

Abstract
The combination of atom transfer radical polymerization (ATRP) and click chemistry has created unprecedented opportunities for controlled syntheses of functional polymers. ATRP of azido-bearing methacrylate monomers (e.g. 2-(2-(2-azidoethyoxy)ethoxy)ethyl methacrylate, AzTEGMA), however, proceeded with poor control at commonly adopted temperature of 50 °C, resulting in significant side reactions. By lowering reaction temperature and monomer concentrations, well-defined pAzTEGMA with significantly reduced polydispersity were prepared within a reasonable timeframe. Upon subsequent functionalization of the side chains of pAzTEGMA via Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry, functional polymers with number-average molecular weights (Mn) up to 22 kDa with narrow polydispersity (PDI < 1.30) were obtained. Applying the optimized polymerization condition, we also grafted pAzTEGMA brushes from Ti6Al4 substrates by surface-initiated ATRP (SI-ATRP), and effectively functionalized the azide-terminated side chains with hydrophobic and hydrophilic alkynes by CuAAC. The well-controlled ATRP of azido-bearing methacrylates and subsequent facile high-density functionalization of the side chains of the polymethacrylates via CuAAC offers a useful tool for engineering functional polymers or surfaces for diverse applications.
Keywords: alloy, atom transfer radical polymerization (ATRP), azido-bearing methacrylate, click chemistry, kinetics (polym.), SI-ATRP
GRAPHICAL ABSTRACT
We demonstrated the poor controllability of ATRP of an azido-bearing methacrylate under commonly adopted conditions, and identified more suitable polymerization temperature and monomer concentration to optimize the polymerization outcome in solution and on metallic surfaces. The wellcontrolled ATRP of azido-bearing methacrylates provides high-quality precursor polymers for subsequent high-density click functionalization of the azido-bearing side chains for macromolecular engineering and surface functionalization.
INTRODUCTION
Click chemistry, such as Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC),1,2 Diels-Alder reaction,3 thiol-ene coupling,4–6 and strain-promoted azide-alkyne cycloaddition (SPAAC),7 has gained significant attention within the polymer chemistry/material sciences community for tethering polymer segments (end group coupling), functionalizing polymer side chains, conjugating drugs to polymers, or encapsulating biological cargos within in situ formed polymeric hydrogels.8–13 Among these click reactions, CuAAC possesses the advantages of high orthogonality (e.g. compared to thiol-ene coupling) and relatively low reagent cost (e.g. compared to SPAAC).2,8
CuAAC has been combined with atom transfer radical polymerization (ATRP), known for excellent control over the molecular weight distributions of the polymer,14,15 for fabricating a wide range of well-controlled polymeric architectures and functional materials.10,16–20 CuAAC and ATRP can share the same catalyst systems (e.g. Cu(I)/ligand), making it possible to carry out the polymerization and subsequent click conjugation in one pot without to the need for isolation of azide/alkyne-containing precursor polymers.21–23
Whereas end-group CuAAC of azide/alkyne-terminated polymers prepared by ATRP,17,24–29 after substituting the terminal halide originated from the ATRP initiator with azide,30,31 can be extended to covalently conjugate drugs, imaging probes or biomolecules of interest, the functional density introduced is limited by one copy per polymer. For high-density functionalization of polymers, combining ATRP of azido-bearing monomers with subsequent CuAAC functionalization of pendant side chains, as first demonstrated by Matyjaszewski et al.,32 is more appealing. For instance, ATRP of 3-azidopropyl methacrylate (AzPMA) followed by CuAAC coupling32 was shown to result in high conversion yield (> 95 %). The combination of facile surface-initiated ATRP (SI-ATRP) of azido-bearing methacrylates and CuAAC (often one-pot) has also been applied for surface functionalization of substrates ranging from polymers, silicon wafers, gold foils to magnetic nanoparticles.19,30,33–37 However, due to the instability of azides (e.g. prone to thermal decomposition) under the high temperature typically applied to ATRP (e.g. 50 °C or higher), ATRP of AzPMA and subsequent CuAAC tended to result in polymers with relative broad molecular weight distributions (e.g. PDI > 1.5).38,39 Similarly, the quality of azide motifs grafted to 2-D surfaces via SI-ATRP, which affects ultimate surface functional group densities enabled by CuAAC, was often overlooked. Overall, optimizing reaction conditions to ensure well-controlled ATRP and SI-ATRP of azido-bearing methacrylate monomers remain a significant interest.
In this study, we first characterized the kinetics of solution ATRP of 2-(2-(2-azidoethyoxy)ethoxy)ethyl methacrylate (AzTEGMA) at 50 °C, a temperature that has been commonly adopted for ATRP of azido-bearing methacrylates, and identified thermal-induced complications contributing to the poorly controlled outcome of the polymerization. We then optimized the reaction conditions to achieve better-controlled ATRP of AzTEGMA in reduced monomer concentrations at or slightly above room temperature, resulting in polymers with higher molecular weight and narrower molecular weight distributions. Implementing the optimized condition, we also grafted pAzTEGMA brushes with varying degrees of polymerization from Ti6Al4V substrates using SI-ATRP. Finally, facile orthogonal functionalization of the azide-terminated side chains of the surface brushes with functional alkynes by CuACC was demonstrated and characterized by water contact angle measurements and x-ray photoelectron spectroscopy (XPS).
EXPERIMENTAL
Materials
Methacryloyl chloride (97 %), 4-methylphenol (99 %), propargyl alcohol (99 %), phenylacetylene (98 %), ethyl α-bromoisobutyrate (EBiB, 98 %), α-bromoisobutyryl bromide (BiBB, 98 %), 2, 2′-bipyridyl (BPY, 97 %), copper(I) bromide (CuBr, 99.999 %), sodium azide (99.5 %), trifluoroethanol (TFE, 99 %), benzene (99.8 %) and dimethylformamide (DMF, 99.5 %) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Diethyl (hydroxymethyl)phosphonate (P-OH, 98 %) and 2-(2-(2-Chloroethyoxy)ethoxy)-ethanol (TEG-Cl, 96 %) were purchased from TCI America (Portland, OR, USA) and used as received. Triethylamine (TEA, 99.5 %, Sigma-Aldrich) was dried by calcium hydride (CaH2, 99.99 %, Sigma-Aldrich) and distilled prior to use. Ti6Al4V plates (1.3 mm thick, Titanium Metal Supply Inc., Poway, CA, USA) was cut into 10 × 10 mm2 square pieces, which were sequentially polished under water with 600, 1500, and 3000 grit silicon carbide sandpapers and ultrasonically cleaned with hexane (10 min), DCM (10 min), and acetone (10 min) prior to use.
Synthesis of 2-(2-(2-Azidoethyoxy)ethoxy)ethanol (AzTEG)
To prepare AzTEG, sodium azide (0.69 mol) was added to a mixture of water (180 mL) and 2-(2-(2-Chloroethyoxy)ethoxy)-ethanol (0.36 mol). The mixture was stirred at 75 °C for 48 h and then cooled to room temperature. Water was evaporated under reduced pressure, and the residue was condensed over NaOH pellets (0.2 g) to trap any HN3 potentially produced. The residue was suspended in ether (300 mL) for filtration and the filtrate was concentrated by rotary evaporation to give the colorless liquid product (yield 90.5 %). 1H NMR (400 MHz, CDCl3, δ): 3.59 (m, 10H; -OCH2-), 3.32 (t, J = 5.06 Hz, 2H; N3CH2-), 2.59 (b, 1H; -OH). 13C NMR (100 MHz, CDCl3, δ): 72.72 (O-CH2-CH2-OH), 70.75, 70.51, 70.15 (-O-CH2-), 61.76 (-CH2-OH), 50.77 (N3-CH2). NMR spectra are shown in Supplementary Figures S2 and S3.
Synthesis of 2-(2-(2-Azidoethyoxy)ethoxy)ethyl methacrylate (AzTEGMA)
To prepare AzTEGMA monomer, AzTEG (40 mmol), TEA (45 mmol) and 4-methylphenol (0.05 g) were added to 80 mL of benzene and cooled to 0 °C in a two-neck round bottom flask by an ice-bath. Methacryloyl chloride (48 mmol) in 20 mL benzene was added drop-wise into the mixture. The reaction was slowly warmed to room temperature under stirring overnight. The resulting mixture was filtered, concentrated and subjected to silica gel flash chromatography (hexane: ethyl acetate/5:1 as eluent). The product fractions were concentrated in vacuum (yield 75.6 %). 1H NMR (CDCl3, 400 MHz, δ): 6.07 (m, 1H; =CH2), 5.52 (m, 1H, =CH2), 4.24 (m, 2H; -CH2-OC=O), 3.70 (m, 2H; -OCH2-CH2-OC=O), 3.61(m, 6H; -OCH2-), 3.32 (t, J = 5.02 Hz, 2H; N3CH2-), 1.89 (m, 3H; -CH3). 13C NMR (CDCl3, 100 MHz, δ): 167.39 (C=O), 136.29 (H2C=C-C=O), 125.80 (=CH2), 70.81, 70.22, 69.32 (-C-O-), 63.98 (-C-O-C=O), 50.78 (N3-C-), 18.41 (-CH3). NMR spectra are shown in Supplementary Figures S4, and S5.
Preparation of poly[2-(2-(2-Azidoethyoxy)ethoxy)ethyl methacrylate] (pAzTEGMA) via ATRP
BPY (0.2 mmol) and TFE (1 mL) were charged into a dry Schlenk flask. After three “freeze-pump-thaw” cycles to remove oxygen, the flask was back filled with argon followed by the addition of CuBr (0.1 mmol) under argon protection. The mixture was stirred until a uniform dark brown catalyst complex was formed. AzTEGMA (10 mmol), EBiB (0.1 mmol) and TFE (1 mL) were charged into another dry Schlenk flask. The flask was then degassed by three “freeze-pump-thaw” cycles, after which the uniform catalyst complex was injected by syringe to start the polymerizations at 50 °C, 23 °C or 34 °C. Small aliquots of the reaction mixture were retrieved at predetermined time points for 1H NMR and GPC monitoring of the polymerization. To terminate the polymerization, the reactor was exposed to air and the reaction solution was diluted by acetone and passed through a pad of silica gel (Alfa Aesar, silica gel 60, mesh 230–400) to remove the deactivated green catalyst complex. Colorless pAzTEGMA polymers were obtained after removing the solvent under reduced pressure.
Monomer conversion calculation
The AzTEGMA monomer conversion (C) was calculated based on real time 1H NMR data according to the following equation:
Where Ip is the integration of the broad proton peak (δ 0.75–1.25 ppm) that belongs to the methyl (−CH3) group on the backbone of the polymer, while Im is the integration of the two proton peaks (δ 5.52, 6.07 ppm) that belongs to the methylene (=CH2) group of the unconsumed monomer.
Functionalization of pAzTEGMA via CuACC
pAzPEGMA polymer (0.608 g) and proparyl alcohol (3.5 mmol) were added into a solution of BPY (1.0 mmol) in dry DMF (5 mL). After three “freeze-pump-thaw” cycles to remove oxygen and back filled with argon, CuBr (0.5 mmol) was added into the flask under argon protection. The resulting mixture was stirred overnight at room temperature before being exposed to air. The reaction mixture was diluted by methanol and passed through silica gel. Colorless pAzTEGMA polymers were obtained upon removing solvent under reduced pressure.
Gel permeation chromatography (GPC)
GPC measurements were performed on a Varian Prostar HPLC system equipped with two 5-mm PLGel MiniMIX-D columns (Polymer Laboratory, Amherst, MA), an UV-vis detector and a PL-ELS2100 evaporative light scattering detector (Polymer Laboratory, Amherst, MA). THF was used as an eluent at a flow rate of 0.3 mL/min at room temperature. The number-average molecular weight (Mn) and the polydispersity index (PDI) of pAzTEGMA were calculated by the Cirrus AIA GPC Software using the narrowly dispersed polystyrenes (ReadyCal kits, PSS Polymer Standards Service Inc. Germany) as the calibration standards.
Surface immobilization of initiator PA-O-Br on Ti6Al4V
Annealed Ti6Al4V substrates were cleaned in an air plasma cleaner (Harrick, PDC-001) for 2 min before being placed in a plastic dish, and submerged under 40 mL of 3-mM anhydrous methanol solution of PA-O-Br (prepared and purified as we previously reported40) at room temperature in dark for 24 h. All retrieved substrates (Ti-Br) were then annealed at 110 °C for 15 min in a vacuum oven to stabilize the bonding of the phosphonic acid group to the thin oxidized metal surface before being subjected to extensive sonication in methanol (10 min each time, twice) and vacuum drying.
SI-ATRP of AzTEGMA from Ti-Br substrates
SI-ATRP of AzTEGMA was conducted in a similar fashion as those described for the solution ATRP except that the freshly formed polymerization mixture was transferred into a flat-bottom reactor containing Ti-Br substrates under argon at room temperature. Upon terminating the nearly simultaneously carried out SI-ATRP and solution ATRP, the pAzTEGMA-grafted Ti6Al4V substrates (e.g. Ti-AzTEGMA-10, where 10 refers to the targeting degree of polymerization/DP of 10) were washed with a copious amount of acetone (30-min stirring each time, 3 times) to remove the free polymers physically absorbed on the surface, and dried under vacuum. pAzTEGMA brushes with different targeting DPs (10 and 100) were grafted from Ti-Br by varying the ratio of monomers relative to initiators accordingly.
Functionalization of the pAzTEGMA brushes on Ti6Al4V substrates by CuAAC
To demonstrate the versatile functionalization of Ti-pAzTEGMA substrates by CuAAC, both hydrophilic proparyl alcohol and hydrophobic phenylacetylene were chosen to be ”clicked” to the azide-terminated pedant chains. Specifically, propargyl alcohol or phenylacetylene (3.5 mmol) was added into a solution of BPY (1.0 mmol) in dry DMF (5 mL). After three “freeze-pump-thaw” cycles to remove oxygen, the flask was back filled with argon before the addition of CuBr (0.5 mmol). The resulting mixture was stirred until forming a uniform dark brown catalyst complex and was then injected into another argon-protected flask containing pAzTEGMA-grafted Ti6Al4V substrates (Ti-pAzTEGMA-100). After stirring the mixture for 17-h at room temperature, the substrates were removed and washed with a copious amount of DMF (30-min stirring each time, 3 times).
XPS analysis
Surface compositional analyses of substrates before and after modification were carried out on a Thermo Scientific K-Alpha XPS equipped with an Al Kα radiation source under the spot size of 400 μm. Survey scan spectra were obtained from two consecutive scans of a randomly chosen area of interest while high resolution scan spectra were obtained from five consecutive scans under the snapshot mode. All binding energies were referenced to the C1s hydrocarbon peak at 285.0 eV.
Water contact angle measurement
The static water contact angles of the substrates before and after surface modifications were recorded on a CAM200 goniometer (KSV Instruments). A droplet (2 μL) of Milli-Q water was placed on the substrate and the contact angles (left and right) of the droplet were recorded after 30 s. The left and right contact angles of each droplet and three substrates of each sample group were averaged, and reported as averages ± standard deviation (SD).
RESULTS AND DISCUSSION
Weak controllability of ATRP of AzTEGMA at 50 °C
Polymers with azido pendants prepared through radical polymerizations are promising candidates for preparing functional materials through orthogonal CuAAC conjugation with alkynes. However, most reported ATRP of azido-bearing monomers, carried out at 50 °C or higher temperatures, were not well-controlled, often resulting in polymers with relatively broad molecular weight distribution (PDI > 1.4).20,32
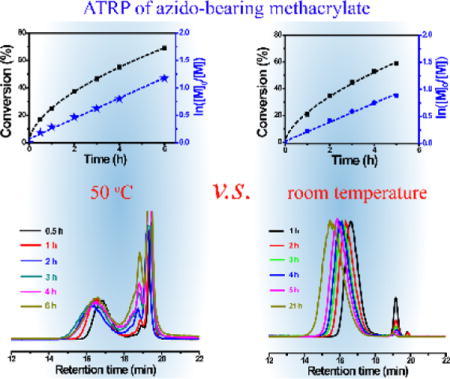
To identify the underlying factors contributing to the weak controllability of the ATRP of azido-bearing monomers, we first investigated the kinetics of ATRP of AzTEGMA at 50 °C in TFE (Table 1, entries 1–3). As shown by the plots of AzTEGMA monomer conversion (black square) and conversion index (blue star) over polymerization time (Fig. 1a), first-order monomer conversion presenting a typical living polymerization was observed. However, the number-average molecular weight (Mn) increases as a function of monomer conversion (Fig. 1b) did not support a well-controlled ATRP as the linear increase of the molecular weight was only observed up to 40% monomer conversion (polymerization time up to 2 h). Prolonged polymerization time did not result in further increase in the molecular weight (Mn in fact decreased after 50% conversion; Fig. 1b, Table 1). It should be noted that the reaction mixture maintained a brown color throughout the ATRP process, eliminating deactivation of the Cu(I)/BPY catalyst system (would have resulted in a green color) as a potential cause for decreased polymerization rate or termination of the ATRP. Accordingly, we hypothesize that chain propagation of the ATRP gradually slowed down (nearly halted by 2 h) at 50 °C, and further conversion of the monomers likely contributed to side reactions giving rise to lower molecular weight oligomers/polymers. It has been reported that the azido and the methacrylate groups from azido-bearing methacrylate monomers can form intermolecular triazoline rings during reversible addition–fragmentation chain transfer (RAFT) polymerization at 50 °C.41 Alkyl azides can also undergo a 1,3-dipolar cycloaddition with electron-deficient olefins (e.g. N-isopropylacrylamide, dimethylacrylamide, methyl acrylate, and methyl methacrylate) to form triazoles and aziridines during RAFT at 60 °C even in the absence of any catalysts.42 The side reaction(s) involving the consumption of vinyl group of the AzTEGMA monomer may also occur in a first-order kinetics similar to that observed with the ATRP. This would not have affected the conventional monomer conversion calculation based on the reduction of the vinyl proton signals by 1H NMR monitoring. Meanwhile, real-time GPC monitoring of the ATRP of AzTEGMA at 50 °C indeed detected a lower molecular weight component (elution time around 18.5–19 min; note that the monomer was eluted at 19–19.5 min) at as early as 30 min after the initiation of the ATRP (Fig. 1c, black line). The signal for this component became stronger and broader (note the increasing shoulder peak eluted around 18.5 min) over time, with its intensity exceeding that of the pAzTEGMA (eluted around 15–18 min) after 2 h. These data combined support that the side reaction occurred early on during the ATRP process at 50 °C and it became a dominant event after 2 h.
Table 1.
ATRP of AzTEGMA in TFE with varied monomer concentrations, targeting DPs and polymerization temperatures.
| entry | DP | temperature (°C) | Concentration mol/L | time (h) | a Conversion (%) | b Mn (Da.) | PDI | polymer solubility |
|---|---|---|---|---|---|---|---|---|
| 1 | 100 | 50 | 3 | 1 | 25.10 | 3,900 | 1.15 | soluble |
| 2 | 100 | 50 | 3 | 6 | 69.10 | 4,300 | 1.17 | soluble |
| 3 | 100 | 50 | 3 | 18 | / | / | / | gelled |
| 4 | 50 | 23 | 3 | 5 | 58.80 | 6,300 | 1.17 | soluble |
| 5 | 50 | 23 | 3 | 21 | 79.73 | 8,200 | 1.27 | soluble |
| 6 | 50 | 23 | 2 | 6 | 45.48 | 7,300 | 1.10 | soluble |
| 7 | 100 | 23 | 2 | 31 | 76.15 | 10,100 | 1.13 | soluble |
| 8 | 100 | 34 | 2 | 3 | 76.64 | 11,800 | 1.13 | soluble |
| 9 | 100 | 34 | 2 | 15 | 96.36 | 13,800 | 1.17 | soluble |
Monomer conversion as determined by 1H NMR.
Mn of the polymers as determined by GPC.
Figure 1.
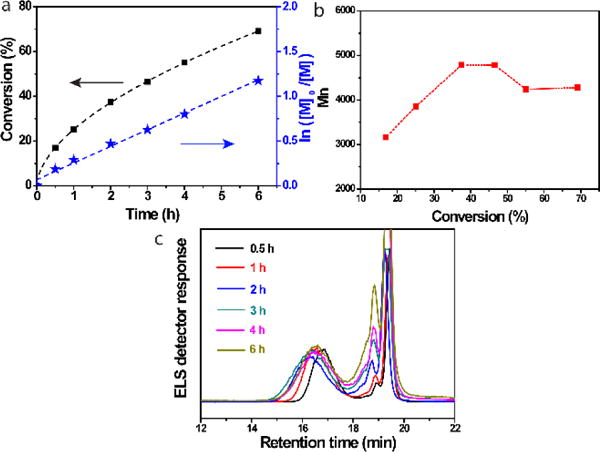
Kinetics of the ATRP of AzTEGMA at 50 °C. (a) Kinetics plots of monomer conversion (%) & conversion index In([M]0/[M]) over polymerization time; (b) Number-average molecular weight (Mn) of the polymers as a function of monomer conversion; (c) GPC curves of the polymerization mixture as a function of reaction time. Polymerization condition: [AzTEGMA] = 3 M, [AzTGMA]/[EBiB]/[CuBr]/[BPY] = 100:1:1:2.
With further prolongation of the polymerization time (18 h) at 50 °C, the brown reaction mixture eventually gelled (Table 1, entry 3) with many bubbles encapsulated. Thermal decomposition of the azide groups43,44 and the insertion reaction43 are likely responsible for releasing nitrogen and crosslinking the polymer chains, respectively, resulting in the gelation of the mixture and the entrapment of the in situ released nitrogen gas.
Optimization of ATRP of AzTEGMA
To mitigate the thermal decomposition of azides and the accompanying side reactions often observed during the preparation of polymers with azide-terminated side chains via radical polymerization processes, shortening polymerization time and lowering polymerization temperature are both logical strategies to pursue. Pierce et al. implemented both strategies to optimize the polymerization of azido-bearing vinyl monomers by RAFT.42 For optimizing the ATRP of azido-bearing monomers, however, shortening polymerization time has been more broadly implemented than lowering polymerization temperature. Here we first investigated the efficacy of lowering the temperature of ATRP of AzTEGMA from 50 °C to room temperature (23 °C) on improving the controllability of the polymerization (Table 1, entries 4 & 5).
The kinetics plots of AzTEGMA monomer conversion (black square) and the conversion index (blue cycle) as a function of polymerization time agreed with first-order monomer conversion (Fig. 2a). Unlike the polymerization conducted at 50 °C, the molecular weight of the polymer increased linearly as a function of the monomer conversion at 23 °C, with the narrow polydispersity (PDI < 1.20) maintained throughout the polymerization process (Fig. 2b). In addition, no low molecular weight components resulting from side reactions (elution time around 18–18.5 min as observed in ATRP at 50 °C) was detected by GPC monitoring during the 21 h polymerization at 23 °C with monomer conversions over 80% (Fig. 2c). The shift of pAzTEGMA peak to earlier elution, consistent with the increase of molecular weight of the polymer, also correlated well with the reduction of monomer peak intensity over time (Fig. 2c). These observations support that the room temperature ATRP of AzTEGMA was well-controlled and that lowering polymerization temperature effectively suppressed the side reaction of azido-bearing monomers and polymers that were observed at 50 °C.
Figure 2.
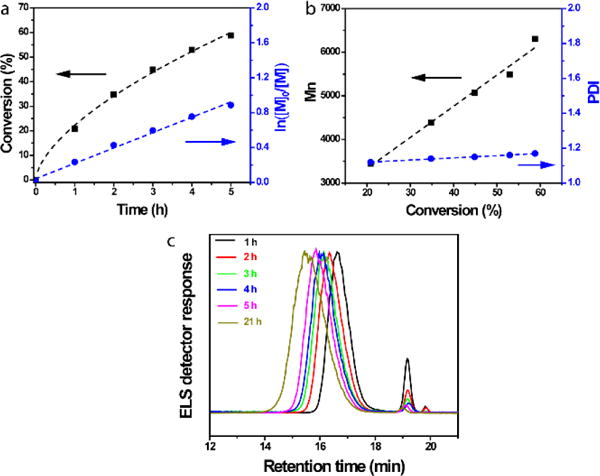
Kinetics of the ATRP of AzTEGMA at room temperature. (a) Kinetic plots of monomer conversion (%) & conversion index In([M]0/[M]), (b) molecular weight & molecular weight distribution, and (c) GPC curve as a function of reaction time. Polymerization condition: [AzTEGMA] = 3 M, [AzTGMA]/[EBiB]/[CuBr]/[BPY] = 50:1:1:2.
To further optimize the controllability of ATRP of AzTEGMA at room temperature, we lowered the monomer concentration from 3 M to 2 M while keeping the initiator concentration the same (Table 1, entries 6 & 7). Consistent with first-order controlled polymerization kinetics,29 the room temperature ATRP proceeded at a slower rate when the monomer concentration decreased from 3 mol/L to 2 mol/L, with the apparent propagation rate constant (kapp) decreasing from 4.89 ×10−5 s−1 to 2.84 ×10−5 s−1 (Fig. 3a). The slower ATRP carried out in the more diluted AzTEGMA solution, however, resulted in pAzTEGMA with narrower polydispersity (PDI ~1.10) at comparable monomer conversions (Table 1, entry 4 vs. entry 6, entry 5 vs. entry 7).
Figure 3.
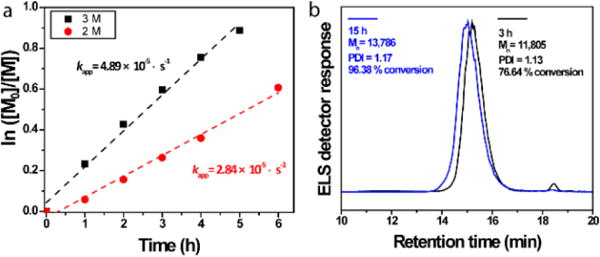
Tailoring kinetics of ATRP of AzTEGMA by monomer concentration and polymerization temperature. (a) Monomer conversion index In([M]0/[M]) of ATRP of AzTEGMA as a function of time with monomer concentration of 2 M (red) vs. 3 M (black) at room temperature. [AzTGMA]/[EBiB]/[CuBr]/[BPY] = 50:1:1:2. (b) GPC traces of pAzTEGMA prepared by ATRP at 34 °C with a monomer concentration of 2 M. [AzTGMA]/[EBiB]/[CuBr]/[BPY] = 100:1:1:2.
To take advantage of better controlled ATRP of AzTEGMA in lower monomer concentration while aiming to accelerate the polymerization without high temperature-induced side effects, we carried out ATRP of AzTEGMA at 2-M monomer concentration at 34 °C (Table 1, entries 8 & 9). The 11 °C increase from room temperature significantly accelerated the polymerization (DP = 100) to reach 76% monomer conversion in 3 h and 96.36% conversion by 15 h, resulting in pAzTEGMA with much higher molecular weights while still maintaining a low polydispersity (Fig. 3b). Overall, the combination of an intermediate temperature of 34 °C and a lower monomer concentration of 2 M resulted in an accelerated yet well-controlled ATRP of AzTEGMA.
Post-functionalization of pAzTEGMA by CuAAC
To examine the efficacy of CuAAC for high-density functionalization of the azide-terminated pendant chains of pAzTEGMA (Mn = 17,300, PDI = 1.29), prepared by the optimized ATRP at 34 °C with an initial monomer concentration of 2 M, proparyl alcohol was allowed to conjugate with the polymer at room temperature in the presence of Cu(I) catalyst (Fig. 4a). After the click conjugation, the molecular weight of the polymer increased from 17,300 to 22,000 while the PDI remained largely unchanged (Fig. 4b). 1H NMR confirmed the complete disappearance and up-filed shifts of the methylene signals alpha and beta to the azide (protons 8 and 7, bottom spectrum, Fig. 4c) consistent with the methylenes alpha and beta to the triazole nitrogen (protons 8′ and 7′, top spectrum, Fig. 4C), supporting quantitative conversion of azides by CuAAC. Signal ascribable to the triazole ring proton (proton 9′) was also observed and its integration relative to that of the methyl group on the polymer backbone (proton 1′) closely matched with the theoretic ratio (1:3 theoretical; 1:2.97 detected), further supporting quantitative coupling by CuAAC.
Figure 4.
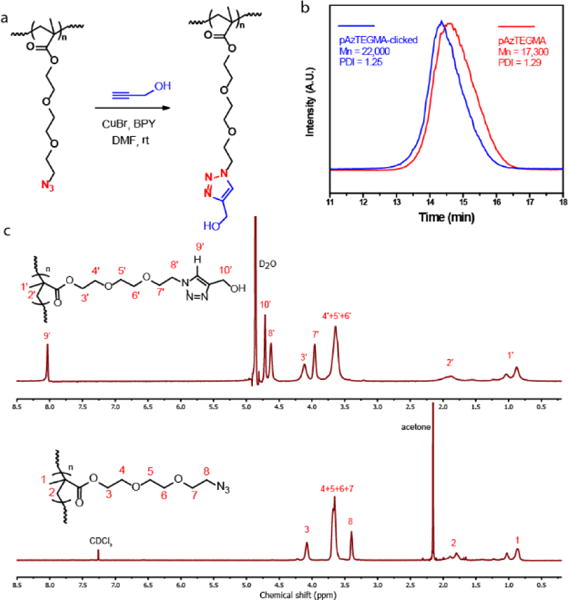
Functionalization of pAzTEGMA by CuAAC. (a) Schematic illustration of the functionalization of pAzTEGMA with propargyl alcohol at room temperature. (b) GPC traces of pAzTEGMA before (red line) and after the click reaction with propargyl alcohol. (c) 1H NMR spectra of the pAzTEGMA before (bottom) and after (top) the click reaction with propargyl alcohol.
Controlled grafting of pAzTEGMA brushes from Ti6Al4V substrates
To investigate the possibility of applying the optimized ATRP conditions and subsequent CuAAC functionalization to controlled surface modifications, we first grafted pAzTEGMA by SI-ATRP (Fig. 5) from Ti6Al4V substrates under the optimized conditions identified above. Phosphonic acid-terminated initiator PA-O-Br (Fig. 5) was synthesized, covalently attached to the metal alloy surfaces and annealed as we previously described.40 The initiator-bound substrate Ti-Br was then subjected to grafting of pAzTEGMA by SI-ATRP at room temperature.
Figure 5.
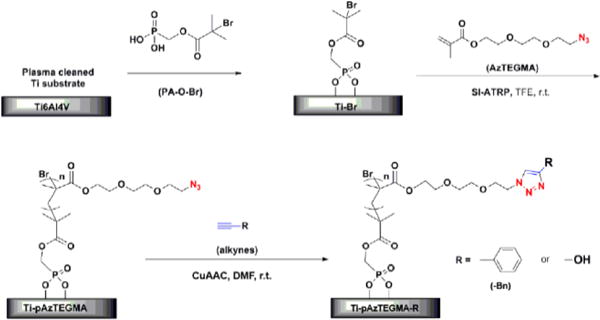
Schematic illustration of the grafting of well-controlled pAzTEGMA brushes from Ti6Al4V and the subsequent functionalization via CuAAC. R = −OH or −Bn.
XPS survey scans of the pAzTEGMA-grafted substrates (Ti-pAzTEGMA, DP = 100) detected a N1s peak (binding energy ~400 eV) along with residue P2p signal ascribable to the azide groups on the pendant side chains and the phosphonic acid anchor that were absent from that of the pristine Ti6Al4V (Fig. 6a, Table 2). Meanwhile, the intensity of the Ti2p signal significantly decreased on Ti-pAzTEGMA compared to Ti6Al4V (Fig. 6a, Table 2) as the metal alloy substrate became masked by the grafted polymer brushes. High-resolution scan of N1s using the snapshot mode that reduces the X-ray irradiation-induced in situ decomposition of azide (see conventional scans in Supplementary Fig. S6) revealed 2 peaks at 405.5 eV and 401.5 eV (Fig. 6b) attributable to the terminal N’s and the center N+ species, respectively.45,46 The detected abundance of the terminal N’s relative to that of the center N+ species of the azide, averaged from three randomly selected Ti-pAzTEGMA substrates, closely approximates that of the theoretical ratio (2.3±0.1 detected vs. 2.0 theoretical; Fig. 6b), further supporting successful grafting of the azide-rich pAzTEGMA brushes by SI-ATRP.
Figure 6.
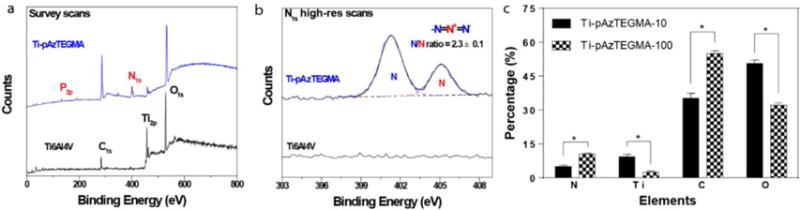
Surface modification of Ti6Al4V by grafting of pAzTEGMA brushes via SI-ATRP. (a) XPS survey scans and (b) N1s high resolution scans of the pristine Ti6Al4V and Ti-pAzTEGMA surfaces. The Ti6Al4V substrates were grafted with pAzTEGMA at a targeted degree of polymerization of 100 (DP =100). (c) Relative contents of N, Ti, C and O elements detected by XPS on Ti-pAzTEGMA surfaces with varying degrees of polymerization (DP =10 vs. DP =100). *p < 0.05 (ANOVA).
Table 2.
Surface elemental composition of surface-modified Ti6Al4V substrates (n = 3) as determined by XPS
| Substrates | Elemental composition (%)
|
|||
|---|---|---|---|---|
| N | Ti | C | O | |
| Ti-Br | / | 9.93 ± 1.81 | 26.19 ± 2.87 | 53.63 ± 1.96 |
| Ti-pAzTEGMA-10 | 4.99 ± 0.48 | 9.25 ± 0.92 | 35.18 ± 1.82 | 50.59 ± 1.24 |
| Ti-pAzTEGMA-100 | 10.45 ± 0.32 | 2.45 ± 0.43 | 55.00 ± 1.02 | 32.10 ± 0.91 |
By introducing free initiator into the reaction system, a method for controlling the chain length of surface grafted polymer brushes,40,47,48 we grafted pAzTEGMA with targeted degrees of polymerization of 10 and 100 from Ti-Br. As revealed by XPS analyses, upon grafting the shorter pAzTEGMA-10, the surface carbon content and nitrogen content of the Ti-Br substrates increased from ~26% to ~35% and 0% to ~5%, respectively, while the surface Ti content decreased by ~1 % (Fig. 6c, Table 2). When longer pAzTEGMA brushes (DP = 100) was grafted, the surface C and N content sharply increased to 55% and >10%, respectively, while the surface Ti content substantially decreased to <3% (Fig. 6c, Table 2), supporting much dense surface coverage by the longer pAzTEGMA brushes.
Facile high-density side chain functionalization of the Ti-pAzTEGMA surface brushes by CuAAC
To demonstrate the general applicability of CuAAC for high-density side chain functionalization of the pAzTEGMA surface brushes, both hydrophobic and hydrophilic alkynes are “clicked” to Ti-pAzTEGMA-100 and the extent of azide to triazole conversion was examined by high-resolution N1s XPS scans under the snapshot mode. As shown in Figure 7a, the N+ signal of the azide (405.5 eV) completely disappeared upon CuAAC while a new N signal of comparable intensity ascribable to the uncharged center N in the triazole was detected (peaking ~402 eV upon curve fitting). This observation was made upon CuAAC by both hydrophilic (benzyl terminated) and hydrophobic (hydroxyl terminated) alkynes, supporting quantitative conversion of azide-terminated pendant chains in both cases. Finally, water contact angle measurements confirmed significant increase in surface hydrophilicity upon “clicking” the proparyl alcohol (Ti-pAzTEGMA-OH), with the water contact angle decreased from 70° for the Ti-pAzTEGMA to 43° for Ti-pAzTEGMA-OH (Fig. 7b). Likewise, expected decrease in surface hydrophilicity upon “clicking” the hydrophobic phenylacetylene was also observed. Overall, these data show that well-controlled grafting of polymer brushes with stable azide-terminated side chains from metallic alloy surface could be accomplished by SI-ATRP of AzTEGMA under the optimal conditions we identified. Subsequent quantitative high-density functionalization of the azide-terminated side chains by CuAAC with small molecule alkynes enabled further surface modifications.
Figure 7.
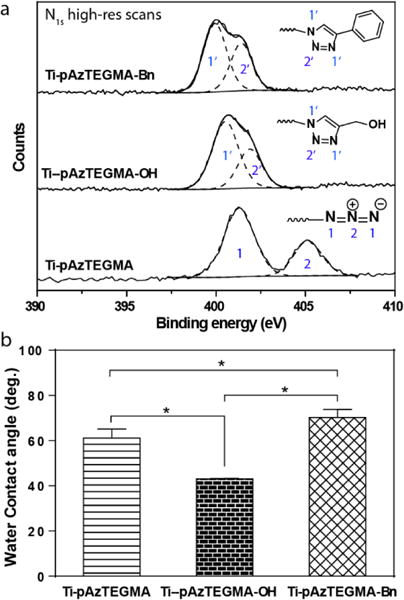
Facile high-density functionalization of Ti6Al4V substrates via the combination of SI-ATRP and CuAAC. (a) XPS N1s high resolution scans of Ti-pAzTEGMA surface vs. those “clicked” with proparyl alcohol (Ti-pAzTEGMA-OH) or phenylacetylene (Ti-pAzTEGMA-Bn). (b) Water contact angle of the Ti-pAzTEGMA substrates before and after conjugation with hydrophilic and hydrophobic alkynes by CuAAC. *p < 0.05 (Student’s t-test).
CONCLUSIONS
In summary, we demonstrated that the poor controllability of ATRP of azido-bearing methacrylate monomer AzTEGMA at commonly adopted temperature of 50 °C could be addressed by lowering polymerization temperature and reducing the monomer concentrations. Lowing the polymerization temperature to room temperature effectively suppressed the side reaction induced by the thermal decomposition of azido groups, thereby significantly increased the molecular weight (up to 22 kD) and reduced the polydispersity (PDI < 1.3) of pAzTEGMA. By carrying out the ATRP of AzTEGMA at a reduced monomer concentration while at an intermediate temperature 34 °C, the ATRP was accelerated without compromising the controllability of the polymerization. The optimized ATRP of AzTEGMA was readily extended to graft pAzTEGMA surface brushes with controlled lengths from Ti6Al4V substrates. The azide-terminated side chains of these polymers and surface polymer brushes could be readily functionalized by functional alkynes in a quantitative manner by facile CuAAC. The optimized ATRP and SI-ATRP of AzTEGMA combined with orthogonal CuAAC provides a useful tool for facile preparations of functional polymers and controlled modification of surfaces for a wide range of applications.
Supplementary Material
Acknowledgments
The authors acknowledge grant support from the National Institutes of Health (R01AR055615, R01AR068418). XPS was performed at the Center for Nanoscale Systems (CNS) at Harvard University supported by the National Science Foundation award ECS-0335765.
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES AND NOTES
- 1.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Edit. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JMJ, Sharpless KB, Fokin VV. Angew Chem Int Edit. 2004;43:3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 3.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem Int Edit. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 4.Hoyle CE, Bowman CN. Angew Chem Int Edit. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 5.Kade MJ, Burke DJ, Hawker CJ. J Polym Sci, Polym Chem. 2010;48:743–750. [Google Scholar]
- 6.Killops KL, Campos LM, Hawker CJ. J Am Chem Soc. 2008;130:5062–5064. doi: 10.1021/ja8006325. [DOI] [PubMed] [Google Scholar]
- 7.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 8.Lallana E, Sousa-Herves A, Fernandez-Trillo F, Riguera R, Fernandez-Megia E. Pharm Res-Dordr. 2012;29:1–34. doi: 10.1007/s11095-011-0568-5. [DOI] [PubMed] [Google Scholar]
- 9.Thirumurugan P, Matosiuk D, Jozwiak K. Chem Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 10.Golas PL, Matyjaszewski K. Qsar Comb Sci. 2007;26:1116–1134. [Google Scholar]
- 11.Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Chem Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Feng E, Song J. J Am Chem Soc. 2014;136:4105–4108. doi: 10.1021/ja4130862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu JW, Filion TM, Prifti F, Song J. Chemistry-an Asian J. 2011;6:2730–2737. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matyjaszewski K, Xia JH. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 15.Wang JS, Matyjaszewski K. J Am Chem Soc. 1995;117:5614–5615. [Google Scholar]
- 16.Gao HF, Matyjaszewski K. Macromolecules. 2006;39:4960–4965. [Google Scholar]
- 17.Lutz JF, Borner HG, Weichenhan K. Macromolecules. 2006;39:6376–6383. [Google Scholar]
- 18.Topham PD, Sandon N, Read ES, Madsen J, Ryan AJ, Armes SP. Macromolecules. 2008;41:9542–9547. [Google Scholar]
- 19.He F, Luo BW, Yuan SJ, Liang B, Choong C, Pehkonen SO. Rsc Adv. 2014;4:105–117. [Google Scholar]
- 20.Olson MA, Braunschweig AB, Fang L, Ikeda T, Klajn R, Trabolsi A, Wesson PJ, Benitez D, Mirkin CA, Grzybowski BA, Stoddart JF. Angew Chem Int Edit. 2009;48:1792–1797. doi: 10.1002/anie.200804558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han DH, Tong X, Zhao Y. Macromolecules. 2011;44:5531–5536. [Google Scholar]
- 22.Zhang YF, Li CH, Liu SY. J Polym Sci Polym Chem. 2009;47:3066–3077. [Google Scholar]
- 23.Mespouille L, Vachaudez M, Suriano F, Gerbaux P, Coulembier O, Degee P, Flammang R, Dubois P. Macromol Rapid Comm. 2007;28:2151–2158. [Google Scholar]
- 24.Vogt AP, Sumerlin BS. Macromolecules. 2006;39:5286–5292. [Google Scholar]
- 25.Golas PL, Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Macromolecules. 2006;39:6451–6457. [Google Scholar]
- 26.Abdellatif MM, Nomura K. Acs Macro Lett. 2012;1:423–427. doi: 10.1021/mz300061a. [DOI] [PubMed] [Google Scholar]
- 27.Zhou C, Qian SS, Li XJ, Yao F, Forsythe JS, Fu GD. Rsc Adv. 2014;4:54631–54640. [Google Scholar]
- 28.Soto-Cantu E, Lokitz BS, Hinestrosa JP, Deodhar C, Messman JM, Ankner JF, Kilbey SM. Langmuir. 2011;27:5986–5996. doi: 10.1021/la2000798. [DOI] [PubMed] [Google Scholar]
- 29.Storms-Miller WK, Pugh C. Macromolecules. 2015;48:3803–3810. [Google Scholar]
- 30.Zhang YR, Ren L, Tu Q, Wang XQ, Liu R, Li L, Wang JC, Liu WM, Xu J, Wang JY. Anal Chem. 2011;83:9651–9659. doi: 10.1021/ac202517x. [DOI] [PubMed] [Google Scholar]
- 31.He XH, Liang LY, Wang K, Lin SL, Yan DY, Zhang YQ. J Appl Polym Sci. 2009;111:560–565. [Google Scholar]
- 32.Sumerlin BS, Tsarevsky NV, Louche G, Lee RY, Matyjaszewski K. Macromolecules. 2005;38:7540–7545. [Google Scholar]
- 33.Li W, Xu YH, Zhou Y, Ma WH, Wang SX, Dai YN. Nanoscale Res Lett. 2012;7:485. doi: 10.1186/1556-276X-7-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song WT, Xiao CS, Cui LG, Tang ZH, Zhuang XL, Chen XS. Colloid Surface B. 2012;93:188–194. doi: 10.1016/j.colsurfb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Saha S, Bruening ML, Baker GL. Macromolecules. 2012;45:9063–9069. doi: 10.1021/ma301556v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu LQ, Wan D, Gong HF, Neoh KG, Kang ET, Fu GD. Langmuir. 2010;26:15376–15382. doi: 10.1021/la102775y. [DOI] [PubMed] [Google Scholar]
- 37.He H, Zhang Y, Gao C, Wu J. Chem Commun. 2009:1655–1657. doi: 10.1039/b821280e. [DOI] [PubMed] [Google Scholar]
- 38.Devaraj NK, Collman JP. Qsar Comb Sci. 2007;26:1253–1260. [Google Scholar]
- 39.Li Y, Yang JW, Benicewicz BC. J Polym Sci Pol Chem. 2007;45:4300–4308. [Google Scholar]
- 40.Liu PS, Domingue E, Ayers DC, Song J. ACS Appl Mater & Interfaces. 2014;6:7141–7152. doi: 10.1021/am501967y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanjarappa MJ, Gujraty KV, Joshi A, Saraph A, Kane RS. Biomacromolecules. 2006;7:1665–1670. doi: 10.1021/bm060098v. [DOI] [PubMed] [Google Scholar]
- 42.Ladmiral V, Legge TM, Zhao YL, Perrier S. Macromolecules. 2008;41:6728–6732. [Google Scholar]
- 43.Ruud CJ, Jia JP, Baker GL. Macromolecules. 2000;33:8184–8191. [Google Scholar]
- 44.Murray KA, Holmes AB, Moratto SC, Friend RH. Synthetic Met. 1996;76:161–163. [Google Scholar]
- 45.Gouget-Laemmel AC, Yang J, Lodhi MA, Siriwardena A, Aureau D, Boukherroub R, Chazalviel JN, Ozanam F, Szunerits S. J Phys Chem C. 2013;117:368–375. [Google Scholar]
- 46.Collman JP, Devaraj NK, Eberspacher TPA, Chidsey CED. Langmuir. 2006;22:2457–2464. doi: 10.1021/la052947q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koylu D, Carter KR. Macromolecules. 2009;42:8655–8660. [Google Scholar]
- 48.Marutani E, Yamamoto S, Ninjbadgar T, Tsujii Y, Fukuda T, Takano M. Polymer. 2004;45:2231–2235. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.