

Abstract
Serotonin N-acetyltransferase (AANAT) converts serotonin to N-acetylserotonin (NAS), a distinct biological regulator and the immediate precursor of melatonin, a circulating hormone that influences circadian processes, including sleep. N-terminal sequences of AANAT enzymes vary among vertebrates. Mechanisms that regulate the levels of AANAT are incompletely understood. Previous findings were consistent with the possibility that AANAT may be controlled through its degradation by the N-end rule pathway. By expressing the rat and human AANATs and their mutants not only in mammalian cells but also in the yeast Saccharomyces cerevisiae, and by taking advantage of yeast genetics, we show here that two “complementary” forms of rat AANAT are targeted for degradation by two “complementary” branches of the N-end rule pathway. Specifically, the Nα-terminally acetylated (Nt-acetylated) Ac-AANAT is destroyed through the recognition of its Nt-acetylated N-terminal Met residue by the Ac/N-end rule pathway, whereas the non-Nt-acetylated AANAT is targeted by the Arg/N-end rule pathway, which recognizes the unacetylated N-terminal Met-Leu sequence of rat AANAT. We also show, by constructing lysine-to-arginine mutants of rat AANAT, that its degradation is mediated by polyubiquitylation of its Lys residue(s). Human AANAT, whose N-terminal sequence differs from that of rodent AANATs, is longer-lived than its rat counterpart and appears to be refractory to degradation by the N-end rule pathway. Together, these and related results indicate both a major involvement of the N-end rule pathway in the control of rodent AANATs and substantial differences in the regulation of rodent and human AANATs that stem from differences in their N-terminal sequences.
Keywords: acetyltransferase, circadian, proteolysis, serotonin, ubiquitin
Introduction
Arylalkylamine N-acetyltransferase (AANAT)2 converts the neurotransmitter serotonin to N-acetylserotonin (NAS) (1–11). Regulatory functions of NAS include its activity as an agonist of TrkB, the receptor for the brain-derived neurotrophic factor (12–14). NAS is the immediate precursor of melatonin, a circulating hormone that regulates sleep and other circadian processes in vertebrates. Melatonin is also present in invertebrates, including insects, as well as in animals that lack recognizable neurons (1, 15–21). The functions of melatonin in mammals include the modulation of circadian rhythms in response to light-dark cycles. Melatonin also contributes to the control of seasonal physiology (20). These mechanisms are a part of a broader range of processes that involve distinct biological oscillators in all organisms (22–24). In the course of daily light-dark cycles, the activity of AANAT and the level of the ∼23-kDa AANAT protein in the brain's pineal gland are high during nocturnal periods and rapidly decrease following exposure to light (1, 20, 25).
In mammals, changes of AANAT levels in the brain's pineal gland are controlled by the circadian oscillator in the suprachiasmatic nucleus of the hypothalamus. At night, axons of rodent superior cervical ganglion neurons release norepinephrine in the pineal gland, in response to circadian signals from the suprachiasmatic nucleus, thereby activating adrenergic receptors to increase intracellular Ca2+ and cAMP. The resulting phosphorylation of CREB, the cAMP-response element-binding protein, up-regulates the Aanat transcriptional promoter, which contains cAMP-response element sequence elements (1, 4, 25–27). Increases in cAMP also stimulate the site-specific phosphorylation of AANAT by PKA, thereby causing the binding of AANAT to 14-3-3 proteins. These transitions augment the enzymatic activity of AANAT and inhibit its degradation (1, 6, 7, 28). Although AANAT is predominantly expressed in the pineal gland and the retina, this enzyme is also present in cells of the gastrointestinal tract and apparently at other sites as well, including the hippocampus, the olfactory bulb, the cerebellum, and the spinal cord (12, 29, 30).
Melatonin is produced from NAS by hydroxyindole-O-methyltransferase. The levels of NAS, synthesized by AANAT, can constrain the rate of melatonin synthesis from NAS during light parts of daily cycles (when NAS levels are low) but appear not to be the rate-limiting step during dark parts of these cycles, when both AANAT and NAS levels are high (21). In rodents such as rats, the activity of AANAT starts to increase 3–4 h after the onset of darkness and begins to decrease before the onset of light in the morning (21, 31). Hence the importance of regulating the expression and the enzymatic activity of AANAT, because these parameters determine the levels of both NAS and melatonin.
The concentration of AANAT, a largely cytosolic enzyme (32), is controlled through both transcriptional and post-translational mechanisms (1, 11, 31, 33–35). In rodents, the onset of darkness leads to an increase in melatonin (requiring a preceding increase of NAS) after a lag period, whereas in sheep and primates, increases in melatonin occur rapidly upon the onset of darkness. The levels of Aanat mRNA in rodents can vary by >100-fold during light-dark cycles (10), whereas in non-rodent mammals such as, for example, sheep, these differences can be as low as ∼2-fold, suggesting a major involvement of translational and/or post-translational AANAT regulation in latter cases (11, 36). In addition to variability in regulation at the level of AANAT-encoding mRNAs, AANAT is also unusual in the extent of variability of its N-terminal sequences during vertebrate evolution (Fig. 1C). In 2010, one of our laboratories showed that N-terminal residues of rat AANAT play a role in the control of its degradation, suggesting the involvement of the N-end rule pathway (37).
FIGURE 1.

The mammalian N-end rule pathway and N-terminal regions of AANAT enzymes. See the Introduction for references and descriptions of the pathway's mechanisms and biological functions. Amino acid residues are denoted by single-letter codes. A, the mammalian Arg/N-end rule pathway. It targets proteins for degradation through their specific unacetylated N-terminal residues. A yellow oval denotes the rest of a protein substrate. Primary, secondary, and tertiary, mechanistically distinct classes of destabilizing N-terminal residues. Ntan1 and Ntaq1, N-terminal amidases (Nt-amidases) that convert the tertiary destabilizing N-terminal residues Asn and Gln to Asp and Glu, respectively. The Ate1 arginyltransferase (R-transferase) conjugates Arg, a primary destabilizing residue, to N-terminal Asp, Glu, and (oxidized) Cys. Type 1 and type 2, two sets of primary destabilizing N-terminal residues, basic (Arg, Lys, and His) and bulky hydrophobic (Leu, Phe, Trp, Tyr, Ile, and Met, if the latter is followed by a bulky hydrophobic residue (Φ)), respectively. These sets of N-terminal residues are recognized by two distinct substrate-binding sites of N-recognins, the pathway's E3 ubiquitin ligases, whose (possibly incomplete) list includes Ubr1, Ubr2, Ubr4, and Ubr5. B, mammalian Ac/N-end rule pathway. It targets proteins through their Nα-terminally acetylated (Nt-acetylated) residues. Red arrow on the left, cotranslational removal of the N-terminal Met residue by Met-aminopeptidases (MetAPs). N-terminal Met is retained if a residue at position 2 is larger than Val. C, the first 10 amino acid residues of animal AANAT enzymes, in vertebrates and an invertebrate, such as Drosophila melanogaster. See the Introduction for descriptions of AANATs.
The N-end rule pathway is a set of intracellular proteolytic systems whose unifying feature is the ability to recognize and polyubiquitylate proteins containing N-terminal degradation signals (degrons) called N-degrons, thereby causing degradation of these proteins by the proteasome (Fig. 1, A and B) (38–46). Ubiquitin (Ub) ligases of the N-end rule pathway, called N-recognins, can recognize not only N-degrons but also specific internal degradation signals (47–50). The main determinant of an N-degron is either an unmodified or chemically modified destabilizing N-terminal residue of a protein. Another determinant of an N-degron is a protein's internal Lys residue(s). It functions as the site of polyubiquitylation and tends to be located in a conformationally disordered region (41, 51, 52). Bacteria also contain the N-end rule pathway, but Ub-independent versions of it (53–58).
Regulated degradation of proteins and their natural fragments by the N-end rule pathway has been shown to mediate a strikingly broad range of biological functions, including the sensing of heme, nitric oxide (NO), oxygen, and short peptides; the control of the input stoichiometries of subunits in oligomeric protein complexes; the elimination of misfolded or otherwise abnormal proteins; the degradation of specific proteins after their retrotranslocation to the cytosol from membrane-enclosed compartments; the regulation of apoptosis and repression of neurodegeneration; the regulation of DNA repair, transcription, replication, and chromosome cohesion/segregation; the regulation of G proteins and cytoskeletal proteins, such as actin and myosin; the regulation of autophagy, peptide import, meiosis, immunity, fat metabolism, cell migration, cardiovascular development, spermatogenesis, and neurogenesis; the functioning of adult organs, including the brain, muscle, testis, and pancreas; and the regulation of leaf and shoot development, leaf senescence, and many other processes in plants (Fig. 1, A and B) (see Refs. 41–46 and references therein).
In eukaryotes, the N-end rule pathway consists of two branches. One branch, called the Ac/N-end rule pathway, targets proteins for degradation through their Nα-terminally acetylated (Nt-acetylated) residues (Fig. 1B) (39, 40, 46, 59–62). Degradation signals and E3 Ub ligases of the Ac/N-end rule pathway are called Ac/N-degrons and Ac/N-recognins, respectively. Nt-acetylation of cellular proteins is apparently irreversible, in contrast to acetylation-deacetylation of proteins' internal Lys residues. About 90% of human proteins are cotranslationally Nt-acetylated by ribosome-associated Nt-acetylases (63). Ac/N-degrons are present in many, possibly most, Nt-acetylated proteins (Fig. 1B). Natural Ac/N-degrons are regulated through their reversible shielding in cognate protein complexes (59).
The pathway's other branch, called the Arg/N-end rule pathway, targets specific unacetylated N-terminal residues (Fig. 1A) (40, 64–68). The “primary” destabilizing N-terminal residues Arg, Lys, His, Leu, Phe, Tyr, Trp, and Ile are directly recognized by N-recognins. The unacetylated N-terminal Met, if it is followed by a bulky hydrophobic (Φ) residue, also acts as a primary destabilizing residue (Fig. 1A) (40). In contrast, the unacetylated N-terminal Asn, Gln, Asp, and Glu (as well as Cys, under some metabolic conditions) are destabilizing due to their preliminary enzymatic modifications, which include N-terminal arginylation (Fig. 1A) (41–43, 69). In the yeast Saccharomyces cerevisiae, the Arg/N-end rule pathway is mediated by the Ubr1 N-recognin, a 225-kDa RING-type E3 Ub ligase and a part of the targeting complex comprising the Ubr1-Rad6 and Ufd4-Ubc4/5 E2-E3 holoenzymes (41, 70). In multicellular eukaryotes, several E3 Ub ligases, including Ubr1, function as N-recognins of the Arg/N-end rule pathway (Fig. 1A).
In S. cerevisiae, the Ac/N-end rule pathway is mediated by (at least) the cytosolic/nuclear E3 Ub ligase Not4 and by Doa10, an endoplasmic reticulum membrane-embedded E3 (59). In mammalian cells, this pathway is mediated by (at least) the Teb4 E3, which is sequelogous (similar in sequence (71)) to yeast Doa10 (60). Human RGS2, a regulator of specific G proteins, is a short-lived substrate of the Ac/N-end rule pathway both in mammalian cells and in the heterologous setting of S. cerevisiae (60, 72, 73). In addition, the naturally occurring (blood pressure-elevating) human RGS2Q2L mutant (in which Gln at position 2 is replaced by Leu) is targeted for degradation by both the Ac/N-end rule pathway and the Arg/N-end rule pathway (60). The Ac/N-end rule pathway recognizes the Nt-acetylated Ac-RGS2Q2L (specifically its N-terminal Ac-Met residue), whereas the non-Nt-acetylated RGS2Q2L is targeted (through its N-terminal Met-Leu sequence) by the Arg/N-end rule pathway (60).
In the present work, we analyzed the proteasome-mediated degradation of rat AANAT (37), whose N-terminal sequence Met-Leu (Fig. 1C) is identical to that of the otherwise unrelated human RGS2Q2L protein (60). We also characterized human AANAT, which is highly sequelogous (71) (84% identical) to rat AANAT but bears a different N-terminal sequence (Fig. 1C). Our analyses employed the previously helpful approach of dissecting degradation of a mammalian protein of interest not only in a homologous (mammalian) setting but also in S. cerevisiae (60, 66), thereby making possible the use of yeast genetics.
We show here that two alternative versions of rat AANAT, its Nt-acetylated and non-Nt-acetylated forms, are targeted for degradation by the Ac/N-end rule pathway and the Arg/N-end rule pathway, respectively. In contrast, human AANAT, whose N-terminal sequence differs from that of rodent AANATs, is significantly longer-lived than its rat counterpart and appears to be largely refractory to degradation by the N-end rule pathway. Together, these and related results indicate both a major involvement of the N-end rule pathway in the control of rodent AANATs and substantial differences in the regulation of rodent and human AANATs that stem from differences in their N-terminal sequences.
Results
Wild-type Rat and Human AANATs and Their Mutants
The start (AUG) codon-encoded N-terminal Met residue of nascent proteins is cotranslationally cleaved off by ribosome-associated Met-aminopeptidases (MetAPs) if a residue at position 2, to be made N-terminal by the cleavage, is not larger than Val (41, 74). Thus, for example, the Met residue of the N-terminal Met-Leu-Ser sequence of wild-type rat AANAT (denoted as MLSrAANAT) (Fig. 1C) is retained in mature MLSrAANAT, inasmuch as Leu is larger than Val. Given the previously demonstrated usefulness of employing genetic tractability of S. cerevisiae for understanding the targeting of mammalian N-end rule substrates (60, 66), we carried out the present study by expressing AANAT test proteins not only in human HEK293T cells but also in S. cerevisiae. Cited below are the examined wild-type and mutant AANAT proteins.
When present, parentheses around a superscript's N-terminal Met residue in the notations of AANAT test proteins denote the fact that this Met is cotranslationally cleaved off by MetAPs.
(i) MLSrAANAT3f, the wild-type rat MLSrAANAT C-terminally tagged with a triple-FLAG epitope (Figs. 1C and 2A).
FIGURE 2.

The wild-type rat MLSrAANAT as a substrate of both branches of the N-end rule pathway. A, the wild-type rat MLSrAANAT and its mutants that were constructed and analyzed in the present study. Amino acid residues are denoted by single-letter codes. The Leu residue, at position 2 in wild-type MLSrAANAT, is highlighted in red to make it easier to follow sequence alterations in specific mutants. B, immunoblotting with antibody to actin was used as a loading control. Lane 1, fluorescently labeled molecular mass markers (LI-COR); their masses, in kDa, are indicated on the left. Lane 2, wild-type S. cerevisiae were transformed with vector (V) alone (control). CHX-chases were performed at 30 °C for the indicated times in wild-type S. cerevisiae with the wild-type rat MLSrAANAT3f (lanes 3–6) and with its mutants (M)SMLSrAANAT3f (lanes 7–10), (M)SIrAANAT3f (lanes 11–14), and (M)PLSrAANAT3f (lanes 15–18). The bands of AANAT3f and actin are indicated on the right. C, immunoblotting with antibody to tubulin was used as a loading control. Lane 1, 37 and 50 kDa molecular mass markers (see lane 1 in B). Shown are CHX-chases with the wild-type rat MLSrAANAT3f in wild-type S. cerevisiae (lanes 2–5) and its mutants naa30Δ (lanes 6–9), naa10Δ (lanes 10–13), ubr1Δ (lanes 14–17), naa30Δ ubr1Δ (lanes 18–21), and naa10Δ ubr1Δ (lanes 22–25). The bands of MLSrAANAT3f and tubulin are indicated on the left. D, quantification of data in B. E, quantification of data in C. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown.
(ii) (M)SMLSrAANAT3f, in which the sequence (Met)-Ser was placed before the wild-type Met-Leu-Ser sequence of rat MLSrAANAT3f, yielding the N-terminal sequence (Met)-Ser-Met-Leu-Ser. Its N-terminal Met residue would be cotranslationally cleaved off by MetAPs (Fig. 2A). The (Met)-Ser sequence was added to wild-type MLSrAANAT3f in order to mimic the (Met)-Ser N-terminal sequence of primate (including human) AANATs (Fig. 1C).
(iii) (M)SIrAANAT3f, in which Leu at position 2 of the wild-type rat MLSrAANAT3f (Fig. 1C) was deleted, thereby making the N-terminal Met of the resulting (M)SIrAANAT3f removable by MetAPs (Figs. 1C and 2A).
(iv) (M)PLSrAANAT3f, in which the Pro residue was inserted between N-terminal Met and second-position Leu of wild-type rat MLSrAANAT3f. This alteration aimed to address the relevance of Nt-acetylation to the degradation of wild-type rat MLSrAANAT3f. Specifically, the N-terminal sequence Met-Leu-Ser is a priori likely to be cotranslationally Nt-acetylated in vivo at its (retained) N-terminal Met (59, 62, 63). In contrast, the N-terminal Met residue of the mutant N-terminal sequence (Met)-Pro-Leu-Ser of (M)PLSrAANAT3f would be cotranslationally cleaved off by MetAPs. The resulting N-terminal Pro is not Nt-acetylated, at least in S. cerevisiae, and is usually not Nt-acetylated in mammalian cells as well (59, 63).
(v) MLSrAANAT3ha, the wild-type rat MLSrAANAT (Fig. 1C) C-terminally tagged with a triple-HA epitope. It was used as a control for the MLSrAANAT3haK8R mutant described in item viii.
(vi) (M)SThAANAT3f, the wild-type human (M)SThAANAT (Fig. 1C) C-terminally tagged with a triple-FLAG epitope.
(vii) (M)PThAANAT3f, in which the second-position Ser of the wild-type human (M)SThAANAT3f (Fig. 1C) was replaced by the Pro residue, yielding (M)PThAANAT3f. The N-terminal Met residue of either the wild-type human (M)SThAANAT3f or the mutant (M)PThAANAT3f would be cotranslationally removed by MetAPs (41, 74). However, in contrast to the resulting N-terminal Ser of wild-type SThAANAT3f, which would be expected to be cotranslationally Nt-acetylated, the N-terminal Pro of the mutant PThAANAT3f is not Nt-acetylated, at least in S. cerevisiae, and is usually not Nt-acetylated in mammalian cells as well (59, 63).
(viii) MLSrAANAT3haK8R, the otherwise wild-type triple-HA-tagged rat MLSrAANAT in which the Lys-8 residue was replaced by Arg.
(ix) MLSrAANAT3haKzero, the otherwise wild-type triple-HA-tagged rat MLSrAANAT in which each of its four Lys residues were replaced by Arg (Fig. 6B).
FIGURE 6.

Degradation and ubiquitylation assays with rat and human AANATs in human HEK293T cells. A, lane 1, HEK293T cells were transformed with vector (V) alone (control) bearing the (weakened) PCMVt1 promoter (see “Results” and Fig. 5, A–C). Immunoblotting with antibody to tubulin was used as a loading control. CHX-chases were performed at 37 °C for the indicated times in HEK293T cells with the wild-type rat MLSrAANAT3f (lanes 2–5) and with its mutants (M)SMLSrAANAT3f (lanes 7–10), (M)SIrAANAT3f (lanes 12–15), and (M)PLSrAANAT3f (lanes 17–20). Lanes 6, 11, 16, and 21, same as lanes 5, 10, 15, and 20 except that the MG132 proteasome inhibitor was present during a 4-h CHX-chase in each case (see “Experimental Procedures”). The bands of AANAT3f and tubulin are indicated on the right. An asterisk on the left denotes a band of protein that cross-reacted with anti-FLAG antibody (the band is also present in lane 1, the vector-only control). B, left, lane 1, same as the control in lane 1 of A. CHX-chases were performed at 37 °C for the indicated times in HEK293T cells with the wild-type rat MLSrAANAT3ha (lanes 2–5) and with its lysine-lacking mutant MLSrAANAT3haKzero (lanes 7–10). Lanes 6 and 11, same as lanes 5 and 10 except that MG132 was present during a 4-h CHX-chase in each case. Right, same as in the left panel, but both the wild-type MLSrAANAT3f and its lysine-lacking counterpart were C-terminally tagged with a triple-FLAG tag, which contains a Lys residue, in contrast to the HA tag, which lacks lysines (see “Results” and “Discussion”). C, quantification of data in A. D, quantification of data in B. Symbols shown separately and framed in black squares or rectangles correspond to altered levels of test proteins in the presence of MG132. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown. E, immunoblot analyses (IB), using anti-Ub antibody, of total, input (before immunoprecipitation) extracts from HEK293T cells that had been transfected either with vector alone (lanes 1 and 2) or with plasmid expressing the wild-type rat MLSrAANAT3ha (lanes 3 and 4) or with plasmid expressing its lysine-lacking counterpart MLSrAANAT3haKzero (lanes 5 and 6). The MG132 proteasome inhibitor was either omitted (lanes 1, 3, and 5) or added to HEK293T cells (to a final concentration of 10 μm) 6 h before preparation of extracts (lanes 2, 4, and 6). The bottom panel shows the results of detecting non-ubiquitylated MLSrAANAT3ha (absent in the control lanes 1 and 2) with anti-HA antibody. F, same as in E but immunoblot analyses (using anti-Ub antibody) of the input samples after their immunoprecipitation (IP) with anti-HA antibody (followed by SDS-PAGE) to isolate and detect ubiquitylated AANAT3ha species.
(x) (M)SThAANAT3haKzero, the otherwise wild-type triple-HA-tagged human (M)SThAANAT in which both of its Lys residues were replaced by Arg (Fig. 7, A and D).
FIGURE 7.
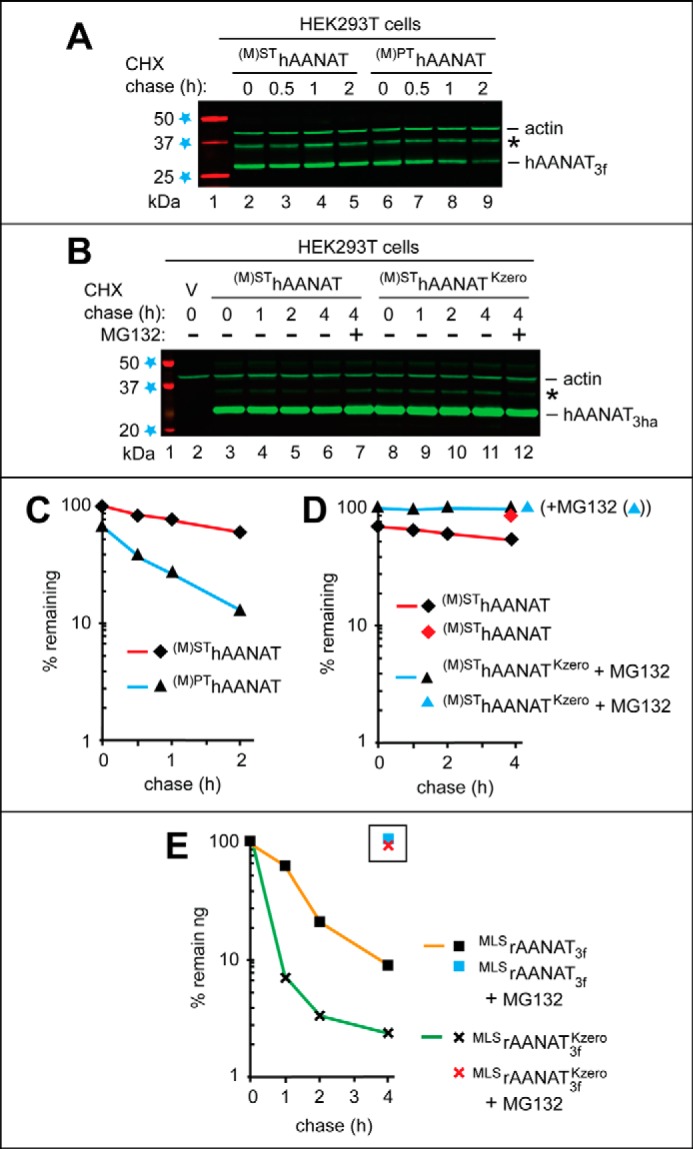
Degradation of the wild-type human (M)SThAANAT and its mutants in human HEK293T cells. The test proteins were expressed in HEK293T cells using a plasmid bearing the (weakened) PCMVt1 promoter (see “Results” and see Fig. 5, A–C). A, lane 1, fluorescently labeled molecular mass markers (LI-COR), with their masses, in kDa, indicated on the left. CHX-chases were performed at 37 °C for the indicated times in HEK293T cells with the wild-type human (M)SThAANAT3f (lanes 2–5) and with its mutant (M)PThAANAT3f (lanes 6–9). B, lane 1, fluorescently labeled molecular mass markers, with their masses, in kDa, indicated on the left. Lane 2, HEK293T cells were transformed with vector (V) alone (control). CHX-chases were performed at 37 °C for the indicated times in HEK293T cells with the wild-type human (M)SThAANAT3ha (lanes 3–6) and with its lysine-lacking mutant (M)SThAANAT3haKzero (lanes 8–11). Lanes 7 and 12, same as lanes 6 and 11, except that the MG132 proteasome inhibitor was present during a 4-h CHX-chase in each case (see “Experimental Procedures”). The bands of actin and AANAT are indicated on the right. Asterisks on the right in A and B denote a protein band (possibly the same protein and possibly a derivative of AANAT) that reacted with both anti-FLAG antibody in A and anti-HA antibody in B. C, quantification of data in A. D, quantification of data in B. E, quantification of data in Fig. 6B (right), similar to the analogous quantifications described in the legend to Fig. 6, C and D. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown.
Degradation of Rat AANAT by the S. cerevisiae N-end Rule Pathway
In cycloheximide (CHX) assays, a protein of interest is analyzed by immunoblotting as a function of time after the inhibition of translation by CHX (39, 59, 60). Both the wild-type rat MLSrAANAT3f (Fig. 1C) and its (M)SMLSrAANAT3f mutant (see item ii above) were rapidly degraded in wild-type S. cerevisiae, with half-lives (t½) < 5 min (Fig. 2, B (lanes 3–10) and D). In striking contrast, (M)SIrAANAT3f (see item iii above), produced from wild-type MLSrAANAT3f by deleting the Leu residue at position 2, was virtually completely stable under the same conditions, with a t½ > 1 h (Fig. 2, B (lanes 11–14) and D).
Because the N-terminal Met residue in the Met-Leu-Ser sequence of the wild-type rat MLSrAANAT3f would be likely to be Nt-acetylated in vivo (59, 62, 63), the rapid degradation of MLSrAANAT3f (t½ < 5 min) (Fig. 2, B and D) suggested the involvement of the Ac/N-end rule pathway (Fig. 1A). To address this possibility, we constructed the non-Nt-acetylatable (M)PLSrAANAT3f mutant (see item iv above). (This mutant would also not be expected to be targetable by the Arg/N-end rule pathway, because the latter does not recognize N-terminal Pro (40, 41).) Remarkably, (M)PLSrAANAT3f was long-lived in S. cerevisiae, in comparison with the short-lived wild-type MLSrAANAT3f (t½ ≈ 1 h versus t½ < 5 min, respectively), strongly suggesting (but not proving) a role for the Ac/N-end rule pathway in the degradation of the wild-type rat MLSrAANAT3f (Fig. 2, B (lanes 15–18) and D; also see below).
To further address the involvement of the Ac/N-end rule and Arg/N-end rule pathways (Fig. 1, A and B) in the degradation of wild-type MLSrAANAT3f, we performed CHX-chases in S. cerevisiae mutants that lacked essential components of either one or both of these pathways. The normally short-lived rat MLSrAANAT3f (t½ < 5 min; Fig. 2, C (lanes 2–5) and E) was partially stabilized in both naa30Δ and ubr1Δ mutants (Fig. 2, C (lanes 6–9 and 14–17) and E). S. cerevisiae naa30Δ cells lack the cognate NatC Nt-acetylase whose substrates include proteins bearing the N-terminal Met-Leu sequence (59, 62, 63, 75, 76). S. cerevisiae ubr1Δ cells lack Ubr1, the E3 Ub ligase (N-recognin) that is essential for the proteolytic activity of the Arg/N-end rule pathway (Fig. 1A) (40, 41, 59).
Crucially, the relative stabilization of MLSrAANAT3f was dramatically higher in the double mutant naa30Δ ubr1Δ than in either one of the single mutants (Fig. 2, C (lanes 18–21) and E). These findings should be considered together with the conceptually independent result that the non-Nt-acetylatable (and also not targetable by the Arg/N-end rule pathway) (M)PLSrAANAT3f mutant (see the preceding paragraph) was much longer-lived in wild-type S. cerevisiae than its wild-type MLSrAANAT3f counterpart (Fig. 2, B (lanes 15–18 versus lanes 3–6) and D). Together, these sets of findings indicated that the wild-type rat MLSrAANAT3f (its two alternative forms) was targeted for degradation by both the Ac/N-end rule pathway and the Arg/N-end rule pathway. Specifically, the Nt-acetylated Ac-MLSrAANAT3f is destroyed through the recognition of its Nt-acetylated N-terminal Met residue by the Ac/N-end rule pathway (Fig. 1B), whereas the non-Nt-acetylated MLSrAANAT3f is targeted by the “complementary” Arg/N-end rule pathway, which recognizes the unacetylated N-terminal Met-Leu sequence of MLSrAANAT3f as the Met-Φ motif (N-terminal Met followed by a bulky hydrophobic residue) (Fig. 1A).
This conclusion was in agreement with the finding that the rapid degradation of MLSrAANAT3f in wild-type S. cerevisiae (t½ < 5 min) remained unchanged in naa10Δ cells, which lacked the non-cognate NatA Nt-acetylase (which does not Nt-acetylate N-terminal Met) (59, 76). Furthermore and also in agreement with the above conclusion, the degradation of MLSrAANAT3f in double-mutant naa10Δ ubr1Δ cells proceeded at a rate similar to that in the single ubr1Δ mutant(Fig. 2, C (lanes 22–25) and E). Thus, a double-mutant background that nearly completely stabilizes the wild-type rat MLSrAANAT3f must be naa30Δ ubr1Δ in that it should also lack the activity of the cognate NatC Nt-acetylase (Fig. 2, C (lanes 18–21) and E). In agreement with this conclusion, the ablation of NAA10, encoding the non-cognate NatA Nt-acetylase, did not have a synergistic effect on the rate of MLSrAANAT3f degradation in double-mutant naa10Δ ubr1Δ cells (Fig. 2, C (lanes 22–25) and E).
Previous work has identified the endoplasmic reticulum membrane-embedded E3 Ub ligase Doa10 as one Ac/N-recognin of the S. cerevisiae Ac/N-end rule pathway (39, 59). We found that the degradation of the wild-type rat MLSrAANAT3f (t½ < 5 min in wild-type S. cerevisiae) was not significantly impaired in doa10Δ cells, in comparison with wild-type cells (Fig. 3, D and F). We did not examine, so far, the other yeast Ac/N-recognin, Not4 (see the Introduction) for its possible role in targeting the Nt-acetylated rat Ac-MLSrAANAT3f in S. cerevisiae. We also do not know, thus far, whether Teb4, the mammalian counterpart of the yeast Doa10 Ac/N-recognin (60), is involved in the degradation of rat MLSrAANAT3f in a homologous (mammalian) setting.
FIGURE 3.

Degradation assays with the wild-type human (M)SThAANAT in S. cerevisiae. A, comparison of degradation rates of the wild-type rat MLSrAANAT3f and the wild-type human (M)SThAANAT3f in S. cerevisiae. Immunoblotting with antibody to actin was used as a loading control. Lane 1, wild-type S. cerevisiae were transformed with vector (V) alone (control). CHX-chases were performed at 30 °C for the indicated times in wild-type S. cerevisiae with the wild-type rat MLSrAANAT3f (lanes 2–5) and with the wild-type human (M)SThAANAT3f (lanes 6–9). The bands of AANAT and actin are indicated on the left. B, quantification of data in A. C, CHX-chases with the wild-type human (M)SThAANAT3f in wild-type S. cerevisiae (lanes 1–4) and its mutants naa10Δ (lanes 5–8), ubr1Δ (lanes 9–12), and naa10Δ ubr1Δ (lanes 13–16). The bands of human (M)SThAANAT3f and actin are indicated on the left. D, lane 1, wild-type S. cerevisiae were transformed with vector (V) alone (control). Shown are CHX-chases with the wild-type human (M)SThAANAT3f in wild-type S. cerevisiae (lanes 2–5) and its mutants naa10Δ (lanes 6–9), doa10Δ (lanes 10–13), and ubr1Δ (lanes 14–17). The bands of human (M)SThAANAT3f and actin are indicated on the left. E, quantification of data in C. F, quantification of data in D. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown.
Degradation of Rat AANAT Is Proteasome-dependent and Does Not Require Lys-8
The demonstrated degradation of two forms of the wild-type rat MLSrAANAT3f (Ac-MLSrAANAT3f and its non-Nt-acetylated counterpart) by the two branches of the N-end rule pathway (Fig. 2) already implied the proteasome dependence of this degradation, given the known organization of the N-end rule pathway (Fig. 1, A and B). To verify this in S. cerevisiae by independent means, CHX-chases with rat MLSrAANAT3f were carried out in wild-type versus pdr5Δ cells in either the presence or absence of the MG132 proteasome inhibitor. (Cells lacking the transmembrane transporter PDR5 are more sensitive to MG132 (77, 78).)
As expected, given the proteasome dependence of the N-end rule pathway (Fig. 1, A and B), the normally rapid degradation of the wild-type rat MLSrAANAT3f was substantially inhibited in wild-type S. cerevisiae in the presence of 50 μm MG132 (Fig. 4, A and B). This degradation was inhibited even more strongly in mutant pdr5Δ cells under the same conditions, confirming the proteasome dependence of at least the bulk of MLSrAANAT3f degradation (Fig. 4, A and B).
FIGURE 4.

Degradation assays with a proteasome inhibitor and AANAT mutants in S. cerevisiae. A, lane 1, wild-type S. cerevisiae were transformed with vector (V) alone (control). Lanes 2–5, CHX-chase was performed at 30 °C for the indicated times in wild-type S. cerevisiae with the wild-type rat MLSrAANAT3ha. Lanes 6–9, same as in lanes 2–5, but the CHX-chase was in the presence of the MG132 proteasome inhibitor (see “Experimental Procedures”). Lanes 10–13, same as in lanes 2–5 but in pdr5Δ S. cerevisiae lacking the efflux pump Pdr5. Lanes 14–17, same as in lanes 10–13 but in the presence of MG132. The bands of actin and MLSrAANAT3ha are indicated on the left. B, quantification of data in A. C, CHX-chases were performed in wild-type S. cerevisiae with the wild-type rat MLSrAANAT3ha (lanes 2–5) and with its Lys-8 → Arg mutant MLSrAANAT3haK8R (see “Results”). The bands of tubulin and AANAT are indicated on the left. An asterisk on the right denotes a band of protein that cross-reacted with anti-HA antibody (the band is also present in lane 1, the vector-only control). D, lane 1, wild-type S. cerevisiae were transformed with vector (V) alone (control). Lanes 2–5, CHX-chase was performed at 30 °C for the indicated times in wild-type S. cerevisiae with the wild-type human (M)SThAANAT3f. Lanes 6–9, same as in lanes 2–5 but with the mutant human (M)PThAANAT3f. The bands of actin and AANAT are indicated on the left. E, quantification of data in A. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown.
N-terminal regions of mammalian AANATs contain a highly conserved Lys residue (e.g. Lys-8 in rat AANAT and Lys-10 in human AANAT) that has been suggested as a potential ubiquitylation site and thus a determinant of the AANAT degron (33, 79–81). To address this possibility, CHX-chases were performed in S. cerevisiae with wild-type MLSrAANAT3ha and a mutant containing a Lys-8 → Arg mutation (MLSrAANAT3haK8R). Wild-type MLSrAANAT3ha and the mutant MLSrAANAT3haK8R were degraded at similar rates, indicating that the conserved lysine at this position (Lys-8 in rat MLSrAANAT) is not, by itself, a significant determinant of MLSrAANAT stability (Fig. 4C).
Degradation of Human AANAT in S. cerevisiae
Fig. 3 (A (lanes 6–9) and B) shows the results of a CHX-chase, in wild-type yeast, with the 207-position residue (not counting the tag) (M)SThAANAT3f, the wild-type human AANAT C-terminally tagged with a triple-FLAG epitope. Parentheses around the superscript's N-terminal Met residue denote the fact that this Met is cotranslationally cleaved off by MetAPs. Whereas human (M)SThAANAT3f was relatively unstable in S. cerevisiae (t½ ≈ 30 min), it was much longer-lived (including its higher zero-time, prechase level) than the wild-type rat MLSrAANAT3f under the same conditions (Fig. 3, A and B).
As was also done with the wild-type rat MLSrAANAT3f (Fig. 2C), we characterized the degradation of wild-type human (M)SThAANAT3f in S. cerevisiae strains deficient in specific components of the N-end rule pathway. The N-terminal Ser-Thr sequence of wild-type human (M)SThAANAT3f (after the cotranslational removal of the initially present N-terminal Met) made it likely that (M)SThAANAT3f was cotranslationally Nt-acetylated in vivo (59, 63). This modification of human (M)SThAANAT3f would make it a potential target for degradation by the Ac/N-end rule pathway (Fig. 1B). In contrast, the non-Nt-acetylated human (M)SThAANAT3f would not be expected to be recognized by either the Ac/N-end rule or the Arg/N-end rule pathway (Fig. 1A), in the latter case due to the absence of both a destabilizing N-terminal residue and a bulky hydrophobic (Φ) second residue. (A Met-Φ N-terminal motif, containing a second-position Φ residue, can act as an N-degron recognized by the Arg/N-end rule pathway (60, 82).
Indeed, the rate of degradation of human (M)SThAANAT3f in ubr1Δ S. cerevisiae, which lacked the Ubr1 N-recognin and therefore lacked the Arg/N-end rule pathway (Fig. 1A), was similar to the rate of (M)SThAANAT3f degradation in wild-type cells (Fig. 3, C (lanes 1–4 versus lanes 9–12), D (lanes 2–5 versus lanes 14–17), E, and F). The rate of (M)SThAANAT3f degradation that was observed with wild-type S. cerevisiae was also not significantly changed in doa10Δ cells, which lacked one of two known Ac/N-recognins of the Ac/N-end rule pathway (Figs. 1B and 3, D (lanes 10–13 versus lanes 2–5) and F). Surprisingly, however, (M)SThAANAT3f was strongly destabilized in naa10Δ cells, which lacked the cognate Nt-acetylase for the N-terminal Ser residue (59, 62); similar results were obtained with double-mutant naa10Δ ubr1Δ cells (Fig. 3, C–F).
Given the much faster degradation of (M)SThAANAT3f in the absence of Naa10 (e.g. Fig. 3, C (lanes 1–4 versus lanes 5–8) and E) (i.e. in cells that would be incapable of Nt-acetylating (M)SThAANAT3f), we also carried out CHX-chases, in wild-type S. cerevisiae, with (M)PThAANAT3f. In the latter mutant, the N-terminal Ser was replaced by the non-Nt-acetylatable Pro residue. In contrast to wild-type (M)SThAANAT3f in naa10Δ cells, in which the (non-Nt-acetylated) (M)SThAANAT became strikingly short-lived, the (non-Nt-acetylatable) (M)PThAANAT mutant was found to be longer-lived, in wild-type cells, than the Nt-acetylatable wild-type (M)SThAANAT3f (Fig. 4, D and E). These results indicated that the observed accelerated degradation of the wild-type human (M)SThAANAT in naa10Δ S. cerevisiae was not caused by the failure to Nt-acetylate (M)SThAANAT in the absence of the NatA Nt-acetylase.
We do not understand the mechanistic cause of the (reproducibly observed) much faster degradation of human (M)SThAANAT in naa10Δ S. cerevisiae (Fig. 3, C–F). One possibility, which remains to be examined, is the apparent inhibition of the yeast Hsp90 chaperone system (centered on the S. cerevisiae Hsc82/Hsp82 proteins) in naa10Δ cells.3 If human (M)SThAANAT expressed in wild-type S. cerevisiae is a protected (from degradation) client of the Hsc82/Hsp82 system, a failure of this protection in naa10Δ mutant cells would make (M)SThAANAT vulnerable to a currently unknown pathway of the Ub system that is distinct from the Arg/N-end rule pathway. (A proteolytic pathway in question would be distinct from the Arg/N-end rule pathway because (M)SThAANAT was equally short-lived in single-mutant naa10Δ and double-mutant naa10Δ ubr1Δ cells (Fig. 3, C–F).) In sum, the above Hsp90-based mechanism (which remains to be addressed in the context of the (M)SThAANAT protein) might underlie the strongly accelerated degradation of (M)SThAANAT in naa10Δ S. cerevisiae.
Degradation of Rat AANAT in Human HEK293T Cells
To compare the relative rates of degradation of N-terminal mutants of rat AANAT in human HEK293T cells, the wild-type rat MLSrAANAT3f and its mutants (M)SMLSrAANAT3f, (M)SIrAANAT3f, and (M)PLSrAANAT3f (see items i–iv at the beginning of “Results”) were expressed in these cells using transient transfection. Preliminary experiments with wild-type MLSrAANAT3f encoded by a pcDNA3-based plasmid and expressed from the full-strength PCMV promoter indicated that the levels of expression of MLSrAANAT3f were high enough to saturate or nearly saturate pathways that targeted MLSrAANAT3f for degradation in HEK293T cells. Specifically, at those (high) levels of expression, wild-type rat MLSrAANAT3f either was stable or was degraded slowly (data not shown).
We addressed this problem by constructing a set of truncated derivatives of the original PCMV promoter, aiming to attenuate its activity. The resulting nested set of truncated promoters, denoted as PCMVt1, PCMVt2, and PCMVt3, comprised progressively shortened versions of the original PCMV. These truncations (Fig. 5A) were similar (although not identical) to a series of PCMV truncations described earlier by Promega Inc. (83). Each of the PCMVt1, PCMVt2, and PCMVt3 promoter variants was cloned into pcDNA3, replacing full-length PCMV.
FIGURE 5.
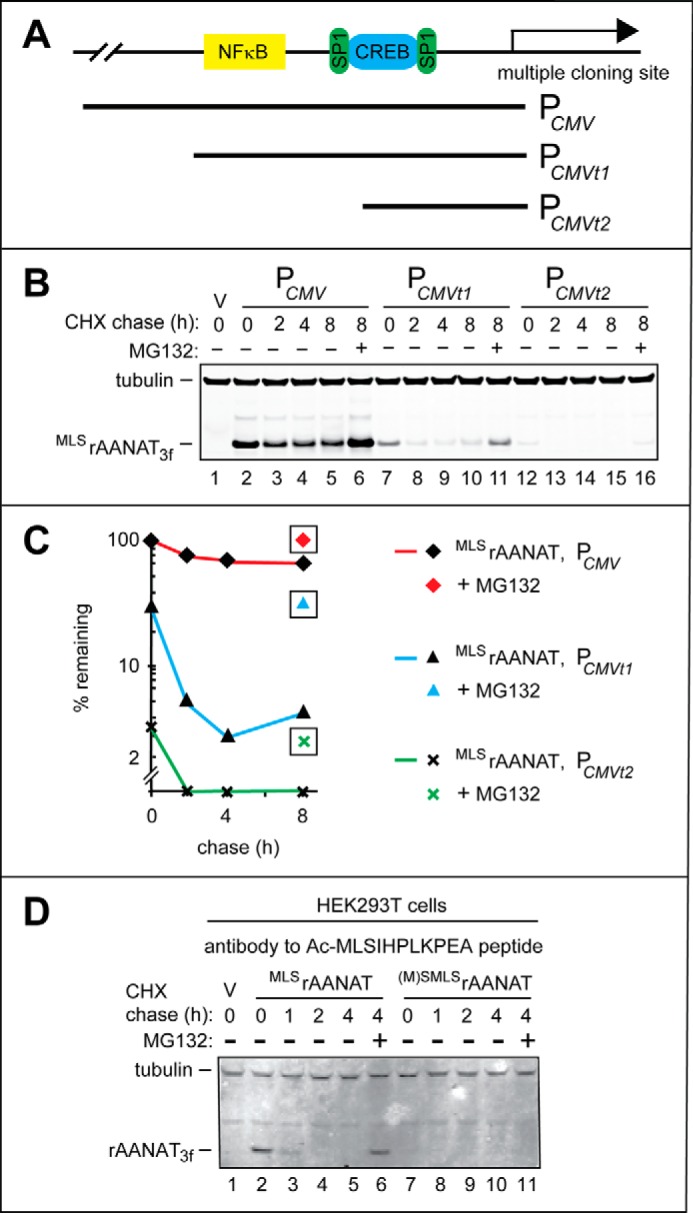
Weakened derivatives of the PCMV promoter in HEK293T cells and degradation assays using antibody to Nt-acetylated rat MLSrAANAT. A, a simplified diagram of the “wild-type” (unmodified) PCMV promoter in the cloning vector, with schematically illustrated binding sites for the NFκB, SP1, and CREB transcriptional regulators. Two 5′-terminal truncations of wild-type PCMV, denoted as PCMVt1 and PCMVt2, are also indicated. B, lane 1, HEK293T cells were transformed with vector (V) alone (control). CHX-chases were performed at 37 °C for the indicated times in HEK293T cells with the wild-type rat MLSrAANAT3f expressed either from the unmodified PCMV promoter (lanes 2–5), from the PCMVt1 promoter (lanes 7–10), or from the PCMVt2 promoter (lanes 12–15). Lanes 6, 10, and 16, same as lanes 5, 9, and 15 except that the MG132 proteasome inhibitor was present during 8-h CHX-chases in each case (see “Experimental Procedures” and “Results”). The bands of tubulin and MLSrAANAT3f are indicated on the left. C, quantification of data in B. Symbols shown separately and framed in black squares correspond to altered (increased) levels of test proteins in the presence of MG132. All quantified CHX-chase assays were carried out at least three times and yielded results within 10% of the data shown. D, immunoblotting with affinity-purified antibody to Nt-acetylated MLSrAANAT (see “Experimental Procedures”). Lane 1, HEK293T cells were transformed with vector (V) alone (control) bearing the (weakened) PCMVt1 promoter. CHX-chases were performed at 30 °C for the indicated times in HEK293T cells with the wild-type rat MLSrAANAT3f (lanes 2–5) and its mutant (M)SMLSrAANAT3f (lanes 7–10). Lanes 6 and 11, same as lanes 5 and 10 except that the MG132 proteasome inhibitor was present during 4-h CHX-chases in each case (see “Experimental Procedures”). The bands of rAANAT and tubulin (the latter a loading control) are indicated on the left.
The wild-type rat MLSrAANAT3f was cloned into each of the above pcDNA3-based plasmids, and CHX-chases were performed in HEK293T cells transiently transfected with these constructs. Expression of MLSrAANAT3f from the PCMVt1 and PCMVt2 promoters reduced time-zero levels of MLSrAANAT3f (measured by immunoblotting at the beginning of CHX-chase) by ∼70% and by ∼97%, respectively, in comparison with the unmodified PCMV promoter (Fig. 5, A–C). With the most truncated promoter, PCMVt3, no MLSrAANAT3f could be detected at the same level of immunoblotting sensitivity (data not shown). The lower levels of MLSrAANAT3f expression that have been attained through the use of the PCMVt1 and PCMVt2 promoters resulted in a strongly increased rate of the post-translational degradation of MLSrAANAT3f (t½ < 2 h versus t½ > 8 h when MLSrAANAT3f was expressed from the full-length PCMV promoter; Fig. 5, A–C). Treatment of HEK293T cells with the proteasome inhibitor MG132 during the chase increased the levels of MLSrAANAT3f expressed from these promoters, thereby confirming that in each case, the bulk of degradation was proteasome-dependent (Fig. 5, A–C). The pcDNA3CMVt1 plasmid, which contained PCMVt1, one weakened version of the PCMV promoter, was chosen for experiments with HEK293T cells in this study.
In agreement with the relative metabolic stabilities of AANAT proteins expressed in S. cerevisiae (Fig. 1, A and B), the mutant rat (M)SIrAANAT3f, bearing a deletion of the Leu residue at position 2 of wild-type MLSrAANAT3f (Fig. 2A), was the longest-lived protein in the set of rat AANAT mutants examined in HEK293T cells (t½ > 4 h; Fig. 6, A (lanes 12–16) and C). The wild-type rat MLSrAANAT3f and its (M)SMLSrAANAT3f mutant (see items i and ii at the beginning of “Results”) were significantly shorter-lived in HEK293T cells than (M)SIrAANAT3f, with t½ ≈ 1 h and t½ < 1 h, respectively, versus t½ > 4 h (Fig. 6, A (lanes 2–11) and C).
We expected, a priori, that (M)SMLSrAANAT3f would initiate from the first encoded Met residue, particularly because of the inclusion of the Kozak “optimal” upstream sequence in the relevant plasmids used for expression of test AANAT proteins. We could verify that expectation through chase-degradation assays using an affinity-purified antibody against the Nt-acetylated N terminus of MLSrAANAT3f (Fig. 5D). We produced and purified this antibody as described under “Experimental Procedures.” As shown in Fig. 5D, immunoblotting of SDS-PAGE-fractionated samples containing MLSrAANAT3f or (M)SMLSrAANAT3f with the anti-Ac- MLSrAANAT antibody generated a positive signal only in samples expressing MLSrAANAT3f. If a construct expressing (M)SMLSrAANAT3f had initiated translation (to a significant extent) from the second Met residue, the resulting translated protein would have been MLSrAANAT3f. However, no signal was detected (using the above anti-Ac-MLSrAANAT antibody) with samples containing (M)SMLSrAANAT3f (Fig. 5D), indicating negligible levels of translation initiation at the position 2 Met.
Although the mutant (M)PLSrAANAT3f was partially stabilized relative to wild-type MLSrAANAT3f, the former protein was still significantly shorter-lived (t½ ≈ 2 h) than the most stable (M)SIrAANAT3f mutant (Fig. 6, A (lanes 17–21) and C). Although N-terminal Pro is not Nt-acetylated (59, 63) (this makes N-terminal Pro-bearing proteins invulnerable to the Ac/N-end rule pathway), the N-terminal Pro residue might still be (weakly) recognized by the Arg/N-end rule pathway in some sequence contexts. Specifically, our SPOT-type, peptide-based binding assays (84) suggested that the Ubr1 N-recognin of the S. cerevisiae Arg/N-end rule pathway (and, by inference, mammalian Ubr1 and Ubr2 as well) may weakly bind to N-terminal Pro when this residue is followed by a hydrophobic residue.4 Thus, one interpretation of the “residual” instability of the rat (M)PLSrAANAT3f mutant (in comparison with the longest-lived (M)SIrAANAT3f mutant) is that the (non-Nt-acetylated) N-terminal sequence Pro-Leu of (M)PLSrAANAT3f is weakly targeted by the Arg/N-end rule pathway, in contrast to the absence of targeting of the N-terminal Ser-Ile sequence of the (M)SIrAANAT3f mutant (Fig. 6, A and C; also see Fig. 2, B and D).
As mentioned above, we found that transient transfection-based expression of AANAT from the full-strength PCMV promoter could be high enough to result in saturation or near saturation of the relevant proteolytic pathways. Although the use of the considerably weaker PCMVt1 promoter at least partially solved this problem (see above), additional studies would be required to make sure that pathway saturation effects were consistently negligible with longer-lived AANAT derivatives that were expressed from PCMVt1. That said, the preponderance of our data makes it nearly certain that the observed differences in time-zero (before chase) levels of AANAT proteins were caused largely by differences in the rates of their degradation.
The Bulk of Degradation of Rat AANAT Is Mediated by Polyubiquitylation of Its Lys Residue(s)
An earlier study, in which the wild-type rat MLSrAANAT was apparently not stabilized by conversion of its Lys residues to Arg, was based on measurements of steady-state levels of MLSrAANAT in HEK293 cells that had been treated or left untreated with the proteasome inhibitor MG132 (37). To address this issue more directly, we generated a triple-HA-tagged mutant, denoted MLSrAANAT3haKzero, of the wild-type rat MLSrAANAT in which all four of its Lys residues were converted to Arg. (A FLAG-based epitope tag could not be employed in these experiments, because FLAG contains a Lys residue that could potentially serve as a target for polyubiquitylation.)
CHX-chases in HEK293T cells (performed as described above) with the wild-type MLSrAANAT3ha versus its lysine-lacking MLSrAANAT3haKzero mutant indicated that MLSrAANAT3haKzero was nearly completely stable during the chase (t½ ≫ 4 h), whereas the wild-type MLSrAANAT3ha was degraded, with t½ ≈ 2 h (Fig. 6, B and D). A treatment with MG132 during CHX-chase strongly stabilized the wild-type MLSrAANAT3ha, confirming that its degradation is proteasome-dependent (Fig. 6, B (left, lanes 2 and 6) and D). In contrast, the MG132 treatment during CHX-chase of the lysine-lacking MLSrAANAT3haKzero mutant produced at most a marginal stabilizing effect, suggesting that even if there is a small (residual) amount of lysine-independent degradation of MLSrAANAT3haKzero, such a degradation would still require the proteasome (Fig. 6, B (left, lanes 7 and 11) and D).
In a different approach to the same problem, HEK293T cells were transiently transfected with the pcDNA3 vector or with plasmids expressing either MLSrAANAT3ha or MLSrAANAT3haKzero. Cells were then treated for 6 h with either MG132 or an equivalent volume of DMSO (in which the stock solution of MG132 was made). Thereafter, cell extracts were immunoprecipitated with anti-HA magnetic beads, and the immunoprecipitates were fractionated by SDS-PAGE, followed by immunoblotting with anti-HA and anti-ubiquitin antibodies. Polyubiquitin chains (apparently linked to MLSrAANAT3ha) could be readily detected in fractionated immunoprecipitates of extracts from cells that expressed MLSrAANAT3ha (but, crucially, not from cells that expressed an empty vector), and the levels of polyubiquitin chains were significantly increased in the presence of MG132 (Fig. 6F, lanes 1–4). In contrast, significantly lower amounts of polyubiquitin chains were observed in fractionated immunoprecipitates of extracts from cells that expressed the lysine-lacking MLSrAANAT3haKzero (Fig. 6F, lane 5). Moreover, the presence of MG132 did not increase the low levels of detected polyubiquitin chains (Fig. 6F, lane 5 versus lane 6). The latter result suggested that (residual) polyubiquitin chains observed in cells that expressed MLSrAANAT3haKzero may be of a kind (e.g. see Ref. 85) that do not contribute to the proteasome-dependent degradation of MLSrAANAT3haKzero. In agreement with this interpretation, the lysine-lacking MLSrAANAT3haKzero mutant was expressed at significantly higher steady-state levels than the wild-type rat MLSrAANAT3ha in the absence of MG132 (in DMSO-treated cells) (Fig. 6E, bottom), suggesting that the absence of Lys residues stabilized MLSrAANAT3haKzero against degradation throughout life histories of MLSrAANAT3haKzero molecules, including, possibly, their degradation during or immediately after their synthesis.
Degradation of Human AANAT in Human HEK293T Cells
Degradation of the wild-type human (M)SThAANAT3f and its N-terminal Pro residue-bearing mutant (M)PThAANAT3f (see items vi and vii at the beginning of “Results”) were examined by CHX-chases in HEK293T cells. In agreement with the findings in S. cerevisiae (Figs. 2 and 3), the wild-type human (M)SThAANAT was longer-lived than the wild-type rat MLSrAANAT3f in HEK293T cells (compare Fig. 7A with Fig. 6A). Interestingly, the mutant human (M)PThAANAT3f was less stable than the wild-type human MLSrAANAT3f during the course of a 2-h chase in HEK293T cells (Fig. 7, A and C).
We also asked whether the (relatively slow) degradation of the wild-type human (M)SThAANAT3f (Fig. 7, A and C) required both its lysine-dependent ubiquitylation and the proteasome. CHX-chases were carried out with (M)SThAANAT3ha and its lysine-lacking mutant (M)SThAANAT3haKzero. The latter protein was stable during the chase (t½ ≫ 4 h), whereas wild-type (M)SThAANAT3ha exhibited a weak but observable instability (Fig. 7, B and D). Treatment with the MG132 proteasome inhibitor increased the level of the wild-type human (M)SThAANAT3ha during CHX-chase, confirming proteasome dependence of its (slow) degradation (Fig. 7, B (lanes 3 and 7) and D).
Discussion
The ∼23-kDa AANAT (also called serotonin N-acetyltransferase) is apparently universal among animals and is present in plants as well. AANAT converts the neurotransmitter serotonin to NAS, a regulatory compound in its own right and the immediate precursor of melatonin, a circulating hormone that regulates sleep and other circadian processes in vertebrates. The levels of melatonin and NAS are modulated by oscillatory circadian circuits and also impact those circuits, in addition to other effects of these compounds (see the Introduction). Hence the importance of temporal control of the AANAT enzyme. The expression of AANAT is regulated through both transcriptional and post-translational mechanisms. Details of these mechanisms differ among vertebrates. In addition, although AANAT enzymes from different species are highly sequelogous (71) throughout their ORFs, the sequences of the first ∼10 residues of AANAT tend to differ even among relatively closely related mammals, let alone other animals (Fig. 1C). This pattern of variation suggests a blend of adaptive (function-linked, natural selection-based) changes in N-terminal sequences of AANATs versus an unknown (extent-wise) but apparently strong flux of quasi-neutral, drift-mediated changes of these N-terminal sequences on evolutionary time scales.
In the present study, we approached the proteolysis-based regulation of rat and human AANATs (Fig. 1C) by expressing them and their mutants not only in mammalian cells but also in the yeast S. cerevisiae. This strategy made possible the use of yeast genetics to illuminate specific proteolytic pathways involved, in parallel with experiments in mammalian cells.
We found that the wild-type rat AANAT (MLSrAANAT) is targeted for degradation by two complementary branches of the N-end rule pathway. Specifically, we showed that the Nt-acetylated Ac-MLSrAANAT is destroyed through the recognition of its Nt-acetylated N-terminal Met residue by the Ac/N-end rule pathway, whereas the non-Nt-acetylated MLSrAANAT is targeted for degradation by the Arg/N-end rule pathway, which recognizes the unacetylated N-terminal Met-Leu sequence of MLSrAANAT.
The N-terminal sequence of the wild-type human (M)SThAANAT ((Met)-Ser-Thr) is different from the Met-Leu-Ser sequence of rat MLSrAANAT (Fig. 1C). We found that the wild-type human (M)SThAANAT is considerably longer-lived than its rat MLSrAANAT counterpart and does not appear to be an efficacious N-end rule substrate. Together, these and related results (Figs. 2–7) indicated both a major involvement of the N-end rule pathway in the control of rodent (specifically rat) AANAT and substantial differences in the regulation of rodent and human AANATs that stem from differences in their N-terminal sequences.
The observed co-targeting of the two “complementary” forms of the rat MLSrAANAT by the two “complementary” branches of the N-end rule pathway (Figs. 2 and 3) is in agreement with the earlier finding that the N-terminal region of MLSrAANAT, and specifically its Leu-2 residue, are an important determinant of the observed metabolic instability of this AANAT (37). The cited study also suggested that either the Lys residues of MLSrAANAT or its surface-exposed Cys residues were not essential for its proteasome-mediated degradation (37). In the present work, we compared the degradation of wild-type MLSrAANAT and its lysine-lacking MLSrAANAT3haKzero mutant using CHX-chases. Our results showed that the C-terminally triple-HA-tagged MLSrAANAT3haKzero mutant was completely stabilized in human HEK293T cells, in contrast to instability of wild-type MLSrAANAT, indicating that at least some Lys residues of MLSrAANAT were required for its degradation (Fig. 6, B and D).
A possible explanation of the above discrepancy stems from different choices of C-terminal epitope tags in the earlier and the present study. Specifically, Huang et al. (37) employed MLSrAANAT C-terminally tagged with c-Myc epitope (EQKLISEEDL), which contains a Lys residue. In the present work, we used a triple-HA tag, which lacks Lys residues (the sequence of single HA is YPYDVPDYA). Previous analyses of N-degrons have shown that the targeting apparatus of the Arg/N-end rule pathway selects a substrate's internal lysine as a polyubiquitylation site through a process in which different (spatially competing, conformationally mobile) Lys residues can be chosen stochastically, depending on their ability to become transiently close to the substrate-bound E2-E3 complex (41, 51, 86). Thus, the presence of a Lys residue in the C-terminal c-Myc epitope of the earlier study (37) might have rendered the otherwise lysine-lacking MLSrAANAT mutant still targetable by the N-end rule pathway. Indeed, tellingly, our initial experiments with lysine-lacking mutants of MLSrAANAT have employed, inadvertently, a lysine-containing C-terminal FLAG tag instead of the lysine-lacking HA tag. Remarkably, the otherwise lysine-lacking but FLAG-tagged MLSrAANAT3fKzero mutant was found to be unstable (Figs. 6B (right) and 7E), in contrast to the actually (completely) lysine-lacking, HA-tagged MLSrAANAT3haKzero mutant (Fig. 6, B (left) and D), in agreement with the above tag-based explanation.
In addition to polyubiquitylation and degradation, AANAT is also regulated through a site-specific phosphorylation, a modification that stimulates the binding of AANAT to at least one of the 14-3-3 chaperone-like proteins (they are encoded by a family of several 14-3-3 genes in mammals). This interaction with 14-3-3 both protects (phosphorylated) AANAT from degradation and enhances its catalytic activity. Rat, human, and other AANATs contain two previously mapped phosphorylation sites, Thr-29 and Ser-203 (position numbers refer to the sequence of rat MLSrAANAT). Phosphorylation at Thr-29 is conserved in all examined mammalian AANATs (31). This phosphorylation, by PKA and/or by PKC, augments the interaction between AANAT and 14-3-3 both in vitro and in vivo (1, 5, 37). By expressing AANATs not only in mammalian cells but also in the heterologous setting of S. cerevisiae, we could analyze the degradation of AANATs in the possible absence of phosphorylation at Thr-29 (or its equivalent) and also in the possible absence of AANAT interactions with 14-3-3 proteins. Whereas S. cerevisiae has two 14-3-3 proteins (87), there is no evidence, so far, that bears on their binding, in vivo, to a mammalian AANAT.
Our attempts to observe a phosphorylation-induced in vivo binding of the wild-type rat MLSrAANAT either to an endogenous yeast 14-3-3 protein or to the (overexpressed) human 14-3-3ζ protein were unsuccessful so far, despite a variety of tried approaches.5 Nevertheless, the likely conceptual benefit of achieving a phosphorylation-inducible interaction between a mammalian AANAT and a specific 14-3-3 protein, in a setting of greatly reduced complexity of 14-3-3s (in comparison with the large set of their mammalian counterparts), justifies further work in this direction. 14-3-3 proteins are abundant, broadly expressed, multifunctional chaperone-like proteins that modulate catalytic activities of enzymes to which 14-3-3s bind and also regulate protein-protein interactions and subcellular targeting of specific proteins (88–90). It is possible, indeed likely, that 14-3-3 proteins may bind, in a regulated manner, not only to, for example, rat MLSrAANAT but to other N-end rule substrates as well, thereby controlling the rates of degradation of such substrates in ways that remain to be explored.
Recently, the naturally occurring mutant human protein RGS2Q2L was shown to be a conditionally short-lived substrate of both the Ac/N-end rule pathway and the Arg/N-end rule pathway (60). This clinically relevant mutant of RGS2 (a negative regulator of specific G proteins) was identified in a cohort of hypertensive Japanese patients (91) (also see the Introduction). In the present study, we demonstrated that the otherwise unrelated rat MLSrAANAT protein, whose N-terminal Met-Leu sequence is identical to that of RGS2Q2L, is also targeted for degradation by both branches of the N-end rule pathway (Figs. 1 and 2). These results further expanded the set of identified mammalian N-end rule substrates of the Met-Φ type.
Interestingly, RGS2, an N-end rule substrate (60), has been identified as a physiologically relevant inhibitor of the AANAT-dependent melatonin production (92). Light-stimulated, norepinephrine-induced increases in the levels and activity of AANAT are mediated by G-protein-coupled adrenergic receptors, whose activation increases intracellular cAMP, and subsequently the levels of AANAT as well, at least in part through an increased phosphorylation of AANAT and the resulting induction of its binding to protective 14-3-3 proteins (5, 28). While acting to increase the levels and activity of AANAT (which is required for the synthesis of NAS and melatonin), cAMP also acts as a negative feedback inhibitor of melatonin production through up-regulation of Rgs2 transcription. The resulting increase in RGS2 inhibits the activity of G proteins that are coupled to the norepinephrine-responsive receptor, a negative feedback circuit that acts to decrease cAMP levels (92). AANAT results of the present study, together with the earlier evidence that RGS2 is both a conditionally short-lived N-end rule substrate and an (indirect) down-regulator of AANAT (60, 92), indicate that the N-end rule pathway is a pleiotropic controller of circuits that either directly or indirectly modulate AANAT.
Previous studies of AANAT demonstrated that its transcriptional regulation can be different in different species, but it has been conjectured that mechanisms and regulation of AANAT degradation would be more conserved in evolution (11, 34–36). As shown in the present study, the rates of degradation of the human and rat AANAT proteins are quite different. Moreover, whereas rat MLSrAANAT is destroyed by the N-end rule pathway (Figs. 2 and 6), human (M)SThAANAT is substantially resistant to this pathway (Figs. 3 and 7). Nevertheless, if the identification of RGS2 as a feedback inhibitor of AANAT expression in the rat pineal gland (92) could also be shown to extend to human RGS2, the N-end rule pathway would be a regulator of AANAT in humans as well, albeit an indirect one.
An earlier study of human (M)SThAANAT described its apparent degradation by the proteasome but did not detect its ubiquitylation (33). In that study, the instability of human (M)SThAANAT was inferred from increases in its steady-state levels in the presence of a proteasome inhibitor. In the present work, we employed, in particular, CHX-chases to show that although human (M)SThAANAT is degraded by the proteasome, it is substantially longer-lived than rat MLSrAANAT under the same conditions, either in mammalian cells or in S. cerevisiae (Figs. 2–7).
In sum, our results indicate both a major involvement of the N-end rule pathway in the control of rodent AANATs and substantial differences in the regulation of rodent and human AANATs that stem from differences in their N-terminal regions (Fig. 1C). What is the cause of the remarkable variability of N-terminal sequences of AANATs during mammalian (and, more generally, animal) evolution? Given the extent of variability (Fig. 1C), it is nearly certain that a substantial fraction of these changes resulted from a quasi-neutral, unselected genetic drift (93). At the same time, the broad and biologically relevant variation in detailed circadian rhythms among different animal species (due to specific ecological and physiological adaptations) and the role of AANAT in these rhythms suggest that the seeming randomness of the N-terminal sequences of AANATs (Fig. 1C) may be obscuring, so far, a set of adaptive (selected) changes in these sequences over evolutionary time scales.
Experimental Procedures
Yeast Strains, Media, and Genetic Techniques
Standard yeast genetic techniques were used (94–96). S. cerevisiae strain BWY29 was constructed by transforming the strain CHY345 (ubr1Δ::LEU2 in the strain background of BY4742) with a PCR-amplified DNA fragment that encoded the selection marker KanMX6 (making cells resistant to kanamycin) and was targeted to the 5′- and 3′-flanking regions of the NAA10 gene, thereby replacing the ORF of NAA10 with KanMX6 (Table 1). S. cerevisiae strain JOY487 was made by transforming BY4742 with a PCR-amplified DNA fragment that encoded the natNT2 marker (making cells resistant to nourseothricin) and was targeted to the 5′- and 3′-flanking regions of PDR5, thereby replacing the ORF of PDR5 with natNT2 (Table 1). The resulting pdr5Δ cells were used in experiments that involved the MG132 proteasome inhibitor (Fig. 4, A and B). Other S. cerevisiae strains used in this study were constructed previously and are cited in Table 1. S. cerevisiae were transformed using the LiAc/PEG method (97). S. cerevisiae media included YPD medium, synthetic complete medium, and synthetic drop-out medium (95, 96).
TABLE 1.
S. cerevisiae strains used in this study
| Strains | Relevant genotypes | Source or reference |
|---|---|---|
| BY4742 | MATα his3-1 leu2-0 lys2-0 ura3-0 can1-100 | Open Biosystems |
| BY15470 | naa30Δ::kanMX6 in BY4742 | Open Biosystems |
| BY10976 | naa10Δ::KanMX6 in BY4742 | Open Biosystems |
| BY17299 | doa10Δ:KanMX6 in BY4742 | Open Biosystems |
| CHY345 | ubr1Δ::LEU2 in BY4742 | Ref. 82 |
| CHY346 | ubr1Δ::LEU2 doa10Δ:KanMX6 in BY4742 | This study |
| BWY29 | naa10Δ::kanMX6 ubr1Δ::LEU2 in BY4742 | This study |
| CHY349 | naa30Δ::kanMX6 ubr1Δ::LEU2 in BY4742 | Ref. 40 |
| JOY487 | naa30Δ::natNT2 in BY4742 | This study |
Plasmids, cDNAs, and Primers
Turbo Escherichia coli (New England Biolabs) was used for cloning and maintaining plasmids. Phusion High-Fidelity DNA polymerase (New England Biolabs) was used for carrying out PCR. Nucleotide sequences of all plasmids were verified by DNA sequencing. The plasmids and PCR primers used in this study are described in Tables 2 and 3, respectively.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pcDNA3 | Invitrogen | |
| pCISII-rAANAT | rAANAT in pCISII | Ref. 98 |
| pRS313Cup1 | pRS313 containing the PCUP1 promoter | Varshavsky laboratory collection |
| pRS416Gal1 | pRS416 containing the PGAL1 promoter | Varshavsky laboratory collection |
| pBW105 | MLSrAANAT3f in pRS416 with PGal1 | This study |
| pBW106 | (M)SIrAANAT3f in pRS416 with PGal1 | This study |
| pBW107 | (M)SMLSrAANAT3f in pRS416 with PGal1 | This study |
| pBW135 | MLSrAANAT3ha in pRS313 with PCup1 | This study |
| pBW173 | pcDNA3 (PCMVt1) | This study |
| pBW206 | MLSrAANAT3f in pcDNA3 (PCMVt1) | This study |
| pBW209 | (M)PLSrAANAT3f in pRS416 with PGal1 | This study |
| pBW211 | (M)SMLSrAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW212 | (M)SIrAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW213 | (M)PLSrAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW394 | MLSrAANAT3haK8R in pRS416 with PGal1 | This study |
| pBW395 | (M)SThAANAT3f in pRS416 with PGal1 | This study |
| pBW419 | (M)PThAANAT3f in pRS416 with PGal1 | This study |
| pBW466 | (M)SThAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW467 | (M)PThAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW477 | MLSrAANAT3ha in pcDNA3 (PCMVt1) | This study |
| pBW478 | MLSrAANAT3haKzero in pcDNA3 (PCMVt1) | This study |
| pBW481 | (M)SThAANAT3f in pcDNA3(PCMVt1) | This study |
| pBW482 | (M)SThAANAT3haKzero in pcDNA3(PCMVt1) | This study |
TABLE 3.
PCR primers used in this study
| Primer | Sequence |
|---|---|
| BW239 | 5′-CCTTGAATTCATACCCATGTTGAGCATCCA-3′ |
| BW240 | 5′-CCTTGAATTCATACCCATGAGCATCCA-3′ |
| BW241 | 5′-CCTTGAATTCATACCCATGAGCATGTTGAGCA-3′ |
| BW243 | 5′-CCTTTCCTTGTAGTCGGATCCACCGCAGCCACTGTT-3′ |
| BW244 | 5′-CCTTAAGCTTACTTGTCATCGTCGTCCTTGTAGTCGG-3′ |
| BW335 | 5′-TTGAATTCATGTTGAGCATCCACCCC-3′ |
| BW338 | 5′-GTATGGGTAGCAGCCACTGTTCC-3′ |
| BW339 | 5′-CAGTGGCTGCTACCCATACGATG-3′ |
| BW340 | 5′-TTCTCGAGTCAAGCGTAATCTGGAACGTC-3′ |
| BW352 | 5′-AATTGAATTCACCATGTTGAGCATCC-3′ |
| BW353 | 5′-AATTGAATTCACCATGAGCATCCACC-3′ |
| BW354 | 5′-AATTGAATTCACCATGAGCATGTTGA-3′ |
| BW355 | 5′-AATTCTCGAGTTACTTGTCATCGTCG-3′ |
| BW375 | 5′-TTAATCTAGATGCATGCTCGAGC-3′ |
| BW376 | 5′-CGTTAATCGCGACCAAAATCAACGGGACTTTC-3′ |
| BW412 | 5′-TTGAATTCATGCCCTTGAGCATCCACCCC-3′ |
| BW705 | 5′-TTGAATTCATACCCATGTCCACGCAGAGCACCCACCCCCTGAAACCTGA GGCCCCACGTCTGCCACCTGGGATC-3′ |
| BW711 | 5′-CTTATAGTCACCAGAACCAGAGCAGCCGCTGTTCCTGCGCAGG-3′ |
| BW712 | 5′-CAGGAACAGCGGCTGCTCTGGTTCTGGTGACTATAAGGATG-3′ |
| BW771 | 5′-TTGAATTCATACCCATGCCAACGCAGAGCACCCACCCCCTGAAACCTG AGGCCCCACGTCTGCCACCTGGGATC-3′ |
| BW784 | 5′-CTGAGTAAGTCTCTCTCTGTCCCAAAGTGAACC-3′ |
| BW785 | 5′-GGTTCACTTTGGGACAGAGAGAGACTTACTCAG-3′ |
| BW786 | 5′-CAGCAGGACGGAGCCTCTGCCCTGCTGCCGGAAG-3′ |
| BW787 | 5′-CTTCCGGCAGCAGGGCAGAGGCTCCGTCCTGCTG-3′ |
| BW788 | 5′-GGCCTGGAAACCAAATCTCTCATAGAAGGGCAC-3′ |
| BW789 | 5′-GTGCCCTTCTATGAGAGATTTGGTTTCCAGGCC-3′ |
| BW866 | 5′-TTGAATTCACCATGTCCACGCAGAGCACCCACCCCC-3′ |
| BW867 | 5′-TTGAATTCACCATGTCCACGCAGAGCACCCACCCCCTGAGACCTGAGGC-3′ |
| BW868 | 5′-TTGAATTCACCATGCCAACGCAGAGCACCCACCCC-3′ |
| BW884 | 5′-TTAAGAATTCACCATGTCCACGCAGAGCACC-3′ |
| BW885 | 5′-GTCAGGAACATCGTATGGGTAGCAGCCGCTGTTCCTGCGCAGG-3′ |
The low copy pBW105, pBW106, pBW107, and pBW209 plasmids expressed the wild-type rat MLSrAANAT3f (wild-type rat AANAT bearing the N-terminal sequence Met-Leu-Ser and C-terminally tagged with a triple-FLAG epitope) and its N-terminal mutants (M)SIrAANAT3f, (M)SMLSrAANAT3f, and (M)PLSrAANAT3f, respectively, from the PGAL1 promoter. To construct these plasmids, the rat Aanat ORF was amplified from pCISII-rAANAT (98), using primers BW2394, BW240, BW241, BW412, BW243, and BW244 (Table 3). The resulting PCR products were digested with EcoRI and HindIII and cloned in EcoRI/HindIII-cut pRS416GAL1.
The low copy pBW135 and pBW394 plasmids expressed the wild-type rat MLSrAANAT C-terminally tagged with the triple-HA epitope (MLSrAANAT3ha) and its Lys-8 → Arg mutant MLSrAANAT3haK8R, respectively, from the PCUP1 promoter. To construct these plasmids, the rat Aanat ORF was amplified from pCISII-rAANAT (98) using primers BW339, BW704, BW338, BW339, and BW340 (Table 3). The amplified DNA fragments were digested with EcoRI and XhoI and cloned in EcoRI/XhoI-cut pRS313CUP1 (59, 99).
The low copy pBW395 and pBW419 plasmids expressed the wild-type human (M)SThAANAT C-terminally tagged with a triple-FLAG epitope ((M)SThAANAT3f) and its N-terminal mutant (M)PThAANAT3f, respectively, from the PGAL1 promoter. To construct these plasmids, the human AANAT ORF from Genscript clone OHu55705 (Genscript, Piscataway, NJ) was amplified using primers BW705, BW711, BW712, and BW244 or primers BW771, BW711, BW712, and BW244, respectively (Table 3). The PCR-amplified DNA fragments were digested with EcoRI and HindIII and were ligated into EcoRI/HindIII-cut pRS416GAL1 (99). All final DNA constructs were verified by DNA sequencing. See items i–x at the beginning of “Results” for information about rat and human AANAT notations.
For expression of the above AANAT-encoding ORFs in human HEK293T cells, these ORFs were cloned into a modified expression vector derived from pcDNA3. That (modified) vector, termed pcDNA3CMVt1, contained a truncated (weakened) version of the PCMV promoter, termed PCMVt1. The pcDNA3CMVt1 plasmid was constructed by amplifying, using PCR, the 276-bp fragment that encompassed 104 bp at the 3′-end of the PCMV promoter region in pcDNA3, its T7 promoter site, and its multiple cloning site, using primers BW375 and BW376 (Table 3). The amplified DNA fragment was digested with NruI and XbaI and ligated into NruI/XbaI-cut pcDNA3, yielding the plasmid pBW173 (Table 2). Its truncated PCMVt1 promoter retained several proximal promoter elements, including the consensus NFκB site, the CREB-binding site, and two SP1/SP3-binding sites, but lacked more distal enhancer elements. This resulted in a ∼70% lower activity of the PCMVt1 promoter in HEK293T cells, in comparison with PCMV (Fig. 5, A–C).
The pcDNA3CMVt1-based plasmids pBW206, pBW211, pBW212, and pBW213 expressed, in HEK293T cells, either the wild-type rat MLSrAANAT3f or its N-terminal mutants (M)SIrAANAT3f, (M)SMLSrAANAT3f, and (M)PLSrAANAT3f from the PCMVt1 promoter. To construct these plasmids, Aanat ORFs were amplified from the plasmids pBW105, pBW106, pBW107, and pBW209 using primers BW352, BW353, BW354, BW413, and BW355, respectively (Table 3). The amplified DNA fragments were digested with EcoRI and XhoI and ligated in EcoRI/XhoI-cut pBW173 (Table 2). All pcDNA3-based constructs designed for expression in HEK293T cells contained the Kozak sequence 5′-ACC immediately upstream of the start codon.
pBW477, which expressed the MLSrAANAT3ha from the PCMVt1 promoter, was constructed by using pBW135 as a PCR template and the primers BW352/BW340. The resulting PCR-amplified DNA fragment was digested with EcoRI and XhoI and ligated into EcoRI/XhoI-cut pBW173 (Table 2). pBW478 expressed MLSrAANAT3haKzero (i.e. the lysine-lacking derivative of MLSrAANAT3ha). It was constructed by using pBW135 as a PCR template to generate four separate DNA fragments containing lysine-to-arginine mutations. The primer pairs were 704/784, 785/786, 787/788, and 789/340 (Table 3). These fragments were then assembled into a contiguous ORF using PCR. The resulting DNA fragment, encoding lysine-to-arginine mutations at each of the four lysines contained in the wild-type rat MLSrAANAT3f, was digested with EcoRI and XhoI, and ligated into EcoRI/XhoI-cut pBW173.
pBW466 and pBW467, the pcDNA3-based counterparts of pBW395 and pBW419 (Table 2) were generated by PCR using pBW395 and pBW419 as templates and the primers BW866/BW355 and BW868/BW355, respectively (Table 3). PCR-amplified DNA fragments were digested with EcoRI and XhoI and ligated into EcoRI/XhoI-cut pBW173 (Table 2).
Cell Culture
The HEK293T cell line (derived from human embryonic kidney cells) was obtained from American Type Culture Collection (ATCC, Manassas, VA) and was grown at 37 °C in 5% CO2 in DMEM supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA) and penicillin/streptomycin (100 units/ml; Hyclone). Cells were transfected using Lipofectamine-2000 (Invitrogen) according to the manufacturer's protocol.
Antibody Specific for the Nt-acetylated Form of Rat AANAT (MLSrAANAT)
Unpurified rabbit polyclonal antisera against the synthetic peptide Ac-MLSIHPLKPEAC were produced by Abgent (San Diego, CA) essentially as described for other peptide-mediated polyclonal antibodies produced in studies by the Varshavsky laboratory (e.g. see Refs. 59 and 66). The sequence of the Nt-acetylated peptide immunogen comprised the first 11 residues of MLSrAANAT (Fig. 1C; see below), followed by the C-terminal Cys residue that was used to conjugate the peptide to keyhole limpet hemocyanin carrier protein. The antiserum was affinity-purified in two sequential steps. First, the antibody sample was incubated with the C-terminally immobilized original immunogen Ac-MLSIHPLKPEAC. The peptide-bound fraction was eluted as described previously (59, 66). That fraction was thereafter “negatively” purified against the otherwise identical but non-Nt-acetylated immobilized MLSIHPLKPEAC peptide (the peptides were immobilized using a sulfhydryl-coupling resin (Abgent)), collecting, this time, the unbound fraction. The resulting antibody sample was used to detect, selectively, the Nt-acetylated form of rat AANAT (see “Results” and Fig. 5D). The fraction of once purified antibody that was bound to the non-Nt-acetylated MLSIHPLKPEAC peptide was also eluted and retained and was used as an antibody that could recognize the N-terminal region of rat AANAT irrespective of its Nt-acetylation state.
Cycloheximide Chase Assay in S. cerevisiae
CHX-chase assays in S. cerevisiae were performed largely as described (39, 59). Briefly, S. cerevisiae cells were grown in overnight cultures, and expression of epitope-tagged test proteins was induced by the addition of 2% galactose (for plasmids containing the PGAL1 promoter) or 0.1 mm CuSO4 (for plasmids containing the PCUP1 promoter (100)). 3 h post-induction, CHX was added to a final concentration of 0.1 mg/ml. Samples were collected at the indicated time points, centrifuged at 11,200 × g for 2 min to pellet the cells, and snap-frozen in liquid nitrogen. Proteins were extracted by resuspending the pellets in 1 ml of 0.2 m NaOH and incubating on ice for 20 min (101). Cells were then pelleted and resuspended in 50 μl of SUMEB loading buffer (1% SDS, 8 m urea, 10 mm EDTA, 0.01% bromphenol blue, 10 mm MOPS, pH 6.8), followed by heating at 95 °C for 10 min. Samples were then centrifuged at 11,200 × g for 5 min, followed by SDS-PAGE (with 10 μl loaded per well) using 4–12% NuPAGE gels (Invitrogen). Fractionated proteins were transferred onto nitrocellulose membranes for immunoblotting analyses (see below).
Cycloheximide Chase Assay in Mammalian Cell Lines
HEK293T cells were transfected with 4 μg of plasmid DNA per well (10-cm2 surface area) in 6-well plates using Lipofectamine-2000 (Invitrogen) according to the manufacturer's protocol. 24 h after transfection, cells were treated with CHX (0.1 mg/ml) to initiate a chase. All cells from one well were collected at each indicated time point by washing cells quickly in ice-cold PBS and then rapidly scraping cells into 1.5-ml tubes, pelleting them by centrifugation at 4 °C (at 10,000 × g for 1 min), and snap-freezing the pellets in liquid nitrogen. Cells in these samples were later lysed by the addition of 0.2 ml of “mammalian lysis buffer” (1% Nonidet P-40, 0.15 m NaCl, 50 mm Tris-HCl, pH 7.5) containing 1× Roche Complete Protease Inhibitor Mixture and brief sonication, followed by centrifugation at 11,200 × g for 10 min. Total protein concentration in the supernatants was measured by the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). 30 μg of total protein in a thus prepared extract in lithium dodecyl sulfate (LDS)-sample buffer (in a volume of 45 μl) were heated at 70 °C for 10 min, followed by LDS-PAGE on a 4–12% BisTris NuPAGE gel (Invitrogen) and subsequent transfer onto nitrocellulose membranes for immunoblotting.
Immunoblotting
Following electrophoresis, proteins separated by LDS-PAGE or SDS-PAGE, as indicated above, were electroblotted on nitrocellulose membranes by iBlot (Invitrogen; Program 3; 7-min transfer). Membranes were blocked and thereafter incubated with the appropriate primary antibody, followed by LI-COR IRDye-conjugated secondary antibodies. IRDye fluorescence was detected using an Odyssey 9120 system (LI-COR, Lincoln, NE), facilitating quantification of immunoblots. All quantification was done on LI-COR Odyssey software. Antibodies used include anti-FLAG mouse monoclonal antibody (clone M2; 1:2,000 dilution; Sigma-Aldrich), anti-α-tubulin mouse monoclonal antibody (clone B-5-1-2; 1:10,000 dilution; Sigma-Aldrich), anti-HA mouse monoclonal antibody (clone HA-7; 1:2,000 dilution; Sigma-Aldrich), anti-β-actin mouse monoclonal antibody Ab8224 (1:4,000 dilution; Abcam, Cambridge, UK), and anti-14-3-3ζ rabbit monoclonal antibody EPR6379 (1:100,000 dilution; Abcam).
Immunoprecipitation of Polyubiquitylated AANAT
HEK293T cells were transfected with 4 μg of plasmid DNA per well (10-cm2 surface area) in 6-well plates using Lipofectamine-2000 (Invitrogen) according to the manufacturer's protocol. 24 h after transfection, cells were treated with the proteasome inhibitor MG132 (final concentration of 10 μm; AG Scientific, San Diego, CA) or with an equivalent volume of DMSO in which the 10 mm stock solution of MG132 was made, as indicated. 6 h post-treatment, cells were collected by washing them quickly on a plate in ice-cold PBS and then rapidly scraping cells into 1.5-ml tubes and pelleting them by centrifugation at 4 °C at 10,000 × g for 1 min. Pellets were resuspended in 0.1 ml of buffer containing 1% SDS and 50 mm Tris (pH 7.5) and heated at 95 °C for 10 min. After boiling, samples were diluted with 1 ml of buffer TNN (0.5% Nonidet P-40, 0.25 m NaCl, 5 mm EDTA, 50 mm Tris, pH 7.5) containing 1× Roche Complete Protease Inhibitor Mixture, 20 mm N-ethylmaleimide (Sigma-Aldrich), and 50 μm PR-619 (LifeSensors, Malvern, PA) as inhibitors of deubiquitylation. 25 μl of anti-HA magnetic beads were then added to each sample, and samples were incubated at 4 °C overnight, followed by three washes in TNN, 1 wash in 10 mm Tris-HCl, pH 8.5, and elution with 50 μl of 0.1 m glycine, pH 2.0, by incubation at room temperature for 10 min. After elution, the samples were neutralized and then heated at 70 °C in LDS-PAGE sample buffer, followed by LDS-PAGE on a 4–12% NuPAGE gel. Fractionated proteins were then electroblotted onto nitrocellulose membranes. The resulting membranes (containing transferred proteins) were autoclaved as described previously (102) before “downstream” immunoblotting procedures with either anti-HA or anti-ubiquitin antibodies to increase the sensitivity of detection of polyubiquitin chains.
Author Contributions
B. W., A. V., J. B., Z. H., J.-H. O., and C-S. H. designed the experiments. B. W. performed the experiments, with participation by J.-H. O. B. W., A. V., and J. B. wrote the paper. All authors discussed the results and commented on the manuscript.
Acknowledgments
We thank the present and former members of the Varshavsky laboratory, particularly K. Piatkov, for helpful discussions during this study.
This study was supported by National Institutes of Health Grants R01-DK039520 and R01-GM031530 (to A. V.) and R01-NS057583 (to J. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
J.-H. Oh and A. Varshavsky, unpublished data.
J.-H. Oh, B. Wadas, and A. Varshavsky, unpublished data.
B. Wadas and A. Varshavsky, unpublished data.
- AANAT
- arylalkylamine N-acetyltransferase
- Ub
- ubiquitin
- NAS
- N-acetylserotonin
- CHX
- cycloheximide
- N-degron
- an N-terminal degradation signal recognized by the N-end rule pathway
- N-recognin
- an E3 ubiquitin ligase that can recognize at least some N-degrons
- CREB
- cAMP-response element-binding protein
- Nt-
- Nα-terminally/Nα-terminal
- MetAP
- Met-aminopeptidase
- LDS
- lithium dodecyl sulfate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Klein D. C. (2006) Evolution of the vertebrate pineal gland: the AANAT hypothesis. Chronobiol. Int. 23, 5–20 [DOI] [PubMed] [Google Scholar]
- 2. Coon S. L., and Klein D. C. (2006) Evolution of arylalkylamine N-acetyltransferase: emergence and divergence. Mol. Cell. Endocrinol. 252, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li J., You X., Bian C., Yu H., Coon S. L., and Shi Q. (2016) Molecular evolution of aralkylamine N-acetyltransferase in fish: a genomic survey. Int. J. Mol. Sci. 17, E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borjigin J., Zhang L. S., and Calinescu A. A. (2012) Circadian regulation of pineal gland rhythmicity. Mol. Cell. Endocrinol. 349, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi B. H., Chae H. D., Park T. J., Oh J., Lim J., Kang S. S., Ha H., and Kim K. T. (2004) Protein kinase C regulates the activity and stability of serotonin N-acetyltransferase. J. Neurochem. 90, 442–454 [DOI] [PubMed] [Google Scholar]
- 6. Szewczuk L. M., Tarrant M. K., Sample V., Drury W. J. 3rd, Zhang J., and Cole P. A. (2008) Analysis of serotonin N-acetyltransferase regulation in vitro and in live cells using protein semisynthesis. Biochemistry 47, 10407–10419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Obsil T., Ghirlando R., Klein D. C., Ganguly S., and Dyda F. (2001) Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell 105, 257–267 [DOI] [PubMed] [Google Scholar]
- 8. Hickman A. B., Namboodiri M. A., Klein D. C., and Dyda F. (1999) The structural basis of ordered substrate binding by serotonin N-acetyltransferase: enzyme complex at 1.8 Å resolution with a bisubstrate analog. Cell 97, 361–369 [DOI] [PubMed] [Google Scholar]
- 9. Hickman A. B., Klein D. C., and Dyda F. (1999) Melatonin biosynthesis: the structure of serotonin N-acetyltransferase at 2.5 Å resolution suggests a catalytic mechanism. Mol. Cell 3, 23–32 [DOI] [PubMed] [Google Scholar]
- 10. Borjigin J., Wang M. M., and Snyder S. H. (1995) Diurnal variation in mRNA encoding serotonin N-acetyltransferase in pineal gland. Nature 378, 783–785 [DOI] [PubMed] [Google Scholar]
- 11. Coon S. L., Roseboom P. H., Baler R., Weller J. L., Namboodiri M. A., Koonin E. V., and Klein D. C. (1995) Pineal serotonin N-acetyltransferase: expression cloning and molecular analysis. Science 270, 1681–1683 [DOI] [PubMed] [Google Scholar]
- 12. Tosini G., Ye K., and Iuvone P. M. (2012) N-Acetylserotonin: neuroprotection, neurogenesis, and the sleepy brain. Neuroscientist 18, 645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jang S. W., Liu X., Pradoldej S., Tosini G., Chang Q., Iuvone P. M., and Ye K. (2010) N-Acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. U.S.A. 107, 3876–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sompol P., Liu X., Baba K., Paul K. N., Tosini G., Iuvone P. M., and Ye K. (2011) N-Acetylserotonin promotes hippocampal neuroprogenitor cell proliferation in sleep-deprived mice. Proc. Natl. Acad. Sci. U.S.A. 108, 8844–8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schippers K. J., and Nichols S. A. (2014) Deep, dark secrets of melatonin in animal evolution. Cell 159, 9–10 [DOI] [PubMed] [Google Scholar]
- 16. Zhdanova I. V., Wang S. Y., Leclair O. U., and Danilova N. P. (2001) Melatonin promotes sleep-like state in zebrafish. Brain Res. 903, 263–268 [DOI] [PubMed] [Google Scholar]
- 17. Gandhi A. V., Mosser E. A., Oikonomou G., and Prober D. A. (2015) Melatonin is required for the circadian regulation of sleep. Neuron 85, 1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. González S., Moreno-Delgado D., Moreno E., Pérez-Capote K., Franco R., Mallol J., Cortés A., Casadó V., Lluís C., Ortiz J., Ferré S., Canela E., and McCormick P. J. (2012) Circadian-related heteromerization of adrenergic and dopamine D(4) receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol. 10, e1001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rawashdeh O., and Maronde E. (2012) The hormonal Zeitgeber melatonin: role as a circadian modulator in memory processing. Front. Mol. Neurosci. 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arendt J. (2006) Melatonin and human rhythms. Chronobiol. Int. 23, 21–37 [DOI] [PubMed] [Google Scholar]
- 21. Liu T., and Borjigin J. (2005) N-Acetyltransferase is not the rate-limiting enzyme of melatonin synthesis at night. J. Pineal Res. 39, 91–96 [DOI] [PubMed] [Google Scholar]
- 22. Tu B. P., and McKnight S. L. (2006) Metabolic cycles as an underlying basis of biological oscillations. Nat. Rev. Mol. Cell Biol. 7, 696–701 [DOI] [PubMed] [Google Scholar]
- 23. Masri S., and Sassone-Corsi P. (2013) The circadian clock: a framework linking metabolism, epigenetics and neuronal function. Nat. Rev. Neurosci. 14, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar R. S., Green E. W., Zhao Y., van Ooijen G., Olmedo M., Qin X., Xu Y., Pan M., Valekunja U. K., Feeney K. A., Maywood E. S., Hastings M. H., Baliga N. S., Merrow M., Millar A. J., Johnson C. H., Kyriacou C. P., O'Neill J. S., and Reddy A. B. (2012) Peroxiredoxins are conserved markers of circadian rhythms. Nature 485, 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borjigin J., Li X., and Snyder S. H. (1999) The pineal gland and melatonin: molecular and pharmacologic regulation. Annu. Rev. Pharmacol. Toxicol. 39, 53–65 [DOI] [PubMed] [Google Scholar]
- 26. Stehle J. H., Saade A., Rawashdeh O., Ackermann K., Jilg A., Sebestény T., and Maronde E. (2011) A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 51, 17–43 [DOI] [PubMed] [Google Scholar]
- 27. Mohawk J. A., and Takahashi J. S. (2011) Cell autonomy and synchrony of suprachiasmatic nucleus circadian oscillators. Trends Neurosci. 34, 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ganguly S., Gastel J. A., Weller J. L., Schwartz C., Jaffe H., Namboodiri M. A., Coon S. L., Hickman A. B., Rollag M., Obsil T., Beauverger P., Ferry G., Boutin J. A., and Klein D. C. (2001) Role of a pineal cAMP-operated arylalkylamine N-acetyltransferase/14-3-3-binding switch in melatonin synthesis. Proc. Natl. Acad. Sci. U.S.A. 98, 8083–8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mukherjee S., and Maitra S. K. (2015) Gut melatonin in vertebrates: chronobiology and physiology. Front. Endocrinol. 6, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hardeland R. (2010) Melatonin metabolism in the central nervous system. Curr. Neuropharmacol. 8, 168–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chattoraj A., Liu T., Zhang L. S., Huang Z., and Borjigin J. (2009) Melatonin formation in mammals: in vivo perspectives. Rev. Endocr. Metab. Disord. 10, 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schomerus C., Laedtke E., Olcese J., Weller J. L., Klein D. C., and Korf H. W. (2002) Signal transduction and regulation of melatonin synthesis in bovine pinealocytes: impact of adrenergic, peptidergic and cholinergic stimuli. Cell Tissue Res. 309, 417–428 [DOI] [PubMed] [Google Scholar]
- 33. Schomerus C., Korf H. W., Laedtke E., Weller J. L., and Klein D. C. (2000) Selective adrenergic/cyclic AMP-dependent switch-off of proteasomal proteolysis alone switches on neural signal transduction: an example from the pineal gland. J. Neurochem. 75, 2123–2132 [DOI] [PubMed] [Google Scholar]
- 34. Bernard M., Iuvone P. M., Cassone V. M., Roseboom P. H., Coon S. L., and Klein D. C. (1997) Avian melatonin synthesis: photic and circadian regulation of serotonin N-acetyltransferase mRNA in the chicken pineal gland and retina. J. Neurochem. 68, 213–224 [DOI] [PubMed] [Google Scholar]
- 35. Gastel J. A., Roseboom P. H., Rinaldi P. A., Weller J. L., and Klein D. C. (1998) Melatonin production: proteasomal proteolysis in serotonin N-acetyltransferase regulation. Science 279, 1358–1360 [DOI] [PubMed] [Google Scholar]
- 36. Roseboom P. H., Coon S. L., Baler R., McCune S. K., Weller J. L., and Klein D. C. (1996) Melatonin synthesis: analysis of the more than 150-fold nocturnal increase in serotonin N-acetyltransferase messenger ribonucleic acid in the rat pineal gland. Endocrinology 137, 3033–3045 [DOI] [PubMed] [Google Scholar]
- 37. Huang Z., Liu T., and Borjigin J. (2010) N-terminal residues regulate proteasomal degradation of AANAT. J. Pineal Res. 48, 290–296 [DOI] [PubMed] [Google Scholar]
- 38. Bachmair A., Finley D., and Varshavsky A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 [DOI] [PubMed] [Google Scholar]
- 39. Hwang C. S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim H. K., Kim R. R., Oh J. H., Cho H., Varshavsky A., and Hwang C. S. (2014) The N-terminal methionine of cellular proteins as a degradation signal. Cell 156, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Varshavsky A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gibbs D. J., Bacardit J., Bachmair A., and Holdsworth M. J. (2014) The eukaryotic N-end rule pathway: conserved mechanisms and diverse functions. Trends Cell Biol. 24, 603–611 [DOI] [PubMed] [Google Scholar]
- 43. Tasaki T., Sriram S. M., Park K. S., and Kwon Y. T. (2012) The N-end rule pathway. Annu. Rev. Biochem. 81, 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dougan D. A., Micevski D., and Truscott K. N. (2012) The N-end rule pathway: from recognition by N-recognins to destruction by AAA+ proteases. Biochim. Biophys. Acta 1823, 83–91 [DOI] [PubMed] [Google Scholar]
- 45. Varshavsky A. (2008) Discovery of cellular regulation by protein degradation. J. Biol. Chem. 283, 34469–34489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee K. E., Heo J. E., Kim J. M., and Hwang C. S. (2016) N-terminal acetylation-targeted N-end rule proteolytic system: the Ac/N-end rule pathway. Mol. Cells 39, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turner G. C., Du F., and Varshavsky A. (2000) Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 405, 579–583 [DOI] [PubMed] [Google Scholar]
- 48. Hwang C. S., Shemorry A., and Varshavsky A. (2009) Two proteolytic pathways regulate DNA repair by cotargeting the Mgt1 alkylguanine transferase. Proc. Natl. Acad. Sci. U.S.A. 106, 2142–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heck J. W., Cheung S. K., and Hampton R. Y. (2010) Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eisele F., and Wolf D. H. (2008) Degradation of misfolded proteins in the cytoplasm by the ubiquitin ligase Ubr1. FEBS Lett. 582, 4143–4146 [DOI] [PubMed] [Google Scholar]
- 51. Bachmair A., and Varshavsky A. (1989) The degradation signal in a short-lived protein. Cell 56, 1019–1032 [DOI] [PubMed] [Google Scholar]
- 52. Inobe T., and Matouschek A. (2014) Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 24, 156–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tobias J. W., Shrader T. E., Rocap G., and Varshavsky A. (1991) The N-end rule in bacteria. Science 254, 1374–1377 [DOI] [PubMed] [Google Scholar]
- 54. Mogk A., Schmidt R., and Bukau B. (2007) The N-end rule pathway of regulated proteolysis: prokaryotic and eukaryotic strategies. Trends Cell Biol. 17, 165–172 [DOI] [PubMed] [Google Scholar]
- 55. Rivera-Rivera I., Román-Hernández G., Sauer R. T., and Baker T. A. (2014) Remodeling of a delivery complex allows ClpS-mediated degradation of N-degron substrates. Proc. Natl. Acad. Sci. U.S.A. 111, E3853–E3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Piatkov K. I., Vu T. T., Hwang C.-S., and Varshavsky A. (2015) Formyl-methionine as a degradation signal at the N-termini of bacterial proteins. Microbial Cell 2, 376–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Humbard M. A., Surkov S., De Donatis G. M., Jenkins L. M., and Maurizi M. R. (2013) The N-degradome of Escherichia coli: limited proteolysis in vivo generates a large pool of proteins bearing N-degrons. J. Biol. Chem. 288, 28913–28924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Graciet E., Hu R. G., Piatkov K., Rhee J. H., Schwarz E. M., and Varshavsky A. (2006) Aminoacyl-transferases and the N-end rule pathway of prokaryotic/eukaryotic specificity in a human pathogen. Proc. Natl. Acad. Sci. U.S.A. 103, 3078–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shemorry A., Hwang C. S., and Varshavsky A. (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 50, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park S. E., Kim J. M., Seok O. H., Cho H., Wadas B., Kim S. Y., Varshavsky A., and Hwang C. S. (2015) Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 347, 1249–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aksnes H., Hole K., and Arnesen T. (2015) Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell. Mol. Biol. 316, 267–305 [DOI] [PubMed] [Google Scholar]
- 62. Dörfel M. J., and Lyon G. J. (2015) The biological functions of Naa10: from amino-terminal acetylation to human disease. Gene 567, 103–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Starheim K. K., Gevaert K., and Arnesen T. (2012) Protein N-terminal acettyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 64. Piatkov K. I., Brower C. S., and Varshavsky A. (2012) The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. U.S.A. 109, E1839–E1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Piatkov K. I., Oh J.-H., Liu Y., and Varshavsky A. (2014) Calpain-generated natural protein fragments as short-lived substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 111, E817–E826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brower C. S., Piatkov K. I., and Varshavsky A. (2013) Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol. Cell 50, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamano K., and Youle R. J. (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cha-Molstad H., Sung K. S., Hwang J., Kim K. A., Yu J. E., Yoo Y. D., Jang J. M., Han D. H., Molstad M., Kim J. G., Lee Y. J., Zakrzewska A., Kim S. H., Kim S. T., Kim S. Y., et al. (2015) Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 17, 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang H., Piatkov K. I., Brower C. S., and Varshavsky A. (2009) Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol. Cell 34, 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hwang C. S., Shemorry A., Auerbach D., and Varshavsky A. (2010) The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat. Cell Biol. 12, 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Varshavsky A. (2004) Spalog and sequelog: neutral terms for spatial and sequence similarity. Curr. Biol. 14, R181–R183 [DOI] [PubMed] [Google Scholar]
- 72. Hassink G., Kikkert M., van Voorden S., Lee S.-J., Spaapen R., van Laar T., Coleman C. S., Bartee E., Früh K., Chau V., and Wiertz E. (2005) TEB4 is a C4HC3 RING-finger containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 388, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Foresti O., Ruggiano A., Hannibal-Bach H. K., Ejsing C. S., and Carvalho P. (2013) Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife 2, e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xiao Q., Zhang F., Nacev B. A., Liu J. O., and Pei D. (2010) Protein N-terminal processing: substrate specificity of Escherichia coli and human methionine aminopeptidases. Biochemistry 49, 5588–5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Varland S., Osberg C., and Arnesen T. (2015) N-terminal modifications of cellular proteins: the enzymes involved, their substrate specificities and biological effects. Proteomics 15, 2385–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Perrot M., Massoni A., and Boucherie H. (2008) Sequence requirements for Nα-terminal acetylation of yeast proteins by NatA. Yeast 25, 513–527 [DOI] [PubMed] [Google Scholar]
- 77. Lee D. H., and Goldberg A. L. (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271, 27280–27284 [DOI] [PubMed] [Google Scholar]
- 78. Fleming J. A., Lightcap E. S., Sadis S., Thoroddsen V., Bulawa C. E., and Blackman R. K. (2002) Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc. Natl. Acad. Sci. U.S.A. 99, 1461–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee S. J., Liu T., Chattoraj A., Zhang S. L., Wang L., Lee T. M., Wang M. M., and Borjigin J. (2009) Posttranscriptional regulation of pineal melatonin synthesis in Octodon degus. J. Pineal Res. 47, 75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Falcón J., Coon S. L., Besseau L., Cazaméa-Catalan D., Fuentès M., Magnanou E., Paulin C. H., Boeuf G., Sauzet S., Jørgensen E. H., Mazan S., Wolf Y. I., Koonin E. V., Steinbach P. J., Hyodo S., and Klein D. C. (2014) Drastic neofunctionalization associated with evolution of the timezyme AANAT 500 Mya. Proc. Natl. Acad. Sci. U.S.A. 111, 314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ganguly S., Mummaneni P., Steinbach P. J., Klein D. C., and Coon S. L. (2001) Characterization of the Saccharomyces cerevisiae homolog of the melatonin rhythm enzyme arylalkylamine N-acetyltransferase. J. Biol. Chem. 276, 47239–47247 [DOI] [PubMed] [Google Scholar]
- 82. Kim J. M., and Hwang C. S. (2014) Crosstalk between the Arg/N-end and Ac/N-end rule. Cell Cycle 13, 1366–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Slater M., Hartzell D., Hartnett J., Wheeler S., Stecha P., and Karassina N. (2008) Achieve the protein expression level you need with the mammalian HaloTag 7 Flexi vectors. Promega Notes 100, 16–18 [Google Scholar]
- 84. Wu C., and Li S. S. (2009) CelluSpots: a reproducible means of making peptide arrays for the determination of SH2 domain binding specificity. Methods Mol. Biol. 570, 197–202 [DOI] [PubMed] [Google Scholar]
- 85. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 86. Inobe T., Fishbain S., Prakash S., and Matouschek A. (2011) Defining the geometry of the two-component proteasome degron. Nat. Chem. Biol. 7, 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. van Heusden G. P., and Steensma H. Y. (2006) Yeast 14-3-3 proteins. Yeast 23, 159–171 [DOI] [PubMed] [Google Scholar]
- 88. Obsil T., and Obsilova V. (2011) Structural basis of 14-3-3 protein functions. Semin. Cell Dev. Biol. 22, 663–672 [DOI] [PubMed] [Google Scholar]
- 89. Kakiuchi K., Yamauchi Y., Taoka M., Iwago M., Fujita T., Ito T., Song S. Y., Sakai A., Isobe T., and Ichimura T. (2007) Proteomic analysis of in vivo 14-3-3 interactions in the yeast Saccharomyces cerevisiae. Biochemistry 46, 7781–7792 [DOI] [PubMed] [Google Scholar]
- 90. Obsilova V., Kopecka M., Kosek D., Kacirova M., Kylarova S., Rezabkova L., and Obsil T. (2014) Mechanisms of the 14-3-3 protein function: regulation of protein function through conformational modulation. Physiol. Res. 63, Suppl. 1, S155–S164 [DOI] [PubMed] [Google Scholar]
- 91. Yang J., Kamide K., Kokubo Y., Takiuchi S., Tanaka C., Banno M., Miwa Y., Yoshii M., Horio T., Okayama A., Tomoike H., Kawano Y., and Miyata T. (2005) Genetic variations of regulator of G-protein signaling 2 in hypertensive patients and in the general population. J. Hypertens. 23, 1497–1505 [DOI] [PubMed] [Google Scholar]
- 92. Matsuo M., Coon S. L., and Klein D. C. (2013) RGS2 is a feedback inhibitor of melatonin production in the pineal gland. FEBS Lett. 587, 1392–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lynch M. (2007) The Origins of Genome Architecture, Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- 94. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Smith J. A., Seidman J. G., and Struhl K. (2010) Current Protocols in Molecular Biology, Wiley-Interscience, New York [Google Scholar]
- 95. Guthrie C., and Fink G. R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol. 194, 1–863 [PubMed] [Google Scholar]
- 96. Andrews B., Boone C., Davis T. N., and Fields S. (2016) Budding Yeast: A Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 97. Gietz R. D., and Woods R. A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 [DOI] [PubMed] [Google Scholar]
- 98. Huang Z., Deng J., and Borjigin J. (2005) A novel H28Y mutation in LEC rats leads to decreased NAT protein stability in vivo and in vitro. J. Pineal Res. 39, 84–90 [DOI] [PubMed] [Google Scholar]
- 99. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in S. cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ghislain M., Dohmen R. J., Levy F., and Varshavsky A. (1996) Cdc48p interacts with Ufd3p, a WD repeat protein required for ubiquitin-mediated proteolysis in Saccharomyces cerevisiae. EMBO J. 15, 4884–4899 [PMC free article] [PubMed] [Google Scholar]
- 101. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 102. Swerdlow P. S., Finley D., and Varshavsky A. (1986) Enhancement of immunoblot sensitivity by heating of hydrated filters. Anal. Biochem. 156, 147–153 [DOI] [PubMed] [Google Scholar]