

Seven hundred ninety-three gliomas (grades II–IV) were analyzed by single nucleotide polymorphism array and for TP53 mutations. Homodisomy revealed by copy number-neutral loss of heterozygosity (CNLOH) was observed in 156 cases (19.7%), was tightly associated with TP53 mutation, and was mutually exclusive with 1p19q codeletion. In IDH-mutated 1p19q non-codeleted gliomas, CNLOH 17p was associated with longer survival (86.3 vs. 46.2 months), particularly in grade III gliomas (>100 vs. 37.9 months).
Keywords: Gliomas, Copy number neutral loss of heterozygosity, TP53 mutation
Abstract
Background.
The 1p19q non-codeleted gliomas with IDH mutation, defined as “molecular astrocytomas,” display frequent TP53 mutations and have an intermediate prognosis. We investigated the prognostic impact of copy number-neutral loss of heterozygosity (CNLOH) in 17p in this population.
Methods.
We analyzed 793 gliomas (206 grade II, 377 grade III, and 210 grade IV) by single nucleotide polymorphism array and for TP53 mutations.
Results.
Homodisomy revealed by CNLOH was observed in 156 cases (19.7%). It was more frequent in astrocytomas and oligoastrocytomas (98/256, 38%) than oligodendrogliomas (28/327, 8.6%; p < .0001) or glioblastoma multiforme (30/210, 14.3%; p < .0001), tightly associated with TP53 mutation (69/71 vs. 20/79; p = 2 × 10−16), and mutually exclusive with 1p19q codeletion (1/156 vs. 249/556; p < .0001). In the group of IDH-mutated 1p19q non-codeleted gliomas, CNLOH 17p was associated with longer survival (86.3 vs. 46.2 months; p = .004), particularly in grade III gliomas (overall survival >100 vs. 37.9 months; p = .007). These data were confirmed in an independent dataset from the Cancer Genome Atlas.
Conclusion.
CNLOH 17p is a prognostic marker and further refines the molecular classification of gliomas.
Implications for Practice:
Homodisomy of chromosome 17p (CNLOH 17p) is a frequent feature in IDH-mutated 1p19q non-codeleted gliomas (group 2). It is constantly associated with TP53 mutation. It was found, within this specific molecular group of gliomas (corresponding to molecular astrocytomas), that CNLOH 17p is associated with a much better outcome and may therefore represent an additional prognostic marker to refine the prognostic classification of gliomas.
Introduction
Independently of histological grading, gliomas can be separated into three distinct prognostic subgroups according to the presence of IDH mutation and 1p19q codeletion: group 1, glioma with 1p19q codeletion, has the best survival; group 2, non-codeleted glioma with IDH mutation, has an intermediate prognosis; and group 3, IDH wild-type glioma, has the poorest outcome [1–3]. Groups 1 and 2 also differ by the occurrence of mutually exclusive mutations: TERT promoter (90%), CIC (50%–60%), and FUBP1 (15%–20%) for group 1 and ATRX mutation (associated with the alternative lengthening telomeres phenotype) and TP53 mutation for group 2 [2–4]. Recent single nucleotide polymorphism (SNP) analysis showed several cases of copy neutral loss of heterozygosity (CNLOH) with duplication of the retained allele. The presence of CNLOH in glial tumors has been reported to affect several genomic regions [5–9]. In a recent report on anaplastic oligodendrogliomas, CNLOH frequently affected the short arm of chromosome 17 [5]. Moreover, Yin et al. described eight cases with CNLOH 17p in a series of 55 glioblastomas [9]. To date, the frequency and prognostic significance of this alteration have not been investigated.
In this study, we investigated the presence of CNLOH 17p in a large cohort of grade II–IV glial tumors, analyzed the associations with TP53 mutation and other molecular alterations, and investigated the prognostic impact of CNLOH 17p.
Patients and Methods
Patients and Tissue Samples
Patients were selected according to the following criteria: histologic diagnosis of primary glial tumor, clinical data and follow-up available in the neuro-oncology database (OncoNeurotek, Groupe Hospitalier Pitié Salpêtrière, Paris, France), and written informed consent. Corresponding clinical annotations were collected from the neuro-oncology department database. As a
duplication cohort, we used the DNA sequencing, copy number variant (level 1 copy number data), and survival data (level 3) from lower-grade gliomas (LGGs) of the Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov).
DNA Isolation and SNP Array
Tumor DNA from cryopreserved samples was extracted using the QIAmp DNA Midi Kit (Qiagen, Hilden, Germany, http://www.qiagen.com) according to the manufacturer’s instructions. DNA was extracted from blood samples by conventional saline method, quantified using a NanoVue spectrophotometer, and qualified by agarose gel electrophoresis. Tumor DNA was run on an Infinium Illumina Human 610-Quad SNP array (Illumina, San Diego, CA, http://www.illumina.com). Array processing, using 250 ng tumor DNA, was outsourced to Integragen, Évry, France. Extracted data using Feature Extraction software were imported and analyzed using Nexus 5.1 (Biodiscovery, El Segundo, CA, http://www/biodiscovery.com), as previously described [10]. The confirmatory cohort from LGG TCGA was analyzed using PennCNV-Affy from the PennCNV algorithm [11] to convert raw CEL files from LGG TCGA into log R ratio and B-allele frequency. Log R ratio and B-allele frequency files were used to perform allele-specific copy number analysis with GC correction using ASCAT (version 2.4) [12]. We considered loss of heterozygosity in a given chromosome region when ≥95% of SNP probes in a DNA segment of at least 500 kb exhibited B-allele frequencies ≥0.8 and ≤0.2. Loss of heterozygosity with a copy number of 2 was considered CNLOH. Only terminal CNLOH on chromosome 17p with a minimum size of 5 Mb was considered. Molecular characterization of glioma samples (IDH1/2 mutation, TERT promoter mutation, and MGMT promoter methylation) was performed as previously described [13].
TP53 Pyrosequencing
Coding exons (2–11) of TP53 gene were first amplified using primers detailed in supplemental online Table 1. Amplification conditions were 94°C for 3 minutes followed by 45 cycles of 94°C for 15 seconds, 60°C for 45 seconds, and 72°C for 1 minute, with a final step at 72°C for 8 minutes. Polymerase chain reaction (PCR) products were purified conforming to the Agencourt AMPure XP PCR purification protocol (Beckman-Coulter, Nyon, Switzerland, http://www.beckmancoulter.com) with the Biomek 3000 Automation Workstation. Universal tailed amplicon resequencing approach (454 Sequencing Technology; Roche, Basel, Switzerland, http://www.roche.com) was used for sequencing of coding exons of TP53. This system includes a second PCR, aiming for multiplex identifiers and incorporation of 454 adaptors, an emulsion PCR according to the emPCR Amplification Method Manual Lib-A protocol (GS Junior Titanium Series, Roche), enrichment, and pyrosequencing according to the Sequencing Method Manual (Roche). Sequence analysis was performed using CLC Genomics Workbench software.
TP53 Sanger Sequencing
TP53 mutations identified by pyrosequencing were confirmed by direct Sanger sequencing. Tumor DNA was first amplified and purified using the same primers and conditions described for pyrosequencing. Sequencing reactions were performed in both orientations using Big-Dye Terminator Cycle Sequencing Ready Reaction (PerkinElmer, Waltham, MA, http://www.perkinelmer.com). Extension products were purified with the Agencourt CleanSEQ protocol according to the manufacturer’s instructions (Beckman-Coulter). Purified sequences were analyzed on an ABI Prism 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). Forward and reverse sequences were systematically analyzed using Chromas Lite software.
Statistical Analysis
We used chi-square and Fisher exact test to compare genotype distribution. The association with continuous variables was calculated with the Mann-Whitney test. Overall survival (OS) was defined as the time between diagnosis and death or last follow-up. Patients who were alive at last follow-up were considered as a censored event in analysis. Progression-free survival (PFS) was defined as the time between diagnosis and recurrence or last follow-up. Patients who were recurrence-free at last follow-up were considered as a censored event in analysis. To find clinical or genomic factors related to OS or PFS, survival curves were calculated according to the Kaplan-Meier method, and differences between curves were assessed using the log-rank test. Variables with a significant p value were used to build a multivariate Cox model. Two-sided p values < .05 were considered significant.
Results
We screened the genomic profiles of 793 gliomas (206 grade II, 377 grade III, and 210 grade IV) for the presence of CNLOH 17p. In the whole cohort, we identified 156 cases with CNLOH 17p (19.7%), affecting the whole chromosome 17 in 14 cases (9.0%), the whole short arm of chromosome 17 in 15 cases (9.6%), and only the telomeric portion of 17p in 127 cases (81.4%), including in all cases the TP53 locus. The mean size of the affected region was 21.6 ± 1.1 Mb (range 7.7–80.9 Mb) (supplemental online Fig. 1A, 1B). We also screened a series of 96 constitutional DNA samples. We did not find any CNLOH 17p in blood DNA, confirming this as a somatic event.
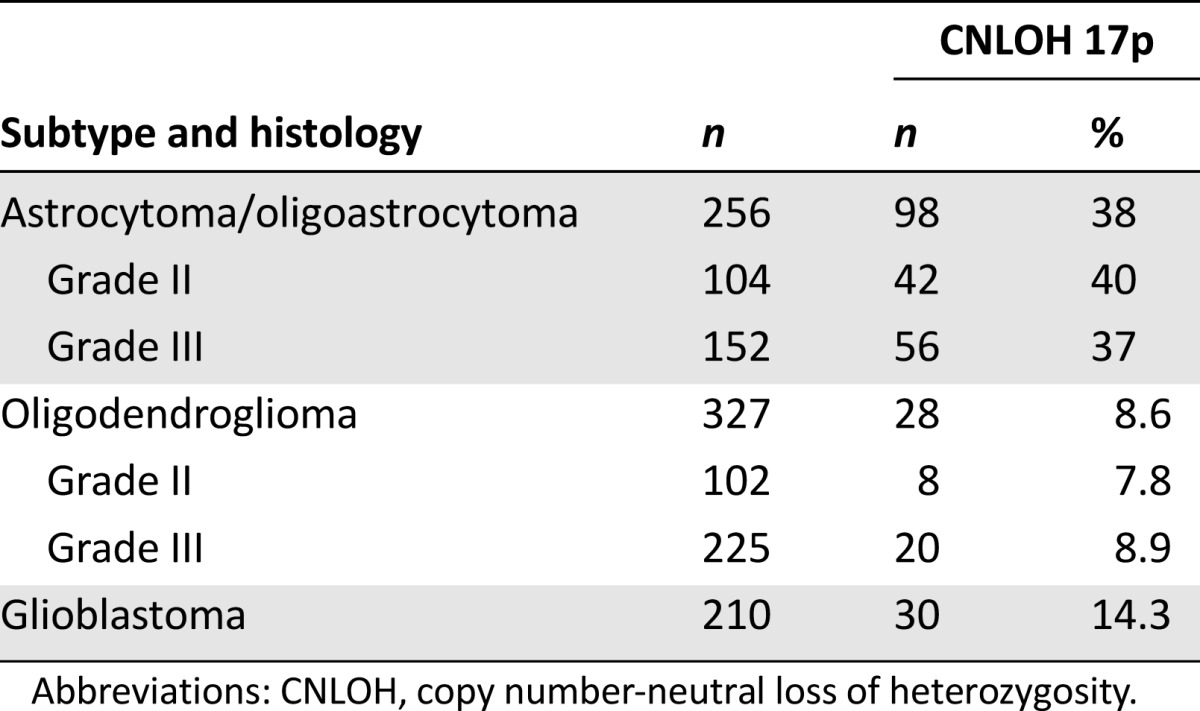
CNLOH 17p affected 50 of 206 grade II (24.3%), 76 of 377 grade III (20.2%), and 30 of 210 grade IV gliomas (14.3%). CNLOH 17p was more frequent in astrocytomas and oligoastrocytomas (98/256, 38%) than oligodendrogliomas (28/327, 8.6%; p < .0001) or glioblastoma multiforme (30/210, 14.3%; p < .0001) (Table 1).
Table 1.
Frequency of CNLOH 17p according to glioma histologic subtype
We investigated the presence of TP53 mutation by pyrosequencing. Each nonsilent variation was then validated by Sanger sequencing. Of the 71 tumors with CNLOH 17p and available DNA, 97.2% (69/71) were mutated on the TP53 gene. Electropherograms showed a pattern of homozygous mutation (supplemental online Fig. 2A) in all cases. Missense mutations were the most frequent (58/71, 81.7%), compared with nonsense mutations (8/71, 11.3%) and frameshifts (5/71, 7.0%). Strikingly, one of the two nonmutated tumors had a focal homozygous deletion of TP53 locus (supplemental online Fig. 3). In all, the TP53 gene was altered in all but one tumor with CNLOH 17p (70/71, 98.6%). Interestingly, P53 was overexpressed by immunohistochemistry in the remaining nonaltered case, suggesting abnormal P53 sequestration (data not shown).
In non-CNLOH 17p gliomas, TP53 mutational status was available in 79 tumors. We identified 24 TP53 mutations (25.3%; p < .0001) on 20 tumors, with four tumors having a double variant consisting of 21 (80.8%) missense mutations, four (15.5%) nonsense mutations, and one (3.8%) frameshift. In all these non-CNLOH 17p gliomas, electropherograms showed a heterozygous pattern of TP53 mutation (supplemental online Fig. 2B). Based on the TP53 database reported by Edlund et al. [14], we found that 86 of 97 (89%) of these mutations affected the TP53 DNA binding domain (65/71 in the CNLOH 17p group and 21/26 in the control group; not significant). All mutations are predicted to be transcriptionally inactive.
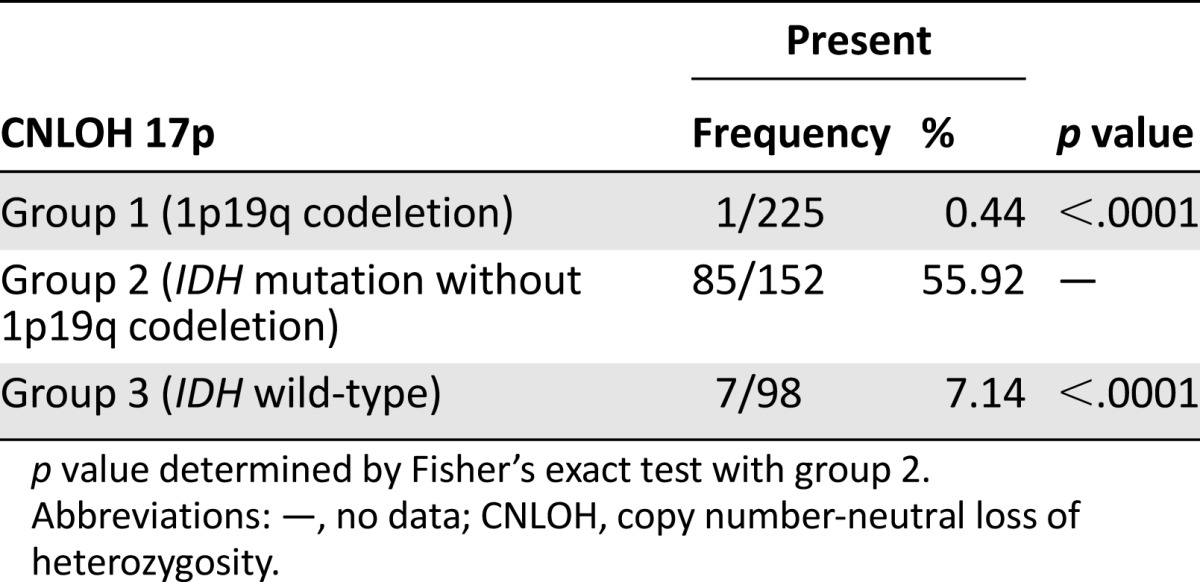
We next investigated the association of CNLOH 17p with other molecular alterations commonly found in gliomas (Table 2). CNLOH 17p was mutually exclusive with 1p19q codeletion (1/156 vs. 249/556; p < .0001) and was associated with IDH mutation (114/141 vs. 309/556; p < .0001). In grade II and III gliomas, CNLOH 17p was associated with the 1p19q non-codeleted IDH-mutated gliomas (group 2) (55.9% of group 2 tumors compared with groups 1 and 3) (Table 3).
Table 2.
Association of CNLOH 17p with common molecular alterations in gliomas

Table 3.
Relative frequency of CNLOH 17p in molecular groups 1, 2, and 3 of grade II–III gliomas
We then evaluated the prognostic impact of CNLOH 17p. We did not find any impact on PFS or OS for grade II–IV gliomas with available clinical data (supplemental online Fig. 4). This is not surprising, because CHLOH 17p is strongly associated with the TP53 mutation, which itself is associated with group 2 gliomas, which have an intermediate prognosis (Fig. 1A). We therefore considered specifically the prognostic impact of CNLOH 17p in group 2 and found an association with a much better outcome (OS 86.3 vs. 46.2 months; p = .004) (Fig. 1B). The difference was particularly clear in grade III gliomas (OS >100 vs. 37.9 months; p = .007) (Fig. 2) but was not found in grade II and IV gliomas.
Figure 1.

(A) Prognostic classification of grade II–IV gliomas according to 1p19q and IDH status (groups 1, 2, and 3). (B) Prognostic impact of CNLOH 17p in group 2. Survival times were compared using log-rank test (Mantel-Cox). The presence of CNLOH 17 p in group 2 was associated with better outcome (OS 86.3 vs. 46.2 months for group 2 with and without CNLOH 17p, respectively; p = .004).
Abbreviations: CNLOH, copy number-neutral loss of heterozygosity; OS, overall survival; w/o, without.
Figure 2.

(A) Prognostic classification of grade III gliomas according to 1p19q and IDH status (groups 1, 2, and 3). (B) Prognostic impact of CNLOH 17p in group 2. Survival times were compared using log-rank test (Mantel-Cox). The presence of CNLOH 17p in group 2 was associated with better outcome (OS >100 vs. 37.9 months for group 2 with and without CNLOH 17p, respectively; p = .007).
Abbreviations: CNLOH, copy number-neutral loss of heterozygosity; OS, overall survival; w/o, without.
We then entered into the Cox model the major histological and biological prognostic markers, i.e., the grading and the molecular subgroup (1p19q codeletion, IDH mutation, IDH wild-type): both were strongly predictive of outcome (hazard ratios 2.094 and 1.840, p = 7 × 10−7 and 2 × 10−5, respectively), but the negative prognostic impact of CNLOH 17p remained significant (hazard ratio 1.641; p = .04). Because CNLOH 17p is specifically found in group 2 (IDH-mutated non-codeletion gliomas), we performed multivariate analysis specifically in this group, entering CNLOH 17p, grade, EGFR amplification, CDKN2A deletion, and TP53 mutation. We found that CNLOH 17p was the strongest (odds ratio [OR] for non-CNLOH p17 = 3.58) and the most significant (p = .014) prognostic marker.
To confirm this result, we analyzed survival data from 142 LGGs from TCGA with IDH1/IDH2 mutations and no 1p19q codeletion. Despite the high rate of censured data, we found that CNLOH 17p, including the TP53 locus, was associated with better outcome (OR = 0.27; p = .026) (supplemental online Fig. 5) [11].
Discussion
Using SNP array, we found that CNLOH 17p is a frequent alteration in gliomas. A similar mechanism has also been reported in other malignancies [15]. Strikingly, CNLOH affects selectively 17p and not (or only marginally) the other chromosome segments, as shown by a recent whole-exome sequencing analysis [2, 16]. We found CNLOH 17p to be almost systematically associated with TP53 mutation or deletion (70 of 71 samples). The sequence analysis showed a homozygous mutation in all cases, suggesting that during the mechanism of tumorigenesis, the normal arm of chromosome 17p is lost and the altered chromosome arm is duplicated, leading to a homozygous mutation of TP53 [9, 17–19].
In our series, CNLOH 17p is mutually exclusive with 1p19q codeletion and is associated with IDH mutation. Regarding the three molecular subgroups [1–3], CNLOH 17p samples were mostly found in group 2, the 1p19q non-codeleted IDH-mutated group, which is associated with TP53 mutation (85/152 vs. 1/225 in the 1p19q codeleted group and 7/98 in the non-1p19q codeleted, non-IDH mutated group).
We therefore analyzed the prognostic impact of CNLOH 17p in this particular subgroup (IDH mutated, non-1p19q codeleted). We found that tumors harboring CNLOH 17p had a better OS than tumors without CNLOH 17p and similar to that of 1p19q codeleted tumors (Fig. 2B). The upcoming World Health Organization classification of gliomas will integrate molecular markers; in this setting, the replication of this finding in the independent TCGA series allows generalization of our conclusion; thus we propose CNLOH 17p as a stratification marker in this subgroup defined as molecular astrocytomas [20].
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
Supported by grants from the Ligue Nationale contre le Cancer and the Association pour la Recherche sur les Tumeurs Cérébrales. This work is part of the national program Cartes d’Identité des Tumeurs (http://cit.ligue-cancer.net/) funded and developed by the Ligue Nationale Contre le Cancer. The research leading to these results has received funding from the program “Investissements d’Avenir” (ANR-10-IAIHU-06). The results published here are based in part on data generated by the Cancer Genome Atlas Research Network (http://cancergenome.nih.gov/).
Author Contributions
Conception/Design: Marianne Labussière, Ahmed Idbaih, Marc Sanson
Provision of study material or patients: Marianne Labussière, Amithys Rahimian, Marine Giry, Blandine Boisselier, Marc Polivka, Karima Mokhtari, Agusti Alentorn, Marc Sanson
Collection and/or assembly of data: Marianne Labussière, Amithys Rahimian, Marine Giry, Blandine Boisselier, Yohann Schmitt, Agusti Alentorn, Marc Sanson
Data analysis and interpretation: Marianne Labussière, Amithys Rahimian, Marc Polivka, Karima Mokhtari, Jean-Yves Delattre, Ahmed Idbaih, Karim Labreche, Marc Sanson
Manuscript writing: Marianne Labussière, Jean-Yves Delattre, Ahmed Idbaih, Karim Labreche, Agusti Alentorn, Marc Sanson
Final approval of manuscript: Marianne Labussière, Amithys Rahimian, Marine Giry, Blandine Boisselier, Yohann Schmitt, Marc Polivka, Karima Mokhtari, Jean-Yves Delattre, Ahmed Idbaih, Karim Labreche, Marc Sanson
Disclosures
Marc Sanson: Roche (C/A). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Labussière M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74:1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 3.Brat DJ, Verhaak RG, Aldape KD, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu XY, Gerges N, Korshunov A, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124:615–625. doi: 10.1007/s00401-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 5.Idbaih A, Ducray F, Dehais C, et al. SNP array analysis reveals novel genomic abnormalities including copy neutral loss of heterozygosity in anaplastic oligodendrogliomas. PLoS One. 2012;7:e45950. doi: 10.1371/journal.pone.0045950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotliarov Y, Kotliarova S, Charong N, et al. Correlation analysis between single-nucleotide polymorphism and expression arrays in gliomas identifies potentially relevant target genes. Cancer Res. 2009;69:1596–1603. doi: 10.1158/0008-5472.CAN-08-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuga D, Mizoguchi M, Guan Y, et al. Prevalence of copy-number neutral LOH in glioblastomas revealed by genomewide analysis of laser-microdissected tissues. Neuro-oncol. 2008;10:995–1003. doi: 10.1215/15228517-2008-064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo KC, Bailey D, Burkhardt T, et al. Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization. Genes Chromosomes Cancer. 2008;47:221–237. doi: 10.1002/gcc.20524. [DOI] [PubMed] [Google Scholar]
- 9.Yin D, Ogawa S, Kawamata N, et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009;7:665–677. doi: 10.1158/1541-7786.MCR-08-0270. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Aguilar A, Idbaih A, Boisselier B, et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res. 2012;18:5203–5211. doi: 10.1158/1078-0432.CCR-12-0845. [DOI] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hadley D, et al. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labussière M, Di Stefano AL, Gleize V, et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br J Cancer. 2014;111:2024–2032. doi: 10.1038/bjc.2014.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edlund K, Larsson O, Ameur A, et al. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci USA. 2012;109:9551–9556. doi: 10.1073/pnas.1200019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: A novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–2739. doi: 10.1182/blood-2009-10-201848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai H, Harmanci AS, Erson-Omay EZ, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48:59–66. doi: 10.1038/ng.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinrichs S, Li C, Look AT. SNP array analysis in hematologic malignancies: Avoiding false discoveries. Blood. 2010;115:4157–4161. doi: 10.1182/blood-2009-11-203182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jasek M, Gondek LP, Bejanyan N, et al. TP53 mutations in myeloid malignancies are either homozygous or hemizygous due to copy number-neutral loss of heterozygosity or deletion of 17p. Leukemia. 2010;24:216–219. doi: 10.1038/leu.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med. 2009;15:120–128. doi: 10.1016/j.molmed.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Sahm F, Reuss D, Koelsche C, et al. Farewell to oligoastrocytoma: In situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014;128:551–559. doi: 10.1007/s00401-014-1326-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.