

Abstract
Anaerobic ammonium-oxidizing (anammox) bacteria derive their energy for growth from the oxidation of ammonium with nitrite as the electron acceptor. N2, the end product of this metabolism, is produced from the oxidation of the intermediate, hydrazine (N2H4). Previously, we identified N2-producing hydrazine dehydrogenase (KsHDH) from the anammox organism Kuenenia stuttgartiensis as the gene product of kustc0694 and determined some of its catalytic properties. In the genome of K. stuttgartiensis, kustc0694 is one of 10 paralogs related to octaheme hydroxylamine (NH2OH) oxidoreductase (HAO). Here, we characterized KsHDH as a covalently cross-linked homotrimeric octaheme protein as found for HAO and HAO-related hydroxylamine-oxidizing enzyme kustc1061 from K. stuttgartiensis. Interestingly, the HDH trimers formed octamers in solution, each octamer harboring an amazing 192 c-type heme moieties. Whereas HAO and kustc1061 are capable of hydrazine oxidation as well, KsHDH was highly specific for this activity. To understand this specificity, we performed detailed amino acid sequence analyses and investigated the catalytic and spectroscopic (electronic absorbance, EPR) properties of KsHDH in comparison with the well defined HAO and kustc1061. We conclude that HDH specificity is most likely derived from structural changes around the catalytic heme 4 (P460) and of the electron-wiring circuit comprising seven His/His-ligated c-type hemes in each subunit. These nuances make HDH a globally prominent N2-producing enzyme, next to nitrous oxide (N2O) reductase from denitrifying microorganisms.
Keywords: electron paramagnetic resonance (EPR), enzyme kinetics, enzyme purification, nitrogen metabolism, reactive nitrogen species (RNS), anaerobic ammonium oxidation, hydrazine, octaheme proteins
Introduction
An estimated 30–70% of all N2 that is released into the atmosphere is produced by anaerobic ammonium-oxidizing (anammox)4 bacteria (1, 2), which represent one of the latest scientific discoveries in the biogeochemical nitrogen cycle. These organisms gain their energy for growth from the oxidation of ammonium, with nitrite as the electron acceptor, to produce N2. With the employment of advanced molecular tools, these bacteria have been detected in nearly every anoxic environment where fixed nitrogen compounds are degraded (3). Besides its biogeochemical and ecological relevance, the anammox process has found worldwide application in ammonium removal from wastewater as an environment-friendly and cost-effective alternative to conventional systems (4).
In our current understanding, anammox catabolism is composed of three consecutive, coupled reactions with two intermediates, nitric oxide (NO) and hydrazine (N2H4): 1) the one-electron reduction of the substrate nitrite to NO (Reaction 1); 2) the activation of the second substrate ammonium with NO and the concomitant input of three electrons to synthesize N2H4 (Reaction 2); and 3) the oxidation of hydrazine, the most powerful reductant in nature, to N2 (Reaction 3) (5–7).
 |
The four electrons that are released during hydrazine oxidation then drive the reduction reactions (Reactions 1 and 2).
The enzyme that catalyzes hydrazine oxidation and thus effectively produces about half of all N2 emitted into the atmosphere is called hydrazine dehydrogenase (HDH) or hydrazine-oxidizing enzyme. In our previous work, we identified HDH from Kuenenia stuttgartiensis (KsHDH) as the gene product of kustc0694 and determined some of its catalytic properties (5). A hydrazine-oxidizing enzyme highly related to kustc0694 was isolated before from the anammox enrichment culture KSU-1, but the exact reaction catalyzed by it has so far remained unknown (8). In the genome of K. stuttgartiensis, kustc0694 is one of the 10 paralogs of hydroxylamine oxidoreductase (HAO)-like proteins (3, 6). HAO-like proteins have two structurally well characterized representatives, NeHAO from Nitrosomonas europaea (9–11) and hydroxylamine oxidase from K. stuttgartiensis (kustc1061; here denoted as KsHOX) (12). NeHAO is a key enzyme in aerobic ammonium-oxidizing bacteria, which catalyzes the four-electron oxidation of hydroxylamine to nitrite (Reaction 4), whereas KsHOX is a dominant protein in the anammox bacterium catalyzing the three-electron oxidation of hydroxylamine to NO (Reaction 5).
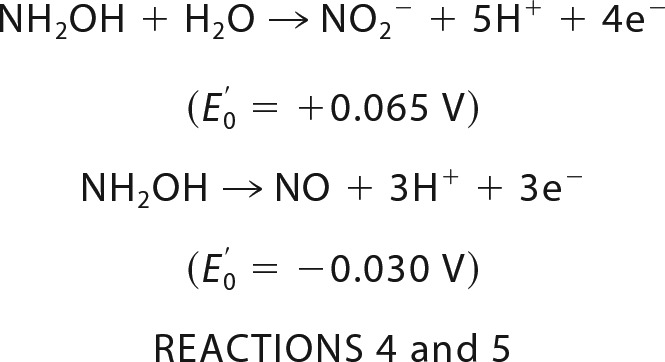 |
Despite a limited sequence identity (∼30% at the amino acid level), NeHAO and KsHOX are structurally highly similar (10–12). Both are homotrimeric proteins in which each monomer binds eight c-type hemes (Fig. 1). The arrangement of 24 hemes of both proteins is fully superimposable. Within a subunit, seven His/His-ligated hemes constitute an electron-wiring circuit toward an external electron acceptor (12, 13). To facilitate efficient electron transport, these hemes may be electronically coupled, resulting in highly convoluted EPR spectra of as-isolated, fully oxidized (all ferric) NeHAO (14, 15). One heme (heme 4) forms part of a structurally conserved catalytic center (Fig. 1). This heme 4 is covalently bound to a tyrosine residue from a neighboring subunit. This unusual cross-link induces a pronounced ruffling of the porphyrin plane and gives rise to a characteristic absorption band at about 460 nm in the UV-visible spectrum of the reduced protein (9, 10, 12, 14). After this absorption band, catalytic heme 4 is termed the P460 cofactor. Due to small structural changes around the P460 catalytic site (Fig. 1C), KsHOX oxidizes hydroxylamine to NO rather than to nitrite as NeHAO does (12). Interestingly, both HAO and KsHOX can use hydrazine as a substrate too (Reaction 3), although with lower catalytic efficiency (kcat/Km) than their physiological substrate, hydroxylamine (9, 12).
FIGURE 1.

Common structural elements of hydroxylamine oxidoreductase-like proteins. A, overall architecture of the homotrimeric KsHOX (PDB code 4N4L). Its three subunits are displayed in different colors. Localization of the catalytic site in one of the three subunits is indicated by the square. B, x-ray structure of the heme 4 (P460) catalytic center of KsHOX soaked with hydrazine seen from the same axis as in A. A dinitrogen species, a putative diazene (HN=NH) (blue) is seen on top of the heme. C, the table shows corresponding amino acids found in the crystal structure of NeHAO (PDB codes 1FGJ, 4FAS, 4N4N, and 4N4O) and in the amino acid sequence of hydrazine dehydrogenase from K. stuttgartiensis, together with putative functions of the amino acids during catalysis (12). D, arrangement of the c-type hemes in homotrimeric KsHOX. The figure shows the outline of the structure of KsHOX (PDB code 4N4J) seen from the bottom along the 3-fold symmetry axis featuring the 24 hemes. The 8 hemes present in the same monomer have the same color and are numbered as indicated. Note that hemes are arranged in a ringlike structure. The spatial arrangement of these hemes is fully conserved in NeHAO (10–12). Following oxidation of the substrate at the catalytic heme 4 (filled numbered circles), the generated electrons pass through heme 6, from which they may take two different routes to an exit heme 1 (dashed arrows): 1) via hemes 3 and 5 toward heme 2 and 2) via hemes 7 and 8. In the latter route, electrons are directed to heme 2 of the next subunit, enabling electron transfer between different subunits. Structural images were made using PyMOL.
Here, we characterized KsHDH in detail and identified it as a homotrimeric octaheme protein in which the three subunits are covalently bound to form a P460-like prosthetic group as found in NeHAO and KsHOX. In contrast to the two latter enzymes, the homotrimers of KsHDH themselves were found to form octamers in solution. KsHDH specifically oxidized hydrazine to N2 in accordance with Equation 3 and was inhibited by NH2OH and NO. The genome of K. stuttgartiensis harbors a close paralog of kustc0694, namely kustd1340, whereas close orthologs of kustc0694 and kustc1340 were detected in all anammox genomes sequenced thus far. Considering a close structural relationship with other HAO-like proteins, we addressed which features determine the specificity of HDH from anammox bacteria.
Results
HDH Is a Homotrimeric Protein with Covalently Bound Subunits Forming Octamers in Solution
Hydrazine dehydrogenase from K. stuttgartiensis was purified as a bright red protein that, when resolved by native PAGE, displayed one prominent band with an estimated molecular mass of ∼200–220 kDa (Fig. 2A). When resolved by SDS-PAGE, the protein remained at the top of the gel. Occasionally, a smaller band was observed that migrated with a size of ∼67 kDa (Fig. 2B, arrow). MALDI-TOF MS verified that all visible bands were derived from the same protein that had been annotated as the octaheme protein kustc0694. Notably, 18 of 83 predicted peptides in the m/z 500–4,000 range were detected after tryptic digestion, including two peptides (molecular masses 2,173.9 and 2,615.2 Da) that distinguished kustc0694 from its close paralog kustd1340 (96% sequence identity at the amino acid level; supplemental Fig. S1). Peptides comprising a CX2,4CH motif for covalent heme c binding remained beyond detection. Linear MALDI-TOF MS established a molecular mass of 201,800 ± 300 Da of the holoenzyme. This mass matched the theoretical value (201,610.5 Da) of a homotrimer with each monomer (62,271.5 Da) possessing eight bound heme c (Mr = 616.5) molecules. Thus, the high molecular mass of native KsHDH indicated that the three subunits were covalently bound to each other. Calculation of the subunit theoretical mass took into account N-terminal cleavage (after Val-107) following an alternative translation start site at Met-75 (see below; supplemental Fig. S1). Cleavage of the N-terminal leader sequence (32 amino acids) would facilitate export of HDH into the anammoxosome, an anammox-specific cell organelle where catabolism resides and where processed HDH is specifically localized, as recently shown by immunogold labeling (16).
FIGURE 2.
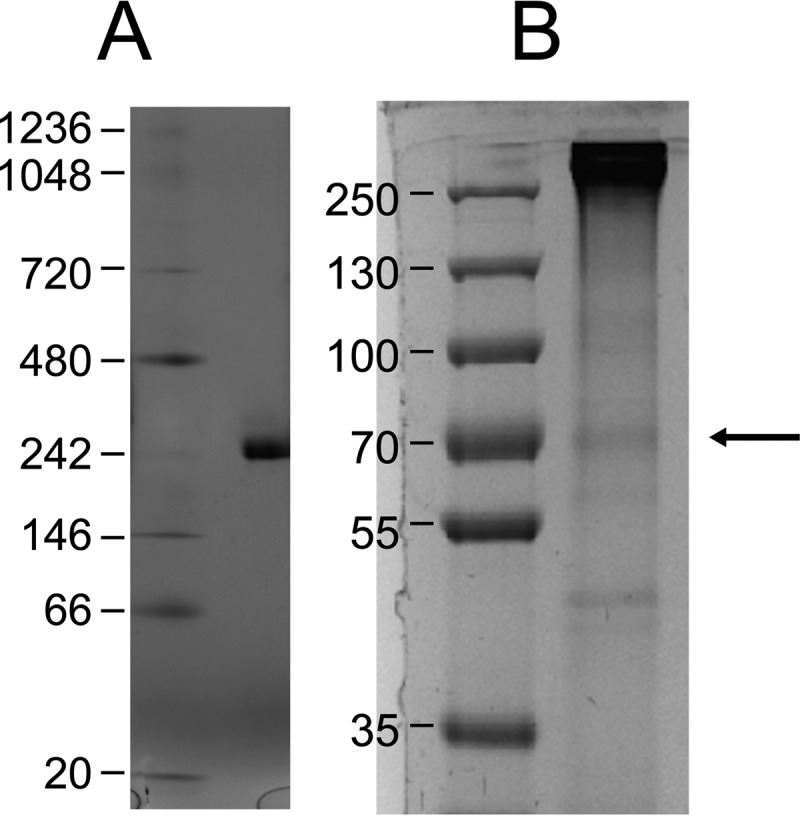
Native and SDS-denaturing PAGE of purified KsHDH. A, protein (∼30 μg) was separated on a 4–16% linear gradient blue native gel. B, 8% SDS-PAGE of KsHDH. Numbers refer to the molecular masses of ruler proteins. Note that the protein did not migrate into the SDS gels. The thin band marked by the arrow was identified as the KsHDH monomer (67 kDa).
The oligomeric state of KsHDH was corroborated by the results from sedimentation velocity analytical ultracentrifugation (AUC SV) and analytical size exclusion chromatography coupled to multiangle light scattering (SEC/MALS) analyses (Table 1 and Fig. 3, A and B). Both methods showed a prominent species corresponding to the homotrimer (α3, calculated mass of 201.6 kDa) and a monomeric species (Mr = 61,000–75,000) in minor amounts. Strikingly, both methods also revealed aggregates with molecular masses of 1,760.7 ± 7.0 and 1,731.7 kDa as determined by SEC/MALS and AUC SV, respectively (Table 1). In the AUC sedimentation profile, these aggregates represented the major peak (Fig. 3A). As judged from their molecular mass, aggregates could account for octamers of homotrimeric KsHDH (α24 = (α3)8, 1,612.9 kDa). Fractionation of the α3 and α24 peaks from SEC and separate reinjection onto the column again resulted in chromatograms featuring two major peaks with elution volumes as observed for the original sample. This indicated a dynamic equilibrium between the α3 and α24 species in solution. Transmission electron microscopy (TEM) of negatively stained KsHDH samples supported the AUC SV and SEC/MALS results and revealed assemblies with an approximate size of 16 × 16 nm, next to smaller (∼8 × 8-nm) globular particles, accounting for the homotrimers (Fig. 4A). After glutaraldehyde cross-linking and repurification by SEC, these 16 × 16-nm particles having a 4-fold symmetry were the most abundant species observed besides particles with a 5-fold symmetry in minor amounts (Fig. 4, B and D–F). The 16 × 16-nm particles were visible as tetrameric configurations and as two parallel dumbbells, probably representing orthogonal views of the same particle (Fig. 4, D and E). These large particles would then be composed of eight egg-like protein molecules, their tops pointing to each other. One may note that one such assembly would carry a calculated 192 heme c molecules. KsHOX did not migrate as higher molecular weight aggregates, neither on SEC/MALS nor on AUC SV chromatograms. TEM showed a homogeneous distribution of 8 × 8-nm particles only (Fig. 4C). Their size and shape fitted the dimensions (9 × 9 × 9 nm) determined for the tulip-shaped KsHOX homotrimeric structures (Fig. 1A) (12).
TABLE 1.
Characterization of KsHDH oligomers in solution
AUC SV coefficients are given in Svedberg units (1 S = 10−13 s). smax, maximal sedimentation coefficients at which smax = 0.00361 m⅔. The ratio of smax/s20,w in the range of 1.2–1.3 indicates a globular shape of the particles. To calculate molecular masses from the sedimentation coefficients, globular particles (smax/s20,w = 1.2) were assumed. MALS analyses accounted for any shape.
| Stoichiometry | Calculated mass | AUC*s20,w | smax | smax/s20,w | Mass from AUC SV | Mass from MALS |
|---|---|---|---|---|---|---|
| kDa | kDa | kDa | ||||
| α | 67.2 | 5 | 6.0 | 1.20 | 68 | 75.0 ± 2.0 |
| α3 | 201.6 | 10.97 | 12.4 | 1.13 | 220.7 | 182.1 ± 1.8 |
| α24 = (a3)8 | 1,612.9 | 43.33 | 49.7 | 1.15 | 1,731.7 | 1760.2 ± 7.0 |
FIGURE 3.

AUC SV and SEC/MALS. A, sedimentation coefficient distribution of KsHDH in 25 mm HEPES/KOH, pH 7.5, 25 mm KCl. The three peaks correspond to the HDH monomer (α, 5 S), the covalently bound homotrimer (α3, 10.97 S), and the non-covalently bound octamer of the homotrimers (α24, 43.33 S). B, SEC/MALS chromatogram of KsHDH on a Superose 6 (10/300 GL) column in 50 mm HEPES-KOH, pH 7.5, 150 mm KCl. The absorbance at 280 nm (dashed line) and 409 nm (solid line) were recorded. The two major peaks correspond to the covalently bound homotrimer (α3, 15.5 ml) and the non-covalently bound octamer thereof (α24, 11.2 ml). The HDH monomer (α, ∼16.7 ml) elutes only as a minor peak. The molecular mass distribution obtained by MALS analysis is shown in gray.
FIGURE 4.

Negative stain TEM. A, negatively stained particles of native KsHDH. Particle sizes corresponding to the homotrimers (8 × 8 nm) as well as tetrameric assemblies (16 × 16 nm) are visible. B, negatively stained particles of KsHDH after glutaraldehyde cross-linking and repurification by SEC. Large tetrameric assemblies are the major species. C, negatively stained particles of KsHOX. D–F, close-up views of particles from B (labeled accordingly) showing particles with 4-fold symmetry as well as particles with a more elongated shape, probably representing two orthogonal views of an octamer of trimers (α24) assembly of KsHDH. We also observe particles with an apparent 5-fold symmetry (F), which could represent an intermediate state in oligomerization. They are probably not due to the cross-linking, given their occurrence in native protein as well (see A, top left).
Sequence Analysis of KsHDH and Its Homologs Related to HAO-like Octaheme Proteins from Other Anammox Species
MALDI-TOF MS indicated KsHDH to be a homotrimeric protein possessing eight c-type hemes per subunit and identified the protein as the gene product of kustc0694 rather than of the close paralog kustd1340. Close homologs of kustc0694 and kustd1340 were readily detected in other sequenced anammox genomes (79–96% sequence identities of the N-terminally cleaved gene products; supplemental Fig. S1). However, kustc0694 differed from all others by the presence of a 75-amino acid N-terminal extension. This extension was most likely an artifact caused by an erroneously annotated start codon, the actual translation start being Met-75. In agreement with this, 21 nucleotides upstream of Met-75, a Shine-Dalgarno sequence (AGGAGG) was present. Met-75 was followed by an N-terminal leader sequence (32 amino acids) seen at this position in all other HDH homologs. Moreover, the first peptide (Mr = 2,691.2 Da) that we could detect in tryptic digests was the one immediately following Val-107 in the KsHDH sequence.
Multiple-protein sequence alignment affiliated the HDH-like proteins with octaheme proteins with known crystal structures and functions (NeHAO and KsHOX) (supplemental Fig. S1). This affiliation was supported by sequence conservation of the eight heme c binding motifs, including the positions of the seven histidines that act as distal ligands to the seven His/His-ligated heme c moieties involved in electron transfer. An unusual feature of the HDH proteins was the presence of a CX4CH, rather than a CX2CH, binding motif for heme 3. Taking into account the conserved position of this motif and of the distal histidine (H3′), this heme 3 would be His/His-ligated, making it an electron-transferring one, but possibly with a different spatial orientation as compared with heme 3 in NeHAO and KsHOX.
Notwithstanding their limited amino acid sequence identity (∼30%), protein folds and the arrangement of heme cofactors of NeHAO and KsHOX are virtually identical (Fig. 1) (10–12). This similarity particularly concerns the structure of the catalytic heme 4 and its surroundings. Sequence comparison suggested that the structural similarity might include the HDH members. Notably, key amino acids involved in the covalent binding of an adjacent subunit to heme 4 (a tyrosine) and in proton withdrawal from the substrates (an aspartate-histidine pair) could be detected in the HDH sequences, next to the amino acids forming part of a hydrogen-bonding network that transports protons to the water-filled cavity at the substrate entrance site (Fig. 1, B and C). A critical difference between NeHAO and KsHOX is the presence a tyrosine (Tyr-358) that assists in the addition of a water molecule to a nitrosyl intermediate during nitrite formation in the former protein (see Reaction 4) (12). In KsHOX, this tyrosine is moved away from the catalytic site by a two-amino acid contraction, and the position of the NeHAO Tyr-358 is occupied by a methionine (Met-323). This very same contraction and methionine replacement were also found in the HDH members. One may note that neither hydroxylamine oxidation to NO (Reaction 5) nor hydrazine oxidation to N2 (Reaction 3) includes a water addition. Opposite to the catalytic site near the proximal histidine of heme 4 (P460), there is a distinction between the three proteins (Fig. 1, B and C) that might be of relevance in determining substrate specificity. Here, the carboxylate oxygen of Asp-231 in NeHAO is at hydrogen-bonding distance to the proximal histidine of heme 4 (P460), whereas the carbonyl oxygen of Ala-201 occupies this position in KsHOX. Remarkably, in the HDH members, a cysteine was observed at this site, possibly affecting the heme 4 redox potential (see below).
KsHDH Is an N2-producing Enzyme
KsHDH catalyzed the four-electron oxidation of hydrazine to N2 according to Reaction 3. This four-electron stoichiometry was apparent when following the reaction with excess ferric cytochrome c as the electron acceptor (Fig. 5A). Experiments with double-labeled hydrazine (H215N-15NH2) confirmed that double-labeled N2 was the end product of this reaction, fitting the expected 1:1 stoichiometry (Fig. 5B). Hydrazine oxidation followed Michaelis-Menten kinetics with a Vmax = 11 ± 1.2 μmol min−1 mg−1 of protein and a Km = 10 ± 2.2 μm for hydrazine (Table 2). Hydroxylamine did not serve as a substrate, neither in oxidative nor in reductive directions, when assays were performed in the presence of oxidized or reduced cytochrome c, respectively. In the absence of cytochrome c, hydroxylamine disproportionation into ammonium and nitrogen species of higher oxidation states (NO2−, NO, N2O, N2) did not occur. Other nitrogenous compounds tested (NH4+, NO, NO2−, NO3−) were not converted in assays with oxidized or reduced cytochrome c as co-substrate either. Instead of being substrates, hydroxylamine (KiNH2OH = 7.9 ± 1.8 μm) and NO (KiNO = 2.5 ± 0.9 μm) were strong competitive inhibitors of hydrazine oxidation activity. These findings demonstrated that KsHDH indeed was a dedicated hydrazine dehydrogenase that makes N2.
FIGURE 5.

Hydrazine oxidation to N2 by KsHDH. A, hydrazine oxidation (initial concentration, 10 μm) by KsHDH (0.65 μg) was followed by recording the reduction of 50 μm cytochrome c at 550 nm. No reaction was observed when followed in the presence of 50 μm hydroxylamine (dashed line). The concentration of reduced cytochrome c was calculated on the basis of the increase of absorbance at 550 nm (Δϵ550 = 19,600 m−1·cm−1). B, formation of labeled 30N2 from double-labeled hydrazine (H215N-15NH2, 16 nmol) was followed over time in the presence of 50 μm cytochrome c and 63 ng of KsHDH.
TABLE 2.
Catalytic and structural properties of hydrazine dehydrogenase from K. stuttgartiensis and other hydrazine-oxidizing enzymes
Kinetic values are given for mammalian cytochrome c as electron acceptor. KSU-1, anammox strain KSU-1. NA, not applicable; NR, not reported.
| K. stuttgartiensis HDH | K. stuttgartiensis HOX | KSU-1 HZO/HDH | KSU-1 HOX | Brocadia anammoxidans HOX | Nitrosomonas europaea HAO | |
|---|---|---|---|---|---|---|
| Vmax N2H4 (μmol min−1 mg−1) | 11 ± 1.2 | 1.6 ± 0.0 | 6.2 | 0.54 | 1.1 | 14 |
| kcat N2H4 (s−1) | 36 | 4.9 | 13.4 | 1.1 | 3.4 | 47 |
| Km N2H4 (μm) | 10 ± 2.2 | 54 ± 3.3 | 5.5 | 25 | 18 | 4 |
| kcat/Km N2H4 (s−1 μm−1) | 3.5 | 0.1 | 2.4 | 0.042 | 0.19 | 12 |
| Vmax NH2OH (μmol min−1 mg−1) | NA | 4.8 ± 0.2 | NA | 9.6 | 21 | 28.5 |
| kcat NH2OH (s−1) | NA | 15 | NA | 19 | 64 | 95 |
| Km NH2OH (μm) | NA | 4.4 ± 0.9 | NA | 33 | 26 | 3.6 |
| kcat/Km NH2OH (s−1 μm−1) | NA | 3.4 | NA | 0.58 | 2.5 | 26 |
| Ki NH2OH (μm) | 7.9 ± 1.8 | NA | 2.4 | NA | NA | NA |
| Ki NO (μm) | 2.5 ± 0.9 | NA | NR | NR | NR | NA |
| Total size (kDa) | 201.5 (∼1,600) | 184 | 130 | 118 | 183 | 200 |
| Subunit size (kDa) | 67.2 | 61.3 | 62 | 53 | 58 | 66 |
| Subunit composition | α3 ((α3)8) | α3 | α2 | α2 | α3 | α3 |
| Catalytic heme optical maximum (nm) | 473 | 468 | 472 | 468 | 468 | 463 |
| Catalytic heme midpoint potential (mV) | −420 | −300 | NR | NR | NR | −260 |
| Reference | This paper | 12 | 8 | 17 | 18 | 19–21 |
The catalytic properties of KsHDH were similar to those reported for its homolog (HDH/hydrazine-oxidizing enzyme) purified from the anammox enrichment culture KSU-1 (Table 2). Octaheme KsHOX from K. stuttgartiensis (kustc1061) and from other anammox bacteria oxidizes hydrazine as well, albeit with lower catalytic efficiency (kcat/Km). Hydrazine also served as a substrate for NeHAO, surprisingly with kcat and Km values that were superior to genuine HDH, although hydrazine does not play a role in N. europaea metabolism; this organism and other aerobic ammonium-oxidizing microorganisms are not able to synthesize hydrazine, which is an exclusive property of anammox bacteria (3, 5–7).
KsHDH Harbors a P460 Prosthetic Group
As expected from its bright red color, linear MALDI-TOF MS, and protein sequence analyses, KsHDH was a multiheme protein. The UV-visible electronic absorbance spectrum of as-isolated, fully oxidized KsHDH displayed features typical for ferric heme c proteins, namely a dominant absorption peak having a maximum at 408 nm and a broad absorption band in the 550 nm region (Fig. 6). After reduction by dithionite, pronounced absorption peaks emerged with maxima at 420, 524, and 554 nm, corresponding to the Soret, β, and α bands, respectively, that are typically for ferrous c-type hemes. However, an absorption band at 473 nm revealed the presence of a P460-type ferrous heme group in KsHDH as is typical for NeHAO and in KsHOX and in agreement with the presence of a conserved Tyr in the sequence alignments (9, 10, 12, 14). The sequential reduction of KsHDH by the addition of small aliquots of dithionite demonstrated that the His/His-ligated hemes were reduced first (Fig. 7, A and B). Only after their reduction, the P460 band emerged, implying that its redox potential would be lowest. Equilibration of KsHDH at a series of defined potentials supported this conclusion (Fig. 7C) and suggested an Em of about −420 mV for P460 in KsHDH, whereas the His/His-ligated hemes reduced at midpoint redox potential (Em) values ranging between −70 and −410 mV. The P460 reduction potentials of NeHAO and KsHOX (Em = −260 and −300 mV versus NHE, respectively) are substantially higher (12–14).
FIGURE 6.
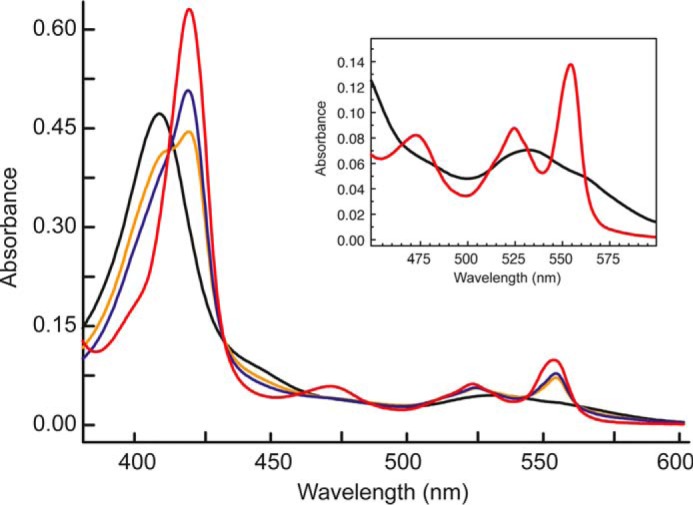
Effect of hydrazine on the electronic absorbance spectrum of KsHDH. Spectra are as follows: as-isolated KsHDH (45 μg·ml−1; 0.67 μm) (black line); fully (dithionite) reduced spectrum (red line); and partially reduced KsHDH in the presence of 1 μm hydrazine (orange line) and in the presence of 3 μm hydrazine (blue line). From the increase in the Soret band at 419 nm, it is inferred that the presence of 1 and 3 μm hydrazine resulted in the reduction of ∼3 and 4 His/His-ligated c-type hemes, respectively, assuming that each heme contributes equally to a spectral change. The inset details the spectrum of as-isolated KsHDH in the 450–600 nm region.
FIGURE 7.

Optical redox determinations of KsHDH. A, reduction of as-isolated KsHDH (43 μg ml−1; 0.64 μm) by the sequential addition of dithionite aliquots under anaerobic conditions. Spectra were corrected for the effect of dilution by the additions. AU, absorbance units. Arrows, directions of the spectral changes. The absorbance band at 473 nm derived from the P460 prosthetic group appeared only after the complete reduction of the His/His-ligated c-type hemes featured in their reduced state by their absorption maxima at 420, 524, and 554 nm. The spectrum of fully reduced KsHDH is highlighted by the red line. B, reduced minus oxidized absorbance spectra during dithionite reduction. Note that spectral changes occur around distinct isosbestic points at 412, 433, 464, 485, 508, 533, and 562 nm, indicating that the spectral changes as the result of the reduction of the P460 catalytic heme do not interfere with those of the His/His-ligated c-type hemes. C, redox titration of KsHDH (∼30 μm) in an optical transparent thin layer electrochemical cell. Normalized redox potential-dependent spectral changes of the His/His-ligated hemes of KsHDH were followed in oxidative (ox) and reductive (red) directions at 554 nm (black squares, oxidative; blue triangles, reductive) and at 420 nm (red circles, oxidative; green diamonds, reductive). Spectral changes at 473 nm as a result of the reduction of the P460 prosthetic group are shown as red triangles. Data were fitted (black line) by a Nernstian curve, assuming a one-electron reduction resulting in an Em = −420 mV.
When as-isolated KsHDH was incubated with its substrate, hydrazine, the protein became partially reduced, as concluded from the increase in the absorbances at 420, 524, and 554 nm derived from the His/His-ligated c-type hemes (Fig. 6). The degree of reduction was dependent on the concentration of hydrazine added. Neither hydroxylamine nor NO influenced the absorption spectrum of oxidized KsHDH, but both compounds instantaneously oxidized the dithionite-reduced protein.
EPR Spectroscopy of HAO-like Octaheme Proteins
The sequence analysis and biophysical data presented above suggested that the HDH members shared the common architecture of NeHAO and KsHOX enzymes (Fig. 1). This was assessed by inspection of EPR spectra arising from the oxidized forms of all three enzymes.
Previously, Hendrich et al. (15) were able to delineate the complex spectra of fully oxidized NeHAO into four distinct EPR species covering the expected eight Fe3+ centers in the eight hemes per subunit (Table 3). Two of the species (species 3 and 4) could be unambiguously assigned to specific hemes. EPR species 4 was the result of spin-coupled heme 4 (P460) and heme 6 (see Fig. 1D for the numbering and spatial orientations of the hemes). Importantly, this species was only observed when EPR spectra were recorded in the parallel mode. EPR species 3 was derived from magnetically interacting heme 3 and heme 5. The analyses by the authors (15) left four magnetically isolated low spin hemes unassigned, which were observed as two species (species 1 and 2) covering one and three heme centers, respectively (Table 3). Thus, none of these showed appreciable interaction with each other. A one-electron reduction of NeHAO caused the partial reduction and a notable change of spin species 2 (15). Such one-electron reduction would concern the heme with the highest redox potential, which is heme 2 in NeHAO (13). Heme 2 is a key electron sink because it can accept electrons passing via hemes 5 and 3 within the same subunit or passing via hemes 7 and 8 from an adjacent subunit before transferring these to solvent-exposed exit heme 1 (Fig. 1D). In the following, we started from the assumption that heme 2 also has the highest redox potential in KsHOX and KsHDH. Supported by our analyses of KsHOX and KsHDH (see below), we then assigned spin species 2 to non-coupled hemes 7 and 8 together with heme 2, leaving the isolated EPR species 1 to heme 1 (Table 3).
TABLE 3.
EPR spectroscopic properties of NeHAO, KsHOX, and KsHDH
EPR species are numbered according to Ref. 15 with our refinement.
| Protein EPR species (iron/monomer)a | gxyz values | Comment/Assignment |
|---|---|---|
| NeHAOb | ||
| 1 (1) | 1.20; 2.26; 3.03 | Uncoupled heme 1 |
| 2 (3) | 1.35; 2.19; 3.11 (3.09)c | Uncoupled hemes 7 and 8; includes the easily reducible heme 2 |
| 3 (2) | 1.27; 2.28; 3.06 | Coupled hemes 3 and 5 |
| 4 (2) | 8 | Coupled hemes 6 and 4 (P460); only observed in parallel mode EPR |
| KsHOX | ||
| 1 (1) | 1.452; 2.305; 2.955 | Heme 1; g values become 1.452, 2.260, and 2.980 after NH2OH incubation |
| 2a (1) | 1.41; 2.145; 3.1412 | Reduced after NH2OH incubation; heme 2 |
| 2b (2) | 1.41, 2.217, 2.955 | Uncoupled hemes 7 and 8; g values become 1.37, 2.217, and 2.980 after NH2OH incubation |
| 3 (2) | 1.40; 2.18; 3.247 | Coupled hemes 3 and 5 (Euler angles: 45, −180, and 135°); after NH2OH incubation, each heme reduces by 60%, gz changes to 3.265, and the exchange splitting increases from 0.031 to 0.035 cm−1 |
| 4 (2) | NDd | |
| 5 (minor) | 6.07; 5.81; 1.98 | High spin heme, but amount is preparation-dependent (0.03–0.14/monomer; the higher value shown in Figs. 8 (A and B) and 9A |
| KsHDH | ||
| 1 (1) | 1.6; 2.05; 3.14 | Uncoupled heme (heme 1) with broad resonances that sharpen and shift (gx,y,z = 1.51, 2.256, 3.10) upon reduction by hydrazine |
| 2a (1) (3.8%) (6.7%) (6.1%) | 1.772; 2.06; 2.579 | Heme 2; N2H4-reducible; same heme in slightly different environments |
| 2b (2) | 1.772; 2.06; 2.543 | |
| 1.772; 2.06; 2.497 | ||
| 1.5; 2.3; 3.0 | Coupled hemes 7 and 8 (Euler angles: 45, 180, and 87°); no reduction by hydrazine, but gz changes to 3.05; the exchange splitting is 0.035 cm−1 | |
| 3 (2) | 1.5; 2.3; 3.0 | Coupled hemes 3 and 5 (Euler angles: 45, −180, and 135°); reduction by hydrazine (70%), and gz changes to 3.04; the exchange splitting is 0.074 cm−1 and 0.087 cm−1 after reduction |
| 4 (2) | ND | |
| 5 (minor) | 6.0; 6.0; 2.0 | Minor high spin heme (<0.02/monomer) |
| 6 (minor) | 2.009 | Possible tyrosine radical coupled to heme 4 |
a In column 1, numbers in parentheses indicate the integral number of spins, and percentage values represent the percentage of the total of integrated spins.
b Data from Hendrich et al. (15).
c In parentheses, gz after 1-electron reduction.
d ND, not determined. Signal accounting for hemes 6 and 4, which only would be observed in parallel mode EPR.
EPR Spectroscopy of KsHOX
Following the same approach as Hendrich et al. (15) and using the same theoretical spin Hamiltonian (Equation 1), we could deconvolute the EPR spectrum of KsHOX into the same spin species and allocate those species to all hemes (Table 3 and Figs. 8A and 9 (A and B)). Whereas spin species 2 now could be resolved into two subspecies (species 2a (heme 2) and species 2b (hemes 7 and 8); Table 3), the overall EPR spectrum of as-isolated, fully oxidized KsHOX (Fig. 8A) was highly similar to the one described for NeHAO (14, 15). This was expected, because the heme arrangement in both proteins was fully conserved, regarding both the spatial orientations and positions of the hemes and their distances with respect to each other (Fig. 1D) (12). Simulation of the normal mode EPR spectra of KsHOX and the individual heme centers is shown in Fig. 9 (A and B); simulation parameters are listed in Table 3. In the normal mode EPR spectra recorded in this work, the coupled heme pair 6 and 4 remained beyond detection, and because we had no access to parallel mode EPR, these hemes were not determined. Consequently, a total of six of eight hemes per monomer would be observed. In agreement herewith, quantitation of the EPR spectrum versus a copper perchlorate standard (31) of oxidized, as-isolated, KsHOX (665 μm hemes, as determined optically) yielded a value of 480 ± 35 μm S = ½ low spin hemes, which is consistent with a total of six EPR-visible hemes. Except for small differences in g values, the main difference between KsHOX and NeHAO was the strength of the magnetic coupling (D′zz - D′xx) between hemes 3 and 5. For KsHOX, a value of 0.031 cm−1 was determined for the oxidized enzyme; for NeHAO, the value was 0.08 cm−1 (15). For analyses, the same relative orientation of the two heme centers given by Euler angles between the g-tensor axes of 45, −180, and 135° were used. These angles differ by only 15 ± 5° from the angles between the crystallographic axis (55.5, −173.3, and 115.0°), similar to the orientation in NeHAO. As extensively discussed by Hendrich et al. (15), the lack of precise coincidence of the two sets of axes is, among other reasons, due to the relative orientation of the imidazole planes of the coordinating His residues, being co-planar or not, and/or to the orientation of the imidazole planes relative to the heme meso-position. A peculiar feature of the EPR spectra of KsHOX was the presence of a high spin heme signal at gx ∼6 (Table 3 and Figs. 8 (A and B) and 9A). Although prominent, the intensity of the signal varied between different enzyme preparations, never taking integral values. This may suggest that the residual high spin heme signal originated from a heme residue that either was pentacoordinated or was hexacoordinated with water (or OH−), a straightforward candidate being heme 4 lacking the covalent tyrosine binding with the neighboring subunit as is the case for monomeric KsHOX.
FIGURE 8.

EPR spectroscopy of KsHOX and KsHDH. A and B, EPR spectrum of as-isolated, oxidized KsHOX (83 μm) (A) and KsHOX after incubation with 100 μm hydroxylamine (B). C and D, EPR spectrum of as-isolated, oxidized KsHDH (33 μm) (C) and KsHDH after incubation with ∼100 μm hydrazine (D).
FIGURE 9.

EPR spectroscopy of KsHOX. A, X-band EPR spectra of oxidized (Ox) KsHOX (83 μm) and KsHOX after reduction by NH2OH (100 μm). Experimental spectra are in red, simulated total spectra are in black, and the simulations of the individual heme centers are in blue. Note that this particular preparation contained 0.14 high spin heme per monomer (Table 3); other preparations had generally lower amounts. B, simulations of EPR spectra (red) and the effect of reduction by NH2OH (blue). In the spectrum of the heme pair 3-5, the magnetic coupling is slightly increased (e.g. see gz peak); for heme 1, the gy resonance is broadened; and for hemes 7 and 8, the gx resonance sharpens after reduction by NH2OH. Small shifts in g values upon reduction are reported in Table 3. C, Q-band EPR spectrum (red) of KsHOX and simulation (blue) using the same parameters as for the X-band spectrum (Table 3).
From UV-visible light optical changes, it was deduced that incubation of KsHOX with its substrate hydroxylamine caused the two-electron reduction of the c-type heme centers (12). This result was corroborated by EPR spectroscopy. After incubation with NH2OH, hemes 3 and 5 were each reduced by ∼60%, whereas one signal had completely disappeared from the EPR spectrum of KsHOX, which is attributed to heme 2 (Figs. 8B and 9A). This observation suggested that hemes 3 and 5 have very similar high midpoint redox potentials. The same conclusion regarding the redox properties of the heme couple 3 and 5 was reached in the study on NeHAO (14). After reduction, the magnetic coupling strength between hemes 3 and 5 increased somewhat to 0.035 cm−1, and the g values of the remaining oxidized species differed slightly from those in the oxidized enzyme (Table 3). In addition, some resonances sharpened slightly (e.g. the gx attributed to hemes 7 and 8) or broadened slightly (e.g. the gy of heme 1) (Table 3 and Fig. 9B). Apparently, the magnetic properties of hemes 1, 7, and 8 were affected by the reduction of a common neighboring heme, which is heme 2 (Fig. 1D), justifying their assignments. We may note that the same EPR spectral changes were found when KsHOX was incubated with hydrazine, giving further support to our previous finding (12) that binding of hydrazine is associated with the two-electron reduction of His/His-ligated c-type hemes.
Overall, the X-band EPR spectra of KsHOX could be accurately simulated using the parameters listed in Table 3 (Fig. 9, A and B). Using these same parameters, the simulated Q-band spectrum of as-isolated KsHOX agreed well with the experimental spectrum (Fig. 9C), supporting the multicomponent analysis.
EPR Spectroscopy of KsHDH
At first glance, the EPR spectrum of as-isolated KsHDH (Fig. 8C) was quite different from the one of KsHOX (Fig. 8A). In addition, analysis of the EPR spectrum of oxidized KsHDH was complicated by the fact that the majority of the EPR signals appeared to be broadened due to magnetic interactions. This was, for example, evidenced by the absence of separate gx resonances (∼1.5 < g < 1.2) or sharp gy resonances (at g ∼2–2.2) in oxidized KsHDH. Due to the broad signals obscuring baseline sloping, quantitation of the EPR spectra was less accurate. Consequently, quantitation of the normal mode EPR spectrum of KsHDH (265 μm hemes as determined optically) showed a larger spread (210 ± 50 μm). Nevertheless, the latter value was in agreement with the presence of six of eight EPR-visible hemes; as before, the coupled hemes 6 and 4 (P460) were not detected. Only after reduction by hydrazine did a magnetically isolated low spin heme center become visible (gx = 1.51, gy = 2.256, and gz = 3.10), which amounted to ∼0.45 hemes/monomer as the result of a partial reduction (Table 3 and Figs. 8 (C and D) and 10 (A and B)). The main difference between the spectra of oxidized KsHDH and of oxidized KsHOX/NeHAO was that an adequate simulation of the spectrum of KsHDH required that not only hemes 3 and 5 but also the signal allocated to hemes 7 and 8 contribute as a magnetically coupled pair of hemes. Other special features were the presence of a trio of relatively isotropic low spin heme species and of a highly anisotropic low spin heme with gx,x,z = 1.6, 2.05, 3.14 (Table 3 and Fig. 11 (A and B)). In total, the trio accounted for one heme per monomer at which individual species integrated to approximately one-third of an integral value.
FIGURE 10.

EPR spectroscopy of KsHDH. A, X-band EPR spectrum of as-isolated oxidized KsHDH (33 μm) and the simulation (simul) of the complete spectrum and of the individual components with integral intensities used in the simulation. Stick diagrams (black) represent magnetic couplings of 0.035 and 0.074 cm−1 in the gz resonance of heme pair 7 and 8 and heme pair 3 and 5, respectively. Heme 1 and heme 2 account for one spin per monomer each; the heme pairs 3-5 and 7-8 represent two spins per monomer each. The radical represents 0.005 spin per monomer. B, X-band EPR spectrum of KsHDH (33 μm) after incubation with hydrazine (∼0.1 mm) and the simulation (simul) of the complete spectrum and the individual components with the intensity factor used in the simulation shown in parentheses. Stick diagrams (in black) represent magnetic couplings of 0.035 and 0.087 cm−1 in the gz resonance of heme pair 7 and 8 and heme pair 3 and 5, respectively. The radical represents 0.002 spin per monomer. C, X-band EPR spectrum of oxidized KsHDH in the g = 2 region.
FIGURE 11.

Proposed catalytic cycle of hydrazine oxidation to dinitrogen gas by HDH. As-isolated KsHDH is fully oxidized and is catalytically competent. Reaction with hydrazine accompanied by the release of two electrons results in the formation of a diazene (HN=NH) derivative bound to the heme 4 (P460) catalytic site. In the structure of KsHOX, which is capable both of hydroxylamine and hydrazine oxidation to NO and N2, respectively, such diazene could be modeled after soaking with hydrazine (Fig. 1B) (PDB code 4N4L) (12). The next steps then would comprise the oxidation of diazene, without an intermediate nitrogenous species, directly to N2, again yielding two electrons. These two electrons have to be stored and discharged one by one through the c-type hemes wiring circuit. It is very conceivable that reduced (ferrous; Fe(II)) heme 4 is an intermediate in this sequence. Such ferrous heme 4 would bind NH2OH and NO, both strong competitive inhibitors of the reaction, explaining their inhibitory effect (dashed line). It should be noted that neither hydroxylamine nor NO interact with oxidized KsHDH. In as-isolated KsHDH, the P460 cofactor most likely binds a distal water molecule, as observed in the KsHOX (PDB code 4N4J) and NeHAO (PDB code 4N4O) crystal structures (12). The absence of a clear high spin signal in the KsHDH EPR spectrum (Figs. 9 (C and D) and 11 (A and B)), indicative of a penta-coordinated heme, is consistent with the presence of distal water in as-isolated KsHDH P460. Following the release of the product (N2) of the four electrons formed during hydrazine oxidation, replacement of water to the catalytic site would prepare HDH for the next reaction cycle. In the figure, the P460 cofactor is represented by the ellipse, including the His proximal ligand and a distal water molecule in the as-isolated resting state.
In KsHDH, the magnetic coupling strength between hemes 3 and 5 (0.074 and 0.087 cm−1 after reduction) and the Euler angles were found to be nearly the same as in NeHAO (Table 3). For heme pair 7 and 8, the magnetic coupling was weaker (0.035 cm−1), and, importantly, the Euler angles were different (45, 180, and 87°), suggesting different relative heme orientations (Table 3). In the KsHOX structure, the difference in orientations of hemes 7 and 8 is evident (Fig. 1D). In the structure of KsHDH, which has yet to be resolved, these Euler angles could be different, resulting in the observed spin-spin interaction between hemes 7 and 8. Thus, spin species 2b in KsHDH (Table 3) is derived from two interacting, neighboring hemes, the only candidates being hemes 7 and 8. Consequently, the corresponding spin species in the KsHOX and NeHAO EPR spectra should comprise these hemes 7 and 8 as well, although they are not spin-coupled in these proteins.
Incubation of KsHDH (∼35 μm) with excess hydrazine (0.1 mm) resulted in a dramatic change of the EPR spectrum (Figs. 8D and 10 (A and B)). In total, each enzyme monomer became reduced by 2.95 ± 0.4 electrons according to the EPR simulations, whereas a value of 3.2 ± 0.5 electrons was determined from direct double integration of the experimental spectra. More specifically, heme pair 3 and 5 became reduced by ∼70%, and the exchange splitting increased to 0.087 cm−1 (Table 3). Reduction of the protein by hydrazine led to the complete disappearance of the trio of relatively isotropic low spin heme species, whereas the highly anisotropic low spin heme became reduced by 55%. Upon reduction, the broad resonances of the latter sharpened and shifted. In contrast, heme pair 7 and 8 remained fully oxidized. Assuming as before that heme 2 has the highest redox potential, this heme 2 was allocated to the trio of relatively isotropic low spin heme species. It is tempting to speculate that within the homotrimeric protein, the three hemes 2 take somewhat different positions, directing electron transfer within and between the subunits and resulting in the three slightly different EPR signals observed. The assignment would leave the isolated highly anisotropic low spin signal to heme 1. It should be noted that treatment of KsHDH with various concentrations of hydrazine resulted in the reduction of 3–4 His/His-ligated hemes as deduced from the UV-visible spectral changes (Fig. 6). The three-electron reduction calculated from the EPR spectra agrees with this.
Except for the signals discussed above, the EPR spectrum of KsHDH showed the presence of two more signals in minor amounts (Table 3). The first one concerned a high spin heme (<0.02 per monomer), highly resembling the one observed in KsHOX. As in the latter protein, this high spin heme could be the result of small contamination of monomeric KsHDH. In the g = 2 region, the EPR spectrum of oxidized KsHDH showed an unusual signal (Fig. 10C) characterized by a peak-to-peak width of 36 Gauss, no resolved hyperfine structure, and an apparent g value of 2.009. Upon reduction by hydrazine, this signal decreased nearly 3-fold in intensity. The signal relaxed rapidly. Its low amount (0.005 spins/monomer) might point to an impurity (but not a high-potential iron-sulfur protein (HIPIP) 4Fe-4S center giving its overall line shape). However, its reduction by hydrazine suggested it to be a component of KsHDH. If so, the signal might derive from a tyrosine radical, more specifically from Tyr-536′ crosslinked to heme 4. This would explain both its rapid relaxation and unusual line shape affected by the magnetism of the S = 5/2 Fe3+ of heme 4.
Overall, the EPR spectra of as-isolated KsHDH, both in the absence and presence of its substrate hydrazine (Fig. 8, C and D), could be adequately simulated (Fig. 10, A and B) by the parameters listed in Table 3, as was the case with KsHOX. Although quite different at first appearance, our analyses showed that the EPR spectra of KsHDH were well described by the same framework as those of NeHAO and KsHOX (Table 3). This provided strong evidence that HDH indeed possessed the same architecture and arrangement of His/His-ligated c-type hemes as NeHAO and KsHDH. Again, differences in the EPR spectra were the likely result of only minute differences in the relative orientation of the two imidazole planes of the two histidines that coordinate the low spin hemes and of the precise location of these imidazole planes relative to, for example, the meso-positions of the porphyrin rings. These differences might affect the heme redox potentials, enabling a fine tuning of electron transfer in order to optimize catalytic activities.
Discussion
Here we purified and characterized KsHDH, the gene product of kustc0694. HDH is a key enzyme in anammox metabolism, which oxidizes the intermediate hydrazine to the end product N2. Biophysical properties, detailed sequence analysis, and UV-visible and EPR spectral data strongly suggested that HDH and its close homologs in other anammox genomes were structurally quite similar or nearly identical to two also structurally highly related octaheme proteins, NeHAO and KsHOX. Both NeHAO and KsHOX are capable of hydrazine oxidation, but HDH was specific in this activity (Table 2). This raises the question of how HDH is tuned for its specific action. An answer to this question may be found in differences in the way HDH is structured, in the way hydrazine becomes bound to its catalytic site, and in the way the four electrons and four protons derived from hydrazine oxidation (Equation 3) are abstracted and transferred during catalysis.
A special property of KsHDH was that it assembled into octamers of homotrimers (Table 1 and Figs. 3–5). Such association would require specific amino acid sequences that promote the interaction between the homotrimers. Interestingly, HDH members contained a conserved stretch of 15 amino acids in their C-terminal region, which was absent in HAO and HOX (supplemental Fig. S1). From the crystal structures of the latter two proteins, it is inferred that the stretch could be localized at the tip of each monomer, where it would be well situated to promote the interaction between two egg- or tulip-shaped, covalently bound homotrimers.
As-isolated, catalytically competent KsHDH reacted with hydrazine. This reaction was accompanied with reduction of His/His-ligated c-type hemes. In the case of KsHOX, incubation with hydrazine caused the reduction of two such hemes, whereas a dinitrogen species bound to the P460 catalytic center was observed in its crystal structure soaked with hydrazine, which could be a diazene (HN=NH) derivative (Fig. 1B) (12). Similarly, binding of hydrazine to the catalytic site, immediately followed by a two-electron oxidation would be the first step in the HDH catalytic cycle proposed in Fig. 11. Analogously, hydroxylamine oxidation by KsHOX and NeHAO is initiated by the concomitant binding of this substrate and its two-electron oxidation, yielding a nitroxyl (HNO) species in a {FeNO}7 configuration as an intermediate (22, 23). An intrinsic problem of such two-electron oxidations is that the first heme that accepts these electrons (heme 6; see Fig. 1D) can carry only one electron at a time. Here, the tyrosine attached to the catalytic heme 4 could function by temporarily storing the second electron as put forward previously (12). This would explain the presence of the P460 group in this type of HAO-like proteins that favor oxidative reactions (24). KsHDH was incapable of hydroxylamine oxidation (Table 2), and this compound did not interact with the fully oxidized, as-isolated enzyme. We note that the redox potential of the P460 prosthetic group (actually P473) in KsHDH (Em ≈ −420 mV; Fig. 8C) was 120–150 mV lower than in KsHOX (E′o = −300 mV) (12) and in NeHAO (E′o = −260 mV) (13, 14). The lower Em value for the catalytic heme 4 in KsHDH could arise from the cysteine predicted to be in the second coordination sphere of the catalytic heme 4 in KsHDH, but not the HAO and HOX, enzymes (Fig. 1, B and C). If present as a thiolate anion, this cysteine would significantly affect the electronic properties of heme 4, including the bond length with the proximal histidine and, as a result thereof, the redox potential of this heme (25). In the case of KsHDH, the electron-donating (reducing) power of NH2OH might become insufficient for bond formation with the ferric P460, preventing turnover of this compound. However, hydroxylamine and also NO were strong competitive inhibitors, and these compounds oxidized reduced KsHDH instantaneously, indicating an interaction with ferrous heme P460 as a catalytic cycle intermediate (Fig. 11). In hindsight, the serendipitous finding that hydrazine is an intermediate in the anammox process (26) was apparently due to the inhibition of the HDH reaction by hydroxylamine added to the assays.
In anammox bacteria, hydrazine is the ultimate electron donor in energy conservation, whereas hydroxylamine is probably a side product of the preceding hydrazine synthase reaction (5, 6). Apparently, these bacteria have evolved dedicated enzymes for hydrazine and hydroxylamine metabolism, at which KsHOX recycles hydroxylamine efficiently into hydrazine synthase substrates (NO and three electrons; see Reaction 2). The above oxidation reactions require the well timed transfer of single electrons to an exit heme 1. Branched electron transfer chains in homotrimeric NeHAO and KsHOX enable electron export both to heme 1 of the subunit where substrate oxidation takes place and to heme 1 of a neighboring subunit (Fig. 1D). Possibly, HDH kinetically fine tunes this electron transfer by (reversible) octamerization and by a rearrangement of the electron transfer circuit, including the modified binding of heme 3 by a CX4CH motif.
HDHs from anammox bacteria, which belong to the HAO-like octaheme proteins, are responsible for the production of a significant amount of N2 that is released into the atmosphere. Besides HDH (kustc0694 and kustd1340) and KsHOX (kustd1061), the genome of K. stuttgartiensis harbors seven more HAO paralogs with as yet unknown functions. Most of these are also found in other anammox genomes (6), whereas genes coding for a plethora of related HAO-like proteins can be found in genomic databases (e.g. see Ref. 24). Together, these HAO-like proteins may represent an environmentally important but still poorly understood potential of enzyme activities, all taking advantage of common structural properties but each one tuned for a specific function by subtle modifications of their catalytic sites and multiheme electron transport module.
Experimental Procedures
Protein Purification
HPLC grade chemicals were purchased from Mallinckrodt Baker; all other chemicals were of the highest grade available. All purification steps took place under air and, apart from FPLC, at 4 °C. KsHDH was isolated from K. stuttgartiensis grown continuously as planktonic cells in a 10-liter membrane bioreactor (5). Routine purification included the following. Biomass (4 liters, A600 = 1.0–1.2) was harvested by centrifugation at 8,000 × g for 15 min. Harvested cells were resuspended in 5 ml of 20 mm potassium phosphate buffer (pH 7). Cells were broken by three subsequent passages through a French pressure cell operating at 138 megapascals. The cell lysate was incubated with 1% (w/v) sodium deoxycholate on a rotating incubator (20 rpm) for 1 h. After incubation, cell debris was removed by centrifugation (3,000 × g, 15 min), and the obtained supernatant was subjected to ultracentrifugation (150,000 × g, 1 h) in a Discovery 100 ultracentrifuge equipped with a T-1270 rotor (Sorvall, Newton, CT) to pellet cell membranes. The intense dark red supernatant after ultracentrifugation constituted the cell-free extract. An Äkta purifier (GE Healthcare) was used for the subsequent FPLC steps. In both steps, the columns were eluted at a flow rate of 2 ml min−1; the eluate was monitored at 280 nm and collected in 2-ml fractions. To purify HDH, cell-free extract was loaded onto a 30-ml column packed with Q Sepharose XL (GE Healthcare), which was equilibrated with 2 column volumes of 20 mm Tris-HCl buffer (pH 8). After application of the sample, the column was washed with 2 volumes of this Tris-HCl buffer to remove unbound proteins. Hereafter, the sample was eluted isocratically in three steps (200, 400, and 1,000 mm NaCl in the 20 mm Tris-HCl buffer, pH 8). Hydrazine dehydrogenase activity was recovered in the 400 mm NaCl step. Three column fractions (6 ml) that were devoid of hydroxylamine oxidase activity were collected and desalted in the above-mentioned potassium phosphate buffer using spin filters (cut-off 100 kDa; Vivaspin 20, Sartorius Stedim Biotech, Aubagne, France) to prepare the sample for the following FPLC step. In this step, the desalted fractions were applied to a 10-ml ceramic hydroxyapatite (Bio-Rad) column, which was equilibrated with 5 column volumes of 20 mm potassium phosphate buffer, pH 7. Following sample loading, HDH activity was eluted from the column as a broad symmetrical peak upon isocratic elution with 20 mm potassium phosphate buffer (pH 7) for approximately three column volumes. Active fractions were collected and concentrated using spin filters as described above. The purity and identity of the fractions were determined by non-denaturing PAGE and SDS-PAGE as well as MALDI-TOF analysis. Enzyme preparations kept in ice were immediately used for further experiments or rapidly frozen in liquid nitrogen for storage. By this procedure, the enzyme was purified 15-fold with a yield of 21% (Table 4). Both the recovery and purification factor were likely to be underestimates, because the cell-free extract contained a substantial amount of hydrazine dehydrogenase activity derived from hydroxylamine oxidase (KsHOX, kustc1061). KsHOX was purified as described previously (12).
TABLE 4.
Purification of hydrazine dehydrogenase from K. stuttgartiensis
| Step | Protein | Volume | Total protein | Specific activity | Total activity | Yield | Purification |
|---|---|---|---|---|---|---|---|
| mg ml−1 | ml | mg | μmol min−1 mg protein−1 | μmol min−1 | % | -fold | |
| Cell extract | 53 | 10 | 530 | 0.55 | 292 | 100 | 1 |
| Q Sepharose | 11 | 6 | 66 | 1.1 | 73 | 25 | 2 |
| Hydroxyapatite | 0.6 | 12 | 7.2 | 8.4 | 60 | 24 | 15 |
AUC
Protein samples were concentrated in 25 mm HEPES-KOH, pH 7.5, 25 mm KCl to A280 nm1 cm = 0.46. Sedimentation velocity analytical ultracentrifugation was performed in a Beckman ProteomeLab XL-I (Beckmann Coulter GmbH, Krefeld, Germany) analytical ultracentrifuge equipped with an An60Ti rotor at 30,000 rpm and 20 °C in a two-sector cell with a 1.2-cm optical path length. Absorption scan data were collected at 280 nm and evaluated using SEDFIT (27). The maximal sedimentation coefficients (smax) were calculated as described (28); smax = 0.00361 m2/3. The ratio of smax/s20,w in the range of 1.2–1.3 indicates a globular shape of the particles. To calculate molecular masses from the sedimentation coefficients obtained by AUC SV, globular particles (smax/s20,w = 1.2) were assumed.
SEC/MALS
KsHDH oligomers in solution were analyzed by analytical size exclusion chromatography at a flow rate of 0.5 ml min−1 using a Waters HPLC system (Waters Corp., Milford, MA), equipped with a Superose 6 (10/300 GL) gel filtration column (GE Healthcare), a filter of 100-nm pore size, and UV-visible and refractive index detectors. Before the experiments, the column had been pre-equilibrated with gel filtration buffer (150 mm KCl, 50 mm HEPES-KOH, pH 7.5) at room temperature. 40-μl samples with A280 nm1 cm ≈ 5 were injected using an autosampler. Static light scattering analysis was performed in line using a DAWN HELEOS multiangle scattered light photometer (Wyatt Technology Corp., Santa Barbara, CA) (laser wavelength λ = 658 nm). Data were processed using the ASTRA software. MALS analysis accounted for any shape.
TEM
Samples of native KsHOX and KsHDH in 25 mm HEPES-KOH, pH 7.5, 25 mm KCl were used directly for negative staining. In addition, a KsHDH sample was subjected to glutaraldehyde cross-linking as follows. KsHDH was diluted to A280 nm1 cm ≈ 0.5 in gel filtration buffer (150 mm KCl, 50 mm HEPES-KOH, pH 7.5), and 0.1% (v/v; ∼10 mm) glutaraldehyde was added. After incubation at 37 °C, the reaction was quenched by the addition of Tris-HCl, pH 8.0, to a final concentration of 167 mm. Afterward, the sample was concentrated and applied to a Superose 6 (10/300 GL) gel filtration column (GE Healthcare, Uppsala, Sweden) at 0.5 ml min−1 and 4 °C. Fractions of the peak eluting at 11 ± 1 ml were pooled, concentrated, and buffer-exchanged to 25 mm HEPES-KOH, pH 7.5, 25 mm KCl using a 100 kDa molecular mass cut-off Amicon ultrafiltration device (Millipore Bioscience, Schwalbach, Germany).
For negative staining, 10 μl of a dilute protein solution (A280 nm1 cm ≈ 1) in 25 mm HEPES/KOH, pH 7.5, 25 mm KCl was pipetted onto a glow-discharged copper grid with carbon-coated Formvar (Plano GmbH, Wetzlar, Germany) and removed after 1 min by gentle blotting from one side with filter paper. The grid was then immediately rinsed with 100 μl of water, blotted again, and treated with 10 μl of 2% (w/v) uranyl acetate solution for 1 min. After removing the staining solution thoroughly by blotting with filter paper, the grid was dried in air for 5–10 min and then inserted in the vacuum port of a FEI Tecnai G2 T20 Twin transmission electron microscope (FEI, Eindhoven, The Netherlands) running at 200 kV accelerating voltage. Electron micrographs were recorded with an FEI Eagle 4k HS, 200-kV CCD camera.
Enzyme Assays
Reaction mixtures (1 ml) in 20 mm potassium phosphate buffer (pH 7) were prepared in 2-ml glass cuvettes (1-cm path length) and contained 50 μm bovine heart cytochrome c (Sigma-Aldrich) and enzyme as required. Cuvettes were sealed with rubber stoppers. Reactions were monitored at 550 nm in a Cary 50 (Agilent, Santa Clara, CA) spectrophotometer at 37 °C. After recording the absorbance at 550 nm for 1 min, the reaction was started by the addition of a helium-sparged hydrazinium sulfate (Merck, Darmstadt, Germany) stock solution or other substrates to be tested. Inhibitors (1, 10, 50, or 100 μm hydroxylamine; 3, 6, or 9 μm NO) were included before the addition of the enzyme. Hydroxylamine was added from a helium-sparged hydroxylammonium chloride (Merck) stock solution. An NO-containing stock solution (0.9 mm) was prepared by sparging anoxic potassium phosphate buffer with an NO-He gas mixture (1:1, v/v) for 10 min. This stock solution was injected to the assays with a 50-μl glass syringe. To minimize the headspace, the volume of reaction mixtures containing NO was increased to 1.5 ml. Reactions in the reductive direction were assayed by adding partially reduced cytochrome c as an electron donor. Reduction was achieved by mixing cytochrome c (50 μm) with 20 μm ascorbic acid, yielding 40 μm reduced and 10 μm oxidized cytochrome c, respectively. Reaction rates were determined from the initial linear part of the reaction curves using the spectrophotometer (Cary) software package. Quantification was performed on the basis of the increase of the molar absorbance at 550 nm (Δϵ550 = 19,600 m−1·cm−1) upon reduction of cytochrome c (29). Rates were evaluated by non-linear regression analysis using the Origin version 8.5.1 program (OriginLab Corp., Northampton, MA) applying Michaelis-Menten kinetics. Apparent inhibition constants (Ki) of hydroxylamine and NO were evaluated by applying the Michaelis-Menten equation, and best fits were obtained when adapted for competitive rather than for other modes of inhibition.
To determine 30N2 formation from double-labeled hydrazine (H215N-15NH2), reaction mixtures were prepared in 3-ml Exetainers (Labco, High Wycombe, UK) inside a glove box (97.5% argon and 2.5% H2; <0.2 ppm O2). Reaction mixtures (2 ml) in 20 mm potassium phosphate buffer (pH 7) contained 50 μm cytochrome c, 8 μm double-labeled hydrazine (98% pure; Cambridge Isotope Laboratories, Cambridge, UK), and 33 ng of purified HDH. After closing the exetainers with a rubber stopper, 1 ml of argon gas was injected by a syringe to establish a 1-atm overpressure. Incubations were performed inside the glove box at 37 °C, and reactions were followed by GC-MS analysis of headspace samples that were taken at regular time intervals with a gas-tight syringe.
Optical Redox Determinations
For reduction by dithionite, small aliquots of a freshly prepared anaerobic stock solution of dithionite were sequentially added to 1 ml of enzyme as specified in anoxic 50 mm HEPES buffer (pH 7). Spectra were recorded at 4 °C in a 1-cm path length cuvette using a V650 spectrophotometer (Jasco Analytical Instruments, Easton, MD) placed inside an anaerobic glove box. Spectra were corrected for the effect of dilution by the subsequent additions of dithionite.
Electrochemical redox titrations of the purified HDH were performed at room temperature in a home-built optical transparent thin layer electrochemical cell (30 μl) connected to a potentiostat (PGSTAT204, Metrohm Autolab, Utrecht, The Netherlands). Enzyme samples (∼30 μm) were analyzed in 50 mm MOPS (pH 7) containing 50 mm KCl and in the presence of 2,5-dimethyl-1,4-benzoquinone, 1,2-naphtoquinone, 5-methylphenazinium methyl sulfate, p-naphtoquinone, phenazine ethosulfate, 5-hydroxy-p-naphtoquinone, 1,2-dimethyl naphtoquinone, 2,5-dihydroxy-p-benzoquinone, 5,8-dihydroxy-1,4-naphtoquinone, 9,10-anthraquinone, 9,10-anthraquinone-2-sulfonate, benzyl viologen, and methyl viologen as redox mediators at 20 μm final concentration each. All titrations were performed in oxidative and reductive directions in steps of 50 mV. The solution potential was changed once no further optical changes were observed and the system was assumed to have reached redox equilibrium. Optical spectra were recorded between 400 and 600 nm on a Cary 60 spectrophotometer (Agilent, Santa Clara, CA). Spectral changes within a single experiment were evaluated on the basis of the Soret band at 420 nm and the α band at 554 nm for the seven His/His-ligated low spin hemes and at 473 nm for the active site P460 chromophore. It should be noted that the P460 signal that only arose at very low redox potentials was not always stable, hampering a precise determination of its midpoint redox potential.
EPR Measurements
X-band EPR spectra were measured on Bruker ER200D (microwave frequency, 9.39 GHz) and Varian E-9 (frequency, 9.188 GHz) spectrometers. The Q-band spectrum was recorded on the Varian E-9 (frequency, 35 GHz). Modulation amplitudes and microwave power were 1 milliteslas and 2 milliwatts, respectively. Samples were kept at 12 K by a homemade helium flow cryostat. Before the measurements, samples of as-isolated, oxidized KsHDH and KsHOX, both in the absence and presence of their substrates, were filled into quartz tubes and shock-frozen in liquid nitrogen. Analysis, spectral simulations, and quantification of the signals against a copper perchlorate standard were performed as described (30, 31).
To simulate EPR spectra of spin-coupled heme centers, we used the spin Hamiltonian in Equation 1 derived by Bencini and Gatteschi (32), which was also used by Hendrich et al. (15) in their analyses of the EPR spectra of NeHAO.
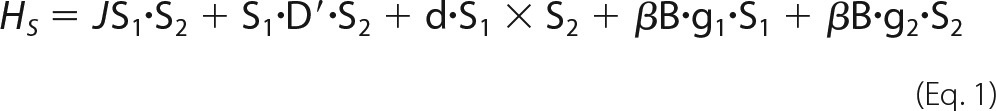 |
In the equation, J is the isotropic exchange coupling between two (S = ½) low spin heme centers, D′ is the symmetric traceless tensor of the exchange interaction, d is the polar vector of the antisymmetric exchange contribution, and the last two terms represent the Zeeman interaction of each S = ½ system with the external magnetic field. In our home-written EPR simulation program, which is available upon request and which was incorporated into the IGOR Pro version 6.0 program, the antisymmetric exchange contribution was omitted from the simulation because it hardly makes a contribution to the spectral shape at the X- and Q-band frequencies used here. Next, our simulations were second order in the spin Hamiltonian parameters rather than using diagonalization of the spin Hamiltonian, as was done in (15). Further, simulation and quantitation of the EPR spectra of magnetically isolated (S = ½) low spin heme centers only used the Zeeman terms of Equation 1, as described (19, 20).
Other Analytical Methods
Proteins were identified from polyacrylamide gels by MALDI-TOF mass spectrometry on a Bruker Biflex III mass spectrometer (Bruker Daltonik, Bremen, Germany) operated in reflectron mode. Samples for MALDI-TOF were prepared as described previously (33). Each spectrum (500–4,000 m/z) was analyzed using the Mascot Peptide Mass Fingerprint (Matrix Science, London, UK) against the K. stuttgartiensis database, allowing methionine oxidation as variable modification, 0.2-Da peptide tolerance, and at most one miscleavage. The molecular mass of the KsHDH holoenzyme was determined on the same Bruker Biflex III mass spectrometer, operating in the linear mode.
The formation of gaseous nitrogen compounds and their masses (NO, NO2, N2O, N2) were quantified by gas chromatography (Agilent 6890 equipped with a Poropak Q column at 80 °C; Agilent, Santa Clara, CA) combined with a mass spectrometer (Agilent 5975c quadruple inert MS) (5). Protein concentrations were measured with the Bio-Rad protein assay, based on the Bradford method (34), using BSA as a standard. Alternatively, the concentration of purified KsHDH was determined from the Soret absorbance peak of the oxidized protein at 408 nm (ϵ408 = 700 mm−1 cm−1). The value is presented in terms of subunit concentration (67.2 kDa).
Protein Sequence and Structure Analyses
Protein sequences were retrieved from genomic databases (K. stuttgartiensis, PRJNA16685; Jettenia caeni strain KSU-1, PRJDA163683, PRJDB68; Scalindua profunda, taxon object IDs 2017108002 and 2022004002 at the Joint Genome Institute; Brocadia sinica, PRJDB103). Multiple-amino acid sequence alignments were made with the ClustalW2 program at the EMBL-EBI web site. Atomic coordinates and structure factors of KsHOX (kustc1061) and its substrate soaks (12) were retrieved from the Protein Data Bank (PDB) (accession numbers PDB 4N4J–4N4M). The crystal structure of NeHAO (10, 11) was obtained from the PDB under accession numbers 1FGJ, 4FAS, 4N4N, and 4N4O.
Author Contributions
W. J. M., J. R., S. d. V., J. N. B., M. S. M. J., J. T. K., and B. K. designed research; W. J. M. and J. R. purified the protein; W. J. M. and N. K. determined its catalytic properties; J. T. K. made the protein sequence analyses; W. J. M., J. N. B., and J. R. investigated the electronic absorbance and redox characteristics; A. D. and T. R. M. B. performed AUC SV and SEC/MALS analyses; U. M. performed the TEM experiments; S. d. V. and J. T. K. performed the EPR analyses; W. J. M., S. d. V., J. N. B., T. R. M. B., and J. T. K. analyzed data; B. K. and J. T. K. wrote the paper.
Supplementary Material
Acknowledgments
We thank Thomas Lowe for the dithionite reduction experiments performed in Norwich. T. R. M. B. is grateful to Ilme Schlichting for continuous support. We acknowledge Christina Ferousi, Rob Schmitz, and Wouter Versantvoort for technical assistance.
This work was supported by Netherlands Organization for Scientific Research (NWO) VENI Grant 863.11.003 (to B. K.), Darwin Grant 142.16.1201 (to W. J. M.), and ALW-NWO Open Programme Grant 822.02.005 (to J. R.); by the Dutch Government “Zwaartekrachtsubsidie” (Gravitation Grant) to the Soehngen Institute for Anaerobic Microbiology (SIAM 024 002 002) (to M. S. M. J.); by European Research Council advanced Grants ERC 232937 (to M. S. M. J.), ERC 339880 (to M. S. M. J.), and ERC 640422 (to B. K.); and by the Max-Planck Society (to T. R. M. B., U. M., and A. D.). The authors declare that they have no conflicts of interest with the contents of this paper.
This work is dedicated to Simon de Vries, who passed away unexpectedly shortly after finishing his contribution to this paper.

This article contains supplemental Fig. S1.
- anammox
- anaerobic ammonium-oxidizing/anaerobic ammonium oxidation
- AUC
- analytical ultracentrifugation
- SV
- sedimentation velocity
- HAO
- hydroxylamine oxidoreductase
- NeHAO
- Nitrosomonas europaea hydroxylamine oxidoreductase
- HDH
- hydrazine dehydrogenase
- KsHDH
- Kuenenia stuttgartiensis hydrazine dehydrogenase
- HOX
- hydroxylamine oxidase
- KsHOX
- K. stuttgartiensis hydroxylamine oxidase
- SEC
- size exclusion chromatography
- MALS
- multiangle static light scattering
- TEM
- transmission electron microscopy
- PDB
- Protein Data Bank.
References
- 1. Devol A. H. (2015) Denitrification, Anammox, and N2 production in marine sediments. Ann. Rev. Mar. Sci. 7, 403–423 [DOI] [PubMed] [Google Scholar]
- 2. Lam P., and Kuypers M. M. M. (2011) Microbial nitrogen cycling processes in oxygen minimum zones. Ann. Rev. Mar. Sci. 3, 317–345 [DOI] [PubMed] [Google Scholar]
- 3. Kartal B., van Niftrik L., Keltjens J. T., Op den Camp H. J., and Jetten M. S. (2012) Anammox: growth physiology, cell biology, and metabolism. Adv. Microb. Physiol. 60, 211–262 [DOI] [PubMed] [Google Scholar]
- 4. Kartal B., Kuenen J. G., and van Loosdrecht M. C. M. (2010) Sewage treatment with Anammox. Science 328, 702–703 [DOI] [PubMed] [Google Scholar]
- 5. Kartal B., Maalcke W. J., de Almeida N. M., Cirpus I., Gloerich J., Geerts W., Op den Camp H. J., Harhangi H. R., Janssen-Megens E. M., Francoijs K.-J., Stunnenberg H. G., Keltjens J. T., Jetten M. S., and Strous M. (2011) Molecular mechanism of anaerobic ammonium oxidation. Nature 479, 127–130 [DOI] [PubMed] [Google Scholar]
- 6. Kartal B., de Almeida N. M., Maalcke W. J., Op den Camp H. J., Jetten M. S., and Keltjens J. T. (2013) How to make a living from anaerobic ammonium oxidation. FEMS Microbiol. Rev. 37, 428–461 [DOI] [PubMed] [Google Scholar]
- 7. Dietl A., Ferousi C., Maalcke W. J., Menzel A., de Vries S., Keltjens J. T., Jetten M. S., Kartal B., and Barends T. R. (2015) The inner workings of the hydrazine synthase multiprotein complex. Nature 527, 394–397 [DOI] [PubMed] [Google Scholar]
- 8. Shimamura M., Nishiyama T., Shigetomo H., Toyomoto T., Kawahara Y., Furukawa K., and Fujii T. (2007) Isolation of a multiheme protein with features of a hydrazine-oxidizing enzyme from an anaerobic ammonium-oxidizing enrichment culture. Appl. Environ. Microbiol. 73, 1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hooper A. B., Vannelli T., Bergmann D. J., and Arciero D. M. (1997) Enzymology of the oxidation of ammonia to nitrite by bacteria. Antonie Van Leeuwenhoek 71, 59–67 [DOI] [PubMed] [Google Scholar]
- 10. Igarashi N., Moriyama H., Fujiwara T., Fukumori Y., and Tanaka N. (1997) The 2.8 Å structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat. Struct. Biol. 4, 276–284 [DOI] [PubMed] [Google Scholar]
- 11. Cedervall P., Hooper A. B., and Wilmot C. M. (2013) Structural studies of hydroxylamine oxidoreductase reveal a unique heme cofactor and a previously unidentified interaction partner. Biochemistry 52, 6211–6218 [DOI] [PubMed] [Google Scholar]
- 12. Maalcke W. J., Dietl A., Marritt S. J., Butt J. N., Jetten M. S., Keltjens J. T., Barends T. R., and Kartal B. (2014) Structural basis of biological NO generation by octaheme oxidoreductases. J. Biol. Chem. 289, 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurnikov I. V., Ratner M. A., and Pacheco A. A (2005) Redox equilibria in hydroxylamine oxidoreductase: electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry 44, 1856–1863 [DOI] [PubMed] [Google Scholar]
- 14. Arciero D. M., Golombek A., Hendrich M. P., and Hooper A. B. (1998) Correlation of optical and EPR signals with the P460 heme of hydroxylamine oxidoreductase from Nitrosomonas europaea. Biochemistry 37, 523–529 [DOI] [PubMed] [Google Scholar]
- 15. Hendrich M. P., Petasis D., Arciero D. M., and Hooper A. B. (2001) Correlations of structure and electronic properties from EPR spectroscopy of hydroxylamine oxidoreductase. J. Am. Chem. Soc. 123, 2997–3005 [DOI] [PubMed] [Google Scholar]
- 16. de Almeida N. M., Neumann S., Mesman R. J., Ferousi C., Keltjens J. T., Jetten M. S., Kartal B., and van Niftrik L. (2015) Immunogold localization of key metabolic enzymes in the anammoxosome and on the tubule-like structures of Kuenenia stuttgartiensis. J. Bacteriol. 197, 2432–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shimamura M., Nishiyama T., Shinya K., Kawahara Y., Furukawa K., and Fujii T. (2008) Another multiheme protein, hydroxylamine oxidoreductase, abundantly produced in an anammox bacterium besides the hydrazine-oxidizing enzyme. J. Biosci. Bioeng. 105, 243–248 [DOI] [PubMed] [Google Scholar]
- 18. Schalk J., de Vries S., Kuenen J. G., and Jetten M. S. (2000) Involvement of a novel hydroxylamine oxidoreductase in anaerobic ammonium oxidation. Biochemistry 39, 5405–5412 [DOI] [PubMed] [Google Scholar]
- 19. Kostera J., McGarry J., and Pacheco A. A. (2010) Enzymatic interconversion of ammonia and nitrite: the right tool for the job. Biochemistry 49, 8546–8553 [DOI] [PubMed] [Google Scholar]
- 20. Hooper A. B., and Nason A. (1965) Characterization of hydroxylamine-cytochrome c reductase from the chemoautotrophs Nitrosomonas europaea and Nitrosocystis oceanus. J. Biol. Chem. 240, 4044–4057 [PubMed] [Google Scholar]
- 21. Terry K. R., and Hooper A. B. (1981) Hydroxylamine oxidoreductase: a 20-heme, 200,000 molecular weight cytochrome c with unusual denaturation properties which forms a 63,000 molecular weight monomer after heme removal. Biochemistry 20, 7026–7032 [DOI] [PubMed] [Google Scholar]
- 22. Fernández M. L., Estrin D. A., abd Bari S. E. (2008) Theoretical insight into the hydroxylamine oxidoreductase mechanism. J. Inorg. Biochem. 102, 1523–1530 [DOI] [PubMed] [Google Scholar]
- 23. Attia A. A., and Silaghi-Dumitrescu R. (2014) Computational investigation of the initial two-electron, two-proton steps in the reaction mechanism of hydroxylamine oxidoreductase. J. Phys. Chem. B 118, 12140–12145 [DOI] [PubMed] [Google Scholar]
- 24. Klotz M. G., Schmid M. C., Strous M., op den Camp H. J., Jetten M. S., and Hooper A. B. (2008) Evolution of an octahaem cytochrome c protein family that is key to aerobic and anaerobic ammonia oxidation by bacteria. Environ. Microbiol. 10, 3150–3163 [DOI] [PubMed] [Google Scholar]
- 25. Kleingardner J. G., and Bren K. L. (2015) Biological significance and applications of heme c proteins and peptides. Acc. Chem. Res. 48, 1845–1852 [DOI] [PubMed] [Google Scholar]
- 26. van de Graaf A. A., de Bruijn P., Robertson L. A., Jetten M. S. M., and Kuenen J. G. (1997) Metabolic pathway of anaerobic ammonium oxidation on basis of N-15 studies in a fluidized bed reactor. Microbiology 143, 2415–2421 [DOI] [PubMed] [Google Scholar]
- 27. Brown P. H., and Schuck P. (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90, 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yonetani T. (1965) Studies on cytochrome c peroxidase. II. Stoichiometry between enzyme, H2O2, and ferrocytochrome c and enzymic determination of extinction coefficients of cytochrome c. J. Biol. Chem. 240, 4509–4514 [PubMed] [Google Scholar]
- 30. de Vries S., and Albracht S. P. J (1979) Intensity of highly anisotropic low-spin heme EPR signals. Biochim. Biophys. Acta 546, 334–340 [DOI] [PubMed] [Google Scholar]
- 31. von Wachenfeldt C., de Vries S., and van der Oost J. (1994) The CuA site of the caa3-type oxidase of Bacillus subtilis is a mixed-valence binuclear copper center, FEBS Lett. 340, 109–113 [DOI] [PubMed] [Google Scholar]
- 32. Bencini A., and Gatteschi D. (1990) Electron Paramagnetic Resonance of Exchange Coupled Systems, Springer-Verlag, New York [Google Scholar]
- 33. Farhoud M. H., Wessels H. J., Steenbakkers P. J., Mattijssen S., Wevers R. A., van Engelen B. G., Jetten M. S., Smeitink J. A., van den Heuvel L. P., and Keltjens J. T. (2005) Protein complexes in the archaeon Methanothermobacter thermautotrophicus analyzed by blue native/SDS-PAGE and mass spectrometry. Mol. Cell Proteomics 4, 1653–1663 [DOI] [PubMed] [Google Scholar]
- 34. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.