

Abstract
Cytochrome P450 (P450) reactions can involve C–C bond cleavage, and several of these are critical in steroid and sterol biosynthesis. The mechanisms of P450s 11A1, 17A1, 19A1, and 51A1 have been controversial, in the context of the role of ferric peroxide (FeO2−) versus perferryl (FeO3+, compound I) chemistry. We reinvestigated the 17α-hydroxyprogesterone and 17α-hydroxypregnenolone 17α,20-lyase reactions of human P450 17A1 and found incorporation of one 18O atom (from 18O2) into acetic acid, consonant with proposals for a ferric peroxide mechanism (Akhtar, M., Lee-Robichaud, P., Akhtar, M. E., and Wright, J. N. (1997) J. Steroid Biochem. Mol. Biol. 61, 127–132; Akhtar, M., Wright, J. N., and Lee-Robichaud, P. (2011) J. Steroid Biochem. Mol. Biol. 125, 2–12). However, the reactions were supported by iodosylbenzene (a precursor of the FeO3+ species) but not by H2O2. We propose three mechanisms that can involve the FeO3+ entity and that explain the 18O label in the acetic acid, two involving the intermediacy of an acetyl radical and one a steroid 17,20-dioxetane. P450 17A1 was found to perform 16-hydroxylation reactions on its 17α-hydroxylated products to yield 16,17α-dihydroxypregnenolone and progesterone, suggesting the presence of an active perferryloxo active species of P450 17A1 when its lyase substrate is bound. The 6β-hydroxylation of 16α,17α-dihydroxyprogesterone and the oxidation of both 16α,17α-dihydroxyprogesterone and 16α,17α-dihydroxypregnenolone to 16-hydroxy lyase products were also observed. We provide evidence for the contribution of a compound I mechanism, although contribution of a ferric peroxide pathway in the 17α,20-lyase reaction cannot be excluded.
Keywords: cytochrome P450, enzyme catalysis, enzyme mechanism, mass spectrometry (MS), nuclear magnetic resonance (NMR), steroid, steroidogenesis, oxygenase
Introduction
Cytochrome P450 (P450)3 enzymes catalyze oxidations of more chemicals than any other group of proteins (1). The list of reactions includes aliphatic and aromatic hydroxylations, heteroatom oxidations, epoxidations, and reactions involving both ring formation and cleavage (2–4). Many P450 reactions are important in the biosynthesis and degradation of steroids and sterols (4, 5), including several critical C–C bond cleavage reactions, i.e. those catalyzed by P450s 11A1, 17A1 (Fig. 1), 19A1, and 51A1 (6, 7).
FIGURE 1.

Steroid 17α-hydroxylation and 17α,20-lyase reactions catalyzed by P450 17A1.
The mechanisms of the C–C cleavage reactions have been the subject of considerable interest and debate. One of the questions with P450s 17A1, 19A1, and 51A1 has been whether the active oxidant is a ferric peroxide (FeO2−), which is an early intermediate following oxygen addition to the iron (Fig. 2, step 4) or the FeO3+ species (Fig. 2, step 6), often referred to as compound I (4, 10, 11). With P450s 17A1 and 19A1, a variety of approaches has been applied, including theoretical calculations, biomimetic models, spectroscopy, substrate atom labeling, and kinetics (12–32).
FIGURE 2.

Classic catalytic cycle of P450 enzymes (4). Paths for oxygen surrogates (PhI=O, H2O2) are also included. Note the FeO2− (ferric peroxide) and FeO3+ (compound I) forms discussed in the text. In the literature there exists different nomenclature for the same iron intermediates in this P450 catalytic cycle (i.e. FeIIIO2−, FeIIIO2H, FeIVO+., and FeIVOH) (8, 9). For clarity throughout the text, compound I is referred to interchangeably with FeO3+, and ferric peroxide is referred to interchangeably with FeO2−. The electron transfers from the reductase are simplifications in that the course of electron flow is probably from FMNH2/FADH• to FMNH•/FADH• in the first reduction (step 2) and (assuming that the reductase contributes the second electron to the P450) from FMNH•/FAD• to FMNH•/FAD in the second reduction step 4.
These C–C bond cleavage reactions are complex, and many of the results are ambiguous; also, a “mixed” mechanism would not be discerned in many of these experiments. One powerful approach originally used by Akhtar and co-workers (27–31) analyzes the actual reaction and can provide discrimination between the nucleophilic FeO2− and electrophilic FeO3+ reactions (Fig. 2), based on the incorporation of 18O label from O2 into the carboxylic acid products (Fig. 3) (7). However, these experiments are complicated due to the ubiquitous presence of formic acid (P450 19A1 and 51A1 reactions) and acetic acid (P450 17A1) in laboratory settings. Thus, the data from such experiments are interpreted with the most confidence when the steroid substrates are labeled with 2H or 13C isotopes to facilitate analysis (15, 33). Even then, the mass spectrometry results can be problematic, particularly if a shift of only one atomic mass unit is introduced and isotopologues derived from 18O incorporation are not discriminated from molecules containing natural abundance 13C atoms (33).
FIGURE 3.

Possible mechanisms of P450 17A1-catalyzed 17α,20-lyase reaction and expected 18O labeling (7). The course of 18O (from 18O2) and deuterium (D) labels are indicated with an asterisk. A, ferric peroxide mechanism (27–31); B, compound I mechanism with hydrogen atom abstraction from the 17α alcohol followed by C17-C20 bond scission to yield an acetyl radical; C, compound I mechanism with hydrogen atom abstraction from the C16 carbon; D, compound I mechanism with hydrogen atom abstraction from the 17α alcohol followed by C17-C20 bond scission to yield a hydrated acetyl radical (gem-diol); E, compound I mechanism with hydrogen atom abstraction from the C21 methyl group followed by C17-C20 bond scission to yield a C17 radical; F, addition of the 17α-hydroxyl group to compound I to yield an iron peroxide-C17 complex, which can decompose via either (a) a C20 gem-diol or (b) a C17-C20 dioxetane. See text for discussion and also Fig. 19. Mechanisms B–D result in an acetyl radical that undergoes oxygen rebound with Fe-*OH (compound II), with an oxygen atom from molecular oxygen (*O2) into the acetic acid product.
The incorporation of one atom of 18O label from O2 into formic acid (Fig. 3A) had been considered one of the most critical pieces of evidence in support of an FeO2− mechanism for P450 19A1 (14, 15, 34). Because of the importance of this evidence in the existing dogma, we re-examined this experiment using several technical improvements including the following: (i) purified recombinant P450 19A1; (ii) a new diazo reagent with a pyridine nitrogen to facilitate positive ionization for liquid chromatography-mass spectrometry (LC-MS); and (iii) the use of high resolution mass spectrometry (HRMS) (33). The results for P450 19A1 unambiguously ruled out 18O incorporation of 18O label from O2 into formic acid by distinguishing 2H from 13C isotope composition and are only consistent with an FeO3+ mechanism for P450 19A1 (33).
Because of the impact of the new studies (33), we re-examined the 18O experiments with P450 17A1 (27–31) with the newer methodologies. Although our 18O labeling results could be interpreted as support of an FeO2− mechanism for human P450 17A1, at least three possible FeO3+ mechanisms are still consistent with the data (Fig. 3, B, C, and F). We also employed artificial oxygen surrogates that might distinguish among mechanisms, i.e. iodosylbenzene, a single oxygen atom donor, and H2O2. Finally, we measured kinetic solvent isotope effects for the reactions, in light of inconsistencies in the field (23, 35). Our evidence now suggests that an FeO3+ mechanism is likely, at least in part, for the 17α,20-lyase reaction, and we also demonstrate the ability of the enzyme to catalyze additional 6β- and 16-hydroxylation reactions.
Results
Experimental Design for 18O Experiments
The P450 17A1 17α,20-lyase reaction produces DHEA from 17α-hydroxypregnenolone (Fig. 1). The product acetic acid is of particular interest in determining the mechanism of P450 17A1 catalysis (Fig. 3). To unambiguously distinguish the acetic acid formed as a product of the P450 17A1 reaction, the 17α-hydroxy substrate was d3-labeled at position C21 because of concerns about the level of endogenous acetic acid interfering with that formed in the enzyme reaction, based on our experience with 1- and 2-C carboxylic acids (36–39). Based on possible mechanisms shown in Fig. 3, the acetic acid products of 18O2 incubations with the 17α-hydroxy steroids are as follows: CD3CO18OH (mechanisms A, B, C, and Fb), a 1:2 molar ratio of CD3COOH and CD3CO18OH (mechanism D), only CD2HCOOH (mechanism E), or only CD3COOH (mechanism Fa). Because of the small amounts of acetic acid produced (1:1 stoichiometry with steroid, ∼ 25 μmol) during incubations, the acetic acid was converted into an ester using diazoethylpyridine to facilitate characterization. In addition to the increase in mass, the ester is designed for efficient ionization attributable to the nitrogen in the pyridine ring, i.e. 2-(pyridin-2-yl)ethyl acetate (33) (m/z 166.1, “MH+”) with one 16O incorporated (d2-labeled,“MH+ + 2”) or one 16O incorporated (d3-labeled,“MH+ + 3”), and one 18O incorporated (d3-labeled, “MH+ + 5”). A similar approach was used for 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone.
17α-Hydroxypregnenolone and 17α-Hydroxyprogesterone 18O Experiments
One 18O atom was incorporated into acetic acid without deuterium loss (Fig. 4, B–E) when 17α-hydroxy-[21,21,21-2H3]pregnenolone was used as the substrate, ruling out the mechanism in Fig. 3E.
FIGURE 4.
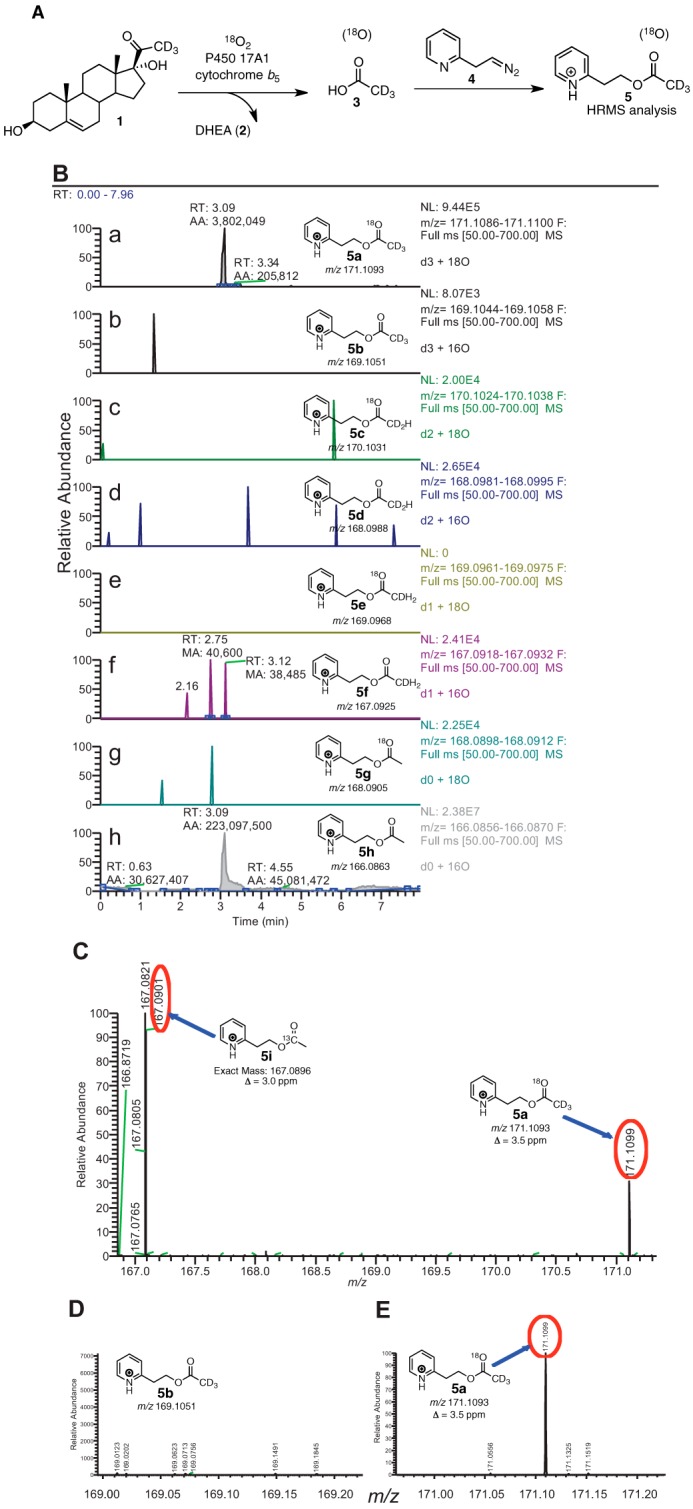
P450 17A1 incubation with [21,21,21-2H3]17α-hydroxypregnenolone (1) in the presence of 18O2 followed by derivatization and analysis by HRMS. A, scheme showing the incubation of deuterated lyase substrate (1) with P450 17A1 and cytochrome b5 in the presence of 18O2. The acetic acid product (3) was derivatized with the diazoethylpyridine reagent (4) and analyzed by liquid chromatography-HRMS. B, ion chromatograms monitoring the various isotopically labeled acetate products that were derivatized to the pyridylethyl esters (5a–h), with 4 ppm mass tolerance parameter. a, m/z 171 window (d3, 18O); b, m/z 169 window (d3, 16O); c, m/z 170 window (d2, 18O); d, m/z 168 window (d2, 16O); e, m/z 169 window (d1, 18O); f, m/z 167 window (d1, 16O); g, m/z 168 window (d0, 18O); h, m/z 166 window (d0, 16O). C, mass spectrum of the m/z 166.5–171.3 range by selecting the tR 3.01–3.12-min time interval in the ion chromatogram corresponding to the pyridine ester retention time. Shown at m/z 167.0901 is the peak corresponding to the acetate from background acetic acid from the natural abundance of 13C isotope (5i, expected mass, m/z 167.0896, Δ 3.0 ppm). The peak at m/z 171.1099 corresponds to the acetate derived from the enzymatic product (5a, expected mass, m/z 171.1093, Δ 3.5 ppm). D, expansion of the mass spectrum (m/z 168.95–169.22) from C showing the absence of d3-labeled acetate with no 18O incorporation (5b, expected mass, m/z 169.1051). E, expansion of the mass spectrum (m/z 170.95–171.22) from C showing the presence of d3-labeled acetate with 18O incorporation (5a, expected mass, m/z 171.1093). p, profile (peaks are shown in profile mode and not “centroid”). ESI, electrospray ionization; RT, retention time; NL, normalized level. More information about the meaning of the settings can be obtained from the Xcalibur Qual Browser User Guide (Thermo Scientific).
In the case of 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone as the substrate (Fig. 5), the signal-to-noise ratio of 18O-incorporated acetate was three times greater than when the 17α-hydroxy-[21,21,21-2H3]pregnenolone substrate was used. This improvement in sensitivity is attributed to the extra centrifugation step to remove the emulsion when extracting the acetic acid product (cf. “Experimental Procedures”). Additionally, we observed a trideutero-pyridine acetate product with no 18O incorporation at 6 ppm mass tolerance (Fig. 5B, 5b); however, the intensity was small compared with the 18O-incorporated acetate product (∼0.1% of 18O-incorporated product), and this isotopologue is likely derived from the residual 16O2 in the 18O2 cylinder (99% 18O abundance).
FIGURE 5.
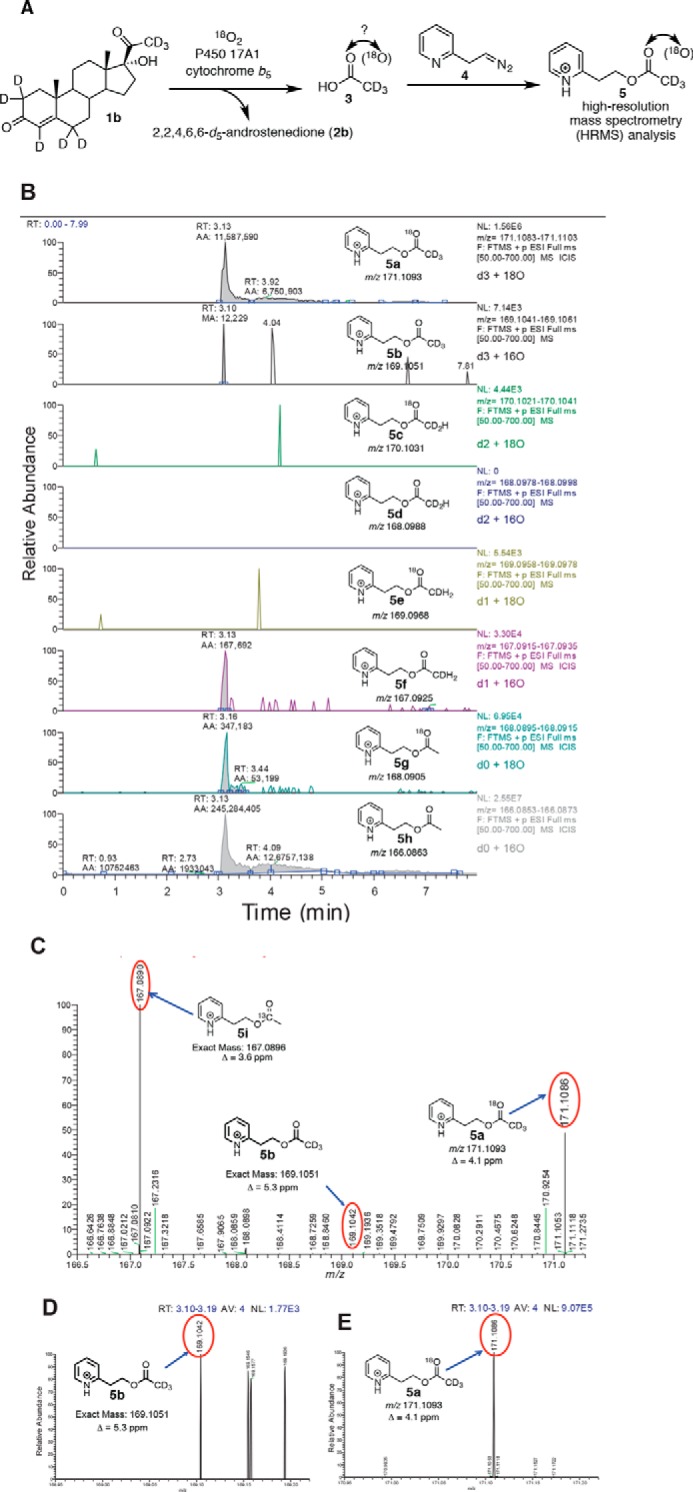
P450 17A1 incubation with 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone (1b) in the presence of 18O2 followed by derivatization and analysis by HRMS. A, scheme showing the incubation of deuterated lyase substrate (1b) with P450 17A1 and cytochrome b5 in the presence of 18O2. The acetic acid product (3) was derivatized with the diazoethylpyridine reagent (4) and analyzed by liquid chromatography-HRMS. B, ion chromatograms monitoring the various isotopically labeled acetate products that were derivatized to the pyridylethyl esters (5a–h), with 6 ppm mass tolerance parameter. a, m/z 171 window (d3, 18O); b, m/z 169 window (d3, 16O); c, m/z 170 window (d2, 18O); d, m/z 168 window (d2, 16O); e, m/z 169 window (d1, 18O); f, m/z 167 window (d1, 16O); g, m/z 168 window (d0, 18O); h, m/z 166 window (d0, 16O). C, mass spectrum of the m/z 166.5–171.3 range by selecting the tR 3.10–3.19-min time interval in the ion chromatogram corresponding to the pyridine ester retention time. Shown at m/z 167.0890 is the peak corresponding to the acetate from background acetic acid from the natural abundance of 13C isotope (5i, expected mass, m/z 167.0896, Δ 3.6 ppm). The peak at m/z 171.1099 corresponds to the acetate derived from the enzymatic product (5a, expected mass, m/z 171.1093, Δ 4.1 ppm). D, expansion of the mass spectrum (m/z 168.95–169.22) (from C) showing the detection of d3-labeled acetate with no 18O incorporation (5b, expected mass, m/z 169.1051, Δ 5.3 ppm). E, expansion of the mass spectrum (m/z 170.95–171.22) from C showing the presence of d3-labeled acetate with 18O incorporation (5a, expected mass, m/z 171.1093). p, profile (peaks are shown in profile mode and not “centroid”). ESI, electrospray ionization. RT, retention time. NL, normalized level. More information about the meaning of the settings can be obtained from the Xcalibur Qual Browser User Guide (Thermo Scientific).
Reactions with Oxygen Surrogates, Background and Previous Studies
If the ferric hydroperoxide mechanism is operative, then one might expect the reaction to be supported by the direct addition of H2O2 to ferric P450 (Fig. 2). However, Auchus and Miller (40) reported that no 17α,20-lyase activity was observed with recombinant human P450 17A1 plus H2O2 in yeast microsomes. Iodosylbenzene is a single oxygen donor and cannot support a reaction that requires two oxygens, i.e. a ferric peroxide complex (41). Iodosylbenzene also did not support the 17α,20-lyase reaction in a P450 17A1 yeast microsomal system (40).
P450 17A1 Reactions with Iodosylbenzene
Preliminary experiments indicated that the most effective concentration to use was 300 μm (results not presented).
Two products were formed from 17α-hydroxyprogesterone in both the iodosylbenzene and NADPH-based systems (Fig. 6). The expected product androstenedione (Fig. 1) was characterized by co-elution with a standard and by both LC-UV and LC-MS comparisons with a standard (data not shown). The other product, which eluted just before androstenedione, was identified as 16,17α-dihydroxyprogesterone by co-elution with a standard and by both LC-UV and LC-MS comparisons with a reference standard (Fig. 7). Although the dihydroxy product co-eluted with the 16α,17α-diastereomer, we cannot exclude the presence of the 16β-stereoisomer.
FIGURE 6.

Formation of 16,17α-dihydroxyprogesterone and androstenedione from 17α-hydroxyprogesterone by P450 supported by the oxygen surrogate iodosylbenzene. Retention times (tR) and integration units are indicated on the chromatograms. A, standard compounds. B, reaction (0.5 μm P450 17A1) supported by NADPH-P450 reductase (2.0 μm), cytochrome b5 (0.5 μm), and NADPH (30-s incubation). C, reaction (0.5 μm P450 17A1 and cytochrome b5 (0.5 μm)) with 0.30 mm iodosylbenzene (30-s incubation). In control experiments with only cytochrome b5 and iodosylbenzene (2 mm) mixed with the 17α-hydroxysteroids, the amounts of androstenedione detected were <15% of the amounts observed in this and similar studies with both 17α-hydroxysteroids.
FIGURE 7.

Identification of 16,17α-dihydroxyprogesterone as a product of 17α-hydroxyprogesterone. HRMS spectrum of 16,17-dihydroxyprogesterone formed in a reaction with NADPH-P450 reductase, cytochrome b5, and NADPH. Exact mass 346.2217 (protonated species): observed for MH+, m/z 347.2184 (Δ 9.5 ppm).
The products formed from 17α-hydroxypregnenolone were converted to Δ4 steroids by the action of cholesterol oxidase. These were identified as 16,17α-dihydroxyprogesterone and androstenedione, thus indicating that the products formed from 17α-hydroxypregnenolone were 16,17α-dihydroxypregnenolone and DHEA.
The rates of formation of 16,17α-dihydroxyprogesterone and androstenedione from 17α-hydroxyprogesterone in the NADPH- and iodosylbenzene-based systems were comparable in the absence of cytochrome b5 (Fig. 8A). The iodosylbenzene-dependent reaction was stimulated 2-fold by cytochrome b5, but the stimulation of the reaction that used NADPH-P450 reductase was much greater (10-fold), so that the iodosylbenzene versus NADPH-P450 reductase comparison (with cytochrome b5 present) is more disparate (Fig. 8A).
FIGURE 8.

Time course and effect of cytochrome b5 on 19-carbon steroid formation in the presence of iodosylbenzene (PhIO) or the typical NADPH-supported reaction. A, oxidation of 17α-hydroxyprogesterone. B, oxidation of 17α-hydroxypregnenolone. The insets show the NADPH-supported reactions in the presence of cytochrome b5. The points are means of duplicate assays, shown as means ± range.
With 17α-hydroxypregnenolone as substrate, a similar conclusion was reached regarding comparisons of the rates of the NADPH/reductase- and iodosylbenzene-supported reactions (Fig. 8B). When the 16-hydroxylation of the 17α-hydroxy steroids was considered, the iodosylbenzene-supported reactions were faster (Fig. 9). It is also notable that these reactions were stimulated by cytochrome b5.
FIGURE 9.

Time course and effect of cytochrome b5 on steroid 16-hydroxylation in the presence of iodosylbenzene (PhIO) or the typical NADPH-supported reaction. A, oxidation of 17α-hydroxyprogesterone. B, oxidation of 17α-hydroxypregnenolone. The points are means of duplicate assays, shown as means ± range.
Reactions with H2O2
H2O2 was added to purified P450 17A1 (with cytochrome b5 present), and no detectable 17α,20-lyase activity was found toward 17α-hydroxypregnenolone or 17α-hydroxyprogesterone, using varying concentrations of H2O2 (up to 10 mm) (Fig. 9). Under these conditions, the usual reconstituted P450 17A1/NADPH-P450 reductase/cytochrome b5 system yielded the expected products (Fig. 10, B and E).
FIGURE 10.

Reaction products formed from 17α-hydroxyprogesterone and 17α-hydroxypregnenolone in P450 17A1 reactions supported by various factors. Retention times (tR) and integration units are indicated on the chromatograms. A–C, 17α-hydroxyprogesterone; D–F, 17α-hydroxypregnenolone. A and D, NADPH-P450 reductase, cytochrome b5, and NADPH; B and E, H2O2 (10 mm) (with cytochrome b5); C and F, iodosylbenzene (PhI=O, 300 μm) (with cytochrome b5). In these studies the Δ5 products (formed from 17α-hydroxypregnenolone) were oxidized to Δ4 products to facilitate LC-UV analysis.
Additional Oxidation Products
With both 17α-hydroxyprogesterone and 17α-hydroxypregnenolone, the rates of formation of the lyase products (androstenedione and DHEA) were no longer linear after 5 min (300 s) (Fig. 8). The change was more obvious in the latter case, with the amount of accumulated product decreasing (Fig. 8B). The phenomenon was found to be the result of further 16-hydroxylation of the lyase products, in that we were able to identify these products (tR, UV spectra, and mass spectra) in the longer term reactions with both substrates (with 16-hydroxy-DHEA being converted to 16-hydroxyandrostenedione by cholesterol oxidase in the assays with 17α-hydroxypregnenolone) (Fig. 11). The time course of formation of these products from androstenedione and DHEA is shown in Fig. 12.
FIGURE 11.

Identification of 16-hydroxy steroids as reaction products formed from DHEA and androstenedione. A, authentic steroid standards: 16α-hydroxyandrostenedione, algestone (16α,17α-dihydroxyprogesterone), androstenedione, and 17α-hydroxyprogesterone; B, 10-min DHEA incubation (with products treated with cholesterol oxidase); C, 10-min androstenedione incubation; D, mass spectrum of peak identified as 16-hydroxyandrostenedione (formed from androstenedione); E, MS/MS analysis of m/z 303.2 peak of D.
FIGURE 12.

Time course of 16-hydroxylation of androstenedione and DHEA by P450 17A1.
We also noted a decrease in the level of 16,17α-dihydroxy steroids with extended time (in the NADPH-supported reactions, Fig. 9). We analyzed the products formed from (commercial) 16α,17α-dihydroxyprogesterone. One product was 16α-hydroxyandrostenedione, identified above (Fig. 13). The major product formed from 16α,17α-dihydroxypregnenolone was 16α-hydroxy-DHEA, identified by its mass and NMR spectra (Fig. 14).
FIGURE 13.
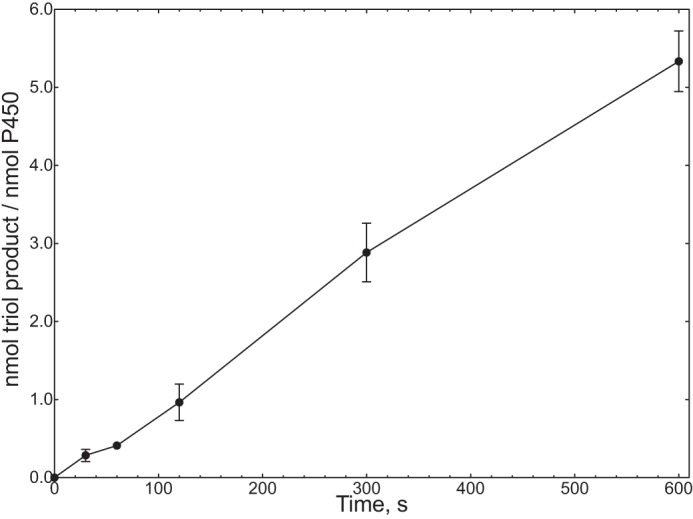
Rate of conversion of 16α,17α-dihydroxyprogesterone to 6β,16α,17α-trihydroxyprogesterone (Fig. 15) by P450 17A1. The points are means of duplicate assays, shown as means ± range.
FIGURE 14.

Characterization of 16α-hydroxy-DHEA. The product was formed in an incubation of an NADPH-reconstituted P450 17A1 system with 16α,17α-dihydroxypregnenolone and isolated by preparative HPLC. A, HRMS spectrum of [DHEA + 16]+ peak, 16α-hydroxy-DHEA (theoretical m/z for MH+ 305.2111, found m/z 305.2094). B, NMR spectra of 16α,17α-dihydroxypregnenolone (a) and product (b) in CDCl3 (600 MHz). See text for discussion.
Identification of Steroid B Ring Hydroxylation Product
The other major product in the incubation of 16α,17α-dihydroxyprogesterone with P450 17A1 was a triol, as judged by HRMS (m/z 363.2160, calculated MH+ m/z 363.2166, Δ 1.7 ppm) (Fig. 15A) (42). The UV spectrum was similar to those of other Δ4 3-keto steroids except blue-shifted ∼5 nm (Fig. 15B), consistent with an intact Δ4 3-keto steroid having some modification near the chromophore. The site of hydroxylation was identified as C-6 by 1H NMR (Fig. 15C and Table 1), i.e. 6β,16α,17α-trihydroxyprogesterone (43, 44). The 18, 19, and 21 methyl groups were intact (δ 0.79, 1.40, and 2.28 ppm), but there was a new multiplet at δ 4.38 ppm. The NOESY spectrum (supplemental Fig. S1) showed a spatial correlation between the new hydroxymethine proton (δ 4.38 ppm) and the Δ4-proton (δ 5.70 ppm). Moreover, the HMBC spectrum (heteronuclear multiple-bond correlation (NMR) spectroscopy) (supplemental Fig. S2) indicated a 3-bond coupling interaction between the C4-carbon (δ 125 ppm) and the hydroxymethine proton (δ 4.38 ppm) suggesting either the C2-position or the C6-position for the newly identified hydroxymethine proton (δ 4.38 ppm). The C6-position for the hydroxymethine proton (δ 4.38 ppm) was established because the C2-methylene protons (δ 2.53 and 2.42 ppm), which had a 2-bond correlation to the C3-keto carbon atom (δ 205.1 ppm) in the HMBC spectrum, were present. Moreover, the COSY spectrum (supplemental Fig. S3) indicated a 3-bond coupling between the C6-proton (δ 4.38 ppm) and the C7-protons (δ 1.98 and 1.34 ppm) (see also HSQC spectrum, supplemental Fig. S4).
FIGURE 15.

Characterization of 6β,16α,17α-trihydroxyprogesterone. The product was formed in an incubation of an NADPH-reconstituted P450 17A1 system with 16α,17α-dihydroxyprogesterone and isolated by preparative HPLC. A, HRMS spectrum (theoretical m/z for MH+ 363.2166, found m/z 363.2160). B, UV spectra of product (b) compared with 16α,17α-dihydroxyprogesterone (a). C, 1H NMR spectra of product (b) and 16α,17α-dihydroxyprogesterone (a) in CDCl3 (600 MHz). Note that the C-18, C-19, and C-21 methyl signals are intact and the chemical shifts of the H-7 protons appear to be moved upfield, as predicted (Table 1). See text and Ref. 42 for discussion, and see supplemental Figs. S-1–S-4 for two-dimensional NMR spectra.
TABLE 1.
NMR shift assignments for 6β,16α,17α-trihydroxyprogesterone

We have assigned the stereochemistry of the C6-hydroxyl group as β, based on the chemical shifts of the 7-protons of the steroid. In considering the chemical shifts of literature compounds (see Refs. 61 and 62 and see Table 2C in Ref. 42 and references therein), corresponding to 6α- and 6β-hydroxyprogesterone), the chemical shifts of the 7α- and 7β-protons are very informative. In the isolated P450 17A1 product, the chemical shifts of the 7α- and 7β-protons were (δ) 1.34 and 1.98 ppm, respectively, and in Ref. 42, the 7α- and 7β-protons were at (δ) 1.28 and 2.02 for 6β-hydroxyprogesterone, whereas the protons had chemical shifts of (δ) 1.11 and 2.19 for 6α-hydroxyprogesterone. Thus, 6β-hydroxy is the most likely stereochemistry of the new product. The NOESY spectrum showed spatial correlation between the C6-proton (δ 4.38 ppm) and both of the 7α- and 7β-protons (δ 1.34 and 1.98 ppm). Considering a Newman projection down the C6–C7 bond axis, this NOESY interaction is supported by the 6β-hydroxy configuration (supplemental Fig. S1).
We also analyzed the products formed from 16α,17α-dihydroxypregnenolone. One product was 16α-hydroxy-DHEA. The other product was either a tetraol or an epoxytriol (5,6-epoxy-3β,16α,17α-trihydroxypregnan-20-one), as judged by HRMS (m/z 365.2305, calculated mass, m/z 365.2323, Δ 4.9 ppm). The site of oxygen incorporation was not identified. The major product isolated from this reaction was 16α-hydroxy-DHEA, as can be seen from the 1H NMR spectrum of the purified product (Fig. 14B). There is a loss of the C21-methyl protons (δ 2.25 ppm) and an upfield shift of the 16β-proton (5.1 ppm to 4.4 ppm) (Fig. 14B). The proton NMR spectrum of the isolated P450 17A1 product also matched a previously reported NMR spectrum of synthetic 16α-hydroxy-DHEA (45).
Solvent Kinetic Isotope Effects on 17α,20-Lyase Reactions
No C–H bond-breaking steps are involved in any steps proposed in Fig. 3 except for Fig. 3D, which is not supported by the 18O work. Thus, no C-H kinetic deuterium isotope effect studies can be applied, but solvent kinetic isotope effect experiments can be informative.
No solvent kinetic isotope effect was found for the 17α,20-lyase reaction with 17α-hydroxyprogesterone (measured rates of 1.47 ± 0.03 min−1 in H2O and 1.40 ± 0.03 min−1 in 95% D2O (v/v), n = 4, calculated isotope effect of 1.05 ± 0.09 (S.D.)). In contrast, we observed a small but repeatable inverse isotope effect (0.83 ± 0.05 (S.D.), n = 4) for the 17α,20-lyase reaction with 17α-hydroxypregnenolone under the usual conditions with cytochrome b5, NADPH-P450 reductase, and substrate concentration of 30 μm (measured rates of 4.10 ± 0.22 min−1 in H2O, 4.94 ± 0.13 min−1 in 95% D2O (v/v), calculated from four independent experiments (± S.D.)) (Figs. 16 and 17). The solvent kinetic deuterium isotope effects for the 16-hydroxylation reactions were 1.35 ± 0.15 for 17α-hydroxyprogesterone and 1.34 ± 0.23 for 17α-hydroxypregnenolone (n = 4, ± S.D., data not shown).
FIGURE 16.

Kinetic solvent isotope effects on 17α-hydroxyprogesterone and 17α-hydroxypregnenolone 17α,20-lyase reactions catalyzed by P450 17A1 (in the presence of NADPH-P450 reductase, NADPH, and cytochrome b5). Results are shown as means of four individual experiments ± S.D.
FIGURE 17.

Solvent kinetic deuterium isotope effects on 17α-hydroxyprogesterone and 17α-hydroxypregnenolone reactions catalyzed by P450 17A1 (in the presence of NADPH-P450 reductase and cytochrome b5). Retention times (tR) and integration units are indicated on the chromatograms. The substrate concentration was 30 μm in all cases, and the reactions were done in either H2O or 95% D2O (v/v) at pH or pD 7.4. A and B, 17α-hydroxyprogesterone; C and D, 17α-hydroxypregnenolone. A and C, H2O; B and D, 95% D2O (v/v).
Discussion
Our results indicate that acetic acid recovered in the human P450 17A1 reactions with either 17α-hydroxyprogesterone or 17α-hydroxypregnenolone contained 18O atom from molecular oxygen. These results are consonant with the original analysis of Akhtar and co-workers on P450 17A1 (27, 28) and, on their own, are consistent with the FeO2− mechanism presented in Fig. 3A (40, 46). Alternative mechanisms that are still consistent with the 18O labeling results, but involving FeO3+, are presented in Fig. 3, B, C, and F, arrow b (7, 40, 47). The mechanisms in Fig. 3, B and C involve formation of an acetyl radical, which has adequate chemical precedent (32, 48–50). The dioxetane mechanism is similar to one that has been proposed for tryptophan and indole dioxygenases (51). One of these two FeO3+ mechanisms is proposed to contribute to the lyase reaction in that (i) iodosylbenzene can support the lyase reaction, and (ii) we report that P450 17A1-17α-hydroxysteroid complexes are poised for multiple hydroxylation reactions in addition to lyase reactions.
Incubations with 18O2
Previous studies of 18O2 incubations with P450 17A1 concluded that 18O incorporation into the acetic acid product was the major isotopologue detected, leading to the conclusion that the ferric peroxide was the iron-active species for C–C bond cleavage (26, 52). However, these studies (i) used low resolution mass spectrometry and (ii) used microsomes from porcine testes as the source of enzyme (i.e. non-purified enzyme); and (iii) the report that used the direct lyase substrate 17α-hydroxy-[21,21,21-2H3]pregnenolone did not present raw data (26), whereas a subsequent report used [16α,17α,21,21,21-2H5]pregnenolone as the substrate and not the direct substrate that results in the formation of DHEA, i.e. 17α-hydroxypregnenolone (52). Furthermore, the low resolution mass spectrometry used in these studies yielded an ambiguous determination of 18O-, 2H-, and 13C-labeled content of the acetate products (52), which resulted in the interpretation of multiple possible mechanisms for the lyase step of P450 17A1 (Fig. 3). We performed the incubation with purified P450 17A1 and labeled lyase substrates (17α-hydroxy-[21,21,21-2H3]pregnenolone and 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone) in the presence of 18O2 and analyzed the acetate product by HRMS after derivatization with the new diazo reagent. Our results unambiguously established that the acetate produced from the enzymatic incubation incorporated one oxygen atom from molecular oxygen in both cases, with the C-21 deuteriums retained (Figs. 4 and 5). Moreover, we did not detect significant amounts of any other acetate isotopologues from the enzyme incubation (i.e. loss of one deuterium or lack of 18O incorporation). These experiments agree with the observation of oxygen incorporation from the reports of Akhtar and co-workers (26, 52). However, although these data can support a ferric peroxide mechanism for C–C bond cleavage (Fig. 3A), they do not rule out a compound I mechanism (Fig. 3, B, C, and F, arrow b).
Oxygen Surrogate Studies, Iodosylbenzene
The use of iodosylbenzene and NADPH-P450 reductase to form compound I with P450 17A1 resulted in two different activities when the 17α-hydroxysteroid was used as the substrate. In both conditions, 16α-hydroxylation and C–C bond cleavage activities toward 17α-hydroxyprogesterone yielded 16,17α-dihydroxyprogesterone and androstenedione, respectively (Figs. 6, 8A, and 9A). However, the product distributions were different; iodosylbenzene yielded more 16-hydroxylation relative to C–C bond cleavage (∼9:1, Fig. 6C) compared when NADPH-P450 reductase was used (∼0.1:1, Fig. 6B). The switch in reactivities depending on the oxidation system used (iodosylbenzene versus NADPH-P450 reductase) suggests a conformational change in the enzyme-substrate complex when the reductase binds to the P450 enzyme. Moreover, when 17α-hydroxypregnenolone was used as the substrate, the 16-hydroxylation activity was diminished (Fig. 10, D and F) relative to when 17α-hydroxyprogesterone was used as the substrate. Similarly, this substrate-dependent switch in reactivity is reminiscent of the different 16- versus 17-hydroxylation regioselectivities observed when two different substrates, progesterone and pregnenolone, are used for P450 17A1 (53), which is explained by the 3-keto-Δ4 versus 3β-hydroxy-Δ5 moieties in the AB-ring systems of these steroid substrates.
Moreover, the fact that P450 17A1 is catalyzing a C-H hydroxylation with its lyase substrate, 17α-hydroxypregnenolone, supports the presence of a compound I species, which either hydroxylates the C16-position or cleaves the C17,C20-bond as shown in Fig. 3. This observation may contradict the conclusions about the active iron hydroperoxo species observed by resonance Raman spectroscopy (9, 25). However, it is possible that the iron peroxohemiketal species reported in the resonance Raman study (9), i.e. Fig. 3A, tetrahedral intermediate, was a structural misassignment and that the actual observed species was indeed an iron peroxo intermediate attached through the C17-position of the steroid (Fig. 3F). This iron peroxo intermediate can be formed from a nucleophilic attack of the C17-hydroxy group of the lyase substrate (i.e. 17α-hydroxypregnenolone or 17α-hydroxyprogesterone, Fig. 3F) onto compound I.
It should be pointed out that the iodosylbenzene mechanism may be more complex than just a direct oxygen transfer, as pointed out by Ortiz de Montellano (46). A possible intermediate is shown in Fig. 18, which may even have oxidant capacity of its own.
FIGURE 18.

Possible oxidizing alternative to compound I in the iodosylbenzene (PhI=O)-supported reactions (46).
Oxygen Surrogate Studies, Hydrogen Peroxide
Many studies in the literature involve the use of alkyl hydroperoxides as oxygen surrogates for P450 reactions, beginning with Kadlubar et al. (54). However, although peracids can be used as reagents to generate P450 compound I (55), studies with alkyl hydroperoxides are problematic due to the production of radicals and their ensuing chemistry (46, 56). Some bacterial family 152 P450s appear to use H2O2 as a physiological cofactor (37, 57–59), and bacterial P450 101A1 (P450cam) was mutated to a species that could efficiently utilize H2O2 in reactions (60). H2O2 can support some mammalian P450 reactions after direct addition (61–66) (although generally not as well as alkyl hydroperoxides (54)), but the role of a ferric peroxide in each oxygenation reaction can only be postulated, in that the ferric peroxide can subsequently convert to compound I.
In principle, the FeO2− complex could proceed to compound I (FeO3+) through appropriate acid-base catalysis, but there are also side reactions that may diminish the progress of a putative Fe3+-H2O2 complex on to FeO3+ (Fig. 2). We made attempts to observe compound I or other intermediates by mixing P450 17A1 with H2O2 (10 mm) or iodosylbenzene (300 μm) in a stopped-flow spectrophotometer (dead time ∼2 ms, rapid scanning) but were unsuccessful in seeing any distinct complexes (data not presented). However, given the difficulties encountered by others in observing these transient species even with bacterial P450s (55), negative results are inconclusive.
Cytochrome b5 Effects
Another issue that is still not resolved is the stimulatory effect of cytochrome b5, which is known to promote the 17α,20-lyase reaction of P450 17A1. Results with apo-cytochrome b5, devoid of heme, have shown that cytochrome b5 does not donate the second electron in the catalytic cycle of this P450 (67). The stimulation of 17α,20-lyase activity by cytochrome b5 in the iodosylbenzene-supported reaction (Fig. 8) is consistent with this view. We also note that cytochrome b5 stimulated the 16-hydroxylation reactions with both 17α-hydroxyprogesterone and 17α-hydroxypregnenolone (Fig. 8). In other unpublished data,4 we have also noted the stimulation of the 17α-hydroxylation of both progesterone and pregnenolone by cytochrome b5. Our conclusion about the stimulatory role of cytochrome b5 in the iodosylbenzene reactions is that it is acting in an allosteric manner to facilitate these reactions (e.g. due to more ideal juxtaposition in reaction intermediates), which is the reason proposed for the normal physiological reaction (6, 7, 67).
Hydroxylations Catalyzed by P450 17A1
The 16α-hydroxylation of DHEA has previously been reported to be catalyzed by P450 3A4 (68, 69). Upon monitoring the production of DHEA from 17α-hydroxypregnenolone by P450 17A1 over time (Fig. 8B, inset), there was a decrease in DHEA formation in the time points greater than 5 min. This observation suggested that DHEA was further being oxidized to another product. We hypothesized that this new product would correspond to 16-hydroxy-DHEA based on the other activities of P450 17A1 (16-hydroxylation of progesterone (53) and 16-hydroxylation of its 17α-hydroxylated products). The new product, which was converted to its 3-keto-Δ4 counterpart by cholesterol oxidase, co-chromatographed with standard 16α-hydroxyandrostenedione (Fig. 10). The 16-hydroxylation product of P450 17A1 was also observed when androstenedione was used as the substrate (Fig. 11). The ability of P450 17A1 to form 16-hydroxylated androgens is physiologically relevant in that estriol, an abundant and characteristic estrogen during human pregnancies, arises from the aromatization of 16α-hydroxy androgens (70, 71).
The current working hypotheses we favor are shown in some detail in Fig. 19. Two involve an acetyl radical and one a steroid dioxetane intermediate, which are both considered viable entities.
FIGURE 19.

Mechanisms of P450 17A1-catalyzed 17α,20-lyase reaction consistent with 18O labeling (7), oxygen surrogate results, and solvent kinetic isotope results. The course of an 18O label (from 18O2) is indicated with an asterisk (7, 40). A, compound I mechanism with hydrogen atom abstraction from the 17α alcohol followed by C17-C20 bond scission to yield an acetyl radical; B, addition of the 17α hydroxyl group to compound I to yield an iron peroxide-C17 complex, followed by decomposition via a C17-C20 dioxetane; C, compound I mechanism with hydrogen atom abstraction from the C16 carbon. See text for discussion and also Fig. 3.
B Ring Hydroxylation of 16α,17α-Dihydroxyprogesterone by P450 17A1
Surprisingly the 6β-position was hydroxylated when 16α,17α-dihydroxyprogesterone was used as a substrate for P450 17A1 (Fig. 15). This shift in regioselectively from the D-ring to the B-ring of the steroid by P450 17A1 was due to the presence of two hydroxyl groups on the 16α- and 17α-positions. Interestingly, regioselectivity was switched from the B-ring to the C–D-ring of the steroid in another P450 system when the Δ4-double bond was reduced. P450 3A4 hydroxylates the 6β-position of testosterone (B-ring of the steroid); however, with 5α-dihydrotestosterone as the substrate, P450 3A4 oxygenated the 18β-methyl group (between the C–D-ring of the steroid) (72). The causes for the switch in regioselectivities of the different P450 systems are probably not the same. Moreover, P450 17A1, which normally hydroxylates the α-face of (the D-ring of) its steroid substrates (pregnenolone and progesterone), introduced a hydroxyl group on the β-face of 16α,17α-dihydroxyprogesterone. The hydroxylation of the opposite face can be rationalized from overlaying 17α-hydroxyprogesterone and 16α,17α-dihydroxyprogesterone (Fig. 20). When the C10, C14, and O16 atoms from 16α,17α-dihydroxyprogesterone were aligned with the C14, C10, and O3 atoms of 17α-hydroxyprogesterone, the O17 atom of 17α-hydroxyprogesterone was positioned 1.4 Å away from the C6 atom of 16α,17α-dihydroxyprogesterone, the site where the new oxygen atom is introduced. Additionally, the 17-oxygen of 17α-hydroxyprogesterone was directed at the β-face of 16α,17α-dihydroxyprogesterone. The 3-oxo group of 17α-hydroxyprogesterone has been shown to hydrogen bond to Asn-202 of P450 17A1 in the crystal structure (73). Based on our observations with 6β-hydroxylation of 16α,17α-dihydroxyprogesterone by P450 17A1 and the overlay of the two different substrates, we reason that the 16α-hydroxy group of 16α,17α-dihydroxyprogesterone hydrogen bonds to Asn-202 of P450 17A1, which in turn directs the 6β-hydrogen to the active iron center of the enzyme. Interestingly, the 17α,20-lyase product for the 16α,17α-dihydroxyprogesterone substrate (i.e. 16α-hydroxyandrostenedione) was detected by LC-MS analysis and co-elution with the standard, but this lyase product seems to be a minor product in comparison with the 6β-hydroxylation product.
FIGURE 20.
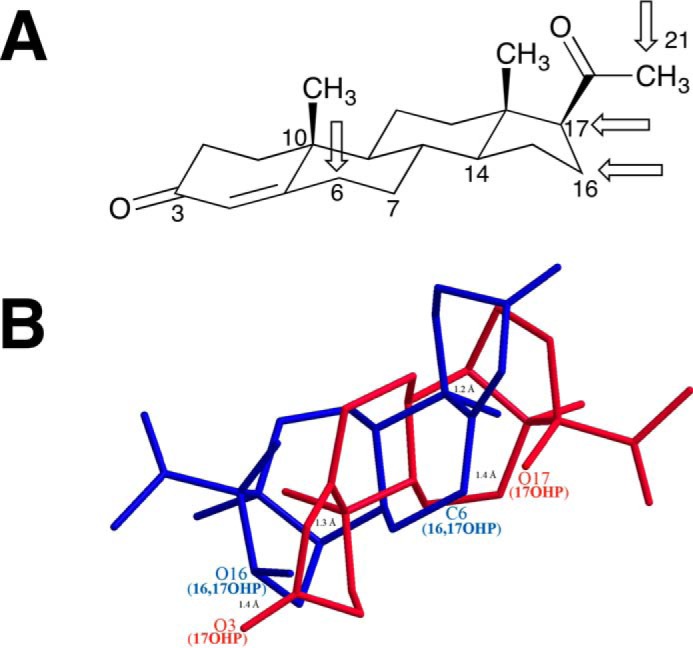
Sites of hydroxylation of progesterone by P450 17A1. A, chair configuration of progesterone, with the four sites of attack indicated by arrows. B, wire diagram of 17α-hydroxyprogesterone (17OHP, red) and 16α,17α-dihydroxyprogesterone (16,17OHP, blue) overlaid, with the latter in an alternative configuration to show the proximity of the C-6 atom of 16α,17α-hydroxyprogesterone with the 17-hydroxy group of 17α-hydroxyprogesterone. The model was made using Chem3D, with a minimum root mean square error of 0.1 and minimum root mean square gradient of 0.01. The C14, C10, and O3 atoms of 17α-hydroxyprogesterone were aligned with the C10, C14, and O16 atoms of 16α,17α-dihydroxyprogesterone, respectively, by displaying the distance measurements of each pair of atoms and then running an overlay minimization calculation. The green lines indicate the pair of atoms that were aligned (after overlay minimization, the distances between C14 of 17-OHP and C10 of 16,17-OHP; C10 of 17-OHP and C14 of 16,17-OHP; and O3 of 17-OHP and O17 of 16,17-OHP were 1.2, 1.3, and 1.4 Å, respectively, and are shown as green lines). The distance between the O17 atom of 17α-hydroxyprogesterone and C6 atom of 16α,17α-dihydroxyprogesterone was 1.4 Å.
17α,20-Lyase Reaction of 16α,17α-Dihydroxypregnenolone by P450 17A1
Conversely, when 16α,17α-dihydroxypregnenolone was used as the substrate for P450 17A1, the major product isolated from the incubation was 16α-hydroxy-DHEA, which is formed from the 17,20-carbon–carbon bond cleavage.
Oxygen Incorporation into 16α,17α-Dihydroxypregnenolone by P450 17A1
An additional oxygenation product (M + 16 of substrate) was detected by LC-HRMS when 16α,17α-dihydroxypregnenolone was used as the substrate. However, there was not enough purified material recovered to determine the location of the oxygen on the steroid ring by 1H NMR. A possible site of oxidation may be the C21-position. Alternatively, from the knowledge of 6β-hydroxylation reactivity of P450 17A1 with 16α,17α-dihydroxyprogesterone as the substrate, the oxygen may be incorporated in two other possible sites as follows: (i) the Δ5,6-double bond of the substrate to form the 5,6-epoxide or (ii) the C7-position may be hydroxylated. Epoxidation activity of P450 17A1 has been previously reported with a Δ16,17-steroid substrate (74). Nevertheless, the shift in favoring C–C bond cleavage reactivity over hydroxylation when using the 3β-hydroxy-Δ5 substrate (16α,17α-dihydroxypregnenolone) instead of the 3-keto-Δ4 substrate (16α,17α-dihydroxypregnenolone) is similar to what occurs with pregnenolone and progesterone (i.e. 17α,20-carbon,carbon bond cleavage versus 16α-hydroxylation). This observation may be related to the hydrogen bonding that occurs between the 3β-hydroxy group of the 3β-hydroxy-Δ5 substrate and Asn-202 of the enzyme.
Kinetic Solvent Isotope Effects Do Not Support a Ferric Peroxide Mechanism
One argument against the proposed acetyl radical mechanism (Fig. 3B) is a reported inverse kinetic solvent deuterium isotope effect (0.39) reported by Sligar and co-workers (23). If the mechanism in Fig. 3, B, C, or F, arrow b, were valid, the abstraction of a hydrogen atom from the 17-hydroxyl group (Fig. 3B) or the heterolytic cleavage of an O–H bond (Fig. 3D) might be expected to be a (partially) rate-limiting step, and an inhibitory effect of hydroxyl deuteration might be expected. In contrast, a similar study by Swinney and Mak (35) reported that (30%) D2O attenuated androgen formation from 17α-hydroxyprogesterone using microsomes from pig testes as the enzyme source (kH/kD ∼1.25 at pH 7), suggesting that the 17α,20-lyase reaction is dependent on compound I formation either through the protonation of the distal oxygen of ferric peroxide (cf. P450 catalytic cycle) or deuterium atom abstraction from the 17-hydroxy group of the substrate (Fig. 3B).
Because of the discrepancy, we reinvestigated the results in our own system (Fig. 16). Running the normal P450 17A1 reaction (with NADPH-P450 reductase and cytochrome b5) in 95% D2O showed no significant change in the rate of conversion of 17α-hydroxyprogesterone to androstenedione and a small but statistically significant change in the rate of oxidation of 17α-hydroxypregnenolone to DHEA, with an apparent isotope effect of 0.83 (Fig. 17), which is much less than the effect (0.39) reported by Gregory et al. (23). Interpretation of solvent kinetic deuterium isotope effects is complex (75), in that protonation and deprotonation can occur throughout the amino acid side chains of an enzyme, not only on an iron-oxygen complex. The reason for the small inverse isotope effect with one lyase substrate but not another (Figs. 16 and 17) is unclear. The opposite pattern between the solvent isotope effects for the 17α-hydroxypregnenolone lyase and the 16-hydroxylation reactions is qualitatively consistent with the report of Gregory et al. (23). One possibility is that the Δ5 substrate (17α-hydroxypregnenolone) 3-hydroxy group exchanges with deuterium and that this has an effect on the juxtaposition of the substrate in the active site. The hydroxyl moiety has been shown by Scott and co-workers (73, 76) to be in hydrogen bonding distance to Asn-202 of human P450 17A1. A substitution of the 3-hydroxyl group by deuteration (i.e. −OD) could shift the substrate to favor the lyase reaction versus 16-hydroxylation. However, the lack of solvent isotope effects does not allow any definite conclusions about the rate-limiting nature of the abstraction of a proton or hydrogen atom from the 17-OH group, due to the multiple complex influences from solvent deuterium on enzyme function.
Although resonance Raman spectra of what is reported to be the human P450 17A1 FeO2−complex have recently been published (9, 25), two caveats are as follows: (i) no cytochrome b5 (for which the 17α,20-lyase reaction is very dependent, e.g. Fig. 8B) was present, and (ii) the observed complex was not tested for its catalytic competence, i.e. to form product(s). Even if the FeO2− complex did form the normal products (androstenedione and DHEA, plus the 16-hydroxylation products, which is unlikely) in these experiments, the simultaneous or subsequent intermediacy of an FeO3+ species as well could not be ruled out.
Conclusions
The ability of iodosylbenzene, but not H2O2, to support the lyase reaction provides what may be the strongest evidence in favor of a compound I mechanism, in that iodosylbenzene cannot possibly form a peroxy intermediate. The apparent rate of the lyase reaction was similar to that of the NADPH-supported reaction (without cytochrome b5) in the case of the 17α-hydroxyprogesterone reaction and was somewhat less than that of the NADPH-supported reaction (without cytochrome b5) in the case of the 17α-hydroxypregnenolone lyase reaction (Figs. 8 and 9). Ideally, the compound I form of P450 17A1 could be prepared using the approaches that Green and co-workers (8, 55, 64, 77) have used with two bacterial P450s (64), and the reaction could be investigated directly. Nevertheless, in considering all of the literature in this field and that presented here in this article, the iodosylbenzene and H2O2 results (Fig. 10) are difficult to dismiss, even if they are not physiological, and are interpreted as evidence for a compound I reaction (Fig. 19).
The multiple hydroxylations are probably catalyzed by FeO3+ intermediates, formed with P450 17A1-17α-hydroxy steroid complexes. It is possible that individual reactions (i.e. hydroxylation, lyase) proceed from different FeO complexes, although it is simpler to explain all as emanating from a single iron-oxygen intermediate. The myriad of reactions is depicted in Fig. 21 and reveals a surprising flexibility in the P450 17A1 enzymes. As indicated, P450 17A1 has been shown to catalyze 21-hydroxylation of progesterone (53). Our observed rates are indicated in the figure. Lyase reactions are not overly dominant. The biological activities of most of the products are, to our knowledge, still unknown.
FIGURE 21.

Summary of current known reactions of human P450 17A1. See also Refs. 7, 26, 53. Rates determined at high substrate concentrations (approximating kcat conditions) in this study are indicated, in units of nanomoles of product formed/min/nmol P450 17A1, when available. A, products formed from progesterone; B, products formed from pregnenolone.
In summary, we have provided evidence that a compound I-type mechanism (Fig. 19) can be involved in the 17α,20-lyase reactions. Our results do not rule out a ferric peroxide mechanism, nor do they define the fraction of the normal reaction that is catalyzed by each of the two mechanisms, if both are operative. If further research implicates compound I in this reaction, then few strong cases for P450 ferric peroxide chemistry will remain, at least in the field of steroid metabolism (33, 64, 78).
Experimental Procedures
General
Bruker instruments (400 and 600 MHz) were used to acquire NMR spectra in the Vanderbilt facility. CD3CN and CDCl3 residual proton peaks were referenced to δ 1.94 and 7.26 ppm, respectively, and the CDCl3 triplet in the carbon spectrum was referenced to δ 77.16 ppm and CD3CN was referenced to 118.26 ppm (79). Unless specified otherwise, all chemicals were purchased from Sigma-Aldrich. A modified version of the nitrosourea reagent was synthesized according to Ref. 33 using 2-(2-pyridyl)ethylamine (instead of 3-(3-pyridyl)propylamine) as the starting material, as described in detail here.
Reagents
17α-Hydroxy-[21,21,21-2H3]progesterone (96.5% atomic excess as judged by 1H NMR, 97.9% atomic excess as judged by LC-MS) was synthesized and characterized as described previously (53). 17α-Hydroxy-[21,21,21-2H3]pregnenolone (nominal 98.4% atomic excess) and 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone (nominal 98.7% atomic excess) were purchased from C/D/N Isotopes (Pointe-Claire, Quebec, Canada). 16α,17α-Dihydroxyprogesterone (algestone) was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). 16α-Hydroxyandrostenedione was obtained from Steraloids (Newport, RI).
Enzymes
Recombinant human P450 3A4 with a C-terminal His5 tag was expressed in Escherichia coli and purified as described previously (80, 81). E. coli recombinant rat NADPH-P450 reductase and human liver cytochrome b5 were prepared as described by Hanna et al. (82) and Guengerich (83), respectively.
Recombinant human P450 17A1 (with a C-terminal His4 tag) was expressed in E. coli and purified by metal-affinity chromatography using a protocol adapted from those previously reported (76, 84, 85). Briefly, an E. coli codon-optimized cDNA, corresponding to the amino acid sequence reported by DeVore and Scott (76), was purchased (Genewiz, South Plainfield, NJ) and inserted into the pCW-Ori(+) expression vector. The construct was used to transform competent E. coli JM109 cells (Agilent), and an isolated colony was used to inoculate 100 ml of Luria-Bertani medium (containing 100 μg/ml ampicillin), which was then incubated at 37 °C with shaking at 250 rpm overnight (12–14 h). Expression ensued by inoculating 1 liter of Terrific Broth medium, containing 100 mg/liter ampicillin and 250 μl/liter of trace elements (86), with 10 ml of the overnight culture and incubating at 37 °C (250 rpm) for ∼4 h (OD600 ∼0.32). The expression culture was then supplemented with 1 mm isopropyl β-d-1-thiogalactopyranoside and 1 mm δ-aminolevulinic acid, and the incubation conditions were changed to 30 °C and 200 rpm. After ∼40 h, the culture was centrifuged at 5000 × g for 10 min, and the bacterial pellet was resuspended in 300 ml of 100 mm Tris-HCl buffer (pH 7.6) containing 500 mm sucrose and 0.5 mm EDTA and placed on ice. The suspension was then treated with 60 μl of a 50 mg/ml lysozyme solution/g of bacterial pellet and incubated on ice for 30 min, with gentle mixing every 10 min. All subsequent steps were conducted on ice or at 4 °C. Next, a spheroplast pellet was obtained by centrifugation at 5000 × g for 10 min and resuspended in 25 ml of 300 mm potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 6 mm Mg(CH3CO2)2, 0.1 mm dithiothreitol (DTT), 0.1 mm phenylmethylsulfonyl fluoride, and a protease inhibitor mixture (cOmpleteTM, EDTA-free, Roche Applied Science). The spheroplasts were lysed by sonication, and debris and unbroken cells were removed by centrifugation at 9000 × g for 20 min. The cytosol was cleared of the membrane fraction by centrifugation at 100,000 × g for 60 min and was supplemented with 300 mm NaCl and 20 mm imidazole prior to loading onto a nickel-nitrilotriacetic acid resin (Qiagen) bed that had been equilibrated with 300 mm potassium phosphate buffer (pH 7.4) containing 300 mm NaCl, 20% glycerol (v/v), 20 mm imidazole, and 0.1 mm DTT. The bound protein was washed with 10 bed volumes of the same buffer and eluted with the same buffer containing 250 mm imidazole. The purified enzyme was then dialyzed four times against 100-fold volumes of 200 mm potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 0.1 mm EDTA, and 0.1 mm DTT and stored at −70 °C until use.
Chemical Synthesis
(2-(Pyridin-2-yl)ethyl)urea (33)
A solution of 2-(2-pyridyl)ethylamine (0.753 g, 6.17 mmol) and benzotriazole-1-carboxamide (1.00 g, 6.17 mmol) in tetrahydrofuran was heated under reflux for 12 h. The reaction mixture was concentrated in vacuo and purified by flash column chromatography (gradient of hexanes to 50% hexanes in ethyl acetate (v/v) to 10% CH3OH in CH2Cl2 (v/v)) to afford (2-(pyridin-2-yl)ethyl)urea as a white solid (1.00 g, 6.06 mmol, 98%). Rf 0.28 (silica gel GF, CH2Cl2/CH3OH, 9:1, v/v); 1H NMR (600 MHz, CD3CN) δ 8.50 (d, J = 4.8 Hz, 1H), 7.66 (ddd, J = 7.7, 7.6, 2.0 Hz, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.17 (dd, J = 7.9, 4.8 Hz, 1H), 5.25 (br s, 1H), 4.54 (br s, 2H), 3.44 (d, J = 6.7 Hz, 1H), 3.42 (d, J = 6.7 Hz, 1H), 2.89 (apparent t, J = 6.8 Hz, 2H); 13C NMR (150 MHz, CD3CN): δ 160.8, 159.4, 150.1, 137.3, 124.2, 122.3, 40.3, 38.9.
1-Nitroso-1-(2-(pyridin-2-yl)ethyl)urea (33)
NaNO2 (0.40 g, 6.1 mmol) was added to a solution of (2-(pyridin-2-yl)ethyl)urea (1.0 g, 6.1 mmol) in 50 ml of 1.2 m aqueous HCl at 0 °C. After 2 h, the reaction was diluted with CH2Cl2 (100 ml), and the resulting mixture was washed with NaHCO3 (saturated aqueous solution, 2× 50 ml). The organic layer was dried with MgSO4 and concentrated under a stream of nitrogen to afford the nitrosourea (0.45 g, 2.3 mmol, 38%) as a yellow oil, which was used without further purification as the precursor to generate the diazo reagent for derivatization of the acetic acid product of P450 17A1. Rf 0.42 (silica gel GF, CH2Cl2/CH3OH, 9:1, v/v); 1H NMR (600 MHz, CD3CN) δ 8.53 (d, J = 4.8 Hz, 1H), 7.70 (ddd, J = 7.7, 7.6, 1.8 Hz, 1H), 7.29 (d, J = 7.7 Hz, 1H), 7.22 (dd, J = 7.7, 4.7 Hz, 1H), 3.71 (apparent t, J = 6.6 Hz, 2H), 3.05 (apparent t, J = 6.6 Hz, 1H).
2-(Pyridin-2-yl)ethyl Acetate
2-(Pyridin-2-yl)ethanol (1.0 ml, 8.9 mmol) and acetic anhydride (840 μl, 8.9 mmol) were stirred overnight in a 4-ml screw cap vial. The reaction was dissolved in CH2Cl2 and treated with aqueous NaHCO3 to remove the by-product CH3CO2H, and the product was purified by preparative TLC (silica gel GF, 20 × 20 cm, 500 μm) using hexane/ethyl acetate (1:1, v/v) as the mobile phase. The product was scraped from the TLC plate, extracted with CH2Cl2, and filtered. The solvent was evaporated under a stream of nitrogen to obtain 2-(pyridin-2-yl)ethyl acetate as a pale yellow liquid (980 mg, yield 67%). Rf 0.61 (silica gel GF, CH2Cl2/CH3OH, 9:1, v/v); 1H NMR (400 MHz, CDCl3): δ 8.55 (d, J = 4.6 Hz, 1H), 7.61 (ddd, J = 7.7, 7.6, 1.8 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7.14 (dd, J = 7.6, 5.0 Hz, 1H), 4.46 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.7 Hz, 2H), 2.02 (s, 3H); 13C-NMR (100 MHz, CDCl3): 171.1, 158.1, 149.5, 136.5, 123.5, 121.7, 63.7, 37.4, 21.0; HRMS (ESI+): calculated for C9H11NO2 [M + H]+ m/z 166.08626, found 166.08678 (Δ 3.1 ppm).
16α,17α-Dihydroxypregnenolone
16α,17α-Dihydroxypregnenolone was synthesized in two steps from commercially available 16,17-dehydropregnenolone-3-acetate. 16,17-Dehydropregnenolone-3-acetate was converted to 16,17-dehydropregnenolone according to a previously reported procedure (74). 16,17-Dehydropregnenolone was subjected to dihydroxylation conditions loosely based on a procedure that was previously reported (87); KMnO4 (105 mg, 0.67 μmol) in acetone/H2O (6:1, v/v, 70 ml) was added over 10 min with an addition funnel to 16,17-dehydropregnenolone (209 mg, 0.67 μmol) in acetone (50 ml) and HCO2H (50 μl) at 0 °C. After the addition was complete, the reaction mixture was stirred for 2 min and then washed with H2O (100 ml) and extracted with ethyl acetate (200 ml). The organic layer was concentrated in vacuo and purified by flash column chromatography (100% hexanes to 50% ethyl acetate/hexanes, v/v), and the most pure fraction (as judged by TLC analysis) was concentrated (10 mg, white solid). The white solid was further purified using an HPLC-UV system with a Beckman Ultrasphere octadecylsilane column (10 × 250 mm, 5 μm) with the following H2O and CH3OH gradients at a flow rate of 4 ml/min: 0–2.3 min, 77% CH3OH; 2.3–11.3 min, linear gradient from 77 to 88% CH3OH; 11.3–11.6 min, hold at 88% CH3OH; 11.6–12.4 min, linear gradient from 88 to 77% CH3OH; 12.4–15 min, hold at 77% CH3OH (all v/v). Elution of the steroid was detected at 215 nm. The CH3OH from the collected fraction was evaporated under a N2 stream, and H2O was removed by lyophilization. The purified material was used for incubation with P450 17A1. Rf 0.3 (silica gel GF, ethyl acetate/hexanes, 1:1, v/v); 1H NMR (600 MHz, CDCl3): δ 5.34–5.30 (m, 1H), 5.08–5.05 (m, 1H), 3.79 (broad s, 1H), 3.56–3.51 (m, 1H), 2.32–2.28 (m, 1H), 2.25 (s, 3H), 2.24–2.21 (m, 1H), 2.07–1.92 (m, 3H), 1.88–1.77 (m, 3H), 1.65–1.58 (m, 2H), 1.53–1.44 (m, 6H), 1.00 (s, 3H), 0.70 (s, 3H).
Iodosylbenzene
Iodosylbenzene was freshly prepared by NaOH hydrolysis of iodobenzene diacetate in 75% yield (88) and stored at −20 °C.
Assays
18O2 Incubations
The standard reconstituted P450 17A1 system contained P450 17A1 (8.4 μm), NADPH-P450 reductase (13 μm), cytochrome b5 (14 μm), 130 μm 17α-hydroxy-[21,21,21-2H3]pregnenolone or 100 μm 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone, and l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (80 μm) in 2.2-ml incubation mixtures containing 50 mm potassium phosphate buffer (pH 7.4). Reaction mixtures were placed in Thunberg tubes, and air was removed on a gas train equipped with a manifold (89, 90) (three exchanges of argon/vacuum, 5 min each cycle). After introduction of 18O2 (Sigma-Aldrich, 99% atomic excess, pressurized cylinder) into a Thunberg tube under vacuum, each reaction was initiated by adding an NADPH-generating system (10 mm glucose 6-phosphate, 0.5 mm NADP+, and 2 μg/ml yeast glucose-6-phosphate dehydrogenase (91); 8% of total reaction volume) tipped from the stopper reservoir, with mixing. Incubations were conducted in a water bath at 37 °C for 30 min for 17α-hydroxy-[21,21,21-2H3]pregnenolone substrate or 60 min for 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone substrate with shaking at 100 rpm.
The reactions were quenched with CH2Cl2 (5 ml), and 500 μl of 3 m HCl (chilled to 0 °C) for the 17α-hydroxy-[21,21,21-2H3]pregnenolone substrate or 1 ml of 3 m HCl (chilled to 0 °C) for the 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone substrate was added to decrease the pH to ∼1 and facilitate extraction of acetic acid into the organic layer. For cases with 17α-hydroxy-[2,2,4,6,6,21,21,21-2H8]progesterone as the substrate, the mixture (after addition of HCl) was mixed with a vortex device and centrifuged at 1500 × g for 1 min to remove the emulsion. Before the reaction of the product acetic acid with the diazo reagent, the organic extracts were collected, and residual water was removed using anhydrous MgSO4 (∼50 mg for each extraction).
Derivatization of Acetic Acid
Diazoethylpyridine was prepared ex tempore from 1-nitroso-1-(2-(pyridin-2-yl)ethyl)urea (5 mg) in diethyl ether (2 ml), after treatment with KOH (1 ml of a 30% (w/v) solution in H2O) (33). The organic layer (containing the diazo reagent) was dried with anhydrous MgSO4 (∼50 mg), filtered with a cotton-plugged Pasteur pipette, and reacted with an organic extract of each P450 17A1-steroid-18O2 incubation. The solvent was evaporated under a stream of nitrogen, and residues were dissolved in CH3CN (70 μl).
LC-MS Analysis
LC-MS analysis of deuterium-labeled 2-(pyridin-2-yl)ethyl acetate from 18O2 incubations was performed using an Acquity UPLC system connected to a Thermo LTQ XL Orbitrap mass spectrometer operating in the electrospray ionization (ESI) positive ion mode. A Phenomenex Kinetex® 2.6-μm C8 100 Å, LC column (100 × 2.1 mm) was used for separation of the acetic acid derivative at a flow rate of 0.3 ml/min with the following gradient: 0–1.0 min, 100% A (v/v); 4.0–5.2 min, 100% B (v/v); 5.3–8.0 min, 100% A (v/v); mobile phase A was 10 mm NH4HCO2 in H2O (v/v); and mobile phase B was 10 mm NH4HCO2 in 95:5 CH3CN/H2O (v/v).
For the 18O2 incubation assays, the LTQ mass spectrometer was tuned in the electrospray ionization positive mode using synthetic 2-(pyridin-2-yl)ethyl acetate (see above). The tune settings were as follows: sheath gas flow rate, 15 (arbitrary units); auxiliary gas flow rate, 5 (arbitrary units); sweep gas flow rate, 0 (arbitrary units); spray voltage, 4 kV; capillary temperature, 300 °C; capillary voltage, 16 V; tube lens, 30 V.
The LTQ Orbitrap XL high resolution mass spectrometer was calibrated with the ESI-positive ion calibration solution by direct infusion (10 μl/min with a 500-μl Hamilton syringe) as done previously (33). The mass spectrometer was first tuned to the standard solution with m/z 524.3 (methionine/arginine/phenylalanine/alanine acetate), and the tube lens voltage was set to 145 V to fragment caffeine (m/z 195 to 138).
17α-Hydroxysteroid Reactions with Oxygen Surrogates
The standard reconstituted system used for comparison included P450 17A1 (0.5 μm for iodosylbenzene, 0.1 μm for H2O2), NADPH-P450 reductase (2.0 μm), cytochrome b5 (0.5 μm), and l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (10 μm) in 0.5-ml incubation mixtures containing 50 mm potassium phosphate buffer (pH 7.4) and the NADPH-generating system. Assays were done as in the case of the 18O-labeling work (see above) except that the incubations were aerobic, as described previously (37), using 10 μm 17α-hydroxyprogesterone or 17α-hydroxypregnenolone with P450 17A1 (0.5 μm) and no reductase, in the absence or presence of cytochrome b5 (0.5 μm). H2O2 (to 10 mm) or iodosylbenzene (to 2 mm) was added at varying concentrations (from aqueous stocks). The incubations were done for 5 min with H2O2 and for 30 s with iodosylbenzene (41, 92), with extraction into CH2Cl2 and analysis of the conversion of 17α-hydroxyprogesterone to androstenedione and of 17α-hydroxypregnenolone to DHEA by UPLC. The Δ5 steroids were converted to Δ4 steroids by treatment with cholesterol oxidase prior to LC-UV analysis (37). Iodosylbenzene reactions were conducted for a short time period because the reagent is very destructive to P450 heme (41).
Product analysis was done on a Waters Acquity UPLC system with a Waters Acquity UPLC Ethylene Bridged Hybrid (BEH) octadecylsilane (C18) column (2.1 × 100 mm, 1.7 μm). LC conditions were as follows: solvent A consisted of 70% CH3OH and 30% H2O (v/v), and solvent B was 100% CH3CN. The products were resolved by a 0.2 ml min−1 gradient with the following steps: 0–1 min, hold at 5% B (v/v), 1–4 min, linear gradient from 5 to 30% B (v/v); 4–4.5 min, linear gradient from 30 to 40% B (v/v); 4.5–4.55 min, 40 to 95% B (v/v); 4.55–6.75 min, hold at 95% B (v/v); 6.75–7 min, 95 to 5% B (v/v); and 7–10 min, hold at 95% B (v/v). The column temperature was maintained at 40 °C, and the Δ4 steroids were quantified by their absorbance at 243 nm.
LC-UV-MS Analysis of New Steroid Products
An LTQ Orbitrap XL mass spectrometer was tuned in the atmospheric pressure chemical ionization-positive mode with commercially available steroid solutions in a 1:1 mixture (v/v) of H2O and CH3CN (16α-hydroxyandrostenedione and 16α,17α-dihydroxyprogesterone). The tune settings were as follows: vaporizer temperature, 350 °C; sheath gas flow rate, 50 (arbitrary units); auxiliary gas flow rate, 5 (arbitrary units); sweep gas flow rate, 0 (arbitrary units); discharge current, 10 μA; capillary temperature, 275 °C; capillary voltage, 10 V; tube lens, 25 V. The same LC method used for UV analysis (see above) was employed for the Δ4 steroid products. The LC conditions for the Δ5 steroids were as follows: solvent A consisted of 95% H2O and 5% CH3OH (v/v), and solvent B was 95% CH3OH and 5% H2O (v/v). The gradient steps were as follows: 0–1.5 min, hold at 60% B (v/v); 1.5–7.5 min, linear gradient from 60 to 85% B (v/v); 7.5–7.75 min, hold at 85% B (v/v); 7.75–8.25 min, 85 to 60% B (v/v); and 8.25–10 min, hold at 60% B (v/v). The column was kept at ambient temperature.
Solvent Kinetic Isotope Effect Assays
These assays were also done as in the case of the oxygen surrogate experiments (see above), with the incubations done aerobically as described previously (37), using 30 μm 17α-hydroxyprogesterone or 17α-hydroxypregnenolone with P450 17A1 (0.5 μm)/cytochrome b5 (0.5 μm); NADPH-P450 reductase (2 μm); and l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (10 μm) in 0.5 ml incubation mixtures containing 50 mm potassium phosphate buffer (pH 7.4), with 1 mm NADPH. In the D2O experiments, the content of D2O was 95% (v/v), with the pD adjusted (pH = pD + 0.4) (75, 93). Incubations were for 60 s at 37 °C, and the products were analyzed as for the oxygen surrogate experiments (see above).
Isolation of 6β,16α,17α-Trihydroxyprogesterone as a Product
A 20-ml reaction mixture consisting of the same components described in the oxygen surrogate experiments (see above), using 280 units/ml of catalase and 40 μm 16α,17α-dihydroxyprogesterone, was run overnight (∼20 h). The product was extracted from the aqueous mixture with 200 ml of CH2Cl2, and the solvent was evaporated. The dried product was purified using the same system outlined for the 16,17-dihydroxypregnenolone purification (see above) using an isocratic HPLC method (63.5% CH3OH) and peak detection at 243 nm.
Isolation of 16α-Hydroxy-DHEA as a Product
The same procedure described for the isolation of the 6β,16α,17α-trihydroxyprogesterone product (see above) was used, with the following exceptions. The enzyme concentrations were increased to 1 μm P450 17A1, 4 μm NADPH-P450 reductase, 1 μm cytochrome b5, and 3700 units/ml catalase, with 50 μm 16α,17α-dihydroxypregnenolone and the NADPH-generating system. The incubation was run for 4 h. Purification was conducted using the same procedure detailed in the LC-UV purification of 16α,17α-dihydroxyprogesterone (see above), except that the wavelength used to detect the Δ5 product was 215 nm.
Author Contributions
F. K. Y. synthesized most of the chemicals, did all of the NMR analysis, and did the 18O analyses. E. G. purified the P450 17A1 and did most of the incubations and HPLC analyses, including some of the mass spectra. R. J. A. supervised part of the work and helped write the paper. F. P. G. oversaw the project, synthesized iodosylbenzene, and assembled the paper. All authors contributed to the writing of the paper and the conclusions.
Supplementary Material
Acknowledgments
We thank L. D. Nagy and T. T. N. Phan for preparing NADPH-P450 reductase; M. Bojić for preparing 2-(pyridine-2-yl)ethyl acetate and performing some of the preliminary experiments; K. M. Johnson for recording the NMR spectra of 2-(pyridine-2-yl)ethyl acetate; and K. Trisler for assistance in preparation of the manuscript.
Note Added in Proof
In the version of this article that was published as a Paper in Press on June 23, 2016, the spectra in Fig. 14B were inadvertently duplicated from Fig. 15C. This error has now been corrected and does not affect the results or conclusions of this work.
This work was supported, in whole or in part, by National Institutes of Health Grants R37 CA090426, R01 GM118122, and R01 GM103937 (to F. P. G.) and R01 GM086596 (to R. J. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S4.
E. Gonzalez and F. P. Guengerich, unpublished data.
- P450
- cytochrome P450
- compound I
- formal FeO3+ form of a hemoprotein
- HMBC
- homonuclear correlation (NMR) spectroscopy
- DHEA
- dehydroepiandrosterone
- ESI
- electrospray ionization
- HRMS
- high resolution mass spectrometry
- LC-MS
- (combined) liquid chromatography-mass spectrometry.
References
- 1. Rendic S., and Guengerich F. P. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 28, 38–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650 [DOI] [PubMed] [Google Scholar]
- 3. Isin E. M., and Guengerich F. P. (2007) Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 1770, 314–329 [DOI] [PubMed] [Google Scholar]
- 4. Ortiz de Montellano P. R. (2015) in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., ed) 4th Ed., pp 111–176, Springer, New York [Google Scholar]
- 5. Miller W. L., and Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guengerich F. P. (2015) in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., ed) 4th Ed., pp 523–785, Springer, New York [Google Scholar]
- 7. Yoshimoto F. K., and Auchus R. J. (2015) The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid Biochem. Mol. Biol. 151, 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krest C. M., Onderko E. L., Yosca T. H., Calixto J. C., Karp R. F., Livada J., Rittle J., and Green M. T. (2013) Reactive intermediates in cytochrome P450 catalysis. J. Biol. Chem. 288, 17074–17081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mak P. J., Gregory M. C., Denisov I. G., Sligar S. G., and Kincaid J. R. (2015) Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U.S.A. 112, 15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Groves J. T. (2006) High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 100, 434–447 [DOI] [PubMed] [Google Scholar]
- 11. Bell S. R., and Groves J. T. (2009) A highly reactive P450 model compound I. J. Am. Chem. Soc. 131, 9640–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morand P., Williamson D. G., Layne D. S., Lompa-Krzymien L., and Salvador J. (1975) Conversion of an androgen epoxide into 17β-estradiol by human placental microsomes. Biochemistry 14, 635–638 [DOI] [PubMed] [Google Scholar]
- 13. Fishman J. (1982) Biochemical mechanism of aromatization. Cancer Res. 42, 3277s–3280s [PubMed] [Google Scholar]
- 14. Akhtar M., Corina D., Pratt J., and Smith T. (1976) Studies on the removal of C-19 in oestrogen biosynthesis using 18O2. J. Chem. Soc., Chem. Commun., 1976, 854–856 [Google Scholar]
- 15. Akhtar M., Calder M. R., Corina D. L., and Wright J. N. (1982) Mechanistic studies on C-19 demethylation in oestrogen biosynthesis. Biochem. J. 201, 569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goto J., and Fishman J. (1977) Participation of a nonenzymatic transformation in the biosynthesis of estrogens from androgens. Science 195, 80–81 [DOI] [PubMed] [Google Scholar]
- 17. Hahn E. F., and Fishman J. (1984) Immunological probe of estrogen biosynthesis. Evidence for the 2β-hydroxylative pathway in aromatization of androgens. J. Biol. Chem. 259, 1689–1694 [PubMed] [Google Scholar]
- 18. Caspi E., Wicha J., Arunachalam T., Nelson P., and Spiteller G. (1984) Estrogen biosynthesis: concerning the obligatory intermediacy of 2β-hydroxy-10β-formyl androst-4-ene-3,17-dione. J. Am. Chem. Soc. 1984, 106, 7282–7283 [Google Scholar]
- 19. Hackett J. C., Brueggemeier R. W., and Hadad C. M. (2005) The final catalytic step of cytochrome P450 aromatase: a density functional theory study. J. Am. Chem. Soc. 127, 5224–5237 [DOI] [PubMed] [Google Scholar]
- 20. Sen K., and Hackett J. C. (2012) Coupled electron transfer and proton hopping in the final step of CYP19-catalyzed androgen aromatization. Biochemistry 51, 3039–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mak P. J., Luthra A., Sligar S. G., and Kincaid J. R. (2014) Resonance Raman spectroscopy of the oxygenated intermediates of human CYP19A1 implicates a compound I intermediate in the final lyase step. J. Am. Chem. Soc. 136, 4825–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cole P. A., and Robinson C. H. (1990) Mechanism and inhibition of cytochrome P-450 aromatase. J. Med. Chem. 33, 2933–2942 [DOI] [PubMed] [Google Scholar]
- 23. Gregory M. C., Denisov I. G., Grinkova Y. V., Khatri Y., and Sligar S. G. (2013) Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc. 135, 16245–16247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khatri Y., Gregory M. C., Grinkova Y. V., Denisov I. G., and Sligar S. G. (2014) Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun. 443, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gregory M., Mak P. J., Sligar S. G., and Kincaid J. R. (2013) Differential hydrogen bonding in human CYP17 dictates hydroxylation versus lyase chemistry. Angew. Chem. Int. Ed. Engl. 52, 5342–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller S. L., Wright J. N., Corina D. L., and Akhtar M. (1991) Mechanistic studies on pregnene side-chain cleavage enzyme (17α-hydroxylase-17,20-lyase) using 18O. J. Chem. Soc. Chem. Commun. 1991, 157–159 [Google Scholar]
- 27. Akhtar M., Corina D. L., Miller S. L., Shyadehi A. Z., and Wright J. N. (1994) Incorporation of label from 18O2 into acetate during side-chain cleavage catalysed by cytochrome P45017α (17α-hydroxylase-17,20-lyase). J. Chem. Soc., Perkin Trans. 1, 263–267 [Google Scholar]
- 28. Lee-Robichaud P., Shyadehi A. Z., Wright J. N., Akhtar M. E., and Akhtar M. (1995) Mechanistic kinship between hydroxylation and desaturation reactions: acyl-carbon bond cleavage promoted by pig and human CYP17 (P-45017α; 17α-hydroxylase-17,20-lyase). Biochemistry 34, 14104–14113 [DOI] [PubMed] [Google Scholar]
- 29. Akhtar M., Lee-Robichaud P., Akhtar M. E., and Wright J. N. (1997) The impact of aromatase mechanism on other P450s. J. Steroid Biochem. Mol. Biol. 61, 127–132 [PubMed] [Google Scholar]
- 30. Akhtar M., Wright J. N., and Lee-Robichaud P. (2011) A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17). J. Steroid Biochem. Mol. Biol. 125, 2–12 [DOI] [PubMed] [Google Scholar]
- 31. Akhtar M., and Wright J. N. (2015) Acyl-carbon bond cleaving cytochrome P450 enzymes: CYP17A1, CYP19A1 and CYP51A1. Adv. Exp. Med. Biol. 851, 107–130 [DOI] [PubMed] [Google Scholar]
- 32. Guengerich F. P., Sohl C. D., and Chowdhury G. (2011) Multi-step oxidations catalyzed by cytochrome P450 enzymes: processive vs. distributive kinetics and the issue of carbonyl oxidation in chemical mechanisms. Arch. Biochem. Biophys. 507, 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshimoto F. K., and Guengerich F. P. (2014) Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J. Am. Chem. Soc. 136, 15016–15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caspi E., Arunachalam T., and Nelson P. A. (1986) Biosynthesis of estrogens: Aromatization of (19R)-, (19S)-, and (19S)-[19-3H,2H,1H]-3β-hydroxyandrost-5-en-17-ones by human placental aromatase. J. Am. Chem. Soc. 108, 1847–1852 [Google Scholar]
- 35. Swinney D. C., and Mak A. Y. (1994) Androgen formation by cytochrome P450 CYP17. Solvent isotope effect and pL studies suggest a role for protons in the regulation of oxene versus peroxide chemistry. Biochemistry 33, 2185–2190 [DOI] [PubMed] [Google Scholar]
- 36. Cai H., and Guengerich F. P. (1999) Mechanism of aqueous decomposition of trichloroethylene oxide. J. Am. Chem. Soc. 1991, 121, 11656–11663 [Google Scholar]
- 37. Pallan P. S., Nagy L. D., Lei L., Gonzalez E., Kramlinger V. M., Azumaya C. M., Wawrzak Z., Waterman M. R., Guengerich F. P., and Egli M. (2015) Structural and kinetic basis of steroid 17α,20-lyase activity in teleost fish cytochrome P450 17A1 and its absence in cytochrome P450 17A2. J. Biol. Chem. 290, 3248–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chowdhury G., Calcutt M. W., Nagy L. D., and Guengerich F. P. (2012) Oxidation of methyl and ethyl nitrosamines by cytochrome P450 2E1 and 2B1. Biochemistry 51, 9995–10007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chowdhury G., Calcutt M. W., and Guengerich F. P. (2010) Oxidation of N-nitrosoalkylamines by human cytochrome P450 2A6: sequential oxidation to aldehydes and carboxylic acids and analysis of reaction steps. J. Biol. Chem. 285, 8031–8044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Auchus R. J., and Miller W. L. (1999) Molecular modeling of human P450c17 (17α-hydroxylase/17,20-lyase): insights into reaction mechanisms and effects of mutations. Mol. Endocrinol. 13, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 41. Lichtenberger F., Nastainczyk W., and Ullrich V. (1976) Cytochrome P-450 as an oxene transferase. Biochem. Biophys. Res. Commun. 70, 939–946 [DOI] [PubMed] [Google Scholar]
- 42. Kirk D. N., Toms H. C., Douglas C., White K. A., Smith K. E., Latif S., and Hubbard R. W. (1990) A survey of the high-field lH NMR spectra of the steroid hormones, their hydroxylated derivatives, and related compounds. J. Chem. Soc., Perkin Trans. 2, 1990, 1567–1594 [Google Scholar]
- 43. Krauser J. A., Voehler M., Tseng L. H., Schefer A. B., Godejohann M., and Guengerich F. P. (2004) Testosterone 1β-hydroxylation by human cytochrome P450 3A4. Eur. J. Biochem. 271, 3962–3969 [DOI] [PubMed] [Google Scholar]
- 44. Hayamizu K., and Kamo O. (1990) Complete assignments of the 1H and 13C NMR spectra of testosterone and 17 α-methyltestosterone and the 1H parameters obtained from 600 MHz spectra. Magn. Reson. Chem. 28, 250–256 [Google Scholar]
- 45. Hamilton N. M., Dawson M., Fairweather E. E., Hamilton N. S., Hitchin J. R., James D. I., Jones S. D., Jordan A. M., Lyons A. J., Small H. F., Thomson G. J., Waddell I. D., and Ogilvie D. J. (2012) Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J. Med. Chem. 55, 4431–4445 [DOI] [PubMed] [Google Scholar]
- 46. Ortiz de Montellano P. R. (1986) in Cytochrome P-450 (Ortiz de Montellano P. R., ed) pp. 217–271, Plenum Press, New York [Google Scholar]
- 47. Betancor C., Francisco C. G., Freire R., and Suarez E. (1988) The reaction of enols with superoxide anion radicals: preparation of tertiary [small α]-ketols. J. Chem. Soc. Chem. Commun. 1998, 947–948 [Google Scholar]
- 48. Pestovsky O., and Bakac A. (2004) Reactivity of aqueous Fe(IV) in hydride and hydrogen atom transfer reactions. J. Am. Chem. Soc. 126, 13757–13764 [DOI] [PubMed] [Google Scholar]
- 49. Bakac A. (2002) Reactions of superoxo and oxo metal complexes with aldehydes. Radical-specific pathways for cross-disproportionation of superoxometal ions and acylperoxyl radicals. J. Am. Chem. Soc. 124, 9136–9144 [DOI] [PubMed] [Google Scholar]
- 50. Paraskevas P. D., Sabbe M. K., Reyniers M. F., Papayannakos N. G., and Marin G. B. (2014) Kinetic modeling of α-hydrogen abstractions from unsaturated and saturated oxygenate compounds by hydrogen atoms. J. Phys. Chem. A 118, 9296–9309 [DOI] [PubMed] [Google Scholar]
- 51. Basran J., Efimov I., Chauhan N., Thackray S. J., Krupa J. L., Eaton G., Griffith G. A., Mowat C. G., Handa S., and Raven E. L. (2011) The mechanism of formation of N-formylkynurenine by heme dioxygenases. J. Am. Chem. Soc. 133, 16251–16257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akhtar M., Corina D., Miller S., Shyadehi A. Z., and Wright J. N. (1994) Mechanism of the acyl-carbon cleavage and related reactions catalyzed by multifunctional P-450s: Studies on cytochrome P45017α. Biochemistry 33, 4410–4418 [DOI] [PubMed] [Google Scholar]
- 53. Yoshimoto F. K., Zhou Y., Peng H. M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., and Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kadlubar F. F., Morton K. C., and Ziegler D. M. (1973) Microsomal-catalyzed hydroperoxide-dependent C-oxidation of amines. Biochem. Biophys. Res. Commun. 54, 1255–1261 [DOI] [PubMed] [Google Scholar]
- 55. Rittle J., and Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science 330, 933–937 [DOI] [PubMed] [Google Scholar]
- 56. Mansuy D., Bartoli J. F., and Momenteau M. (1982) Alkane hydroxylation catalyzed by metalloporphyrins: evidence for different active oxygen species with alkylhydroperoxides and iodosobenzene as oxidants. Tetrahedron Lett. 23, 2781–2784 [Google Scholar]
- 57. Lee D. S., Yamada A., Sugimoto H., Matsunaga I., Ogura H., Ichihara K., Adachi S., Park S. Y., and Shiro Y. (2003) Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J. Biol. Chem. 278, 9761–9767 [DOI] [PubMed] [Google Scholar]
- 58. Rude M. A., Baron T. S., Brubaker S., Alibhai M., Del Cardayre S. B., and Schirmer A. (2011) Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl. Environ. Microbiol. 77, 1718–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujishiro T., Shoji O., Nagano S., Sugimoto H., Shiro Y., and Watanabe Y. (2011) Crystal structure of H2O2-dependent cytochrome P450SPα with its bound fatty acid substrate: Insight into the regioselective hydroxylation of fatty acids at the α position. J. Biol. Chem. 286, 29941–29950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Joo H., Lin Z., and Arnold F. H. (1999) Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 399, 670–673 [DOI] [PubMed] [Google Scholar]
- 61. Renneberg R., Scheller F., Ruckpaul K., Pirrwitz J., and Mohr P. (1978) NADPH and H2O2-dependent reactions of cytochrome P-450LM compared with peroxidase catalysis. FEBS Lett. 96, 349–353 [DOI] [PubMed] [Google Scholar]
- 62. Renneberg R., Damerau W., Jung C., Ebert B., and Scheller F. (1983) Study of H2O2-supported N-demethylations catalyzed by cytochrome P-450 and horseradish peroxidase. Biochem. Biophys. Res. Commun. 113, 332–339 [DOI] [PubMed] [Google Scholar]
- 63. Nordblom G. D., and Coon M. J. (1977) Hydrogen peroxide formation and stoichiometry of hydroxylation reactions catalyzed by highly purified liver microsomal cytochrome P-450. Arch. Biochem. Biophys. 180, 343–347 [DOI] [PubMed] [Google Scholar]
- 64. Renneberg R., Capdevila J., Chacos N., Estabrook R. W., and Prough R. A. (1981) Hydrogen peroxide-supported oxidation of benzo[a]pyrene by rat liver microsomal fractions. Biochem. Pharmacol. 30, 843–848 [DOI] [PubMed] [Google Scholar]
- 65. Holm K. A., Engell R. J., and Kupfer D. (1985) Regioselectivity of hydroxylation of prostaglandins by liver microsomes supported by NADPH versus H2O2 in methylcholanthrene-treated and control rats: formation of novel prostaglandin metabolites. Arch. Biochem. Biophys. 237, 477–489 [DOI] [PubMed] [Google Scholar]
- 66. Guengerich F. P., Vaz A. D., Raner G. N., Pernecky S. J., and Coon M. J. (1997) Evidence for a role of a perferryl-oxygen complex, FeO3+, in the N-oxygenation of amines by cytochrome P450 enzymes. Mol. Pharmacol. 51, 147–151 [DOI] [PubMed] [Google Scholar]
- 67. Auchus R. J., Lee T. C., and Miller W. L. (1998) Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165 [DOI] [PubMed] [Google Scholar]
- 68. Lacroix D., Sonnier M., Moncion A., Cheron G., and Cresteil T. (1997) Expression of CYP3A in the human liver: evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur. J. Biochem. 247, 625–634 [DOI] [PubMed] [Google Scholar]
- 69. Niwa T., Yabusaki Y., Honma K., Matsuo N., Tatsuta K., Ishibashi F., and Katagiri M. (1998) Contribution of human hepatic cytochrome P450 isoforms to regioselective hydroxylation of steroid hormones. Xenobiotica 28, 539–547 [DOI] [PubMed] [Google Scholar]
- 70. Kirschner M. A., Wiqvist N., and Diczfalusy E. (1966) Studies on oestriol synthesis from dehydroepiandrosterone sulphate in human pregnancy. Acta Endocrinol. 53, 584–597 [DOI] [PubMed] [Google Scholar]
- 71. Stevenson D. E., Wright J. N., and Akhtar M. (1988) Mechanistic consideration of P-450 dependent enzymic reactions: Studies on oestriol biosynthesis. J. Chem. Soc. Perkin Trans. 1, 2043–2052 [Google Scholar]
- 72. Cheng Q., Sohl C. D., Yoshimoto F. K., and Guengerich F. P. (2012) Oxidation of dihydrotestosterone by human cytochromes P450 19A1 and 3A4. J. Biol. Chem. 287, 29554–29567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Petrunak E. M., DeVore N. M., Porubsky P. R., and Scott E. E. (2014) Structures of human steroidogenic cytochrome P450 17A1 with substrates. J. Biol. Chem. 289, 32952–32964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yoshimoto F. K., Peng H. M., Zhang H., Anderson S. M., and Auchus R. J. (2014) Epoxidation activities of human cytochromes P450c17 and P450c21. Biochemistry 53, 7531–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Walsh C. (1979) Enzymatic Reaction Mechanisms, p. 121, W. H. Freeman & Co., San Francisco [Google Scholar]
- 76. DeVore N. M., and Scott E. E. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yosca T. H., Rittle J., Krest C. M., Onderko E. L., Silakov A., Calixto J. C., Behan R. K., and Green M. T. (2013) Iron(IV) hydroxide pKa and the role of thiolate ligation in C–H bond activation by cytochrome P450. Science 342, 825–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Davydov R., Strushkevich N., Smil D., Yantsevich A., Gilep A., Usanov S., and Hoffman B. M. (2015) Evidence that compound I is the active species in both the hydroxylase and lyase steps by which P450scc converts cholesterol to pregnenolone: EPR/ENDOR/cryoreduction/annealing studies. Biochemistry 54, 7089–7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gottlieb H. E., Kotlyar V., and Nudelman A. (1997) NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515 [DOI] [PubMed] [Google Scholar]
- 80. Gillam E. M., Baba T., Kim B. R., Ohmori S., and Guengerich F. P. (1993) Expression of modified human cytochrome P450 3A4 in Escherichia coli and purification and reconstitution of the enzyme. Arch. Biochem. Biophys. 305, 123–131 [DOI] [PubMed] [Google Scholar]
- 81. Hosea N. A., Miller G. P., and Guengerich F. P. (2000) Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry 39, 5929–5939 [DOI] [PubMed] [Google Scholar]
- 82. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 [DOI] [PubMed] [Google Scholar]
- 83. Guengerich F. P. (2005) Reduction of cytochrome b5 by NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 440, 204–211 [DOI] [PubMed] [Google Scholar]
- 84. Pallan P. S., Wang C., Lei L., Yoshimoto F. K., Auchus R. J., Waterman M. R., Guengerich F. P., and Egli M. (2015) Human cytochrome P450 21A2, the major steroid 21-hydroxylase: Structure of the enzyme·progesterone substrate complex and rate-limiting C–H bond cleavage. J. Biol. Chem. 290, 13128–13143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pechurskaya T. A., Lukashevich O. P., Gilep A. A., and Usanov S. A. (2008) Engineering, expression, and purification of “soluble” human cytochrome P45017α and its functional characterization. Biokhimiia 73, 806–811 [DOI] [PubMed] [Google Scholar]
- 86. Sandhu P., Baba T., and Guengerich F. P. (1993) Expression of modified cytochrome P450 2C10 (2C9) in Escherichia coli, purification, and reconstitution of catalytic activity. Arch. Biochem. Biophys. 306, 443–450 [DOI] [PubMed] [Google Scholar]
- 87. Vijaykumar D., Mao W., Kirschbaum K. S., and Katzenellenbogen J. A. (2002) An efficient route for the preparation of a 21-fluoro progestin-16α,17α-dioxolane, a high-affinity ligand for PET imaging of the progesterone receptor. J. Org. Chem. 67, 4904–4910 [DOI] [PubMed] [Google Scholar]
- 88. Saltzman H., and Sharefkin J. G. (1973) Organic Synthesis, Collective Volume 5. Iodosobenzene. Org. Syn. 1963, 43, 60 [Google Scholar]
- 89. Foust G. P., Burleigh B. D. Jr., Mayhew S. G., Williams C. H. Jr., and Massey V. (1969) An anaerobic titration assembly for spectrophotometric use. Anal. Biochem. 27, 530–535 [DOI] [PubMed] [Google Scholar]
- 90. Guengerich F. P., Krauser J. A., and Johnson W. W. (2004) Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry 43, 10775–10788 [DOI] [PubMed] [Google Scholar]
- 91. Guengerich F. P. (2014) in Hayes' Principles and Methods of Toxicology (Hayes A. W., and Kruger C. L., eds) 6th Ed., pp. 1905–1964, CRC Press-Taylor & Francis, Boca Raton, FL [Google Scholar]
- 92. Macdonald T. L., Gutheim W. G., Martin R. B., and Guengerich F. P. (1989) Oxidation of substituted N,N-dimethylanilines by cytochrome P-450: Estimation of the effective oxidation-reduction potential of cytochrome P-450. Biochemistry 28, 2071–2077 [DOI] [PubMed] [Google Scholar]
- 93. Jencks W. P. (1969) Catalysis in Chemistry and Enzymology, pp. 253 and 276, McGraw-Hill, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.