

Abstract
Canonical volume-regulated anion channels (VRACs) are crucial for cell volume regulation and have many other important roles, including tumor drug resistance and release of neurotransmitters. Although VRAC-mediated swelling-activated chloride currents (ICl,vol) have been studied for decades, exploration of the structure-function relationship of VRAC has become possible only after the recent discovery that VRACs are formed by differently composed heteromers of LRRC8 proteins. Inactivation of ICl,vol at positive potentials, a typical hallmark of VRACs, strongly varies between native cell types. Exploiting the large differences in inactivation between different LRRC8 heteromers, we now used chimeras assembled from isoforms LRRC8C and LRRC8E to uncover a highly conserved extracellular region preceding the second LRRC8 transmembrane domain as a major determinant of ICl,vol inactivation. Point mutations identified two amino acids (Lys-98 and Asp-100 in LRRC8A and equivalent residues in LRRC8C and -E), which upon charge reversal strongly altered the kinetics and voltage dependence of inactivation. Importantly, charge reversal at the first position also reduced the iodide > chloride permeability of ICl,vol. This change in selectivity was stronger when both the obligatory LRRC8A subunit and the other co-expressed isoform (LRR8C or -E) carried such mutations. Hence, the C-terminal part of the first extracellular loop not only determines VRAC inactivation but might also participate in forming its outer pore. Inactivation of VRACs may involve a closure of the extracellular mouth of the permeation pathway.
Keywords: anion transport; chloride channel; electrophysiology; gating; ion channel; structure-function; ICl,swell; VSOAC; VSOR
Introduction
Volume-regulated anion channels (VRACs)4 are an important part of the molecular machinery involved in cellular volume homeostasis. They open upon cell swelling and allow for the extrusion of intracellular chloride and osmolytes during regulatory volume decrease (1–3). The current ascribed to VRACs, termed volume-activated anion current (ICl,vol), is found in all vertebrate cell types (4). Besides their well described role in cell volume regulation, VRACs have been implicated in many physiological processes, such as cell migration and proliferation, apoptosis, cell cycle maintenance, and release of extracellular signaling molecules (1, 4–6). Further evidence linked VRACs to several pathologies, such as cancer and ischemic stroke (5, 7). However, the lack of knowledge of the underlying protein(s) forming these channels has greatly hampered further exploration and verification of their suggested physiological and pathophysiological roles. Only recently, LRRC8 (leucine-rich repeat-containing 8) family proteins, which display four putative transmembrane domains and a large C terminus with up to 17 leucine-rich repeats, have been identified as essential components of VRACs (8, 9), and LRRC8 heteromers very likely form the channel pore (6, 8, 10). The physiological importance of VRACs is underscored by the drastically increased lethality of constitutive Lrrc8a KO mice that display multiple tissue abnormalities (11). Moreover, recent studies revealed a role of VRACs in the uptake of certain antibiotics (12) and platinum-based anticancer drugs like cisplatin and in tumor drug resistance (6).
The biophysical properties of ICl,vol have been described extensively (4, 5, 13). VRACs are closed under isotonic conditions and open slowly upon hypotonic cell swelling (4). Its permeability sequence (SCN− > I− > NO3− > Br− > Cl− > F− > gluconate− > aspartate−) corresponds to an Eisenmann type I sequence and has been an important criterion to distinguish ICl,vol from other anion currents (4, 13). ICl,vol displays a weak outward rectification, predominantly a property of the pore, as suggested by single channel recordings (14–16). Time- and voltage-dependent current inactivation at membrane potentials above 40 mV is a hallmark of ICl,vol (4, 17). This process is distinct from the spontaneous closure of VRACs upon prolonged hypotonicity and has been ascribed to a decrease in channel open probability (14, 16). Recovery from inactivation is facilitated by negative membrane potentials (18).
Although inactivation could be observed for almost all currents that have been referred to as ICl,vol, vastly different kinetics and voltage dependences were reported in different tissues and cell lines, yet the characteristic selectivity sequence remained unchanged (4, 17). This observation has led to the proposal that differently inactivating currents might be carried by distinct channel isoforms (4). The recent discovery that ICl,vol requires heteromerization of LRRC8A with at least one other LRRC8 isoform and that the subunit composition of VRACs determines their inactivation kinetics and voltage dependence (8) provided compelling vindication of this hypothesis. Co-expression of LRRC8A with LRRC8E in LRRC8−/− cells (in which all five LRRC8 genes are disrupted) yielded currents with fast inactivation kinetics, whereas LRRC8A and LRRC8C elicited slowly inactivating currents. Differences in endogenous expression levels of LRRC8C and LRRC8E correlated well with differences in ICl,vol inactivation kinetics (6, 8). Based on a weak but significant homology of LRRC8 proteins to pannexins (19), it has been proposed that they might assemble into hexamers (8), which, however, remains to be confirmed experimentally. A hetero-hexameric assembly from up to five different LRRC8 isoforms would easily allow for the observed complexity of VRAC inactivation properties.
Here, we sought to elucidate the molecular basis for the differences in VRAC inactivation kinetics. Using chimeras between different LRRC8 isoforms, we identified residues in the first extracellular loop as main determinants for differential inactivation properties. Importantly, charge-reversing mutations in this region significantly reduced I− > Cl− selectivity of ICl,vol, suggesting that the highly conserved C-terminal part of the first extracellular loop of LRRC8 proteins contributes to the outer mouth of the VRAC pore.
Results
In LRRC8−/− HCT116 cells in which all five LRRC8 genes are disrupted, ICl,vol can be rescued by co-expressing LRRC8A together with at least one other LRRC8 isoform (8). When LRRC8A is combined with either LRRC8C or LRRC8E, the differences in current inactivation are striking (8); whereas LRRC8A/C yielded currents that did not inactivate at potentials more negative than ∼60 mV, LRRC8A/E-mediated currents showed pronounced inactivation already at 40 mV (Fig. 1A). We used the ratio of current after 2 s and peak current (I2s/Imax) to assess the voltage dependence of inactivation. This yielded a ∼40-mV difference in the potential of half-maximal inactivation (V50) between LRRC8A/C- and LRRC8A/E-mediated currents (Fig. 1B). Inactivation conferred by LRRC8E saturated after 2 s at potentials of 50 mV or higher (with currents decreased to ∼15% of initial value), whereas the inactivation mediated by LRRC8C apparently did not reach steady state at positive potentials when examined by 2-s pulses (Fig. 1, A and B). However, when studied with much longer depolarizing pulses, ICl,vol mediated by LRRC8A/C inactivated, just like LRRC8A/E currents, to around 15% of peak current at steady state (Fig. 1, D and E). Inactivation kinetics was quantified by fitting exponentials to decaying ICl,vol currents at different voltages. In contrast to wild-type ICl,vol, which is probably mediated by a complex mixture of different LRRC8 heteromers, the current decay of fixed LRRC8 combinations could be adequately described by monoexponential functions (Fig. 1A). The time constants of inactivation (τinactivation) were voltage-dependent and generally much smaller for LRRC8A/E than LRRC8A/C heteromers. τinactivation of LRRC8A/E reached steady-state at voltages more positive than ∼80 mV, whereas LRRC8A/C currents did not reach similar inactivation time constants even at 120 mV (Fig. 1C).
FIGURE 1.

Differences in inactivation of ICl,vol mediated by LRRC8A/C and LRRC8A/E heteromers. A, representative swelling-activated current traces of LRRC8−/− HCT116 cells transfected with LRRC8A and the indicated isoform in response to 2-s voltage steps between −100 and 100 mV in 20-mV increments (top) or to the voltages indicated (bottom). Cells were held at −80 mV for 500 ms before and after voltage steps to allow for complete recovery from inactivation. In the bottom panels, only the outward component of the current is shown. Light lines are from monoexponential fits to the current decay. B, left, ICl,vol inactivation assessed by ratio of current at end (I2s) and beginning (Imax) of 2-s pulse. Solid lines, Boltzmann fits. Right, V50 values from Boltzmann fits. C, inactivation time constants (τinactivation) obtained from monoexponential fits to ICl,vol current traces in response to 2-s pulses. WT currents could not be adequately fitted by monoexponentials and are thus absent. D, representative ICl,vol traces. Cells were held at the indicated voltages until steady-state inactivation was reached. E, relative steady-state ICl,vol (Iss/Imax) as determined from long depolarizing pulses. F, representative currents recovering from inactivation. Currents were inactivated to near steady state with 4-s (LRRC8C) or 1-s pulses (LRRC8E) to 100 mV (not shown) and recovered during 1-s pulses to −120, −100, −80, and −60 mV. Light lines are double-exponential fits to the currents. G, recovery time constants from double exponential fits to currents as shown in F. All experiments were repeated in 5–10 cells. Error bars, S.E.
We next asked whether also hyperpolarization-facilitated recovery from inactivation differs between LRRC8A/C- and LRRC8A/E-mediated currents. Directly after inactivating channels to near steady state with 1-s (LRRC8A/E) or 4-s pulses (LRRC8A/C) to 100 mV, 1-s pulses to voltages from −120 to −60 mV in 20-mV increments were applied. These pulses elicited rapidly increasing inward currents (Fig. 1F), which reflect the reopening of inactivated VRACs and can be fitted by double exponentials (14). Such fits (Fig. 1F) yielded voltage-dependent time constants that were only slightly different between LRRC8A/C and LRRC8A/E (Fig. 1G). Hence, whereas inactivation is drastically faster in LRRC8A/E than in LRRC8A/C channels, both heteromers recover from inactivation on a similar time scale.
To exclude artifacts that might occur upon heterologous overexpression of LRRC8 isoforms in LRRC8−/− cells, we used genomic editing to generate HCT116 cells in which LRRC8B, -D, and -E (LRRC8(B,D,E)−/−) or LRRC8B, -C and -D (LRRC8(B,C,D)−/−) were disrupted in parallel (Tables 1 and 2). These cells can only express LRRC8A/C and LRRC8A/E heteromers, respectively. Both cell lines gave robust swelling-activated currents, but current densities were ∼50% lower in LRRC8(B,D,E)−/− than in WT cells (data not shown). This may relate to relatively high LRRC8E expression levels in HCT116 cells (8). Current inactivation in those triple knock-out cells was strikingly similar to that observed in the corresponding overexpression experiments (Fig. 1, A–C), with ICl,vol of LRRC8(B,C,D)−/− and LRRC8(B,D,E)−/− cells inactivating fast and at mildly positive potentials or slow and at strongly depolarized potentials, respectively (Fig. 1, B and C). However, there was a left shift of the voltage dependence of inactivation parameters in both cell lines when compared with the equivalent overexpression experiments (Fig. 1, B and C). This might be due to different LRRC8A/LRRC8X (with X = B, C, D, or E) stoichiometries in overexpressing cells compared with cells expressing the subunits from endogenous promoters.
TABLE 1.
Clonal cell lines with disrupted LRRC8 genes
a, allele (only given if alleles differed in modifications); fs, frameshift; nt, nucleotide; TMD, transmembrane domain; Δ, deletion. Indicated nucleotide numbers give nucleotide position within the ORF.
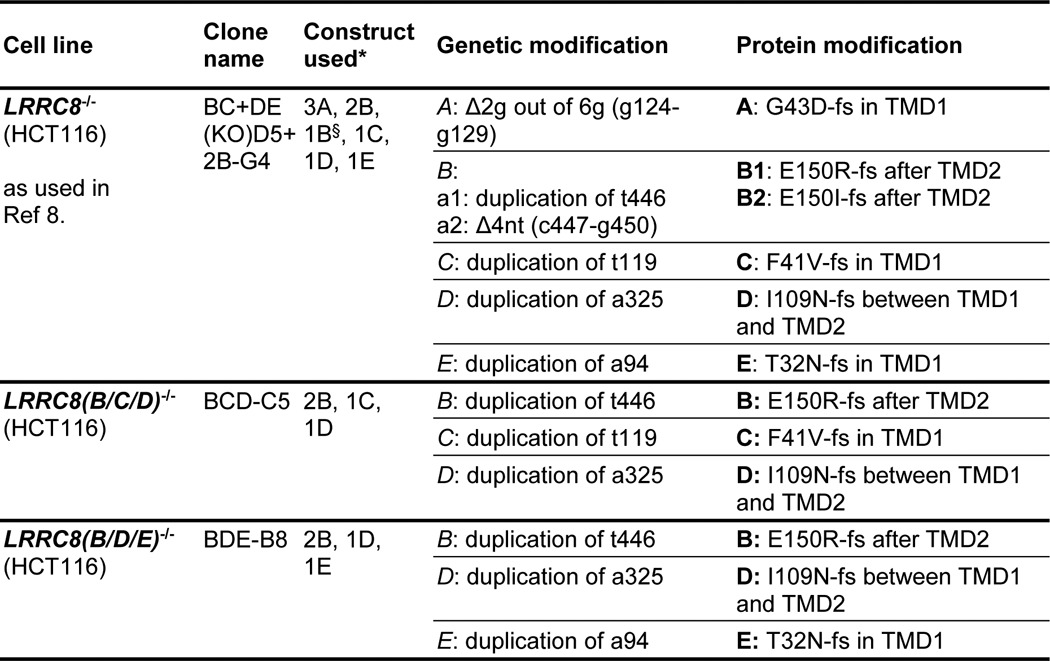
TABLE 2.
Guide sequences used for the generation of knock-out cell lines with the CRISPR/Cas9 system
PAM sequences are underlined. aa, amino acids; TMD, transmembrane domain.
| Target gene | Construct | Guide sequence (5′ → 3′) | Targeting strand | Target location in protein |
|---|---|---|---|---|
| LRRC8A | 1A | ggctgatgtagaaggacgccagg | − | aa 320–328 (beginning of TMD4) |
| 3A | tgatgattgccgtcttcgggggg | + | aa 36–43 (in TMD2) | |
| 4A | tcctgcaatgattcgttccgggg | + | aa 64–71 (between TMD1 and TMD2) | |
| LRRC8B | 1B | tttttctcttaacgcctcaaagg | − | aa 346–353 (after TMD4) |
| 2B | ggccacaaaatgctcgagcctgg | − | aa 147–354 (between TMD2 and TMD3) | |
| LRRC8C | 1C | atgctcatgatcggcgtgtttgg | + | aa 35–42 (in TMD1) |
| LRRC8D | 1D | gtggctctgagaggtatgtcagg | − | aa 107–114 (between TMD1 and TMD2) |
| LRRC8E | 1E | gctggccgagtacctcaccgtgg | + | aa 27–34 (inTMD1) |
To identify regions determining ICl,vol inactivation, we constructed chimeric proteins based on LRRC8C and LRRC8E (Fig. 2A). These chimeras were co-expressed with LRRC8A in LRRC8−/− HCT116 cells. Chimeras containing the N terminus and the first transmembrane (TM) segment of LRRC8E in LRRC8C-background (chimera 1: LRRC8E(1–42)/LRRC8C(43–830)) or the N terminus, first TM segment, and first extracellular loop (ECL1) of LRRC8C (chimera 2: LRRC8C(1–125)/LRRC8E(117–796)) in an LRRC8E background mediated currents that were indistinguishable from wild-type LRRC8A/C currents when co-expressed with LRRC8A (Fig. 2, B and C). Because ECL1 was the only LRRC8C-derived segment that was common to both chimeras, this loop is probably a major determinant of ICl,vol inactivation. A chimera in which only the C-terminal one-third of ECL1 of LRRC8E was substituted by the homologous LRRC8C segment (chimera 3: LRRC8E(1–84,117–796)/LRRC8C(94–125)) also mediated LRRC8A/C-like slow inactivation (Fig. 2, B–D). Remarkably, the converse chimera, in which these residues were replaced in LRRC8C by the corresponding LRRC8E residues (chimera 4: LRRC8C(1–93,126–803)/LRRC8E(85–116)) mediated fast inactivation when co-expressed with LRRC8A, very similar to that observed in LRRC8A/E-mediated currents (Fig. 2, B–D). Hence, LRRC8E residues 85–116 largely account for fast inactivation.
FIGURE 2.

Part of the first extracellular loop determines differences in inactivation between LRRC8C and LRRC8E. A, scheme of LRRC8C/E chimeric constructs. Light blue, LRRC8E. Red, LRRC8C. B, inactivation of ICl,vol mediated by the indicated LRRC8 combinations, assessed by the ratio of current at the end/beginning of a 2-s pulse. Solid lines, Boltzmann fits. C, V50 values from Boltzmann fits shown in B. The number of cells is shown in parentheses. D, inactivation time constants (τinactivation) obtained from monoexponential fits to ICl,vol current traces in response to 2-s pulses. All experiments were repeated in 5–8 cells. Error bars, S.E.
To identify single residues in this highly conserved region (Fig. 3A) that may be responsible for the differences in inactivation, we introduced point mutations into LRRC8C that changed the respective residues to those found in LRRC8E. These mutants were expressed together with LRRC8A in LRRC8−/− cells (Fig. 3, B–D). Whereas the LRRC8C-to-LRRC8E mutations T101N (Fig. 3, B–D) as well as M96V, M114L, and R118T (data not shown) resulted in inactivation indistinguishable from LRRC8A/C, the D102N mutant exhibited faster inactivation and a substantial left shift of V50 (Fig. 3, B–D). Although T101N had no effect by itself, it further accelerated inactivation in the LRRC8C-to-LRRC8E double-mutant T101N,D102N, leading to almost LRRC8E-like inactivation (Fig. 3, B–D) as with chimera 4 (Fig. 2). The critical role of LRRC8C Asp-102 was corroborated by the converse LRRC8E-to-LRRC8C mutation N93D in LRRC8E, which exhibited slow inactivation indistinguishable from that of LRRC8C (Fig. 3, E–G) when co-expressed with LRRC8A. Lack of an effect of the LRRC8E-to-LRRC8C mutation N92T alone or within the double-mutant N92T,N93D suggested a negligible role of this LRRC8E residue.
FIGURE 3.
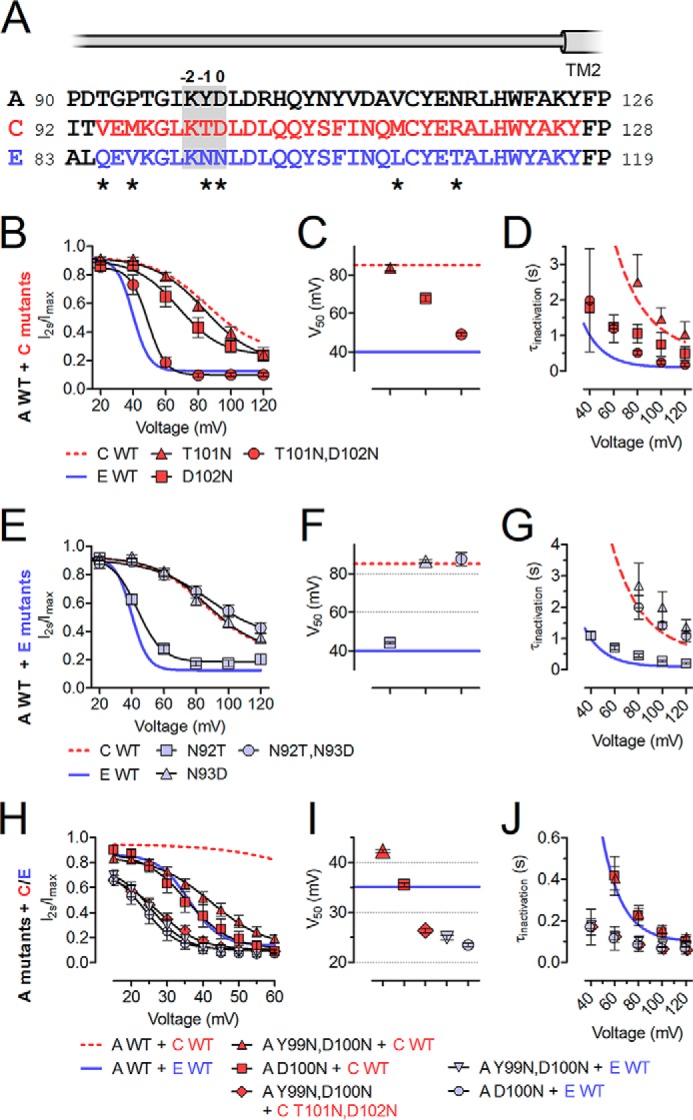
Two non-conserved residues in the C-terminal part of ECL1 affect ICl,vol inactivation. A, protein sequence alignment of the second half of ECL1 of LRRC8A/C/E. The segment swapped in chimeras 3 and 4 is highlighted in red and blue, respectively. The beginning of predicted TM2 is indicated. According to the UniProt database, TM1 in LRRC8A ends at Leu-45, whereas TM2 begins at Tyr-124. LRRC8A residue Asp-102 and the homologous residues in LRRC8C/E are designated as position 0 (indicated above the alignment together with p−1 and p−2). Residues differing between LRRC8C and -E are indicated by asterisks below. B–J, effects of point mutations on inactivation. Shown is inactivation of ICl,vol mediated by LRRC8C mutants (B–D) or LRRC8E mutants co-expressed with WT LRRC8A (E–G) and LRRC8A mutants co-expressed with WT or mutant LRRC8C/E (H–J) in LRRC8−/− HCT116 cells. B, E, and H, ICl,vol inactivation assessed by ratio of current at the end/beginning of a 2-s pulse. Solid lines, Boltzmann fits. C, F, and I, V50 values from Boltzmann fits shown in B, E, and H. D, G, and J, inactivation time constants (τinactivation) obtained from monoexponential fits to ICl,vol current traces in response to 2-s pulses. All experiments were repeated in 4–12 cells. Error bars, S.E.
For clarity, we will from now on refer to the position that had the larger influence on inactivation (LRRC8C 102 and LRRC8E 93) as position 0 (p0, Fig. 3A). We next investigated the role of LRRC8A in inactivation. LRRC8A contains an Asp at p0 (Fig. 3A), which in LRRC8C was associated with slow inactivation. We suspected that LRRC8A-to-LRRC8E mutations at this position should accelerate inactivation independent of the co-expressed isoform. Because only the double mutant LRRC8C(T101N,D102N) led to LRRC8E-like fast inactivation (Fig. 3, B–D), we first generated the LRRC8A-to-LRRC8E double mutant LRRC8A(Y99N,D100N) and co-expressed it with LRRC8E in LRRC8−/− HCT116 cells. Indeed, LRRC8A(Y99N,D100N)/E inactivated even faster than LRRC8A/E, with inactivation apparent already at ∼20 mV (Fig. 3, H–J) and a slight shift of V50 to less positive voltages (Fig. 3I). Likewise, currents mediated by LRRC8A(Y99N,D100N)/C inactivated much faster and at less positive potentials than LRRC8A/C (Fig. 3, H–J). In accord with LRRC8A/C(T101N,D102N) showing LRRC8A/E-like fast inactivation (Fig. 3, B–D), LRRC8A(Y99N,D100N)/C(T101N,D102N) inactivated like LRRC8A(Y99N,D100N)/E channels (Fig. 3, H–J). LRRC8A(D100N) and LRRC8A(Y99N,D100N) displayed almost equal inactivation in heteromers with either LRRC8C or -E (Fig. 3, H–J), revealing the dominant role of the p0 residue.
We next mutated the p0 residue in both LRRC8E and LRRC8A to amino acids that are not present at this position in any LRRC8 isoform and co-expressed both mutants in LRRC8−/− cells. Substitution by Ala drastically accelerated inactivation between 30 and 70 mV, an effect that was even more pronounced when positively charged Lys or Arg were inserted (Fig. 4, A–D). Substitution for negatively charged Glu resulted in slow inactivation comparable with that observed with LRRC8A/C or LRRC8A/E(N93D) (Figs. 4 (A–D) and 3 (E–G), respectively). Qualitatively similar but generally less pronounced effects were obtained when mutants of LRRC8A or LRRC8E were co-expressed with the corresponding wild-type partners or when equivalent mutations in LRRC8A and LRRC8C were co-expressed (data not shown). We conclude that residues at p0 are central determinants of ICl,vol inactivation. Positive and, to a lesser extent, uncharged amino acids in this position led to inactivation that rapidly reached steady state even at mildly positive membrane potentials. In contrast, negative charges caused inactivation to set in only at strongly depolarized potentials, as observed with the LRRC8A/C combination. Similar inactivation properties in Lys and Arg or in Ala and Asn mutants, respectively, indicate that side chain volume is less important (Fig. 4, A–D).
FIGURE 4.

Charged side chains of residues at p0 and p−2 drastically affect ICl,vol inactivation. Shown are the effects of p0 (A–D) and p−2 (E–H) mutations on ICl,vol inactivation. A and E, example ICl,vol currents at the indicated voltages mediated by the indicated LRRC8A/LRRC8E mutants co-expressed in LRRC8−/− HCT116 cells. B and F, ICl,vol inactivation assessed by ratio of current at the end/beginning of a 2-s pulse. Solid lines, Boltzmann fits. C and G, inactivation time constants (τinactivation) obtained from monoexponential fits to ICl,vol current traces in response to 2-s pulses. D and H, current densities of maximally activated ICl,vol (at −60 mV) mediated by the indicated p0 (D) or p−2 (H) mutant combinations. All p0 and p−2 mutants tested gave rise to robust swelling-activated currents of a magnitude similar to that observed in WT. All experiments were repeated in 6–12 cells. Error bars, S.E. n.t., not transfected.
Given the dramatic effects of charged amino acids at p0, we next investigated the role of the conserved Lys at p−2 (Fig. 3A). Charge reversal or neutralization of this residue in LRRC8A and LRRC8E strongly altered inactivation when both mutants were co-expressed in LRRC8−/− cells, whereas a charge-conserving mutation to Arg exerted only a minor decelerating effect (Fig. 4, E–H). In Ala and Asn substitutions, inactivation appeared to be almost absent save for a very fast component that was detected only at the most strongly depolarized potentials (Fig. 4, E–H). Very fast inactivation was also observed when negatively charged Glu was introduced (Fig. 4, E–H). The inactivation of currents from p−2 mutants displayed a very shallow voltage dependence and appeared to saturate at larger remaining currents than observed with WT combinations (>40% of current left versus ∼15% for WT). Co-expression of LRRC8A wild type or p−2 mutants with LRRC8C wild type or p−2 mutants gave results indistinguishable from those obtained with the equivalent LRRC8A/E combinations (data not shown).
We next asked whether p−2 mutations alter the characteristic I− > Cl− selectivity of VRACs. Cells in which ICl,vol was maximally activated were superfused with solutions in which Cl− was substituted by I−, and the resulting shifts in reversal potential (ΔErev) were used to calculate relative permeabilities. Whereas the mutant combinations LRRC8A(K98A)/E(K91A) and LRRC8A(K98N)/E(K91N) did not alter ΔErev compared with the WT combination, ΔErev was significantly smaller for the charge reversal mutants LRRC8A(K98E)/E(K91E), reducing the relative permeability (PI/PCl) from 1.25 to 1.12 (Fig. 5, A–C). None of these mutations significantly changed the NO3−, gluconate, or Na+ permeability of VRACs (Fig. 5B). When only one of the two co-expressed LRRC8 subunits contained the p−2 Lys-to-Glu mutation, ΔErev was only significantly altered in LRRC8A/E(K91E), and not in LRRC8A(K98E)/E, suggesting a larger contribution of LRRC8E to the selectivity of the pore (Fig. 5D). Introduction of p−2 Lys-to-Glu mutations also altered ΔErev in the LRRC8A + C combination (Fig. 5D). As reported by ourselves as well as others (8, 10), ΔErev upon iodide substitution was not significantly different between LRRC8A/C and LRRC8A/E heteromers (Fig. 5D). We also investigated whether p0 mutations affect ICl,vol selectivity but did not find significant changes in PI/PCl or PNMDG/PNa (Fig. 5E).
FIGURE 5.
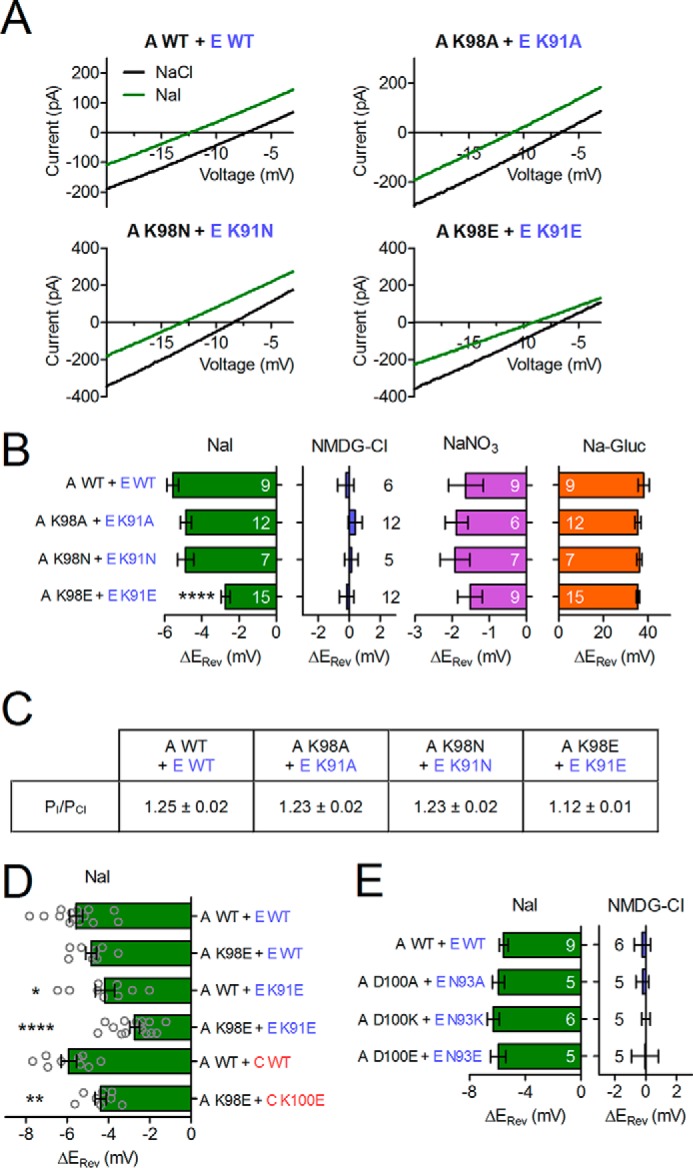
Charge reversal of the conserved Lys in p−2 affects ICl,vol anion selectivity. A, example ICl,vol current traces in response to voltage ramps. B, D, and E, relative ΔErev upon substitution of NaCl with the indicated substances. The number of cells is indicated (B and E), or single experiments are plotted (D). Error bars, S.E. *, p < 0.05; **, p < 0.01; ****, p < 0.0001; one-way analysis of variance, Bonferroni's test. C, relative iodide permeability as calculated from ΔErev.
Discussion
Voltage-dependent inactivation, defined as spontaneous decay of currents after the establishment of a certain transmembrane voltage, is a hallmark of many ion channels. It is most often due to a closure of the channel pore. Whereas fast inactivation is crucial for certain cation channels involved in action potential generation, its biological role is less clear for ion channels like VRACs whose functions are normally not related to fast voltage changes. Voltage-dependent inactivation has been studied in great detail over several decades for various voltage-dependent K+ and Na+ channels. Fast N-type inactivation of Kv channels involves a cytoplasmic N-terminal blocking particle that binds to the intracellular opening of the pore (20, 21), whereas the mechanism of the slower C-type inactivation is less clear but may involve a constriction of the channel pore (22). In contrast, nothing was known about the structural basis of the inactivation of VRACs, the molecular composition of which remained unknown until recently. We now identified amino acids in the highly conserved extracellular stretch preceding the second transmembrane domain as key determinants of inactivation. Mutations in one of these residues also changed the ion selectivity of VRACs, suggesting that this stretch forms part of the extracellular opening of the ion-conducting pore that might constrict with channel inactivation.
We previously attributed differences in voltage-dependent inactivation of native ICl,vol (4) to tissue-specific expression patterns of LRRC8 isoforms (8). Analysis of cultured cells expressing the obligatory LRRC8A together with particular other LRRC8 isoforms showed that differences in inactivation were largest between channels containing LRRC8C or -E, which induced the slowest and fastest inactivation of ICl,vol, respectively (8). Co-expression of different LRRC8C/E chimeras with LRRC8A now identified the C-terminal half of the first extracellular loop (ECL1) as a major determinant of VRAC inactivation. In this stretch, we identified the negatively charged Asp-102 of LRRC8C, which is replaced by uncharged Asn at this position (p0) in LRRC8E, as a key determinant of channel inactivation. Mutations at p−1 modulated the effect of the p0 mutation on inactivation in LRRC8C but not in the other subunits tested. The charge, rather than size, of the residue at p0 strongly influenced the rate and voltage dependence of ICl,vol inactivation. Likewise, the charge at p−2 (Lys in all LRRC8 isoforms but LRRC8B) markedly affected the voltage dependence and kinetics of inactivation but surprisingly also the ion selectivity of VRAC.
The strong influences of charges at positions p−2 and p0 on voltage-dependent inactivation raise the question of whether they directly act as voltage sensors. A positive charge at p0 accelerated channel inactivation and shifted its voltage dependence to the left, whereas negative charges at p−2 shifted the voltage dependence to the right but also accelerated inactivation. The similar effect of positive charges at positions p−2 and p0 on the voltage dependence evokes the hypothesis that a putative, and possibly pore-forming, re-entrant loop in the stretch delimited by the glycosylation site and the predicted extracellular end of the second transmembrane span (Asn-83 and Tyr-126, respectively, in LRRC8A) dips into the electric field of the outer mouth of the channel and causes inactivation by an electrostatically induced movement in response to depolarization. However, the observation that neutralization of the positive charge at p−2 leads to a more pronounced right shift of the voltage dependence of inactivation than charge reversal is difficult to reconcile with this hypothesis, as is the similar effect of opposite charges at p0 and p−2 on inactivation kinetics.
Inactivation of ICl,vol was not complete, but saturated, depending on the particular channel composition, at maximum values of 80–95% that were asymptotically reached at long times and strong depolarization. Even less complete inactivation was observed with p−2 mutants. Previous single-channel recordings of native ICl,vol currents suggested that voltage-dependent inactivation involves the stepwise closure of single VRACs that display a single-channel conductance of 50–80 picosiemens at positive potentials (14, 16, 18). Therefore, the current remaining after maximal inactivation may represent the spontaneous flickering of such channels with a low average open probability. However, the observation that LRRC8A/C and LRRC8A/E channels display similar steady-state inactivation while showing markedly different time constants for entering, but not for leaving inactivated states, argues against the possibility that steady-state inactivation represents an equilibrium of direct transitions between an open and an inactivated state. Moreover, Syeda et al. (10) recently reported that bona fide single VRACs display a spectrum of single channel conductance levels even in cells expressing LRRC8A and only one of the other LRRC8 isoforms. Although not observed in any of the previous studies on VRAC single-channel properties (14, 16, 18), we cannot exclude the possibility that inactivation may also lead to a partial closure of the pore that would result in a reduced single channel conductance.
It is instructive to compare our results with those obtained for pannexins and connexins. LRRC8 proteins display weak but significant sequence homology to pannexin channels (19), which in turn display structural similarity, but no sequence homology, to gap junction-forming connexins. The highest degree of homology between pannexins and LRRC8 proteins is found in TM1 and TM2 (19), which in connexins line the pore, as revealed by high resolution structures (24). The homology between pannexins and LRRC8 channels extends about 10 residues beyond the predicted end of TM1 into ECL1. By contrast, there is no significant homology between both channel classes in the C-terminal part of ECL1 (19), and pannexin hemichannel currents lack conspicuous inactivation (25). Structure-function studies have indicated that besides TM1, ECL1 is important for the permeation of both pannexins (26, 27) and connexins (28, 29). For instance, a change from anion- to cation-preferring hemichannels was observed when ECL1 of connexin32 was replaced by that from connexin46 (29). Moreover, the crystal structure of connexin26 showed that ECL1 contributes to the formation of the outer pore funnel that contacts the hemichannel from the other cell (24). Cysteine-scanning mutagenesis of LRRC8A performed on parts of all transmembrane domains of LRRC8A (9) had identified Thr-44 close to the end of TM1, which, when mutated to cysteine, moderately increased the I− > Cl− permeability of VRACs (9, 10). However, charge-introducing mutations at this position had only negligible effects, and the low currents observed with the T44C mutant (9) increase the risk of a contamination by background currents. By contrast, the Lys to Glu mutations at p−2 described here did not reduce current densities and decreased rather than increased the I− > Cl− permeability of VRACs. As expected for a pore-lining residue, the change in selectivity was larger when both co-expressed subunits carried the mutation. When also considering the permeation of organic substrates, the largest changes in selectivity observed so far were between VRACs containing or lacking the LRRC8D subunit (6). Cells expressing only LRRC8A/D channels displayed a >10-fold higher cisplatin/Cl− transport ratio than LRRC8A/C channels, and LRRC8D also increased the permeation of the mostly zwitterionic sulfo-amino acid taurine, an important osmolyte (6). In comparison with LRRC8A/C channels, LRRC8A/D channels were recently reported to have lower single channel conductances and, conflicting with our previous work (8), a somewhat lower I− > Cl− permeability (10). Although we cannot attribute these changes to the residue at p−2, which is Lys in LRRC8A, -C, and -D, the most salient difference between LRRC8D and the other LRRC8 isoforms is the insertion of a 41-residue-long stretch between the conserved parts of ECL1 in LRRC8D. It is therefore tempting to speculate (23) that the long ECL1 of LRRC8D plays a role in shaping the rather exceptional permeation properties of LRRC8D-containing VRACs.
Our work has revealed that the portion of the first extracellular loop that directly precedes TM2 and that is highly conserved over a length of 30 amino acids in all mammalian LRRC8 isoforms plays a pivotal role in the voltage-dependent inactivation of VRACs. Unexpectedly, mutations in this region also change the ion selectivity of VRAC and hence suggest that it is in contact with the permeation pathway. Hence, VRAC inactivation might involve a constriction of the pore close to its extracellular opening. However, high resolution VRAC structures are necessary to test the feasibility of this and other hypotheses.
Experimental Procedures
Expression Constructs
Human LRRC8A (RefSeq accession number NM_019594; in pcDNA3.1), LRRC8C (RefSeq accession number NM_032270; in pEGFP-N1), and LRRC8E (RefSeq accession number NM_025061; in pEGFP-N1) were used as described previously (8). Mutants and chimeras were generated using PCR-based strategies. All constructs were confirmed by sequencing the complete ORF.
Cell Culture and Transfection
HCT116 cells were maintained in McCoy's 5A medium (PAN Biotech) supplemented with 10% FBS (PAN Biotech) and 1% penicillin/streptomycin at 37 °C and 5% CO2. For experiments, cells were plated onto gelatin-coated coverslips and transfected using the Lipofectamine 2000 (Life Technologies, Inc.) transfection reagent. Cells were transfected with plasmids encoding LRRC8A and LRRC8C-GFP or LRRC8E-GFP and with identical CMV-based promotors at 1:1 ratios to ensure similar expression levels. Because LRRC8A is required for trafficking of LRRC8B through LRRC8E to the plasma membrane, plasma membrane fluorescence indicated correct expression of the untagged LRRC8A (8).
Electrophysiology
Whole-cell voltage clamp experiments were performed as described (6, 8). The hypotonic bath solution used to elicit ICl,vol contained 105 mm NaCl, 6 mm CsCl, 1 mm MgCl2, 1.5 mm CaCl2, 10 mm glucose, 10 mm HEPES, pH 7.4, with NaOH (240 mosm). The pipette solution contained 40 mm CsCl, 100 mm cesium methanesulfonate, 1 mm MgCl2, 1.9 mm CaCl2, 5 mm EGTA, 4 mm Na2ATP, and 10 mm HEPES, pH 7.2, with CsOH (290 mosm). Currents were recorded with an EPC-10 USB patch clamp amplifier and PatchMaster software (HEKA Elektronik). Relative anion permeabilities (PX/PCl) were calculated from the shifts in reversal potential using a modified Goldman-Hodgkin-Katz equation as described previously (8). Time constants of inactivation (τi or τinactivation) were obtained by approximating the current decay over 2 s versus time with an exponential function,
 |
where Iss is the steady-state current after inactivation and Ii is the inactivating current. Time constants of recovery from inactivation (τslow or τfast) were obtained by fitting the recovering current versus time with a double-exponential function,
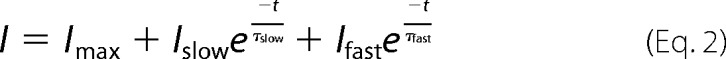 |
where Imax is the fully recovered current, and Islow and Ifast are the currents recovered through the slow and fast components, respectively.
Generation of Monoclonal Knock-out Cell Lines
Cell lines with disruptions in LRRC8 genes were generated using CRISPR/Cas9 as described previously (8). Briefly, target single guide RNAs were cloned into the px330 vector and co-transfected with GFP into HCT116 cells using the Fugene HD transfection reagent. 2–5 days post-transfection, single GFP-positive cells were FACS-sorted into 96-well plates containing McCoy's 5A medium. Monoclonal cell lines were raised and tested for sequence alterations using target site-specific PCR on genomic DNA followed by Sanger sequencing. To generate multiple KOs of several genes, the respective plasmids were transfected together, or cell lines already carrying LRRC8 gene disruptions were targeted again for other LRRC8 genes.
Author Contributions
F. U., S. M. R., and T. J. J. designed the study. F. U., S. M. R., and T. J. J. wrote the paper. F. U. and S. M. R. designed, performed, and analyzed electrophysiological experiments. F. K. V. generated and validated knockout cell lines. F. U., S. M. R., and T. S. designed and cloned mutant and chimeric constructs. All authors approved the final version of the manuscript.
Acknowledgment
We thank J. Liebold for technical assistance.
This work was supported by European Research Council (ERC) Advanced Grant (FP/2007–2013) 294435 “Cytovolion” and the DFG (Exc 257 “Neurocure”) (to T. J. J.). The authors declare that they have no conflicts of interest with the contents of this article.
- VRAC
- volume-regulated anion channel
- ICl,vol
- volume-activated anion current
- TM
- transmembrane
- ECL
- extracellular loop
- ΔErev
- shift in reversal potential.
References
- 1. Hoffmann E. K., Lambert I. H., and Pedersen S. F. (2009) Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277 [DOI] [PubMed] [Google Scholar]
- 2. Pasantes-Morales H., Lezama R., Ramos-Mandujano G., and Tuz K. (2006) Mechanisms of cell volume regulation in hypo-osmolality. Am. J. Med. 119, S4–S11 [DOI] [PubMed] [Google Scholar]
- 3. Jentsch T. J. (2016) VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 17, 293–307 [DOI] [PubMed] [Google Scholar]
- 4. Nilius B., Eggermont J., Voets T., Buyse G., Manolopoulos V., and Droogmans G. (1997) Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol. 68, 69–119 [DOI] [PubMed] [Google Scholar]
- 5. Okada Y., Sato K., and Numata T. (2009) Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 587, 2141–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Planells-Cases R., Lutter D., Guyader C., Gerhards N. M., Ullrich F., Elger D. A., Kucukosmanoglu A., Xu G., Voss F. K., Reincke S. M., Stauber T., Blomen V. A., Vis D. J., Wessels L. F., Brummelkamp T. R., Borst P., Rottenberg S., and Jentsch T. J. (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 34, 2993–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okada Y., Shimizu T., Maeno E., Tanabe S., Wang X., and Takahashi N. (2006) Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J. Membr. Biol. 209, 21–29 [DOI] [PubMed] [Google Scholar]
- 8. Voss F. K., Ullrich F., Münch J., Lazarow K., Lutter D., Mah N., Andrade-Navarro M. A., von Kries J. P., Stauber T., and Jentsch T. J. (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638 [DOI] [PubMed] [Google Scholar]
- 9. Qiu Z., Dubin A. E., Mathur J., Tu B., Reddy K., Miraglia L. J., Reinhardt J., Orth A. P., and Patapoutian A. (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Syeda R., Qiu Z., Dubin A. E., Murthy S. E., Florendo M. N., Mason D. E., Mathur J., Cahalan S. M., Peters E. C., Montal M., and Patapoutian A. (2016) LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell 164, 499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar L., Chou J., Yee C. S. K., Borzutzky A., Vollmann E. H., von Andrian U. H., Park S.-Y., Hollander G., Manis J. P., Poliani P. L., and Geha R. S. (2014) Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. J. Exp. Med. 211, 929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee C. C., Freinkman E., Sabatini D. M., and Ploegh H. L. (2014) The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. J. Biol. Chem. 289, 17124–17131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pedersen S. F., Klausen T. K., and Nilius B. (2015) The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol. 213, 868–881 [DOI] [PubMed] [Google Scholar]
- 14. Voets T., Droogmans G., and Nilius B. (1997) Modulation of voltage-dependent properties of a swelling-activated Cl− current. J. Gen. Physiol. 110, 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackson P. S., and Strange K. (1996) Single channel properties of a volume sensitive anion channel: lessons from noise analysis. Kidney Int. 49, 1695–1699 [DOI] [PubMed] [Google Scholar]
- 16. Okada Y., Petersen C. C., Kubo M., Morishima S., and Tominaga M. (1994) Osmotic swelling activates intermediate-conductance Cl− channels in human intestinal epithelial cells. Jpn. J. Physiol. 44, 403–409 [DOI] [PubMed] [Google Scholar]
- 17. Okada Y. (1997) Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. 273, C755–C789 [DOI] [PubMed] [Google Scholar]
- 18. Jackson P. S., and Strange K. (1995) Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J. Gen. Physiol. 105, 661–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abascal F., and Zardoya R. (2012) LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 34, 551–560 [DOI] [PubMed] [Google Scholar]
- 20. Hoshi T., Zagotta W. N., and Aldrich R. W. (1990) Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538 [DOI] [PubMed] [Google Scholar]
- 21. Hoshi T., Zagotta W. N., and Aldrich R. W. (1991) Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7, 547–556 [DOI] [PubMed] [Google Scholar]
- 22. Cuello L. G., Jogini V., Cortes D. M., and Perozo E. (2010) Structural mechanism of C-type inactivation in K+ channels. Nature 466, 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voets T., Nilius B., and Vennekens R. (2015) VRACs swallow platinum drugs. EMBO J. 34, 2985–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maeda S., Nakagawa S., Suga M., Yamashita E., Oshima A., Fujiyoshi Y., and Tsukihara T. (2009) Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 458, 597–602 [DOI] [PubMed] [Google Scholar]
- 25. Barbe M. T., Monyer H., and Bruzzone R. (2006) Cell-cell communication beyond connexins: the pannexin channels. Physiology 21, 103–114 [DOI] [PubMed] [Google Scholar]
- 26. Wang J., and Dahl G. (2010) SCAM analysis of Panx1 suggests a peculiar pore structure. J. Gen. Physiol. 136, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu F., Wang J., and Dahl G. (2012) Alanine substitution scanning of pannexin1 reveals amino acid residues mediating ATP sensitivity. Purinergic Signal. 8, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trexler E. B., Bukauskas F. F., Kronengold J., Bargiello T. A., and Verselis V. K. (2000) The first extracellular loop domain is a major determinant of charge selectivity in connexin46 channels. Biophys. J. 79, 3036–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kronengold J., Trexler E. B., Bukauskas F. F., Bargiello T. A., and Verselis V. K. (2003) Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J. Gen. Physiol. 122, 389–405 [DOI] [PMC free article] [PubMed] [Google Scholar]