

Abstract
The dysfunction and loss of synapses in Alzheimer disease are central to dementia symptoms. We have recently demonstrated that pathological Amyloid β oligomer (Aβo) regulates the association between intracellular protein mediators and the synaptic receptor complex composed of cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). Here we sought to determine whether Aβo alters the physiological signaling of the PrPC-mGluR5 complex upon glutamate activation. We provide evidence that acute exposure to Aβo as well as chronic expression of familial Alzheimer disease mutant transgenes in model mice prevents protein-protein interaction changes of the complex induced by the glutamate analog 3,5-dihydroxyphenylglycine. We further show that 3,5-dihydroxyphenylglycine triggers the phosphorylation and activation of protein-tyrosine kinase 2-β (PTK2B, also referred to as Pyk2) and of calcium/calmodulin-dependent protein kinase II in wild-type brain slices but not in Alzheimer disease transgenic brain slices or wild-type slices incubated with Aβo. This study further distinguishes two separate Aβo-dependent signaling cascades, one dependent on extracellular Ca2+ and Fyn kinase activation and the other dependent on the release of Ca2+ from intracellular stores. Thus, Aβo triggers multiple distinct PrPC-mGluR5-dependent events implicated in neurodegeneration and dementia. We propose that targeting the PrPC-mGluR5 complex will reverse aberrant Aβo-triggered states of the complex to allow physiological fluctuations of glutamate signaling.
Keywords: Alzheimer disease, amyloid β, Ca2+/calmodulin-dependent protein kinase II (CaMKII), metabotropic glutamate receptor (mGluR), prion, homer, pyk2
Introduction
Alzheimer disease is the most common form of age-related dementia. The Amyloid hypothesis of Alzheimer disease postulates proteolytic processing of the amyloid precursor protein (APP)2 to be disease-relevant (1). This process releases hydrophobic Amyloid β (Aβ) peptides that aggregate in the form of characteristic Amyloid plaques in the brain of patients (1). Aggregation of Aβ is a multistep process, with several intermediate forms of Aβ being generated. The Aβ species that correlates best with the severity of dementia is an oligomeric species of Aβ, referred to as Aβo (2–5). Several independent lines of research have revealed an Aβo-toxic function at synapses (6–10). Thus, a better understanding of intracellular signaling induced by extracellular Aβo could reveal novel ways to prevent neurotoxicity.
Our previous work described cellular prion protein (PrPC) as a high-affinity cell surface receptor for Aβo (11), capable of mediating deficits in plasticity, synapse density, and memory (12–15). Aβo binding to PrPC triggers intracellular signaling via metabotropic glutamate receptor 5 (mGluR5) (12, 16, 17), which includes downstream Fyn activation and synapse loss (12, 13, 18). We recently demonstrated that PrPC is linked to intracellular proteins via mGluR5 and that this coupling is modulated by extracellular Aβo (16). Critically, our work further revealed that the interaction between PrPC and intracellular protein mediators is perturbed in Alzheimer disease (16). Here we focus on comparing how the physiological glutamate analog 3,5-dihydroxyphenylglycine (DHPG) affects the PrPC-mGluR5 complex in the absence and presence of Aβo. We investigate a multiprotein complex consisting of at least PrPC, mGluR5, Homer1b/c, protein-tyrosine kinase 2-β (PTK2B, Pyk2), and calcium/calmodulin-dependent protein kinase II (CamKII). Interestingly, Homer proteins as well as Pyk2 have been associated with susceptibility to Alzheimer disease by imaging quantitative trait loci studies and genome-wide association studies (19–21). These studies provide a genetic link between the PrPC-mGluR5 multiprotein complex and Alzheimer disease. Furthermore, Homer proteins are known to be important postsynaptic protein mediators that link mGluR5 to protein kinases and are able to alter mGluR5-induced signaling independent of glutamate-induced activation (22–25).
Importantly, Aβo has been shown to cluster at excitatory synapses that stain positively for Homer1b/c as well as for CamKII (26, 27). This clustering of pathological Aβo is mGluR5- and PrPC-dependent (26). Renner et al. (26) further observed that Aβo alters the synaptic trafficking of the otherwise laterally mobile mGluR5 within primary neuronal membranes. These data suggest that pathological Aβo bound to PrPC scaffolds mGluR5 into a pathological conformation within the plasma membrane, which might disrupt physiological glutamate signaling.
The aim of this study is to understand whether pathological Aβo alters physiological glutamate-induced stimulation of mGluR5 in neurons. Our data reveal that isolated Aβo as well as APP/PS1 transgene-dependent species prohibit DHPG-induced association changes in the PrPC-mGluR5 multiprotein complex. Furthermore, DHPG is unable to enhance phosphorylation of Pyk2 and CamKII in the presence of Aβo. We conclude that pathological Aβo traps the PrPC-mGluR5 complex in a pathological state that does not allow glutamate-induced regulation of the complex. Thus, the PrPC-mGluR5 complex is a potential target for disease-modifying therapy for Alzheimer disease. Our work advances the understanding of the regulation of the PrPC-mGluR5 complex by physiological versus pathological ligands. This knowledge is important for the development of therapeutics targeting the complex in the context of Alzheimer disease to counteract the Aβo-dependent disruption of physiological glutamate signaling at this metabotropic receptor.
Results
The PrPC-mGluR5 complex isolated by anti-PrPC immunoprecipitation associates with the intracellular protein mediators Homer1b/c, Pyk2, and CamKII in crude synaptoneurosome preparations from acute brain slices in a PrPC-dependent manner (Fig. 1A and Ref. 16). We found that DHPG triggers an enhanced association between PrPC and Homer1b/c but a reduced association between PrPC and Pyk2 as well as between PrPC and CamKII (Fig. 1). Contrarily, Aβo treatment enhances the association of mGluR5 as well as CamKII with PrPC, whereas Aβo stimulation dissociates Homer1b/c as well as Pyk2 from the complex (Fig. 1). We have previously characterized these phenomena to be fully dependent on mGluR5 and PrPC using genetic null brain preparations as well as a HEK cell overexpression system (16). Here we further show that DHPG and Aβo both cause increased mGluR5 but reduced Pyk2 signals in the anti-PrPC precipitates, similar to our previous results showing parallel actions of DHPG and Aβo for Fyn activation and Ca2+ signaling (12). However, DHPG and Aβo have opposite effects on the association of Homer1b/c and CamKII with the complex, demonstrating unique actions of these two ligands. Most critically, DHPG-triggered association changes between PrPC and Homer1b/c or PrPC and CamKII fail to appear in slices pretreated with Aβo for 15 min (Fig. 1, B, D, and F).
FIGURE 1.

DHPG and Aβo differentially regulate the association between PrPC and intracellular protein mediators in acute mouse brain slices. A, representative immunoblots showing co-IP between PrPC and mGluR5, Homer1b/c (Homer), Pyk2, and CamKII in acute brain slice crude synaptoneurosomal preparations. Co-immunoprecipitation between PrPC and mGluR5, Homer1b/c, Pyk2, and CamKII is dependent on the genetic presence of prnp. B, wild-type brain slices were treated as indicated prior to preparation of crude synaptoneurosomes. DHPG treatment, 15 min; Aβo treatment, 30 min; Aβo+DHPG treatment, 15 min of Aβo pretreatment followed by 15 min DHPG treatment. Veh, vehicle. C–F, densitometric analysis of the immunoblots from B, analyzed by one-way ANOVA with Fisher's LSD post hoc pairwise comparisons test (C and F) or post-hoc Tukey's multiple comparisons test (D and E). Data are mean ± S.E. (n = 6 independent experiments for vehicle/DHPG treated slices, n = 14 independent experiments for vehicle/Aβo treated slices, and n = 3 independent experiments for Aβo+DHPG treated slices). Each experiment includes 6–8 slices/treatment condition. C, Aβo as well as Aβo+DHPG treatment of brain slices significantly increases the mGluR5 signal in anti-PrPC immunoprecipitates (*, p < 0.05). D, co-IP between PrPC and Homer1b/c is significantly enhanced after DHPG treatment (*, p < 0.05) and significantly reduced after Aβo treatment (***, p < 0.001). Enhanced DHPG-triggered interaction between PrPC and Homer1b/c is significantly reduced by Aβo pretreatment (*, p < 0.05). E, co-IP between PrPC and Pyk2 is significantly reduced after DHPG treatment, Aβo treatment, as well as DHPG+Aβo treatment (**, p < 0.01; *, p < 0.05). F, co-IP between PrPC and CamKII is significantly reduced after DHPG treatment but significantly enhanced after Aβo as well as DHPG+Aβo treatment (**, p < 0.01; *, p < 0.05).
The brain slice incubations with Aβo occur over a short time period relative to the course of Alzheimer disease. We sought to understand whether chronic accumulation of Aβo in APP/PS1 transgenic brain prevents DHPG-induced association changes of the complex similar to acute Aβo exposure. In aged wild-type preparations, we found DHPG-enhanced association between PrPC and Homer1b/c but reduced association between PrPC and Pyk2 as well as between PrPC and CamKII (Fig. 2). Interestingly, DHPG-triggered alterations of the PrPC-mGluR5 complex are not detectable in APP/PS1 brain slices (Fig. 2). Thus, the APP/PS1 genetic background prevents DHPG-induced association changes in the complex.
FIGURE 2.

The APP/PS1 transgene prevents DHPG-triggered association changes in the PrPC-mGluR5 complex. A, representative immunoblots of co-IP between PrPC and mGluR5, Homer1b/c, Pyk2, and CamKII from acute brain slice crude synaptoneurosomal preparations. The genotype of brain slices and treatment with 100 μm DHPG for 15 min is indicated above each lane. Slices were collected from WT and APP/PS1 mice at 12–16 months of age. B–E, densitometric analysis of the immunoblots from A, analyzed by one-way ANOVA with Fisher's LSD post hoc pairwise comparisons test (B, C, and E) or post-hoc Tukey's multiple comparisons test (D). Data are mean ± S.E. (n = 3 independent experiments). B, the APP/PS1 transgene significantly enhances the mGluR5 signal in anti-PrPC immunoprecipitates (*, p < 0.05). C, co-IP between PrPC and Homer1b/c is significantly enhanced after DHPG treatment in WT slices (*, p < 0.05). DHPG treatment of APP/PS1 slices does not significantly alter the association between PrPC and Homer1b/c compared with vehicle-treated APP/PS1 slices (ns, p > 0.05). D, co-IP between PrPC and Pyk2 is reduced by acute DHPG treatment of WT brain slices as well as the APP/PS1 transgene (*, p < 0.05). DHPG treatment does not significantly modulate the co-IP between PrPC and Pyk2 compared with vehicle-treated APP/PS1 slices (ns, p > 0.05). E, DHPG treatment significantly reduces the co-IP between PrPC and CamKII in WT slices (*, p < 0.05) but not in APP/PS1 slices (ns, p > 0.05).
Alterations in protein kinase activation might participate in Alzheimer disease pathogenesis. Pyk2 and CamKII are two kinases associated with the PrPC-mGluR5 complex that are activated by acute Aβo stimulation in a PrPC- and mGluR5-dependent manner (16, 18). Here we determined that DHPG as well as ionomycin treatments activate both kinases similarly to Aβo-triggered phosphorylation of Pyk2 and CamKII (Fig. 3). A combination of DHPG and Aβo does not lead to a greater increase in phosphorylation of Pyk2 or CamKII compared with either treatment alone (Fig. 3).
FIGURE 3.
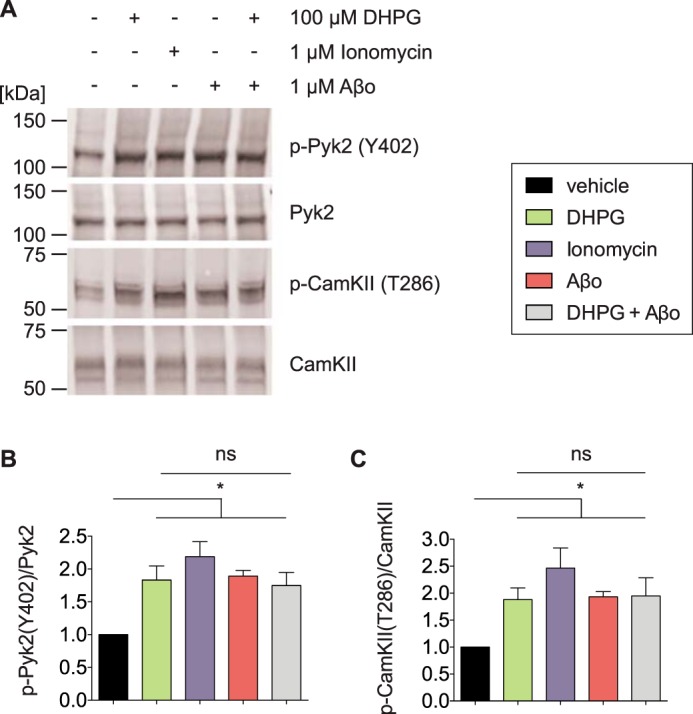
Activation of mGluR5-associated intracellular kinases. A, representative immunoblots showing enhanced Pyk2(Tyr-402) and CamKII(Thr-286) phosphorylation after stimulation with DHPG, ionomycin, or Aβo. The treatment of brain slices is indicated above each lane. DHPG treatment, 15 min; Aβo treatment, 30 min; Aβo+DHPG treatment, 15 min of Aβo pretreatment followed by 15 min of DHPG treatment. DMSO was used as vehicle, and the DMSO content was constant in all lanes. B and C, densitometric analysis of the immunoblots from A, analyzed by one-way ANOVA with Tukey's multiple comparisons test. Data are mean ± S.E. (n = 11 independent experiments for vehicle or DHPG treatment, n = 2 independent experiments for ionomycin treatment, n = 21 independent experiments for Aβo treatment, and n = 8 independent experiments for DHPG+Aβo treatment). 3 slices/condition were averaged for every experiment. All treatment conditions triggered phosphorylation of Pyk2 at Tyr-402 (B) and CamKII at Thr-286 (C) (*, p < 0.05). In the presence of Aβo, Pyk2 and CamKII are fully activated and cannot be stimulated further by DHPG (ns, p > 0.05).
Based on these acute Aβo exposure results, we considered whether chronic APP/PS1 transgene-dependent expression of Aβo similarly prevents DHPG-induced phosphorylation of Pyk2 and CamKII. Importantly, DHPG-triggered activation of the mGluR5-associated kinases Pyk2 and CamKII is not observable in aged APP/PS1 brain slices (Fig. 4). Thus, acute Aβo treatment as well as the APP/PS1 background prevent DHPG-induced activation of Pyk2 and CamKII. Of note, acute Aβo stimulation triggers phosphorylation of CamKII, whereas chronic exposure to Aβo in APP/PS1 mouse brain induces a slight deactivation of CamKII. Our previous work described a biphasic temporal effect of Aβo on CamKII by characterizing the activation of CamKII after different Aβo incubation periods (16). Those experiments documented activation of CamKII after acute Aβo stimulation (e.g. 30 min after stimulation) but deactivation of CamKII after longer Aβo incubation periods (e.g. 6 h after stimulation), representing a more chronic state of Aβo exposure (16). The key point here is that chronic Aβo exposure prevents DHPG regulation of Pyk2 and CamKII activation.
FIGURE 4.

The APP/PS1 transgenic background prevents DHPG-induced activation of Pyk2 and CamKII. A, representative immunoblots of Pyk2(Tyr-402) and CamKII(Thr-286) phosphorylation after 15 min of DHPG treatment of brain slices. The genotype and treatment of brain slices are indicated above each lane. Slices were collected from WT and APP/PS1 mice at 12–16 months of age. Veh, vehicle. B and C, densitometric analysis of the immunoblots from A, analyzed by one-way ANOVA with Tukey's multiple comparisons test. Data are mean ± S.E. (n = 3 independent experiments, with 3 slices/condition averaged for every experiment). B, phosphorylation of Pyk2 at Tyr-402 is significantly enhanced in APP/PS1 brain slices compared with the WT (*, p < 0.05). DHPG triggers phosphorylation of Pyk2 at Tyr-402 in WT brain slices (*, p < 0.05) but not in APP/PS1 brain slices (ns, p > 0.05). C, DHPG treatment increases phosphorylation of CamKII at Thr-286 in WT (*, p < 0.05) but not in APP/PS1 brain slices (ns, p > 0.05). The APP/PS1 transgene significantly reduces DHPG-triggered phosphorylation of CamKII at Thr-286 compared with the WT (*, p < 0.05).
It is striking that DHPG and Aβo induce opposite association changes of CamKII with the PrPC-mGluR5 complex. In contrast, DHPG and Aβo both trigger dissociation of Pyk2 from the PrPC-mGluR5 complex. The differential effects suggest separate mechanisms underlying activation of Pyk2 and CamKII. Although both Pyk2 and CamKII activation are regulated by intracellular Ca2+ (28, 29), we sought to assess whether stimulation of Pyk2 and CamKII in acute brain slices might depend on different sources of Ca2+.
Our data demonstrate that removal of extracellular Ca2+ for 30 min prevents activation of Pyk2 at Tyr-402 in acute brain slices (no significant difference of Pyk2 activation between vehicle treatment and DHPG/Aβo/DHPG+Aβo treatment in the absence of ext. Ca2+; Fig. 5, A and B). In contrast, Aβo-induced activation of CamKII at Thr-286 is independent of extracellular Ca2+, and DHPG as well as DHPG+Aβo stimulation follow the same pattern (Fig. 5, A and C). Importantly, DHPG/Aβo/DHPG+Aβo treatment activates Pyk2 independent of endoplasmic reticulum Ca2+ store depletion by thapsigargin for 30 min (Fig. 6, A and B). In contrast, thapsigargin pretreatment of acute brain slices completely abolished the ability of DHPG/Aβo/DHPG+Aβo treatment to activate CamKII at Thr-286 (Fig. 6, A and C). Thus, removal of extracellular Ca2+ and thapsigargin-induced endoplasmic reticulum Ca2+ depletion differentially affect the activation pattern of the intracellular kinases Pyk2 and CamKII.
FIGURE 5.

Extracellular calcium depletion prevents activation of Pyk2 but not CamKII. A, representative immunoblots showing Pyk2 (Tyr-402) and CamKII (Thr-286) phosphorylation levels after exposure to DHPG (15 min), Aβo (30 min), or DHPG+Aβo (15 min of Aβo pretreatment followed by 15-min DHPG treatment). 30 min prior to treatment, slices were moved into a fresh bath of Ca2+-free artificial cerebral spinal fluid (+ 1 mm EDTA) as indicated. DMSO was used as vehicle, and the DMSO content was constant in all lanes. B and C, densitometric analysis of the immunoblots from A, data are mean ± S.E. Under normal calcium conditions, n = 11 independent experiments for vehicle or DHPG treatment, n = 21 independent experiments for Aβo treatment, and n = 8 independent experiments for DHPG+Aβo treatment. In absence of extracellular (ext.) Ca2+, n = 2 independent experiments for vehicle, DHPG, or DHPG+Aβo treatment, and n = 3 independent experiments for Aβo treatment. 3 slices/treatment condition and experiment were averaged. B, Pyk2 phosphorylation at Tyr-402 is enhanced by all treatment conditions under normal ext. Ca2+ levels (**, p < 0.01 by two-way ANOVA with post-hoc Tukey's multiple comparisons test). Pyk2 phosphorylation at Tyr-402 is not significantly altered by any treatment condition in the absence of ext. Ca2+ (ns, p > 0.05). C, CamKII phosphorylation at Thr-286 is enhanced by all treatment conditions under normal ext. Ca2+ levels (**, p < 0.01 by two-way ANOVA with Dunnett's multiple comparisons test). In the absence of ext. Ca2+, CamKII phosphorylation at Thr-286 is enhanced by Aβo-stimulation (*, p < 0.05 by two-way ANOVA with Dunnett's multiple comparisons test).
FIGURE 6.

Intracellular calcium depletion prevents activation of CamKII but not Pyk2. A, representative immunoblots showing Pyk2 (Tyr-402) and CamKII (Thr-286) phosphorylation levels after exposure to DHPG (15 min), Aβo (30 min), or DHPG+Aβo (15 min of Aβo pretreatment followed by 15-min DHPG treatment). Prior to treatment, slices were preincubated with 2 μm thapsigargin for 30 min as indicated. DMSO was used as vehicle, and the DMSO content was constant in all lanes. B and C, densitometric analysis of the immunoblots from A. Data are mean ± S.E. In absence of thapsigargin, n = 11 independent experiments for vehicle or DHPG treatment, n = 21 independent experiments for Aβo treatment, and n = 8 independent experiments for DHPG+Aβo treatment. In the presence of thapsigargin, n = 2 independent experiments for vehicle, DHPG, or DHPG+Aβo treatment, and n = 3 independent experiments for Aβo treatment. 3 slices/treatment condition and experiment were averaged. B, Pyk2 phosphorylation at Tyr-402 is enhanced by all treatment conditions in the absence and presence of thapsigargin (*, p < 0.05; ***, p < 0.091; by two-way ANOVA with Fisher's LSD post hoc pairwise comparisons test). C, CamKII phosphorylation at Thr-286 is enhanced by all treatment conditions in the absence of thapsigargin (*, p < 0.05) but not in the presence of thapsigargin (ns, p > 0.05).
We sought to further segregate pathways involved in the activation of Pyk2 and CamKII through PrPC-mGluR5 complexes. As shown previously, Fyn kinase is an additional mediator of Aβo-induced neurotoxicity (13, 30–32). Preincubation of acute brain slices with AZD0530, a Src family inhibitor that fully blocks Fyn activation, suppresses basal phospho-Pyk2(Tyr-402) levels and prevents activation of Pyk2 at Tyr-402 induced by DHPG or Aβo (Fig. 7, A and B). In contrast, AZD0530 does not alter the activation of CamKII at Thr-286 (Fig. 7, A and C). Thus, both Fyn kinase activity and the source of intracellular Ca2+ distinguish the activation pathways of Pyk2 and CamKII.
FIGURE 7.

Fyn kinase inhibition prevents activation of Pyk2 but does not alter activation of CamKII. A, representative immunoblots showing Pyk2 (Tyr-402) and CamKII (Thr-286) phosphorylation states after treatment of acute brain slices. Slices were pretreated with 2 μm AZD0530 for 2 h prior to exposure to DHPG (15 min), Aβo (30 min), or DHPG+Aβo (15 min of Aβo pretreatment followed by 15-min DHPG treatment). DMSO was used as vehicle, and the DMSO content was constant in all lanes. B and C, densitometric analysis of the immunoblots from A. Data are mean ± S.E. In the absence of AZD0530, n = 11 independent experiments for vehicle or DHPG treatment, n = 21 independent experiments for Aβo treatment, and n = 8 independent experiments for DHPG+Aβo treatment. In the presence of AZD0530, n = 2 independent experiments for vehicle, DHPG, or DHPG+Aβo treatment, and n = 3 independent experiments for Aβo treatment. 3 slices/treatment condition and experiment were averaged. B, Pyk2 phosphorylation at Tyr-402 is enhanced by all treatment conditions (*, p < 0.05 by two-way ANOVA with post-hoc Tukey's multiple comparisons test). AZD0530 pretreatment slightly suppresses basal p-Pyk2 (Tyr-402) levels and prevents DHPG/Aβo-induced activation of Pyk2 (ns, p > 0.05). C, CamKII phosphorylation at Thr-286 is enhanced by all treatment conditions in the absence and presence of AZD0530 (***, p < 0.001; *, p < 0.05; by two-way ANOVA with Fisher's LSD post hoc pairwise comparisons test).
Discussion
We have shown previously that PrPC and mGluR5 function as a complex in the brain and that PrPC interacts with the intracellular protein mediators Homer1b/c, Pyk2, and CamKII via mGluR5 in an Aβo-regulated manner (12, 16, 17). Importantly, our previous study demonstrated a pathological reduction of the PrPC-Homer1b/c association as well as the PrPC-Pyk2 association in transgenic mouse Alzheimer model brain and in human Alzheimer patient brain compared with age-matched healthy controls (16). Thus, a better understanding of the extracellular modulation of association changes in the PrPC-mGluR5 multiprotein complex could provide important insights into Alzheimer disease pathogenesis and might be relevant for therapeutic development. The major finding of this study is that pathological Aβo prevents DHPG-induced changes in the composition of the PrPC-mGluR5 signaling complex. Specifically, Aβo fully activates the intracellular kinases Pyk2 and CamKII. Maximal phosphorylation of these two kinases precludes further activation by the glutamate analog DHPG because of a ceiling effect. Of note, Aβo is a pathological ligand not present in healthy brain, and Aβo exposure in AD is chronic without rapid fluctuation during the synaptic cycle. This study suggests that Aβo exposure traps the PrPC-mGluR5 complex in a conformation that renders it non-responsive to physiological glutamate-induced activation. Moreover, the characteristics of the Aβo-triggered state appear to be distinct from that induced by the glutamate analog DHPG (Fig. 8).
FIGURE 8.

Alzheimer disease Aβo prevents physiological activation of the PrPC-mGluR5 signaling complex. A, schematic demonstrating the interaction between GPI-anchored PrPC with Pyk2, Homer1b/c, and CamKII via mGluR5 in postsynaptic spines. The interaction is modulated by physiological glutamate as well as by pathological Aβo. B, under physiological conditions, the glutamate analog DHPG triggers enhanced association between Homer1b/c and the PrPC-mGluR5 signaling complex. Contrarily, DHPG stimulation initiates dissociation of Pyk2 as well as CamKII from the complex, which coincides with autophosphorylation of both kinases. In presence of acute Aβo, Homer1b/c as well as Pyk2 are released from the complex, whereas CamKII shows enhanced association with the complex. DHPG and acute Aβo triggers phosphorylation of Pyk2 as well as CamKII. Activation of Pyk2 is dependent on ext. Ca2+ and Fyn kinase activity, whereas phosphorylation of CamKII is dependent on the release of Ca2+ from intracellular stores. Chronic Aβo affects Pyk2 similarly to acute Aβo. Contrarily, CamKII is not altered by pathological chronic exposure to Aβo. Interestingly, association changes in the PrPC-mGluR5 complex induced by Alzheimer disease Aβo prevent physiological DHPG-induced signaling events. ↑ indicates enhanced binding between the protein of interest and the PrPC-mGluR5 complex or enhanced phosphorylation. ↓ indicates reduced binding between the protein of interest and the PrPC-mGluR5 complex or a decrease in phosphorylation. − indicates the absence of changes in co-immunoprecipitation or phosphorylation of the protein of interest. The column DHPG in presence of Aβo compares the effect of DHPG after Aβo pretreatment with the effect of DHPG in the absence of Aβo; − implies no further change induced by DHPG stimulation after Aβo pretreatment.
Our previous work showed reduced association of Homer1b/c with the PrPC-mGluR5 complex upon acute Aβo treatment as well as in an Alzheimer disease transgenic model and patient brain (16). Contrary to the Aβo results, we show here that DHPG stimulation triggers enhanced association between Homer1b/c and the PrPC-mGluR5 complex. Interestingly, pretreatment of brain slices with Aβo followed by DHPG stimulation prevents DHPG-induced enhanced association of Homer1b/c with the PrPC-mGluR5 complex, revealing a dominant effect of prior Aβo exposure.
More importantly, DHPG is unable to alter the association state between PrPC and Homer1b/c in Alzheimer disease transgenic model brain slices. Pathologically enhanced Aβo in human patients might similarly create a chronic state where fluctuating glutamate levels are unable to alter Homer interactions with mGluR5. It has been shown that preventing the interaction between Homer proteins and mGluR proteins prohibits mGluR-dependent synaptic plasticity, and Aβo are known to be potent synaptotoxins (6, 33). Thus, we hypothesize that Aβo could prevent signaling mechanisms underlying synaptic plasticity by pathologically altering the conformational relationship between mGluR5 and Homer proteins.
Associated with the intracellular C-terminal tail as well as the intracellular loop 2 of mGluR5, CamKII is well known to be another important mediator of synaptic plasticity (34, 35). Thus, CamKII could be a further candidate that might be involved in mechanisms underlying Aβo-triggered inhibition of synaptic function. Jin et al. (35) revealed DHPG-induced dissociation of CamKII from mGluR5 in primary striatal neurons (35). Our previous work as well as other studies demonstrated Aβo-induced enhanced association of CamKII with the PrPC-mGluR5 complex (16, 34). Here we reveal abrogation of DHPG-induced dissociation of CamKII from the PrPC-mGluR5 complex in the presence of Aβo. Importantly, CamKII is known to dissociate from mGluR upon long-term potentiation induction (35). After dissociation from mGluRs, the kinase associates more strongly with NR2B subunits of NMDA receptors (35). Phosphorylation of NR2B at Tyr-1472 by Fyn kinase is critically involved in long-term potentiation and Aβo toxicity (12, 18, 36). Aβo-induced inhibition of long-term potentiation may depend in part on CamKII being trapped within the Aβo-PrPC-mGluR5 complex and thus prevent regulation of NMDA NR2B subunits by CamKII.
Activation of intracellular kinases could contribute to neurodegenerative phenotypes, and as such, Fyn kinase inhibition is currently under evaluation as an Alzheimer disease therapeutic in a clinical setting (30, 37, 38). In this study, we focus on Aβo-induced activation of Pyk2 and CamKII. We find that acute DHPG as well as Aβo treatment of brain slices equally activates Pyk2 and CamKII. In the presence of both ligands, DHPG and Aβo, no further stimulation is achieved. We tested whether this observation is due to a ceiling effect achieved by either DHPG or Aβo. We used the calcium ionophore ionomycin to achieve a maximal supraphysiological intracellular calcium increase and thereby activate both kinases maximally. The levels of Pyk2 and CamKII phosphorylation in the presence of ionomycin are not significantly different from DHPG- or Aβo-activated Pyk2 and CamKII levels. Thus, our data indicate that activation of either kinase by a first ligand (Aβo) precludes further phosphorylation by a second ligand (DHPG) because both kinases are already fully activated by the first ligand.
Our results are supported by earlier studies reporting DHPG-induced stimulation of Pyk2 in primary cortical neurons as well as dissociation of Pyk2 from mGluR1 upon mGluR stimulation in HEK-293T cells (39, 40). Interestingly, we observed a strong correlation between Pyk2 phosphorylation and dissociation from the PrPC-mGluR5 complex. Thus, Pyk2 phosphorylation at Tyr-402 might participate in the dissociation of Pyk2 from the complex. Alternatively, conformational changes induced during dissociation of Pyk2 from the PrPC-mGluR5 complex might enhance Pyk2 autophosphorylation at Tyr-402.
Our results do not show a correlation between CamKII phosphorylation and CamKII dissociation from the PrPC-mGluR5 complex. These differences in the structural relationship between the PrPC-mGluR5 complex and either Pyk2 or CamKII led us to investigate different mechanisms of Pyk2 and CamKII activation. We assessed intra- and extracellular Ca2+ compartments to determine how Ca2+ modulates the activation of Pyk2 or CamKII. We found that removal of extracellular Ca2+ completely abrogated the activation of Pyk2. These results suggest that the activation of Pyk2 is most likely dependent on extracellular Ca2+ influx through NMDA receptors. In contrast, stimulation of CamKII was not affected by removal of extracellular Ca2+ but was fully abolished by using an endoplasmic reticulum Ca2+ ATPase inhibitor. Importantly, mGluR5 activity is directly linked to endoplasmic reticulum Ca2+ levels, which, in turn, regulate the expression of stromal interaction molecule 2 (STIM2) (41). STIM2 is an endoplasmic reticulum-localized protein that controls neuronal store-operated calcium entry (nSOC) and thereby regulates CamKII activity and stabilization of mushroom spines (42). Interestingly, previous work demonstrated that the STIM2-nSOC-CaMKII pathway is dysregulated in neurons from Alzheimer mouse models as well as in Alzheimer patient brains (41, 42). Of note, thapsigargin-induced endoplasmic reticulum Ca2+ depletion hyperactivates the nSOC pathway via STIM proteins, which leads to reduced activity of CamKII (42, 43). Thus, our results demonstrating the absence of CamKII activation in the presence of thapsigargin might be due to thapsigargin-dependent opening of nSOC with reduced CamKII activity because of enhanced nSOC. Alternatively, activation of CamKII could directly depend on the release of Ca2+ from endoplasmic reticulum stores. Previous work demonstrated similar results for DHPG-induced ERK phosphorylation in rat striatal neurons. In those studies, intracellular Ca2+ store depletion by thapsigargin completely abolished DHPG-triggered activation of ERK (25). Further work is necessary to ultimately clarify the role of different Ca2+ channels in CamKII activation.
We further characterized activation of Pyk2 to be fully dependent on Fyn kinase activity, whereas activation of CamKII is not affected by Fyn kinase inhibition. Taken together, it is clear that two separate signaling pathways trigger the activation of Pyk2 and CamKII (Fig. 7).
Our data reveal signaling mechanisms that might contribute to Alzheimer disease pathogenesis. It is well established that glutamate-induced activation of metabotropic intracellular signaling is essential for physiological brain function. Our data demonstrate that Alzheimer disease transgenic model mice are unable to respond to stimulation by the glutamate analog DHPG. We speculate that Aβo-induced pathological conformations of the PrPC-mGluR5 complex could participate in the development of synaptic and behavioral abnormalities in transgenic mice (14, 44–47). We conclude that physiologically fluctuating levels of glutamate might be unable to modify the interaction between mGluR5 and intracellular signaling mediators in a pathophysiological Aβo-dependent disease state. Similar pathophysiological association states between mGluRs and Homer scaffolding proteins have been proposed in the context of several neurological diseases, including fragile X syndrome and Angelman syndrome (48, 49). Our data suggests that preventing Aβo-induced activation of the PrPC-mGluR5 signaling complex has potential clinical implications for Alzheimer disease. Our previous work revealed PrPC-directed antibodies and mGluR5-directed compounds to potently modulate the interaction between PrPC and mGluR5 (17). Future studies will further explore their potential as Alzheimer disease therapeutics.
Experimental Procedures
Soluble Aβ1–42 Oligomer Preparation
The Keck Large Scale Peptide Synthesis Facility (Yale University) synthesized Aβ1–42 peptide. The preparation of Aβ1–42 oligomers (Aβo) has been described previously (13). Concentrations of Aβo are expressed in monomer equivalents, with 1 μm total Aβ1–42 peptide corresponding to ∼10 nm oligomeric species (11).
Mouse Strains
Mice were cared for by the Yale Animal Resource Center, and all experiments were approved by the Institutional Animal Care and Use Committee of Yale University. Wild-type and APPswe/PS1dE9 (APP/PS1) mice (50) were purchased from The Jackson Laboratory and maintained on a C57/Bl6J background. Mice were group-housed (2–5 mice/cage) in a 12-h light/dark cycle. Male and female mice were used equally.
Preparation of Acute Mouse Brain Slices
Mouse brains were dissected after rapid decapitation and immediately dispersed in ice-cold artificial cerebral spinal fluid (119 mm NaCl; 2.5 mm KCl; 1.3 mm MgSO4; 26.2 mm NaHCO3; 11 mm d-glucose; 1.25 mm NaH2PO4). 400-μm coronal brain slices were cut in ice-cold artificial cerebral spinal fluid using a 1000 Plus vibratome with steel razor blades. Brain slices were incubated at room temperature in artificial cerebral spinal fluid plus 2.4 mm CaCl2 under constant oxygenation with 95% O2 and5% CO2. After a 2-h recovery period, slices were treated with Aβo and/or DHPG. For acute Aβo/DHPG experiments, 4- to 8-week-old wild-type mice were used. For transgenic studies, wild-type and APP/PS1 brain slices were prepared from mice at 12–16 months of age, when memory deficits, synaptic loss, and Aβ accumulation are established (12–14, 18, 51).
Crude Synaptoneurosome Preparation
Acute brain slices were homogenized in buffer A (0.32 m sucrose, 20 mm HEPES (pH 7.4), 1 mm EDTA, 1× PhosSTOP-Roche, and 1× cOmplete mini protease inhibitor mixture (Roche)). Homogenates were centrifuged for 10 min at 875 × g at 4 °C. The supernatant was again centrifuged for 10 min at 16,000 × g at 4 °C to obtain a cytosolic fraction (supernatant) and a crude synaptoneurosomal fraction (P2 pellet). P2 pellets were resuspended and sonicated in buffer A prior to use.
Immunoprecipitation
For each immunoprecipitation (IP) experiment, crude synaptoneurosomal fractions from 6–8 mouse brain slices/treatment condition were combined prior to IP (n = number of separate experiments). The protein concentration in crude synaptoneurosomal fractions was determined by Bradford assay (Bio-Rad protein assay), and fractions were precleared from endogenous antibodies for 4 h at 4 °C. Crude synaptoneurosomal fractions were then incubated overnight with capture antibody (1 μg/1 mg of homogenate protein) at 4 °C. The capture antibody used was Saf32 (Cayman, 189720, mouse anti-prion protein), which was validated by comparing immunoprecipitation from wild-type to prnp-null brain. The preformed antibody-antigen complexes were then incubated with PureProteome protein A/G mix magnetic beads (Millipore, LSKMAGAG10) for 1 h at 4 °C under gentle rotation. Beads were washed five times in buffer A prior to elution of proteins in SDS-PAGE sample loading buffer. The immunoprecipitated complexes were then resolved by SDS-PAGE and immunoblotted.
Analysis of Protein Activation States after Treatment of Acute Mouse Brain Slices
Acute mouse brain slices were prepared and treated as described above. Aβ was oligomerized as described previously in a vehicle composed of DMSO in glutamate-free F-12 medium (13). The vehicle used for DHPG, thapsigargin, as well as AZD0530 was double-distilled H2O. Ionomycin was prepared in DMSO and used at a concentration of 1 μm. DHPG was used at a concentration of 100 μm, and thapsigargin as well as AZD0530 were used at a concentration of 2 μm. For the experiments in Figs. 3 and 5–7, the DMSO content in all lanes was constant.
After incubation of brain slices with compounds as indicated in the figure legends, slices were homogenized in radioimmune precipitation assay lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 0.1% SDS, 0.5% deoxycholic acid, 1× PhosSTOP-Roche, and 1× cOmplete-mini protease inhibitor mixture (Roche)) and centrifuged at 21,000 × g for 20 min at 4 °C. The protein concentration in the radioimmune precipitation assay-soluble fraction was determined by Bradford assay (Bio-Rad protein assay). The radioimmune precipitation assay-soluble fraction was then mixed with SDS-PAGE sample loading buffer, and proteins were resolved by SDS-PAGE followed by immunoblot. Experiments consisted of averaged data from three slices (n = number of separate experiments). No experiments were excluded from the final data.
Immunoblots
Proteins were electrophoresed through precast 4–20% Tris/glycine gels (Bio-Rad) and transferred with an iBlotTM gel transfer device (Novex-Life Technologies) onto nitrocellulose membranes (Invitrogen). Membranes were blocked (blocking buffer for fluorescent Western blotting, Rockland, MB-070-010) for 1 h at room temperature and incubated overnight in primary antibodies at 4 °C. The following antibodies were used: rabbit anti-actin (Sigma-Aldrich, A2066, 1:3000), goat anti-CaMKII (Santa Cruz Biotechnology, sc-5392, 1:500), rabbit anti-Homer1b/c (Santa Cruz Biotechnology, sc-55463, 1:500), rabbit anti-mGluR5/1 (R&D Systems, PPS079, 1:1000), rabbit anti-phospho-CamKII (Abcam, ab5683, 1:1000), rabbit anti-phospho-Pyk2 (Cell Signaling Technology, 3291, 1:1000), mouse anti-Pyk2 (Cell Signaling Technology, 3480, 1:1000), and Saf32 (Cayman, 189720, 1:200, mouse anti-prion protein). Secondary antibodies were applied for 1 h at room temperature (Odyssey donkey anti-mouse or donkey anti-rabbit conjugated to IRDye 680 or IRDye 800, LI-COR Biosciences), and proteins were visualized with a LI-COR Odyssey infrared imaging system. Quantification of band intensities was performed within a linear range of exposure. Actin was used as loading control, and Actin levels were not affected by any treatment condition. For phosphorylation studies, signals detected by phosphoepitope-specific antibodies were normalized to the total protein level as detected by the non-phospho-specific antibody of the target protein. In the case of co-immunoprecipitation experiments, the level of the co-immunoprecipitated target protein was normalized to the input level of that protein. This normalized level was further normalized by the normalized level of the precipitated protein (PrPC level). Only validated antibodies were used. Validation information can be found on the website of the company or on Antibodypedia.
Statistics
All results are presented as mean ± S.E. Prism 6 software was used for statistical analysis. Data were analyzed using either one-way or two-way ANOVA (analysis of variance), followed by post-hoc Tukey's multiple comparisons test, Dunnett's multiple comparisons test, or Fisher's LSD post hoc pairwise comparisons test, as specified in the figure legends. p < 0.05 was considered statistically significant.
Author Contributions
L. T. H. and S. M. S. designed the experiments, analyzed the results, and wrote the manuscript. L. T. H. conducted the experiments. Both authors approved the final version of the manuscript.
Acknowledgments
We thank Stefano Sodi for assistance with mouse husbandry.
This work was supported by grants from the National Institutes of Health, BrightFocus Foundation, Alzheimer's Association, and the Falk Medical Research Trust (to S. M. S.). S. M. S. is a co-founder of Axerion Therapeutics, seeking to develop PrP-based therapeutics for Alzheimer disease. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- APP
- Amyloid precursor protein
- Aβ
- Amyloid β
- Aβo
- Amyloid β oligomer
- PrPC
- cellular prion protein
- DHPG
- 3,5-dihydroxyphenylglycine
- CamKII
- calcium/calmodulin-dependent protein kinase II
- nSOC
- neuronal store-operated calcium
- STIM
- stromal interaction molecule
- IP
- immunoprecipitation
- ANOVA
- analysis of variance
- ext.
- extracellular
- LSD
- least significant difference.
References
- 1. Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 2. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., and Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., and Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 4. Wang J., Dickson D. W., Trojanowski J. Q., and Lee V. M. (1999) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337 [DOI] [PubMed] [Google Scholar]
- 5. Kostylev M. A., Kaufman A. C., Nygaard H. B., Patel P., Haas L. T., Gunther E. C., Vortmeyer A., and Strittmatter S. M. (2015) Prion-protein-interacting amyloid-β oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 290, 17415–17438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., and Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin M., Shepardson N., Yang T., Chen G., Walsh D., and Selkoe D. J. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., and Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 9. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., and Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., and Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 11. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., and Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Um J. W., Kaufman A. C., Kostylev M., Heiss J. K., Stagi M., Takahashi H., Kerrisk M. E., Vortmeyer A., Wisniewski T., Koleske A. J., Gunther E. C., Nygaard H. B., and Strittmatter S. M. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 79, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Um J. W., Nygaard H. B., Heiss J. K., Kostylev M. A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E. C., and Strittmatter S. M. (2012) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., and Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chung E., Ji Y., Sun Y., Kascsak R. J., Kascsak R. B., Mehta P. D., Strittmatter S. M., and Wisniewski T. (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haas L. T., Salazar S. V., Kostylev M. A., Um J. W., Kaufman A. C., and Strittmatter S. M. (2016) Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain 139, 526–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haas L. T., Kostylev M. A., and Strittmatter S. M. (2014) Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem. 289, 28460–28477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaufman A. C., Salazar S. V., Haas L. T., Yang J., Kostylev M. A., Jeng A. T., Robinson S. A., Gunther E. C., van Dyck C. H., Nygaard H. B., and Strittmatter S. M. (2015) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 77, 953–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., DeStafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., Russo G., Thorton-Wells T. A., Jones N., Smith A. V., Chouraki V., et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X., Lopez O. L., Sweet R. A., Becker J. T., DeKosky S. T., Barmada M. M., Demirci F. Y., and Kamboh M. I. (2015) Genetic determinants of disease progression in Alzheimer's disease. J. Alzheimers Dis. 43, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khondoker M., Newhouse S., Westman E., Muehlboeck J. S., Mecocci P., Vellas B., Tsolaki M., Kłoszewska I., Soininen H., Lovestone S., Dobson R., Simmons A., AddNeuroMed Consortium, and Alzheimer's Disease Neuroimaging Initiative (2015) Linking genetics of brain changes to Alzheimer's disease: sparse whole genome association scan of regional MRI volumes in the ADNI and AddNeuroMed cohorts. J. Alzheimers Dis. 45, 851–864 [DOI] [PubMed] [Google Scholar]
- 22. Brakeman P. R., Lanahan A. A., O'Brien R., Roche K., Barnes C. A., Huganir R. L., and Worley P. F. (1997) Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288 [DOI] [PubMed] [Google Scholar]
- 23. Xiao B., Tu J. C., Petralia R. S., Yuan J. P., Doan A., Breder C. D., Ruggiero A., Lanahan A. A., Wenthold R. J., and Worley P. F. (1998) Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 21, 707–716 [DOI] [PubMed] [Google Scholar]
- 24. Ango F., Prézeau L., Muller T., Tu J. C., Xiao B., Worley P. F., Pin J. P., Bockaert J., and Fagni L. (2001) Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 411, 962–965 [DOI] [PubMed] [Google Scholar]
- 25. Mao L., Yang L., Tang Q., Samdani S., Zhang G., and Wang J. Q. (2005) The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J. Neurosci. 25, 2741–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Renner M., Lacor P. N., Velasco P. T., Xu J., Contractor A., Klein W. L., and Triller A. (2010) Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., and Klein W. L. (2004) Synaptic targeting by Alzheimer's-related amyloid β oligomers. J. Neurosci. 24, 10191–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lev S., Moreno H., Martinez R., Canoll P., Peles E., Musacchio J. M., Plowman G. D., Rudy B., and Schlessinger J. (1995) Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376, 737–745 [DOI] [PubMed] [Google Scholar]
- 29. Schulman H., and Greengard P. (1978) Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature 271, 478–479 [DOI] [PubMed] [Google Scholar]
- 30. Kaufman A. C., Salazar S. V., Haas L. T., Yang J., Kostylev M. A., Jeng A. T., Robinson S. A., Gunther E. C., van Dyck C. H., Nygaard H. B., and Strittmatter S. M. (2015) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 77, 953–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chin J., Palop J. J., Puoliväli J., Massaro C., Bien-Ly N., Gerstein H., Scearce-Levie K., Masliah E., and Mucke L. (2005) Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 25, 9694–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larson M., Sherman M. A., Amar F., Nuvolone M., Schneider J. A., Bennett D. A., Aguzzi A., and Lesné S. E. (2012) The complex PrPc-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J. Neurosci. 32, 16857–16871a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ronesi J. A., and Huber K. M. (2008) Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J. Neurosci. 28, 543–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raka F., Di Sebastiano A. R., Kulhawy S. C., Ribeiro F. M., Godin C. M., Caetano F. A., Angers S., and Ferguson S. S. (2015) Ca2+/calmodulin-dependent protein kinase II interacts with group I metabotropic glutamate and facilitates receptor endocytosis and ERK1/2 signaling: role of β-amyloid. Mol. Brain 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin D. Z., Guo M. L., Xue B., Mao L. M., and Wang J. Q. (2013) Differential regulation of CaMKIIα interactions with mGluR5 and NMDA receptors by Ca2+ in neurons. J. Neurochem. 127, 620–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barria A., and Malinow R. (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301 [DOI] [PubMed] [Google Scholar]
- 37. Nygaard H. B., van Dyck C. H., and Strittmatter S. M. (2014) Fyn kinase inhibition as a novel therapy for Alzheimer's disease. Alzheimers Res. Ther. 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nygaard H. B., Wagner A. F., Bowen G. S., Good S. P., MacAvoy M. G., Strittmatter K. A., Kaufman A. C., Rosenberg B. J., Sekine-Konno T., Varma P., Chen K., Koleske A. J., Reiman E. M., Strittmatter S. M., and van Dyck C. H. (2015) A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer's disease. Alzheimers Res. Ther. 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heidinger V., Manzerra P., Wang X. Q., Strasser U., Yu S. P., Choi D. W., and Behrens M. M. (2002) Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J. Neurosci. 22, 5452–5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nicodemo A. A., Pampillo M., Ferreira L. T., Dale L. B., Cregan T., Ribeiro F. M., and Ferguson S. S. (2010) Pyk2 uncouples metabotropic glutamate receptor G protein signaling but facilitates ERK1/2 activation. Mol. Brain 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang H., Wu L., Pchitskaya E., Zakharova O., Saito T., Saido T., and Bezprozvanny I. (2015) Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer's disease. J. Neurosci. 35, 13275–13286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun S., Zhang H., Liu J., Popugaeva E., Xu N. J., Feske S., White C. L. 3rd, and Bezprozvanny I. (2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gruszczynska-Biegala J., Pomorski P., Wisniewska M. B., and Kuznicki J. (2011) Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 6, e19285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lalonde R., Kim H. D., Maxwell J. A., and Fukuchi K. (2005) Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci. Lett. 390, 87–92 [DOI] [PubMed] [Google Scholar]
- 45. Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., Younkin S. G., and Borchelt D. R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170 [DOI] [PubMed] [Google Scholar]
- 46. Kamphuis W., Mamber C., Moeton M., Kooijman L., Sluijs J. A., Jansen A. H., Verveer M., de Groot L. R., Smith V. D., Rangarajan S., Rodríguez J. J., Orre M., and Hol E. M. (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 7, e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koffie R. M., Meyer-Luehmann M., Hashimoto T., Adams K. W., Mielke M. L., Garcia-Alloza M., Micheva K. D., Smith S. J., Kim M. L., Lee V. M., Hyman B. T., and Spires-Jones T. L. (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giuffrida R., Musumeci S., D'Antoni S., Bonaccorso C. M., Giuffrida-Stella A. M., Oostra B. A., and Catania M. V. (2005) A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 25, 8908–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pignatelli M., Piccinin S., Molinaro G., Di Menna L., Riozzi B., Cannella M., Motolese M., Vetere G., Catania M. V., Battaglia G., Nicoletti F., Nisticò R., and Bruno V. (2014) Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling Angelman syndrome. J. Neurosci. 34, 4558–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jankowsky J. L., Xu G., Fromholt D., Gonzales V., and Borchelt D. R. (2003) Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 62, 1220–1227 [DOI] [PubMed] [Google Scholar]
- 51. Park J. H., Widi G. A., Gimbel D. A., Harel N. Y., Lee D. H., and Strittmatter S. M. (2006) Subcutaneous Nogo receptor removes brain amyloid-β and improves spatial memory in Alzheimer's transgenic mice. J. Neurosci. 26, 13279–13286 [DOI] [PMC free article] [PubMed] [Google Scholar]