaum-Carmeli D, Lubinski J, Gronwald J, Lynch H, Moller P, Ghadirian P, Foulkes WD, Klijn J, Friedman E, Kim-Sing C, Ainsworth P, Rosen B, Domchek S, Wagner T, Tung N, Manoukian S, Couch F, Sun P, Narod SA Hereditary Breast Cancer Clinical Study G. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer (Journal international du cancer) 2008;122(9):2017–2022. doi: 10.1002/ijc.23340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hermsen BB, Olivier RI, Verheijen RH, van Beurden M, de Hullu JA, Massuger LF, Burger CW, Brekelmans CT, Mourits MJ, de Bock GH, Gaarenstroom KN, van Boven HH, Mooij TM, Rookus MA. No efficacy of annual gynaecological screening in BRCA1/2 mutation carriers; an observational follow-up study. Br J Cancer. 2007;96(9):1335–1342. doi: 10.1038/sj.bjc.6603725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olivier RI, Lubsen-Brandsma MA, Verhoef S, van Beurden M. CA125 and transvaginal ultrasound monitoring in high-risk women cannot prevent the diagnosis of advanced ovarian cancer. Gynecol Oncol. 2006;100(1):20–26. doi: 10.1016/j.ygyno.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 74.Finch A, Beiner M, Lubinski J, Lynch HT, Moller P, Rosen B, Murphy J, Ghadirian P, Friedman E, Foulkes WD, Kim-Sing C, Wagner T, Tung N, Couch F, Stoppa-Lyonnet D, Ainsworth P, Daly M, Pasini B, Gershoni-Baruch R, Eng C, Olopade OI, McLennan J, Karlan B, Weitzel J, Sun P, Narod SA Hereditary Ovarian Cancer Clinical Study G. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 Mutation. JAMA. 2006;296(2):185–192. doi: 10.1001/jama.296.2.185. [DOI] [PubMed] [Google Scholar]
- 75.Eisen A, Lubinski J, Klijn J, Moller P, Lynch HT, Offit K, Weber B, Rebbeck T, Neuhausen SL, Ghadirian P, Foulkes WD, Gershoni-Baruch R, Friedman E, Rennert G, Wagner T, Isaacs C, Kim-Sing C, Ainsworth P, Sun P, Narod SA. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J Clin Oncol. 2005;23(30):7491–7496. doi: 10.1200/JCO.2004.00.7138. [DOI] [PubMed] [Google Scholar]
- 76.Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, Garber JE, Neuhausen SL, Matloff E, Eeles R, Pichert G, Van t’veer L, Tung N, Weitzel JN, Couch FJ, Rubinstein WS, Ganz PA, Daly MB, Olopade OI, Tomlinson G, Schildkraut J, Blum JL, Rebbeck TR. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304(9):967–975. doi: 10.1001/jama.2010.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Offit K. Personalized medicine: new genomics, old lessons. Hum Genet. 2011;130(1):3–14. doi: 10.1007/s00439-011-1028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.King MC, Wieand S, Hale K, Lee M, Walsh T, Owens K, Tait J, Ford L, Dunn BK, Costantino J, Wickerham L, Wolmark N, Fisher B, Bowel P National Surgical Adjuvant B. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: national Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA. 2001;286(18):2251–2256. doi: 10.1001/jama.286.18.2251. [DOI] [PubMed] [Google Scholar]
- 79.Phillips KA, Milne RL, Rookus MA, Daly MB, Antoniou AC, Peock S, Frost D, Easton DF, Ellis S, Friedlander ML, Buys SS, Andrieu N, Nogues C, Stoppa-Lyonnet D, Bonadona V, Pujol P, McLachlan SA, John EM, Hooning MJ, Seynaeve C, Tollenaar RA, Goldgar DE, Terry MB, Caldes T, Weideman PC, Andrulis IL, Singer CF, Birch K, Simard J, Southey MC, Olsson HL, Jakubowska A, Olah E, Gerdes AM, Foretova L, Hopper JL. Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2013;31(25):3091–3099. doi: 10.1200/JCO.2012.47.8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF, Raffoul W, Fiche M, Dougall W, Schneider P, Yalcin-Ozuysal O, Brisken C. Progesterone/RANKL is a major regulatory axis in the human breast. Science Transl Med. 2013;5(182):182ra155. doi: 10.1126/scitranslmed.3005654. [DOI] [PubMed] [Google Scholar]
- 81.Narod SA, Brunet JS, Ghadirian P, Robson M, Heimdal K, Neuhausen SL, Stoppa-Lyonnet D, Lerman C, Pasini B, de los Rios P, Weber B, Lynch H Hereditary Breast Cancer Clinical Study G. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet. 2000;356(9245):1876–1881. doi: 10.1016/s0140-6736(00)03258-x. [DOI] [PubMed] [Google Scholar]
- 82.Powles T, Eeles R, Ashley S, Easton D, Chang J, Dowsett M, Tidy A, Viggers J, Davey J. Interim analysis of the incidence of breast cancer in the Royal Marsden Hospital tamoxifen randomised chemoprevention trial. Lancet. 1998;352(9122):98–101. doi: 10.1016/S0140-6736(98)85012-5. [DOI] [PubMed] [Google Scholar]
- 83.Veronesi U, Maisonneuve P, Costa A, Sacchini V, Maltoni C, Robertson C, Rotmensz N, Boyle P. Prevention of breast cancer with tamoxifen: preliminary findings from the Italian randomised trial among hysterectomised women. Italian Tamoxifen Prevention Study. Lancet. 1998;352(9122):93–97. doi: 10.1016/s0140-6736(98)85011-3. [DOI] [PubMed] [Google Scholar]
- 84.Bourne TH, Campbell S, Reynolds K, Hampson J, Bhatt L, Crayford TJ, Whitehead MI, Collins WP. The potential role of serum CA 125 in an ultrasound-based screening program for familial ovarian cancer. Gynecol Oncol. 1994;52(3):379–385. doi: 10.1006/gyno.1994.1065. [DOI] [PubMed] [Google Scholar]
- 85.Bourne TH, Campbell S, Reynolds KM, Whitehead MI, Hampson J, Royston P, Crayford TJ, Collins WP. Screening for early familial ovarian cancer with transvaginal ultrasonography and colour blood flow imaging. BMJ. 1993;306(6884):1025–1029. doi: 10.1136/bmj.306.6884.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bourne TH, Reynolds K, Campbell S. Ovarian cancer screening. Curr Opin Radiol. 1991;3(2):216–224. [PubMed] [Google Scholar]
- 87.Kauff ND, Domchek SM, Friebel TM, Robson ME, Lee J, Garber JE, Isaacs C, Evans DG, Lynch H, Eeles RA, Neuhausen SL, Daly MB, Matloff E, Blum JL, Sabbatini P, Barakat RR, Hudis C, Norton L, Offit K, Rebbeck TR. Risk-reducing salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated breast and gynecologic cancer: a multicenter, prospective study. J Clin Oncol. 2008;26(8):1331–1337. doi: 10.1200/JCO.2007.13.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lynch HT, Albano WA, Lynch JF, Lynch PM, Campbell A. Surveillance and management of patients at high genetic risk for ovarian carcinoma. Obstet Gynecol. 1982;59(5):589–596. [PubMed] [Google Scholar]
- 89.Robson ME. Should the presence of germline BRCA1/2 mutations influence treatment selection in breast cancer? J Clin Oncol. 2011;29(28):3724–3726. doi: 10.1200/JCO.2011.37.2540. [DOI] [PubMed] [Google Scholar]
- 90.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 91.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 92.De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The Elephant and the Blind Men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol. 2013;3:228. doi: 10.3389/fonc.2013.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Curtin N. PARP inhibitors for anticancer therapy. Biochem Soc Trans. 2014;42(1):82–88. doi: 10.1042/BST20130187. [DOI] [PubMed] [Google Scholar]
- 94.Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, Klijn C, van der Heijden I, van der Gulden H, Wientjens E, Pieterse M, Catteau A, Green P, Solomon E, Morris JR, Jonkers J. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell. 2011;20(6):797–809. doi: 10.1016/j.ccr.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 95.Rosen EM, Pishvaian MJ. Targeting the BRCA1/2 tumor suppressors. Curr Drug Targets. 2014;15(1):17–31. doi: 10.2174/1389450114666140106095432. [DOI] [PubMed] [Google Scholar]
- 96.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 97.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 98.Hisada M, Garber JE, Fung CY, Fraumeni JF, Jr, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–611. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 99.Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, Druker H, Novokmet A, Finlay J, Malkin D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12(6):559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 100.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392(6674):402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 101.Pharoah PD, Guilford P, Caldas C International Gastric Cancer Linkage C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121(6):1348–1353. doi: 10.1053/gast.2001.29611. [DOI] [PubMed] [Google Scholar]
- 102.Brooks-Wilson AR, Kaurah P, Suriano G, Leach S, Senz J, Grehan N, Butterfield YS, Jeyes J, Schinas J, Bacani J, Kelsey M, Ferreira P, MacGillivray B, MacLeod P, Micek M, Ford J, Foulkes W, Australie K, Greenberg C, LaPointe M, Gilpin C, Nikkel S, Gilchrist D, Hughes R, Jackson CE, Monaghan KG, Oliveira MJ, Seruca R, Gallinger S, Caldas C, Huntsman D. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet. 2004;41(7):508–517. doi: 10.1136/jmg.2004.018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suriano G, Yew S, Ferreira P, Senz J, Kaurah P, Ford JM, Longacre TA, Norton JA, Chun N, Young S, Oliveira MJ, Macgillivray B, Rao A, Sears D, Jackson CE, Boyd J, Yee C, Deters C, Pai GS, Hammond LS, McGivern BJ, Medgyesy D, Sartz D, Arun B, Oelschlager BK, Upton MP, Neufeld-Kaiser W, Silva OE, Donenberg TR, Kooby DA, Sharma S, Jonsson BA, Gronberg H, Gallinger S, Seruca R, Lynch H, Huntsman DG. Characterization of a recurrent germ line mutation of the E-cadherin gene: implications for genetic testing and clinical management. Clin Cancer Res. 2005;11(15):5401–5409. doi: 10.1158/1078-0432.CCR-05-0247. [DOI] [PubMed] [Google Scholar]
- 104.Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, Nikkel S, Connolly-Wilson M, Weissman S, Rubinstein WS, Sebold C, Greenstein R, Stroop J, Yim D, Panzini B, McKinnon W, Greenblatt M, Wirtzfeld D, Fontaine D, Coit D, Yoon S, Chung D, Lauwers G, Pizzuti A, Vaccaro C, Redal MA, Oliveira C, Tischkowitz M, Olschwang S, Gallinger S, Lynch H, Green J, Ford J, Pharoah P, Fernandez B, Huntsman D. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297(21):2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 105.Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, Bodurtha J, Crowe C, Curtis MA, Dasouki M, Dunn T, Feit H, Geraghty MT, Graham JM, Jr, Hodgson SV, Hunter A, Korf BR, Manchester D, Miesfeldt S, Murday VA, Nathanson KL, Parisi M, Pober B, Romano C, Eng C, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 106.Stratton MR. Recent advances in understanding of genetic susceptibility to breast cancer. Hum Mol Genet. 1996;5:1515–1519. doi: 10.1093/hmg/5.supplement_1.1515. [DOI] [PubMed] [Google Scholar]
- 107.Manegold BC, Bussmann JF, Furstenberg HS. Clinical contribution to the Peutz-Jeghers syndrome with involvement of the gastrointestinal tract, the upper respiratory tract and both breasts. Med Welt. 1969;25:1435–1439. [PubMed] [Google Scholar]
- 108.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37(11):828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amos CI, Keitheri-Cheteri MB, Sabripour M, Wei C, McGarrity TJ, Seldin MF, Nations L, Lynch PM, Fidder HH, Friedman E, Frazier ML. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41(5):327–333. doi: 10.1136/jmg.2003.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boardman LA, Couch FJ, Burgart LJ, Schwartz D, Berry R, McDonnell SK, Schaid DJ, Hartmann LC, Schroeder JJ, Stratakis CA, Thibodeau SN. Genetic heterogeneity in Peutz-Jeghers syndrome. Hum Mutat. 2000;16(1):23–30. doi: 10.1002/1098-1004(200007)16:1<23::AID-HUMU5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 111.Boardman LA, Pittelkow MR, Couch FJ, Schaid DJ, McDonnell SK, Burgart LJ, Ahlquist DA, Carney JA, Schwartz DI, Thibodeau SN, Hartmann LC. Association of Peutz-Jeghers-like mucocutaneous pigmentation with breast and gynecologic carcinomas in women. Medicine. 2000;79(5):293–298. doi: 10.1097/00005792-200009000-00002. [DOI] [PubMed] [Google Scholar]
- 112.Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, Ahlquist DA, Podratz KC, Pittelkow M, Hartmann LC. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998;128(11):896–899. doi: 10.7326/0003-4819-128-11-199806010-00004. [DOI] [PubMed] [Google Scholar]
- 113.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, Trimbath JD, Giardiello FM, Gruber SB, Offerhaus GJ, de Rooij FW, Wilson JH, Hansmann A, Moslein G, Royer-Pokora B, Vogel T, Phillips RK, Spigelman AD, Houlston RS. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 114.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258–1264. doi: 10.1038/ajg.2009.725. authorreply1265. [DOI] [PubMed] [Google Scholar]
- 115.Lee JS, Collins KM, Brown AL, Lee CH, Chung JH. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000;404(6774):201–204. doi: 10.1038/35004614. [DOI] [PubMed] [Google Scholar]
- 116.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286(5449):2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 117.Meijers-Heijboer H, Wijnen J, Vasen H, Wasielewski M, Wagner A, Hollestelle A, Elstrodt F, van den Bos R, de Snoo A, Fat GT, Brekelmans C, Jagmohan S, Franken P, Verkuijlen P, van den Ouweland A, Chapman P, Tops C, Moslein G, Burn J, Lynch H, Klijn J, Fodde R, Schutte M. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am J Hum Genet. 2003;72(5):1308–1314. doi: 10.1086/375121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schutte M, Seal S, Barfoot R, Meijers-Heijboer H, Wasielewski M, Evans DG, Eccles D, Meijers C, Lohman F, Klijn J, van den Ouweland A, Futreal PA, Nathanson KL, Weber BL, Easton DF, Stratton MR, Rahman N Breast Cancer Linkage C. Variants in CHEK2 other than 1100delC do not make a major contribution to breast cancer susceptibility. Am J Hum Genet. 2003;72(4):1023–1028. doi: 10.1086/373965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25(43):5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- 120.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N Breast Cancer Susceptibility C. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 121.Bernstein JL, Bernstein L, Thompson WD, Lynch CF, Malone KE, Teitelbaum SL, Olsen JH, Anton-Culver H, Boice JD, Rosenstein BS, Borresen-Dale AL, Gatti RA, Concannon P, Haile RW, Group WSC. ATM variants 7271T > G and IVS10-6T > G among women with unilateral and bilateral breast cancer. Br J Cancer. 2003;89(8):1513–1516. doi: 10.1038/sj.bjc.6601289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meyer A, Wilhelm B, Dork T, Bremer M, Baumann R, Karstens JH, Machtens S. ATM missense variant P1054R predisposes to prostate cancer. Radiother Oncol (Journal of the European Society for Therapeutic Radiology and Oncology) 2007;83(3):283–288. doi: 10.1016/j.radonc.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 123.Goldgar DE, Healey S, Dowty JG, Silva LD, Chen X, Spurdle AB, Terry MB, Daly MJ, Buys SM, Southey MC, Andrulis I, John EM, BCFR, Khanna KK, Hopper JL, Oefner PJ, Lakhani S, Chenevix-Trench G kConFab. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13(4):R73. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stredrick DL, Garcia-Closas M, Pineda MA, Bhatti P, Alexander BH, Doody MM, Lissowska J, Peplonska B, Brinton LA, Chanock SJ, Struewing JP, Sigurdson AJ. The ATM missense mutation p.Ser49Cys (c.146C > G) and the risk of breast cancer. Hum Mutat. 2006;27(6):538–544. doi: 10.1002/humu.20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987;316(21):1289–1294. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 126.Thompson D, Antoniou AC, Jenkins M, Marsh A, Chen X, Wayne T, Tesoriero A, Milne R, Spurdle A, Thorstenson Y, Southey M, Giles GG, Khanna KK, Sambrook J, Oefner P, Goldgar D, Hopper JL, Easton D, Chenevix-Trench G Investigators KC. Two ATM variants and breast cancer risk. Hum Mutat. 2005;25(6):594–595. doi: 10.1002/humu.9344. [DOI] [PubMed] [Google Scholar]
- 127.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 128.Angele S, Falconer A, Edwards SM, Dork T, Bremer M, Moullan N, Chapot B, Muir K, Houlston R, Norman AR, Bullock S, Hope Q, Meitz J, Dearnaley D, Dowe A, Southgate C, Ardern-Jones A, Easton DF, Eeles RA, Hall J. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004;91(4):783–787. doi: 10.1038/sj.bjc.6602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dombernowsky SL, Weischer M, Allin KH, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG. Risk of cancer by ATM missense mutations in the general population. J Clin Oncol. 2008;26(18):3057–3062. doi: 10.1200/JCO.2007.14.6613. [DOI] [PubMed] [Google Scholar]
- 130.Gutierrez-Enriquez S, Fernet M, Dork T, Bremer M, Lauge A, Stoppa-Lyonnet D, Moullan N, Angele S, Hall J. Functional consequences of ATM sequence variants for chromosomal radiosensitivity. Genes Chromosomes Cancer. 2004;40(2):109–119. doi: 10.1002/gcc.20025. [DOI] [PubMed] [Google Scholar]
- 131.Mao JH, Wu D, DelRosario R, Castellanos A, Balmain A, Perez-Losada J. Atm heterozygosity does not increase tumor susceptibility to ionizing radiation alone or in a p53 heterozygous background. Oncogene. 2008;27(51):6596–6600. doi: 10.1038/onc.2008.280. [DOI] [PubMed] [Google Scholar]
- 132.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR Breast Cancer Susceptibility C. PALB2, which encodes a BR-CA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39(2):165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, Darnel A, Royer R, Poll A, Fafard E, Robidoux A, Martin G, Bismar TA, Tischkowitz M, Rousseau F, Narod SA. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9(6):R83. doi: 10.1186/bcr1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Antoniou AC, Foulkes WD, Tischkowitz M. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(17):1651–1652. doi: 10.1056/NEJMc1410673. [DOI] [PubMed] [Google Scholar]
- 135.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N Breast Cancer Susceptibility C. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38(11):1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 136.Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew CG, Schindler D, Schmutzler RK, Hanenberg H. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 137.Pelttari LM, Heikkinen T, Thompson D, Kallioniemi A, Schleutker J, Holli K, Blomqvist C, Aittomaki K, Butzow R, Nevanlinna H. RAD51C is a susceptibility gene for ovarian cancer. Hum Mol Genet. 2011;20(16):3278–3288. doi: 10.1093/hmg/ddr229. [DOI] [PubMed] [Google Scholar]
- 138.Thompson ER, Boyle SE, Johnson J, Ryland GL, Sawyer S, Choong DY, Chenevix-Trench G, Trainer AH, Lindeman GJ, Mitchell G, James PA, Campbell IG kConFab. Analysis of RAD51C germline mutations in high-risk breast and ovarian cancer families and ovarian cancer patients. Hum Mutat. 2012;33(1):95–99. doi: 10.1002/humu.21625. [DOI] [PubMed] [Google Scholar]
- 139.Loveday C, Turnbull C, Ruark E, Xicola RM, Ramsay E, Hughes D, Warren-Perry M, Snape K, Eccles D, Evans DG, Gore M, Renwick A, Seal S, Antoniou AC, Rahman N Breast Cancer Susceptibility C. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat Genet. 2012;44(5):475–476. doi: 10.1038/ng.2224. authorreply476. [DOI] [PubMed] [Google Scholar]
- 140.Karppinen SM, Heikkinen K, Rapakko K, Winqvist R. Mutation screening of the BARD1 gene: evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J Med Genet. 2004;41(9):e114. doi: 10.1136/jmg.2004.020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maxwell KN, Nathanson KL. Common breast cancer risk variants in the post-COGS era: a comprehensive review. Breast Cancer Res. 2013;15(6):212. doi: 10.1186/bcr3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, Orr N, Rhie SK, Riboli E, Feigelson HS, Le Marchand L, Buring JE, Eccles D, Miron P, Fasching PA, Brauch H, Chang-Claude J, Carpenter J, Godwin AK, Nevanlinna H, Giles GG, Cox A, Hopper JL, Bolla MK, Wang Q, Dennis J, Dicks E, Howat WJ, Schoof N, Bojesen SE, Lambrechts D, Broeks A, Andrulis IL, Guenel P, Burwinkel B, Sawyer EJ, Hollestelle A, Fletcher O, Winqvist R, Brenner H, Mannermaa A, Hamann U, Meindl A, Lindblom A, Zheng W, Devillee P, Goldberg MS, Lubinski J, Kristensen V, Swerdlow A, Anton-Culver H, Dork T, Muir K, Matsuo K, Wu AH, Radice P, Teo SH, Shu XO, Blot W, Kang D, Hartman M, Sangrajrang S, Shen CY, Southey MC, Park DJ, Hammet F, Stone J, Veer LJ, Rutgers EJ, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Peto J, Schrauder MG, Ekici AB, Beckmann MW, Dos Santos Silva I, Johnson N, Warren H, Tomlinson I, Kerin MJ, Miller N, Marme F, Schneeweiss A, Sohn C, Truong T, Laurent-Puig P, Kerbrat P, Nordestgaard BG, Nielsen SF, Flyger H, Milne RL, Perez JI, Menendez P, Muller H, Arndt V, Stegmaier C, Lichtner P, Lochmann M, Justenhoven C, Ko YD, Gene EI, Breast CN, Muranen TA, Aittomaki K, Blomqvist C, Greco D, Heikkinen T, Ito H, Iwata H, Yatabe Y, Antonenkova NN, Margolin S, Kataja V, Kosma VM, Hartikainen JM, Balleine R, Tseng CC, Berg DV, Stram DO, Neven P, Dieudonne AS, Leunen K, Rudolph A, Nickels S, Flesch-Janys D, Peterlongo P, Peissel B, Bernard L, Olson JE, Wang X, Stevens K, Severi G, Baglietto L, McLean C, Coetzee GA, Feng Y, Henderson BE, Schumacher F, Bogdanova NV, Labreche F, Dumont M, Yip CH, Taib NA, Cheng CY, Shrubsole M, Long J, Pylkas K, Jukkola-Vuorinen A, Kauppila S, Knight JA, Glendon G, Mulligan AM, Tollenaar RA, Seynaeve CM, Kriege M, Hooning MJ, van den Ouweland AM, van Deurzen CH, Lu W, Gao YT, Cai H, Balasubramanian SP, Cross SS, Reed MW, Signorello L, Cai Q, Shah M, Miao H, Chan CW, Chia KS, Jakubowska A, Jaworska K, Durda K, Hsiung CN, Wu PE, Yu JC, Ashworth A, Jones M, Tessier DC, Gonzalez-Neira A, Pita G, Alonso MR, Vincent D, Bacot F, Ambrosone CB, Bandera EV, John EM, Chen GK, Hu JJ, Rodriguez-Gil JL, Bernstein L, Press MF, Ziegler RG, Millikan RM, Deming-Halverson SL, Nyante S, Ingles SA, Waisfisz Q, Tsimiklis H, Makalic E, Schmidt D, Bui M, Gibson L, Muller-Myhsok B, Schmutzler RK, Hein R, Dahmen N, Beckmann L, Aaltonen K, Czene K, Irwanto A, Liu J, Turnbull C, Rahman N, Meijers-Heijboer H, Uitterlinden AG, Rivadeneira F, Olswold C, Slager S, Pilarski R, Ademuyiwa F, Konstantopoulou I, Martin NG, Montgomery GW, Slamon DJ, Rauh C, Lux MP, Jud SM, Bruning T, Weaver J, Sharma P, Pathak H, Tapper W, Gerty S, Durcan L, Trichopoulos D, Tumino R, Peeters PH, Kaaks R, Campa D, Canzian F, Weiderpass E, Johansson M, Khaw KT, Travis R, Clavel-Chapelon F, Kolonel LN, Chen C, Beck A, Hankinson SE, Berg CD, Hoover RN, Lissowska J, Figueroa JD, Chasman DI, Gaudet MM, Diver WR, Willett WC, Hunter DJ, Simard J, Benitez J, Dunning AM, Sherman ME, Chenevix-Trench G, Chanock SJ, Hall P, Pharoah PD, Vachon C, Easton DF, Haiman CA, Kraft P kConFab I, Familial Breast Cancer S, Australian Breast Cancer Tissue Bank I. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45(4):392–398. 398e391–392. doi: 10.1038/ng.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stevens KN, Fredericksen Z, Vachon CM, Wang X, Margolin S, Lindblom A, Nevanlinna H, Greco D, Aittomaki K, Blomqvist C, Chang-Claude J, Vrieling A, Flesch-Janys D, Sinn HP, Wang-Gohrke S, Nickels S, Brauch H, Network G, Ko YD, Fischer HP, Schmutzler RK, Meindl A, Bartram CR, Schott S, Engel C, Godwin AK, Weaver J, Pathak HB, Sharma P, Brenner H, Muller H, Arndt V, Stegmaier C, Miron P, Yannoukakos D, Stavropoulou A, Fountzilas G, Gogas HJ, Swann R, Dwek M, Perkins A, Milne RL, Benitez J, Zamora MP, Perez JI, Bojesen SE, Nielsen SF, Nordestgaard BG, Flyger H, Guenel P, Truong T, Menegaux F, Cordina-Duverger E, Burwinkel B, Marme F, Schneeweiss A, Sohn C, Sawyer E, Tomlinson I, Kerin MJ, Peto J, Johnson N, Fletcher O, Dos Santos Silva I, Fasching PA, Beckmann MW, Hartmann A, Ekici AB, Lophatananon A, Muir K, Puttawibul P, Wiangnon S, Schmidt MK, Broeks A, Braaf LM, Rosenberg EH, Hopper JL, Apicella C, Park DJ, Southey MC, Swerdlow AJ, Ashworth A, Orr N, Schoemaker MJ, Anton-Culver H, Ziogas A, Bernstein L, Dur CC, Shen CY, Yu JC, Hsu HM, Hsiung CN, Hamann U, Dunnebier T, Rudiger T, Ulmer HU, Pharoah PP, Dunning AM, Humphreys MK, Wang Q, Cox A, Cross SS, Reed MW, Hall P, Czene K, Ambrosone CB, Ademuyiwa F, Hwang H, Eccles DM, Garcia-Closas M, Figueroa JD, Sherman ME, Lissowska J, Devilee P, Seynaeve C, Tollenaar RA, Hooning MJ, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, John EM, Miron A, Alnaes GG, Kristensen V, Borresen-Dale AL, Giles GG, Baglietto L, McLean CA, Severi G, Kosel ML, Pankratz VS, Slager S, Olson JE, Radice P, Peterlongo P, Manoukian S, Barile M, Lambrechts D, Hatse S, Dieudonne AS, Christiaens MR, Chenevix-Trench G, Group A, Beesley J, Chen X, Mannermaa A, Kosma VM, Hartikainen JM, Soini Y, Easton DF, Couch FJ kConFab I. 19p13.1 is a triple-negative-specific breast cancer susceptibility locus. Cancer Res. 2012;72(7):1795–1803. doi: 10.1158/0008-5472.CAN-11-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC, Wang X, Ademuyiwa F, Ahmed S, Ambrosone CB, Baglietto L, Balleine R, Bandera EV, Beckmann MW, Berg CD, Bernstein L, Blomqvist C, Blot WJ, Brauch H, Buring JE, Carey LA, Carpenter JE, Chang-Claude J, Chanock SJ, Chasman DI, Clarke CL, Cox A, Cross SS, Deming SL, Diasio RB, Dimopoulos AM, Driver WR, Dunnebier T, Durcan L, Eccles D, Edlund CK, Ekici AB, Fasching PA, Feigelson HS, Flesch-Janys D, Fostira F, Forsti A, Fountzilas G, Gerty SM, Giles GG, Godwin AK, Goodfellow P, Graham N, Greco D, Hamann U, Hankinson SE, Hartmann A, Hein R, Heinz J, Holbrook A, Hoover RN, Hu JJ, Hunter DJ, Ingles SA, Irwanto A, Ivanovich J, John EM, Johnson N, Jukkola-Vuorinen A, Kaaks R, Ko YD, Kolonel LN, Konstantopoulou I, Kosma VM, Kulkarni S, Lambrechts D, Lee AM, Marchand LL, Lesnick T, Liu J, Lindstrom S, Mannermaa A, Margolin S, Martin NG, Miron P, Montgomery GW, Nevanlinna H, Nickels S, Nyante S, Olswold C, Palmer J, Pathak H, Pectasides D, Perou CM, Peto J, Pharoah PD, Pooler LC, Press MF, Pylkas K, Rebbeck TR, Rodriguez-Gil JL, Rosenberg L, Ross E, Rudiger T, Silva Idos S, Sawyer E, Schmidt MK, Schulz-Wendtland R, Schumacher F, Severi G, Sheng X, Signorello LB, Sinn HP, Stevens KN, Southey MC, Tapper WJ, Tomlinson I, Hogervorst FB, Wauters E, Weaver J, Wildiers H, Winqvist R, Van Den Berg D, Wan P, Xia LY, Yannoukakos D, Zheng W, Ziegler RG, Siddiq A, Slager SL, Stram DO, Easton D, Kraft P, Henderson BE, Couch FJ Gene Environment I, Breast Cancer in Germany C. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat Genet. 2011;43(12):1210–1214. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Purrington KS, Slager S, Eccles D, Yannoukakos D, Fasching PA, Miron P, Carpenter J, Chang-Claude J, Martin NG, Montgomery GW, Kristensen V, Anton-Culver H, Goodfellow P, Tapper WJ, Rafiq S, Gerty SM, Durcan L, Konstantopoulou I, Fostira F, Vratimos A, Apostolou P, Konstanta I, Kotoula V, Lakis S, Dimopoulos MA, Skarlos D, Pectasides D, Fountzilas G, Beckmann MW, Hein A, Ruebner M, Ekici AB, Hartmann A, Schulz-Wendtland R, Renner SP, Janni W, Rack B, Scholz C, Neugebauer J, Andergassen U, Lux MP, Haeberle L, Clarke C, Pathmanathan N, Rudolph A, Flesch-Janys D, Nickels S, Olson JE, Ingle JN, Olswold C, Slettedahl S, Eckel-Passow JE, Anderson SK, Visscher DW, Cafourek VL, Sicotte H, Prodduturi N, Weiderpass E, Bernstein L, Ziogas A, Ivanovich J, Giles GG, Baglietto L, Southey M, Kosma VM, Fischer HP, Network G, Reed MW, Cross SS, Deming-Halverson S, Shrubsole M, Cai Q, Shu XO, Daly M, Weaver J, Ross E, Klemp J, Sharma P, Torres D, Rudiger T, Wolfing H, Ulmer HU, Forsti A, Khoury T, Kumar S, Pilarski R, Shapiro CL, Greco D, Heikkila P, Aittomaki K, Blomqvist C, Irwanto A, Liu J, Pankratz VS, Wang X, Severi G, Mannermaa A, Easton D, Hall P, Brauch H, Cox A, Zheng W, Godwin AK, Hamann U, Ambrosone C, Toland AE, Nevanlinna H, Vachon CM, Couch FJ. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014;35(5):1012–1019. doi: 10.1093/carcin/bgt404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S, Khan S, Maranian MJ, O’Reilly M, Hillman KM, Betts JA, Carroll T, Bailey PJ, Dicks E, Beesley J, Tyrer J, Maia AT, Beck A, Knoblauch NW, Chen C, Kraft P, Barnes D, Gonzalez-Neira A, Alonso MR, Herrero D, Tessier DC, Vincent D, Bacot F, Luccarini C, Baynes C, Conroy D, Dennis J, Bolla MK, Wang Q, Hopper JL, Southey MC, Schmidt MK, Broeks A, Verhoef S, Cornelissen S, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Fasching PA, Loehberg CR, Ekici AB, Beckmann MW, Peto J, dos Santos Silva I, Johnson N, Aitken Z, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Bojesen SE, Nordestgaard BG, Nielsen SF, Flyger H, Milne RL, Zamora MP, Arias Perez JI, Benitez J, Anton-Culver H, Brenner H, Muller H, Arndt V, Stegmaier C, Meindl A, Lichtner P, Schmutzler RK, Engel C, Brauch H, Hamann U, Justenhoven C, Network G, Aaltonen K, Heikkila P, Aittomaki K, Blomqvist C, Matsuo K, Ito H, Iwata H, Sueta A, Bogdanova NV, Antonenkova NN, Dork T, Lindblom A, Margolin S, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Lambrechts D, Peeters S, Smeets A, Floris G, Chang-Claude J, Rudolph A, Nickels S, Flesch-Janys D, Radice P, Peterlongo P, Bonanni B, Sardella D, Couch FJ, Wang X, Pankratz VS, Lee A, Giles GG, Severi G, Baglietto L, Haiman CA, Henderson BE, Schumacher F, Le Marchand L, Simard J, Goldberg MS, Labreche F, Dumont M, Teo SH, Yip CH, Ng CH, Vithana EN, Kristensen V, Zheng W, Deming-Halverson S, Shrubsole M, Long J, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Devilee P, Seynaeve C, Garcia-Closas M, Figueroa J, Chanock SJ, Lissowska J, Czene K, Klevebring D, Schoof N, Hooning MJ, Martens JW, Collee JM, Tilanus-Linthorst M, Hall P, Li J, Liu J, Humphreys K, Shu XO, Lu W, Gao YT, Cai H, Cox A, Balasubramanian SP, Blot W, Signorello LB, Cai Q, Pharoah PD, Healey CS, Shah M, Pooley KA, Kang D, Yoo KY, Noh DY, Hartman M, Miao H, Sng JH, Sim X, Jakubowska A, Lubinski J, Jaworska-Bieniek K, Durda K, Sangrajrang S, Gaborieau V, McKay J, Toland AE, Ambrosone CB, Yannoukakos D, Godwin AK, Shen CY, Hsiung CN, Wu PE, Chen ST, Swerdlow A, Ashworth A, Orr N, Schoemaker MJ, Ponder BA, Nevanlinna H, Brown MA, Chenevix-Trench G, Easton DF, Dunning AM kConFab I. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet. 2013;92(4):489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, Thun MJ, Cox DG, Hankinson SE, Kraft P, Rosner B, Berg CD, Brinton LA, Lissowska J, Sherman ME, Chlebowski R, Kooperberg C, Jackson RD, Buckman DW, Hui P, Pfeiffer R, Jacobs KB, Thomas GD, Hoover RN, Gail MH, Chanock SJ, Hunter DJ. Performance of common genetic variants in breast-cancer risk models. N Engl J Med. 2010;362(11):986–993. doi: 10.1056/NEJMoa0907727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sawyer S, Mitchell G, McKinley J, Chenevix-Trench G, Beesley J, Chen XQ, Bowtell D, Trainer AH, Harris M, Lindeman GJ, James PA. A role for common genomic variants in the assessment of familial breast cancer. J Clin Oncol. 2012;30(35):4330–4336. doi: 10.1200/JCO.2012.41.7469. [DOI] [PubMed] [Google Scholar]