

Abstract
Slow deactivation of Kv11.1 channels is critical for its function in the heart. The S4-S5 linker, which joins the voltage sensor and pore domains, plays a critical role in this slow deactivation gating. Here, we use NMR spectroscopy to identify the membrane-bound surface of the S4S5 linker, and we show that two highly conserved tyrosine residues within the KCNH subfamily of channels are membrane-associated. Site-directed mutagenesis and electrophysiological analysis indicates that Tyr-542 interacts with both the pore domain and voltage sensor residues to stabilize activated conformations of the channel, whereas Tyr-545 contributes to the slow kinetics of deactivation by primarily stabilizing the transition state between the activated and closed states. Thus, the two tyrosine residues in the Kv11.1 S4S5 linker play critical but distinct roles in the slow deactivation phenotype, which is a hallmark of Kv11.1 channels.
Keywords: electrophysiology, gating, hERG, ion channel, nuclear magnetic resonance (NMR), potassium channel, site-directed mutagenesis, Kv11.1, S4S5 linker, lipid membrane
Introduction
The rhythm of the heartbeat is primarily controlled by the coordinated activity of a range of voltage-gated ion channels (1, 2). Kv11.1, which is encoded by the human ether-à-go-go-related gene (official name, KCNH2) (3), is responsible for the delayed rectifier current (IKr), which plays a critical role in cardiac repolarization (4, 5). Kv11.1 channels have unusual kinetics of gating, namely slow activation and deactivation but fast inactivation and recovery from inactivation (6). Consequently, these channels pass relatively little current during the plateau phase of the action potential but deliver a large current during repolarization (7). The slow kinetics of deactivation during the early diastolic period also enable Kv11.1 channels to play a critical role in terminating ectopic beats that may otherwise lead to cardiac arrhythmias and sudden cardiac arrest (6, 8).
Kv11.1 channels have the typical topology of voltage-gated K+ channels, which includes cytoplasmic N- and C-terminal domains, a voltage sensor domain (VSD)5 composed of the first four transmembrane segments (S1–S4), and a pore domain composed of the 5th and 6th transmembrane segments (S5 and S6). Pore domains from each of the four subunits co-assemble to form the ion conduction pathway, as well as the gates that control activation and inactivation. The slow activation and deactivation kinetics of Kv11.1 channels are due in part to the slow movement of the VSD (9, 10). Motion of the VSD, in response to changes in the membrane electric field, facilitates opening and closing of the activation gate at the intracellular end of the pore domain by pulling on the S4S5 linker (residues 539–550), which connects the VSD to the activation gate at the cytoplasmic end of the pore (11). Numerous studies have emphasized the importance of the S4S5 linker for Kv11.1 gating (11–17). In addition to providing a direct link between the voltage sensor and pore domains, the S4S5 linker interacts with the cytoplasmic domains and is thought to mediate the influence of cytoplasmic domains on the kinetics of deactivation gating (12, 13, 15). Recently, Lörinczi et al. (18) showed that when the S4S5 linker in Kv11.1 channels was disrupted (i.e. co-expressing a construct composed of residues 1–545 with another composed of residues 546–1159), the channels can still assemble and retain their slow activation kinetics, as well as rapid inactivation and recovery from inactivation kinetics, but they have much faster deactivation kinetics compared with full-length wild-type (WT) channels (18). These data indicate that an intact S4S5 linker is not required for normal activation gating, but once the channels have opened, an intact S4S5 linker is required to stabilize the activated state and thereby contributes to slow deactivation. Another important implication of this study is that activation and deactivation gating are not simply reciprocal processes, but rather the transition from the closed state to the open state likely follows a different pathway to the transition from the open state back to the closed state.
In the crystal structure of the Kv1.2/2.1 chimeric channel, which is thought to have captured the channel in the activated state (19), the S4S5 linker forms an α-helix that is located at the cytoplasmic membrane interface, with one surface interacting with the membrane and/or transmembrane regions of the channel when the activation gates are in the open conformation. In this study, we sought to identify key S4S5 linker residues that interact with the membrane and/or intramembrane protein domains to stabilize the open state of the channel. A combination of nuclear magnetic resonance (NMR) and electrophysiology experiments suggest that the two tyrosine side chains (Tyr-542 and Tyr-545) point toward the membrane interior. Thermodynamic mutant cycle analysis experiments indicate that Tyr-542 interacts with both pore domain and voltage sensor residues to stabilize the open state of the channel. Conversely, mutations to Tyr-545 cause marked changes to the kinetics of deactivation but have much smaller effects on the voltage dependence of the distribution between open and closed states, which suggests that Tyr-545 stabilizes an intermediate transition state in the pathway between the activated and closed states.
Results
An alignment of the S4S5 linkers of different voltage-gated potassium channels is shown in Fig. 1. The most highly conserved residues are the charged arginine residues from the distal S4 and a glycine residue in the middle of the S4S5 linker. There are also two leucine residues in the S4S5 linker that are highly conserved in the Kv1–9 channels but are replaced by tyrosines in Kv10–12 channels. In the crystal structure of the Kv1.2/2.1 chimera, the leucine residues in the S4S5 linker are oriented toward the lipid membrane. It has been shown previously that the S4S5 linker is critical for channel activation and deactivation in Kv11.1 (12, 14, 16, 17) and that many of the S4S5 residues contribute to the slow gating kinetics. Previous studies, however, have largely focused on how the S4S5 linker interacts with cytoplasmic domains. For example, Asp-540 interacts with the bottom of the S6 transmembrane domain to stabilize the closed state of Kv11.1 channel (16), and cysteine scanning studies have suggested that the S4S5 domain interacts with the N-terminal tail (13). The tyrosine residues, however, have not been extensively studied. Our sequence homology analysis, coupled to the location of the S4S5 linker in crystal structures of the activated state, suggest that the two tyrosine residues are likely to contribute to interactions with the membrane and/or intra-membrane domains of the channel.
FIGURE 1.
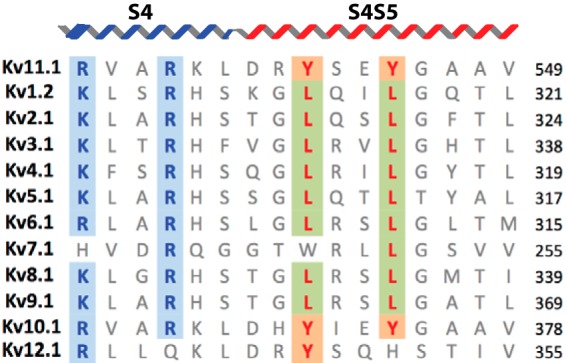
Tyrosine residues are conserved among the KCNH family. Sequence alignment of Kv channels for S4 and S4S5 linker regions. Arginine residues in S4 are highlighted in blue and tyrosine residues, conserved within the KCNH family (Kv10–12), are highlighted in orange. In place of tyrosine, almost all other Kv channels have a leucine at the equivalent sites (green boxes). The topology above the alignment is based on the crystal structure of Kv2.1/1.2 chimeras (Protein Data Bank code 2R9R).
To determine whether the equivalent residues in Kv11.1 (Tyr-542 and Tyr-545) are likely to be oriented toward the lipid membrane, we studied the interaction of a peptide (Leu-532–Phe-551) that contains the S4S5 linker of the Kv11.1 channel with dimyristoylphosphatidylcholine (DMPC) or DMPC/dodecylphosphocholine (DPC) model membranes (Fig. 2). First, a solution NMR was used to gain insight into whether the S4S5 linker interacts with the lipid membrane. We recorded one-dimensional 1H NMR spectra of the S4S5 peptides in water (Fig. 2A, panel i) and in the presence of DMPC/DPC bicelles (Fig. 2A, panel ii). Comparison of the spectra shows changes in the chemical shifts and significant broadening of the peptide resonances, consistent with an interaction between the peptide and the model membranes (20). Phosphorus (31P) and deuterium (2H) solid-state NMR experiments were then employed to further investigate the interaction of the S4S5 peptide with the membrane using DMPC multilamellar vesicles (MLVs). Because phospholipids harbor a phosphate in their polar headgroup, 31P NMR spectra were acquired to monitor change at the membrane surface. Broad power spectra are characteristic of non-oriented MLVs and represent the superposition of all chemical shifts from the distribution of orientations in the magnetic field (Fig. 2B, panel i), with the 90° edge on the right and 0° edge on the left.
FIGURE 2.
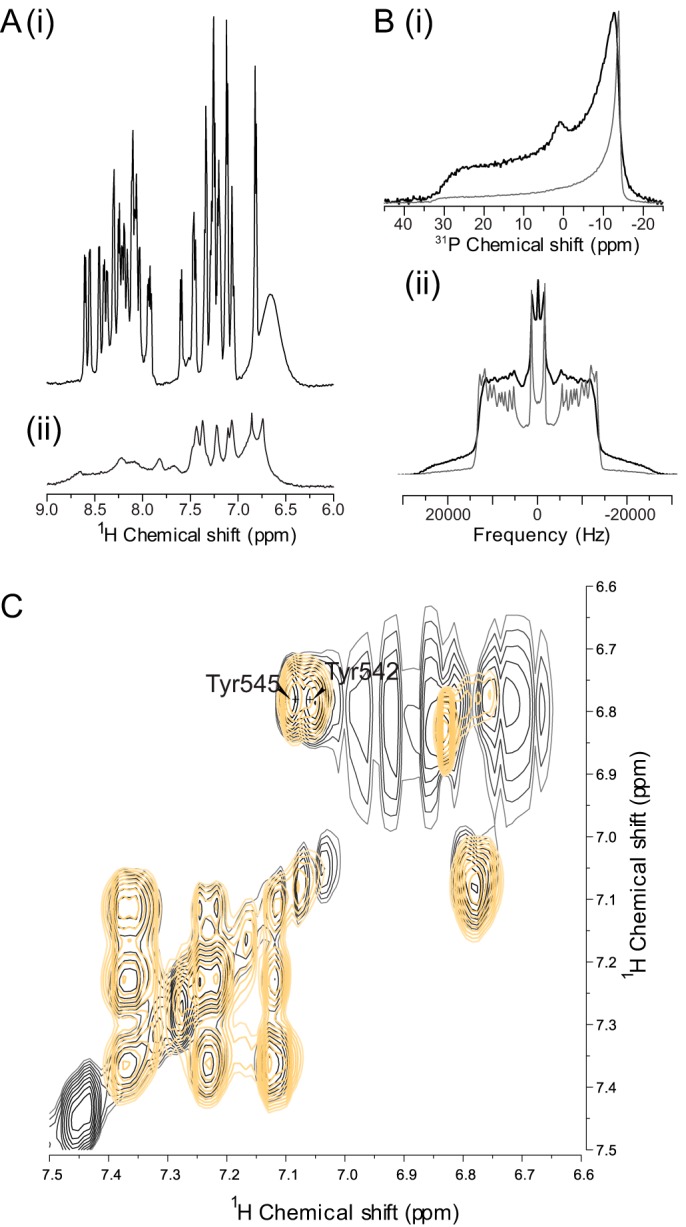
Tyrosine residues within the S4S5 linker of Kv11.1 are oriented toward the lipid membrane. A, S4S5 peptide (Leu-532–Phe-551) interacts with the lipid membrane as the amide and aromatic resonances in the 1H NMR spectrum of the WT peptide in DMPC/DPC-d38 are very broad (panel ii) compared with S4S5 peptide in 10% D2O (panel i). B, pure 31P (panel i) and 2H (panel ii) solid-state NMR spectra of DMPC-d54 MLV model membranes (gray) and with S4S5 peptide (black) showing the S4S5 peptide interacts with the lipid membrane. C, 1H TOCSY spectra of the S4S5 peptide solubilized with DMPC-d54/DPC-d38 in 10% D2O (black) and with 0.5 mm Gd3+ (orange) showing the tyrosine residues (Tyr-542 and Tyr-545) are protected from soluble paramagnetics by the lipids as the aromatic 1H signals for the Tyr-542 and Tyr-545 remain.
To probe the bilayer's hydrophobic core, phospholipids with perdeuterated acyl chains (DMPC-d54) were used. The spacing between each doublet on the 2H NMR spectrum is known as the quadrupolar splitting (ΔνQ) and gives insight on the degree of organization of the acyl chains (Fig. 2B, panel ii). The largest splitting corresponds to the CD2 bonds closer to the headgroup, whereas the smaller splitting corresponds to the most mobile CD3 group at the end of the lipid chain. Pure lipids 31P (Fig. 2B, panel i, gray) and 2H (Fig. 2B, panel ii, gray) spectra were recorded as a reference. The peptide disrupts model DMPC membranes suggesting the formation of fast-tumbling lipid assemblies, as revealed by the isotropic resonance on the 31P and 2H spectra (Fig. 2B, black). Moreover, the 31P spectra for the peptide (Fig. 2B, panel i, black) shows a change in intensity between the 0 and 90° edges of model membranes (Fig. 2B, panel i, gray), with less lipids on the 90° edge, which can be ascribed to a change in vesicle shape or the transverse relaxation (T2) effect caused by an increase in slow motions. The peptide mainly interacts with the surface of the membrane because no changes in ΔνQ of 2H spectra are observed, indicative of no deep insertion in the hydrophobic core of the membrane (21). Although these data suggest that the S4S5 linker is directly involved in membrane interaction, the orientation of the tyrosine residues was further probed by adding soluble paramagnetic gadolinium lanthanide ions (Gd3+) to the samples. The Gd3+ ions attenuate the proton signals of the peptide residues that are solvent-exposed (22). In agreement with solid-state NMR, the 1H total correlation spectroscopy (TOCSY) spectrum of the linker (Fig. 2C) shows that the tyrosine ring protons are protected from the effect of Gd3+. This suggests that Tyr-542 and Tyr-545 are located facing the membrane.
We first performed alanine-scanning mutagenesis on the S4S5 residues (Asp-540–Val-549) to investigate how individual residues in the S4S5 linker contribute to the voltage dependence of the distribution between closed and open states (activation) as well as the rate of activation of Kv11.1 channels. Fig. 3A, panel i, shows the 3-s activation protocol and exemplar traces for WT Kv11.1 channels. The V0.5 of activation for WT is −23.9 ± 1.3 mV, n = 5. Most of the mutants altered the V0.5 of activation when compared with WT with the largest shifts seen for S543A, G546A, and A548V (Fig. 3A, panel ii). The changes observed for Y542A (−7.2 ± 1.5 mV, n = 4) and Y545A (−36.0 ± 1.2 mV, n = 5) were more modest. Fig. 3B, panel i, shows the protocol, and typical family of current traces, used to measure the rate of activation at 20 mV (see under “Experimental Procedures” for more details). The time constants for WT (102.5 ± 6.1 ms, n = 8) and mutants are summarized in Fig. 3B, panel ii, and in Table 1. Notably, both Y542A and Y545A did not significantly perturb the rate of activation at 20 mV compared with WT.
FIGURE 3.
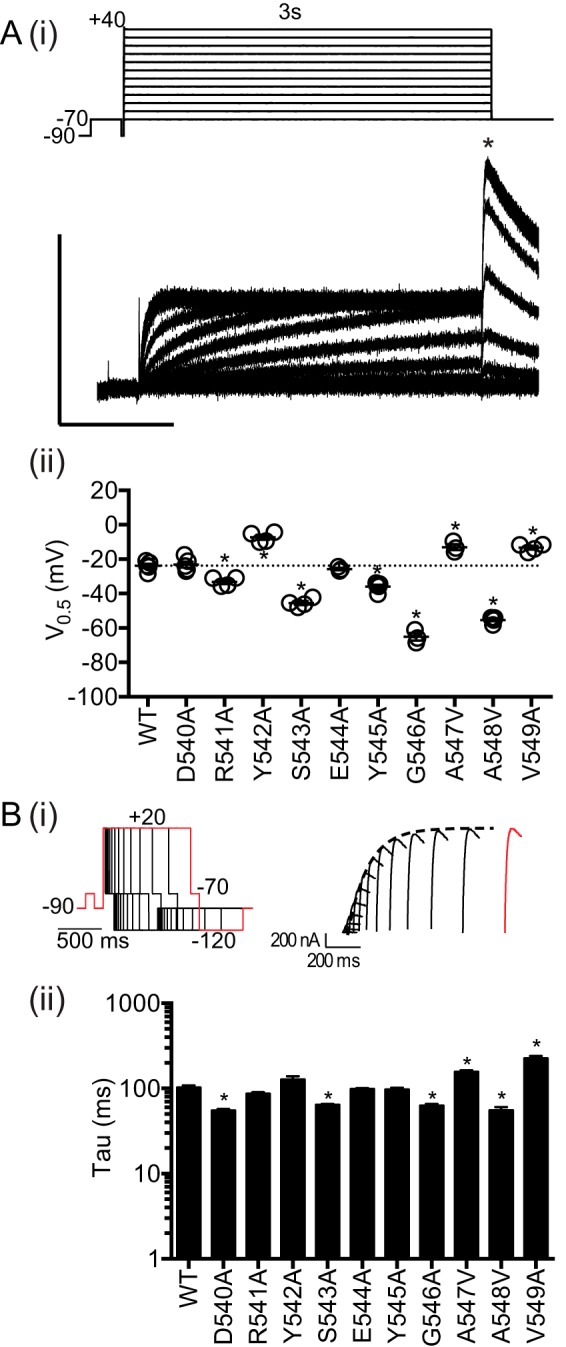
Activation properties for S4S5 mutants. A, 3-s isochronal activation protocol and example traces of WT. x and y axes are 1 s and 0.25 μA, respectively (panel i). The peak tail currents (*) derived from 3-s isochronal activation were fitted with a Boltzmann equation to derive V0.5 of activation (panel ii). The values for all mutants are summarized in Table 1. B, envelope of tail protocol to elicit the tail current at −70 mV after activation at 20 mV. One of the traces and its respective protocol are highlighted (panel i). The time constants of activation at 20 mV for WT and S4S5 mutants are summarized in panel ii. All summary data are presented as mean ± S.E. and statistical comparisons made using one-way ANOVA with Bonferroni post-test. Significant differences (p < 0.05) are highlighted with asterisks. The values for all mutants are summarized in Table 1.
TABLE 1.
Summary of the values for the 3-s isochronal activation V0.5 and time constant of activation at 20 mV for S4S5 alanine mutants
Data are presented as mean ± S.E. (n), and n denotes number of individual recordings. Asterisk indicates p < 0.05 versus WT using one-way ANOVA with Bonferroni post-test.
| Mutant | Activation V0.5 (mV) (n) | Time constant of activation at +20 mV (ms) (n) |
|---|---|---|
| WT | −23.9 ± 1.3 (5) | 102.5 ± 6.1 (8) |
| D540A | −23.1 ± 1.7 (5) | 55.2 ± 2.2* (5) |
| R541A | −33.2 ± 1.3* (4) | 87.0 ± 3.6 (5) |
| Y542A | −7.2 ± 1.5* (4) | 128.0 ± 11.0 (6) |
| S543A | −45.5 ± 1.2* (4) | 64.2 ± 1.6* (7) |
| E544A | −25.7 ± 0.7 (3) | 97.8 ± 2.6 (6) |
| Y545A | −36.0 ± 1.2* (5) | 97.3 ± 4.5 (7) |
| G546A | −65.2 ± 2.2* (3) | 63.0 ± 2.8* (5) |
| A547V | −13.0 ± 1.8* (3) | 157.2 ± 6.0* (6) |
| A548V | −55.3 ± 0.8* (5) | 55.0 ± 5.1* (7) |
| V549A | −13.3 ± 1.1* (4) | 225.8 ± 15.0* (7) |
Previous studies have suggested that activation and deactivation are not simply reciprocal processes but rather occur via different pathways (18, 23). We therefore investigated whether any of the S4S5 mutants preferentially altered the deactivation phenotype. Fig. 4A shows typical examples of 3-s tail current traces recorded at −70 mV (black) and −120 mV (gray) for WT (i), Y545A (ii), and Y542A (iii) following a 1-s step to 40 mV to fully activate the channels. The time constants for the fast component of deactivation in the voltage range from −70 to −150 mV for the three channels shown in Fig. 4B. The rates of deactivation of Y545A (in Fig. 4B, triangle) are 2–3 times faster than WT (circle) at all voltages measured. For Y542A (in Fig. 4B, square), the rate of deactivation is more than 10 times faster than WT at −70 mV, but at −150 mV there is almost no difference in the rates of deactivation for WT and Y542A channels. A summary of the time constant for the fast component of deactivation at −70 mV (black) and −120 mV (gray) for all S4S5 alanine mutants is shown in Fig. 4C and in Table 2. Fig. 4D shows a plot of the voltage dependence of the rates of deactivation for all the S4S5 alanine mutants. Mutation to the three charged residues (D540A, R541A, and E544A) all reduced the voltage dependence of the rates of deactivation. Conversely, mutations to the uncharged residues caused only modest changes to the voltage dependence of the rates of deactivation, with the notable exception of Y542A.
FIGURE 4.
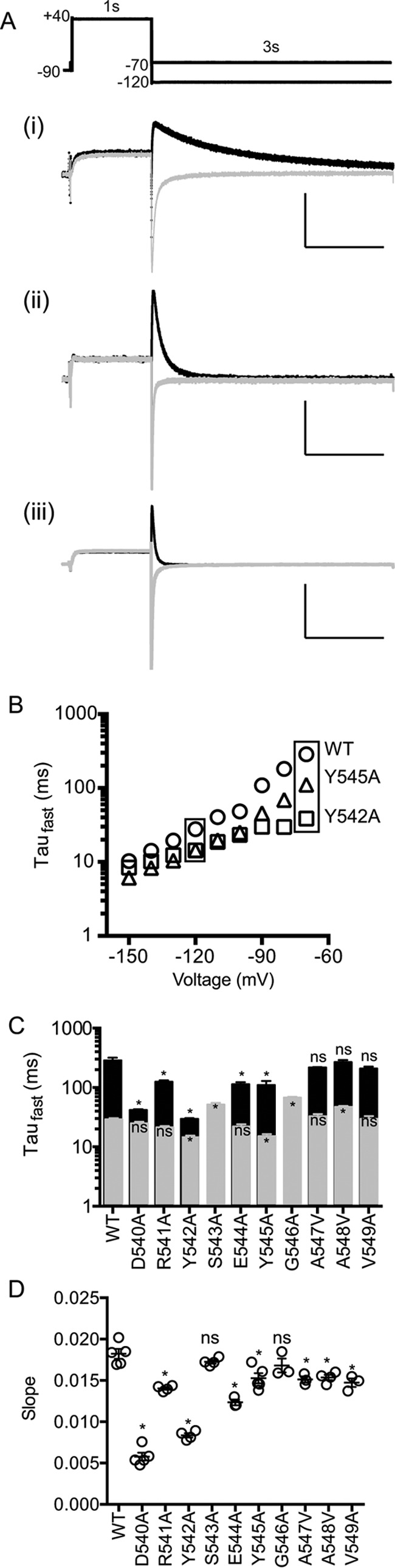
Kinetics of deactivation for S4S5 alanine mutants. A, cells were depolarized to 40 mV for 1-s and followed by repolarization to −70 and −120 mV for 3-s. Example deactivation current traces acquired at −70 (black) and −120 mV (gray) for WT (panel i), Y545A (panel ii), and Y542A (panel iii). x and y axes are 1 s and 0.25 μA, respectively. B, comparison of time constants of deactivation for WT (circle), Y545A (triangle), and Y542A (square) corresponding to the test voltage range of −150 to −70 mV. C, time constants for the fast component of deactivation at −70 mV (black) and −120 mV (gray) for S4S5 alanine mutants (mean ± S.E.). D, summary of the voltage dependence of the rates of deactivation (mean ± S.E.) for WT and S4S5 mutants. Statistical comparisons were made using one-way ANOVA with Bonferroni post test. Significant differences (p < 0.05) are highlighted with asterisks. The values for all mutants are summarized in Table 2.
TABLE 2.
Summary of the values for the 3-s isochronal deactivation V0.5, time constants of deactivation at −70 and −120 mV for S4S5 alanine mutants
Data are presented as mean ± S.E., and n denotes number of individual recordings. Asterisk indicates p < 0.05 versus WT using one-way ANOVA with Bonferroni post-test.
| Mutant (n) | Deactivation V0.5 (mV) | Time constant of deactivation at −120 mV (ms) | Time constant of deactivation at −70 mV (ms) |
|---|---|---|---|
| WT (5) | −61.3 ± 0.5 | 27.8 ± 1.3 | 286.2 ± 32.4 |
| D540A (5) | −25.6 ± 2.0* | 23.8 ± 1.4 | 41.7 ± 1.4* |
| R541A (4) | −46.3 ± 0.8* | 20.4 ± 0.9 | 124.9 ± 7.5* |
| Y542A (5) | −1.6 ± 1.1* | 14.3 ± 0.5* | 29.7 ± 0.7* |
| S543A (4) | −78.6 ± 0.6* | 49.3 ± 3.6* | NA |
| E544A (3) | −40.3 ± 1.4* | 21.1 ± 1.8 | 113.7 ± 9.0 * |
| Y545A (5) | −46.4 ± 1.1* | 14.4 ± 1.1* | 110.4 ± 18.6* |
| G546A (4) | −82.0 ± 0.2* | 64.7 ± 2.2* | NA |
| A547V (3) | −56.5 ± 1.1 | 31.2 ± 2.8 | 217.3 ± 3.4 |
| A548V (5) | −64.8 ± 0.6 | 45.1 ± 1.7 * | 268 ± 21.8 |
| V549A (4) | −53.3 ± 1.1* | 28.4 ± 3.1 | 208.7 ± 15.9 |
We next investigated how S4S5 mutants affected the voltage dependence of the distribution between open and closed states when starting from the open state, i.e. we measured 3-s isochronal deactivation curves to derive the V0.5 of steady-state deactivation (24). Fig. 5A shows WT Kv11.1 channels recorded from a 3-s isochronal deactivation protocol. The midpoints of the voltage dependence of deactivation of V0.5 derived by fitting the normalized amplitude of the −70 mV tail currents for WT (circle), Y542A (square), and Y545A (triangle) using Boltzmann function (Equation 1) were −61.3 ± 0.5 mV (n = 5), −1.6 ± 1.1 mV (n = 5), and −46.4 ± 1.1 mV (n = 5), respectively (Fig. 5B). Y542A caused the largest depolarizing shift in the V0.5 of deactivation of all the S4S5 alanine mutants, whereas Y545A caused a more modest but still statistically significant depolarizing shift in the V0.5 of deactivation (Fig. 5C).
FIGURE 5.
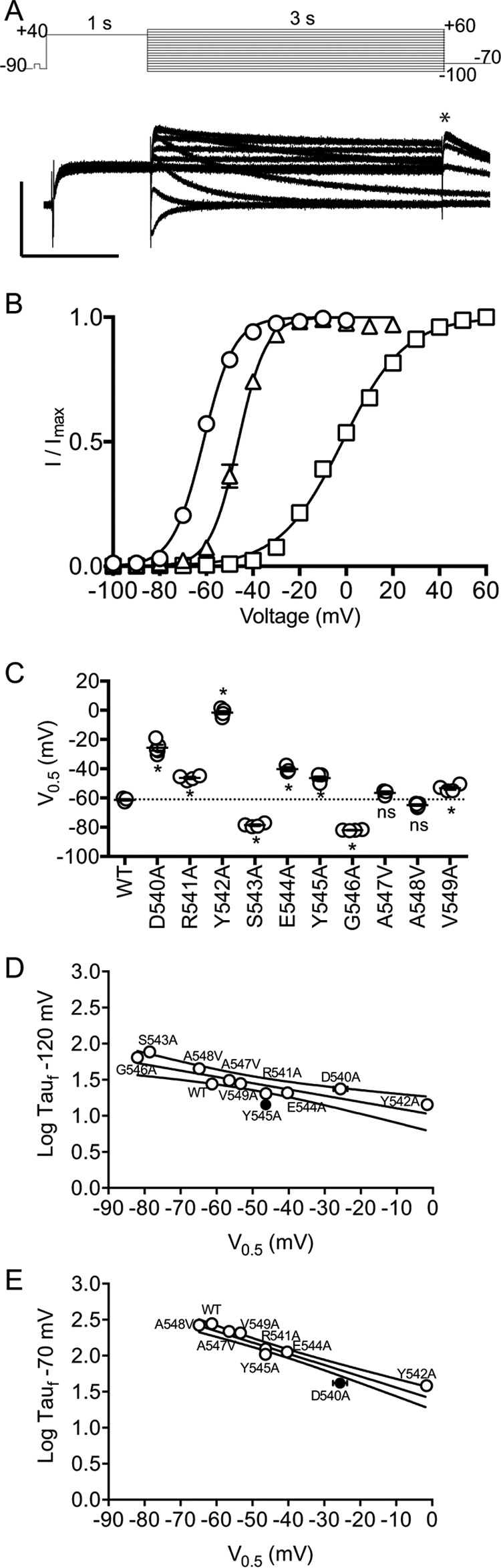
Voltage dependence of the distribution between open and closed states for S4S5 mutants. A, 3-s isochronal deactivation protocol and example traces for WT. x and y axes are 1-s and 0.25 μA, respectively. B, peak tail currents (*) derived from 3-s isochronal deactivation were fitted with Boltzmann equation to derive V0.5 values. The V0.5 values are as follows: WT, −61.3 ± 0.5 mV (mean ± S.E., n = 5; circle); Y545A, −46.4 ± 1.1 mV (mean ± S.E., n = 5; triangle); and Y542A, −1.6 ± 1.1 mV (mean ± S.E., n = 5; square). C, summary of the deactivation V0.5 of all S4S5 alanine mutants. * indicates p < 0.05 versus WT using one-way ANOVA with Bonferroni post-test. The 3-s isochronal deactivation V0.5 values for all mutants are summarized in Table 2. D and E, relationship between time constants of deactivation for S4S5 alanine mutants at −120 and −70 mV, respectively, and their corresponding deactivation V0.5 values from C. Linear regression was applied (straight line) with 95% confidence interval (CI) of the fit. Residues outside the 95% confidence interval are highlighted as filled circles.
Perturbations to the rates of deactivation in the S4S5 alanine mutants could be caused by either a change in the electrochemical driving force for deactivation and/or due to a change in the transition state energy barrier separating the open and closed states, i.e. there can be thermodynamic and/or kinetic components. To explore these two components, we have plotted the time constants for the fast component of deactivation at −120 and −70 mV versus the V0.5 for 3-s isochronal deactivation for all S4S5 alanine mutants (Fig. 5, D and E). When kinetics were measured at −120 mV, most of the alanine mutants, with the notable exception of Y545A, reside within the 95% confidence interval obtained from a regression line fitted to the rate of deactivation versus isochronal deactivation V0.5 data. This suggests that much of the change in the rates of deactivation at −120 mV for these S4S5 alanine mutants can be explained by changes in the thermodynamics of deactivation gating. Conversely, for Y545A, the rate of deactivation is much faster than would have expected from the simple change in the V0.5 of deactivation. Consistent with the data at −120 mV, the change in the rates of deactivation at −70 mV for the S4S5 alanine mutants can also be explained by changes in the thermodynamics of deactivation gating, except for D540A and Y545A, whose mean values lie outside of the 5–95% confidence intervals. Note that S543A and G546A were excluded from analysis as these mutant channels did not deactivate at −70 mV. Our data show that whereas Y542A clearly affected the thermodynamics of deactivation gating, Y545A appeared to have a significant kinetic component on top of the alteration to the thermodynamics of deactivation gating.
As alanine mutagenesis of the two tyrosine residues appear to have quite distinct phenotypes in terms of effects on activation/deactivation, both resulted in accelerated rates of deactivation with minimal perturbation on the rate of activation. We thus focus our investigation on the deactivation phenotypes of Tyr-545 and Tyr-542. Each tyrosine residue was mutated to Ala, Leu, Ile, Phe, or Trp. All of the Tyr-542 and Tyr-545 mutants resulted in accelerated rates of deactivation, with the exception of Y545F (Fig. 6A). Mutations at Tyr-542 tended to cause greater perturbations to the V0.5 of deactivation than mutations at Tyr-545 (Fig. 6B). A plot of the perturbation to the rate of deactivation (measured at −70 mV) versus the change in V0.5 of deactivation, for the groups of mutants at Tyr-542 and Tyr-545, highlights that mutations to Tyr-542 predominantly affect the thermodynamics of channel deactivation, whereas mutations to Tyr-545 cause relatively more perturbation to the kinetics of deactivation gating (Fig. 6C).
FIGURE 6.
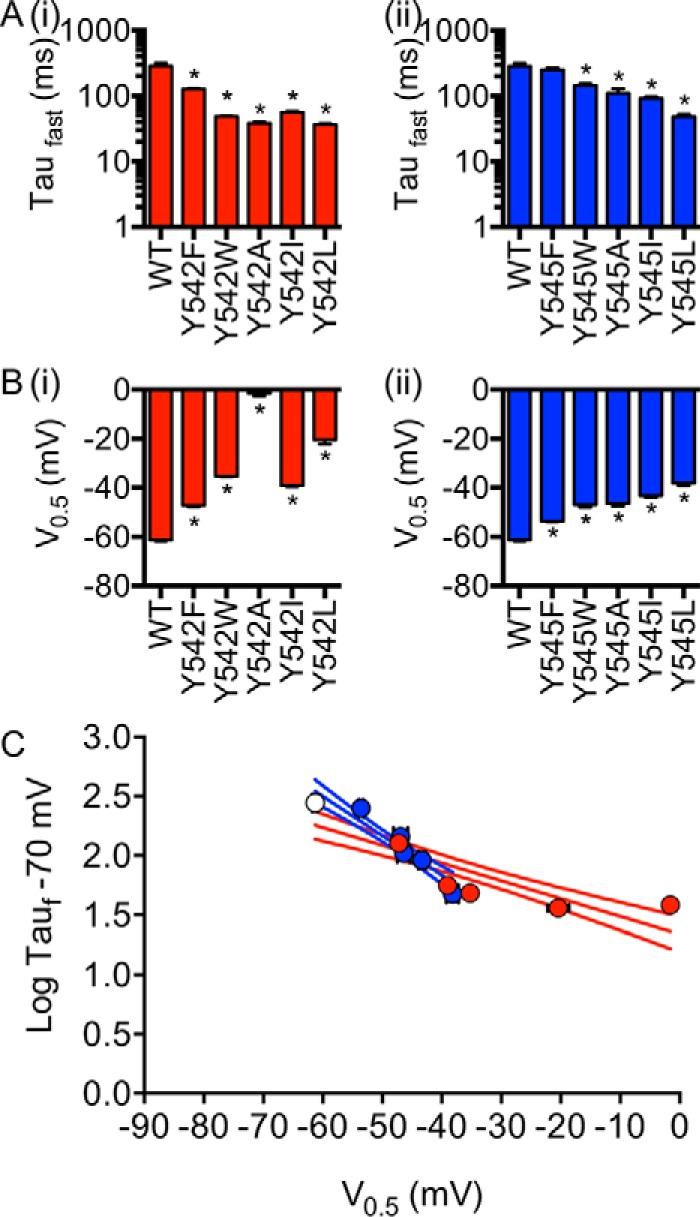
Tyr-545 (blue) is functionally different to Tyr-542 (red). A, time constants of deactivation at −70 mV for different Tyr-542 (panel i) and Tyr-545 (panel ii) side-chain residues. B, 3-s isochronal deactivation V0.5 for Tyr-542 (panel i) and Tyr-545 (panel ii) side-chain residues. Data are presented as mean ± S.E. * indicates p < 0.05 versus WT using one-way ANOVA with Bonferroni post-test. C, relationship between time constants of deactivation for different WT (white), Tyr-542 (red), and Tyr-545 (blue) side-chain residues at −70 mV and their corresponding deactivation V0.5. Linear regression with 95% confidence interval was fitted to each dataset.
As mutations to Tyr-542 significantly perturbed the voltage dependence of the equilibrium distribution of the open and closed states, we probed for residues that might interact with Tyr-542 in the open or closed state by using double mutant cycle analysis. Based on our homology model (Fig. 7A), Tyr-542 lies in close proximity to Val-535 and Arg-537 in S4 and Ile-560 in S5. Fig. 7B, panel i, shows the 3-s isochronal deactivation curves for WT (black circle), I560A (black square), Y542A (white circle), and I560A/Y542A (gray circle). I560A is WT-like, whereas Y542A has a large depolarizing shift. The double mutant I560A/Y542A has an intermediate phenotype between I560A and Y542A. The method for calculating interaction energies and a plot of the ΔΔG0 values for I560A, Y542A, and I560A/Y542A are shown in Fig. 7B, panel ii. The ΔΔG0 value for I560A/Y542A (Fig. 7B panel ii, black bar) is less than the sum of the ΔΔG0 values for the two individual mutants, indicative of an energetic interaction between these two residues. In addition to Ile-560, the double mutant cycle analysis indicates that Tyr-542 is energetically coupled to Val-535 and Arg-537 (Fig. 7C). In contrast, double mutant cycle analysis of Y545A with three residues (Ile-560, Ser-654, and Asn-658) did not reveal any significant energetic interactions (Fig. 7C). A summary of the experimental ΔΔG0 values for all single and double mutants and their interaction energies are shown in Table 3.
FIGURE 7.
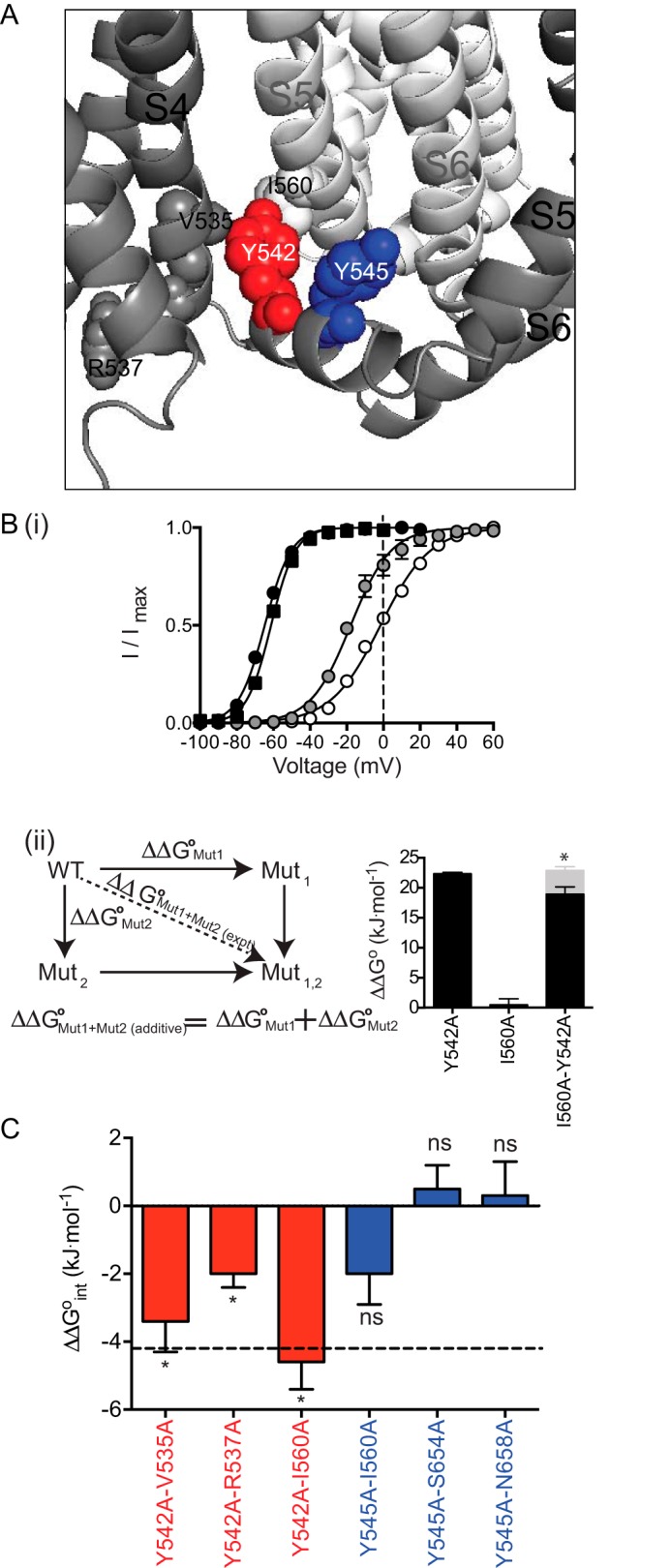
Double mutant cycle analysis of Tyr-542 and Tyr-545. A, orientation of Tyr-542 (red) and Tyr-545 (blue) within the S4S5 linker and the residues that were energetically coupled. B, 3-s isochronal deactivation curves for WT (black circle), I560A (black square), Y542A (white circle), and I560A/Y542A (gray circle). 0 mV was indicated by dotted lines to show the differences between the individual and double mutations (panel i). The additivity effect ΔΔG0Mut1 + Mut2 (additive) is the summation of ΔΔG0 from the individual mutations (panel ii). Comparison between the ΔΔG0 obtained from experiment (black) and additivity effect (gray) for double mutant I560A and Y542A are shown on the right of panel ii. C, summary of the double mutant cycle analysis displayed as the ΔΔG0Mut1 + Mut2 (int) for the Tyr-542 double mutants (red) and Tyr-545 double mutants (blue). * indicates statistical differences, p < 0.05 using an unpaired t test when comparing the experimentally derived ΔΔG0 for the double mutant with the sum of the ΔΔG0 values for the two individual mutants. Only the double mutant I560A/Y542A has an interaction energy greater than 4.2 kJ mol−1, indicated by the dotted line. Data are presented as mean ± S.E.
TABLE 3.
Summary of the values for the ΔΔG0 (mean ± S.E.) and n denotes number of individual recordings
Asterisk means data are statistically different from their corresponding additive effect.
| Mutant (n) | ΔG0(exp) | ΔΔG0(Mut − WT) | ΔΔG0(Mut1 + Mut2) − (ΔΔG0(Mut1) + ΔΔG0 (Mut2)) |
|---|---|---|---|
| kJ·mol−1 | kJ·mol−1 | kJ·mol−1 | |
| WT(5) | −22.7 ± 0.6 | ||
| Y542A(5) | −0.3 ± 0.2 | 22.3 ± 0.2 | |
| V535A(3) | −20.8 ± 1.8 | 1.8 ± 1.8 | |
| R537A(4) | −20.1 ± 0.6 | 2.6 ± 0.6 | |
| I560A(3) | −22.1 ± 1.0 | 0.5 ± 1.0 | |
| Y542A/V535A(5) | −1.8 ± 0.1 | 20.8 ± 0.1 | −3.4 ± 0.9* |
| Y542A/R537A(3) | 0.3 ± 0.1 | 22.9 ± 0.1 | −2.0 ± 0.4* |
| Y542A/I560A(4) | −4.4 ± 1.1 | 18.2 ± 1.1 | −4.6 ± 0.8* |
| Y545A(5) | −19.5 ± 0.6 | 3.2 ± 0.6 | |
| I560A(3) | −22.1 ± 1.0 | 0.5 ± 1.0 | |
| S654A(4) | −23.7 ± 1.0 | −1.1 ± 1.0 | |
| N658A(4) | −17.2 ± 1.1 | 5.5 ± 1.1 | |
| Y545A/I560A(4) | −21.0 ± 1.2 | 1.7 ± 1.2 | −2.0 ± 0.9 |
| Y545A/S654A(3) | −20.1 ± 0.4 | 2.6 ± 0.4 | 0.5 ± 0.7 |
| Y545A/N658A(4) | −13.7 ± 1.2 | 8.9 ± 1.2 | 0.3 ± 1.0 |
Discussion
Kv11.1 channels have unusual kinetics, which include rapid recovery from inactivation and slow deactivation. In the heart, the slow deactivation kinetics play a particularly important role in the suppression of propagation of ectopic beats that occur during the early diastolic interval (6). The S4S5 linker clearly plays a critical role in deactivation gating of voltage-gated K+ channels, including Kv11.1 channels (12, 15, 16). Here, we have shown that two tyrosine residues in the S4S5 linker, Tyr-542 and Tyr-545, play important, but distinct, roles in stabilizing the open state of the Kv11.1 channel and thus in mediating the slow deactivation gating of Kv11.1 channels.
In this study, mutations to Tyr-542 resulted in large shifts in the voltage dependence of the equilibrium distribution between the open and closed states, with all mutants shifting the equilibrium in favor of the closed state (Fig. 6B, panel i). All Tyr-542 mutants also resulted in an acceleration of the rates of deactivation (Fig. 6A, panel i). These results are consistent with a role for Tyr-542 in stabilizing the open state of the channel. Mutating Tyr-542 to alanine caused a marked acceleration of the rates of deactivation with a shallow slope (Fig. 4D). The fact that the slope of the voltage dependence of rates of deactivation for Y542A is much shallower than that of WT or Y545A suggests that the native Tyr-542 interacts with, and so influences, the motion of the VSD during deactivation. This conclusion is consistent with our mutant cycle analysis experiments indicating an energetic interaction between Tyr-542 and Arg-537, as well as with Val-535 and Ile-560 (Fig. 7), although only the interaction with Ile-560 exceeds the 1 kcal mol−1 threshold that is often used to indicate biologically relevant interactions (25–30).
In contrast to Tyr-542, mutations to Tyr-545 caused less perturbation to the voltage dependence of the equilibrium distribution between the open and closed states (Fig. 6B, panel ii) but still caused a marked acceleration of the rates of deactivation (Fig. 6A, panel ii). This suggests that Tyr-545 might form important interactions that stabilize the transition state complex; however, these interactions are destabilized by mutations, i.e. lower the energy barrier for channel deactivation. This suggestion is also consistent with the thermodynamic mutant cycle analysis experiments, which showed that there were no significant energetic interactions involving Tyr-545 and other residues in the stable end states. However, at this stage we cannot determine the nature of such interactions. They could be with transmembrane domain residues and/or with the membrane lipids. Given that the NMR experiments suggest that the peptide mainly interacts with the surface of the membrane (because there were no changes in ΔνQ of 2H spectra), if there are any specific interactions between Tyr-545 and the membrane, these would most likely involve interactions with the lipid headgroups.
The S4S5 linker is thought to be essential to transmit conformational changes of the voltage sensor to the pore domain, as evidenced by its proximity to the S6 helix in the crystal structure of the Kv1.2 channel, as well as its direct interaction with the S6 domain (16). Previous studies of the S4S5 linker in Kv11.1 channels have focused on its role in linking the voltage sensor domain to the activation gate at the intracellular end of the pore domain (11, 16), as well as its role in mediating interactions with cytoplasmic domains that can also modulate deactivation gating (13). Our NMR studies of a peptide that contains the Kv11.1 S4S5 linker in a model membrane indicate that both Tyr-542 and Tyr-545 are likely to point toward the membrane, which is consistent with the homology model based on the structure of the activated Kv1.2/2.1 channel (see Fig. 7A). This suggests that the S4S5 linker could also have important interactions with the membrane directly and/or with residues in the transmembrane domains of the channel. We have clear evidence that Tyr-542 interacts with other transmembrane domain residues. However, the relatively small magnitude of this interaction (e.g. −4.6 kJ mol−1 for Y542A/I560A) suggests that other factors might be involved, which could include interactions between Tyr-542 and the membrane. Tyr-545 interactions with the membrane contributing to the stabilization of the transition state in the pathway connecting the activated state with the closed states are also plausible, but we do not yet have definitive proof of such interactions.
Recently, Lorinczi et al. (18) showed that cutting the S4S5 linker of Kv11.1 channels in half could still produce functional channels with normal activation but accelerated deactivation. This indicates that the major role of S4S5 linker is to stabilize the open state, and it is conceivable that the split S4S5 linker in the Kv11.1 prevents the interaction promoted by the S4S5 linker with other parts of the channel in the activated state. It is also possible that the split S4S5 may disrupt its interaction with the lipid membrane. It is unlikely that a split S4S5 linker would maintain a continuous helix that lies parallel to the membrane in the activated state of the channel (31–36). Our functional studies provide complementary information that is consistent with recent findings (18) by showing that the two conserved tyrosine residues play important but distinct roles in stabilizing the open state of Kv11.1 channels.
Experimental Procedures
NMR Spectroscopy
Materials
Peptide that contains the S4S5 linker (LVRVARKLDRYSEYGAVLF) was synthesized by GL Biochem Ltd. (Shanghai, China) with >98% purity. Protonated and deuterated dimyristoylphosphatidylcholine (DMPC, DMPC-d54) were purchased from Avanti Polar Lipids (Alabaster, AL), and deuterated dodecylphosphocholine (DPC-d38), deuterium-depleted water, and diethylenetriaminepentaacetic acid gadolinium (III) dihydrogen salt hydrate were purchased from Sigma. Deuterium oxide (D2O) was obtained from CDN Isotopes (Pointe-Claire, Quebec, Canada). Phosphatidylcholines are abundantly found in biological membranes; consequently, DMPC is frequently used in model membranes, although DPC has been previously used to determine the Kv11.1 S4S5 linker structure by NMR (12). DMPC/DPC bicelles are a more biologically relevant alternative and have recently been introduced as a new membrane mimicking system for the NMR analysis of membrane proteins (37).
NMR Sample Preparation
Bicelle and MLV samples used for solution- and solid-state NMR, respectively, were prepared by mixing freeze-dried DMPC and/or DPC in water. The mixture was submitted to a series of at least three freeze (liquid N2)/thaw (50 °C)/vortex shaking cycles. In all experiments, the peptide concentration was kept between 2 and 5 mm, and the lipid concentration was maintained well above the critical micelle concentration of the lipids. To ensure the stability of the model membranes, the hydration percentages were kept at 75% for solid-state NMR and 90% for high resolution NMR using deuterium-depleted water and 10% D2O, respectively. Solution NMR experiments used lipid molar ratios (q = DMPC/DPC) of 0.25 and 1, as well as a lipid/peptide (L/P) molar ratio ranging from of 50:1 to 58:1. Solid-state NMR samples were prepared at a L/P molar ratio of 100:1.
NMR Experiments
All experiments, except high resolution solution experiments, were recorded on a hybrid solution/solid-state Varian Inova Unity 600 (Agilent Technologies, Mississauga, Ontario, Canada) spectrometer operating at frequencies of 599.95 MHz for 1H, 246.86 MHz for 31P, and 92.125 MHz for 2H. Solid-state NMR experiments used a 4-mm broadband/1H dual-frequency magic-angle spinning probehead. The 1H chemical shifts were internally referenced by adding 0.5 mm of 2,2-dimethyl-2-silapentane-5-sulfonic acid set to 0.0 ppm, and 31P NMR spectra were externally referenced with respect to the signal of 85% phosphoric acid set to 0 ppm. All data were processed using MestReNova (Mestrelab Research, Santiago de Compostela, Spain).
1H total correlation spectroscopy (TOCSY) spectra were recorded on a hybrid solution/solid-state Bruker Avance III-HD narrow bore spectrometer (Milton, Ontario, Canada) operating at a frequency of 599.95 MHz for 1H with a broadband double resonance 5-mm probe. A 90° pulse of 13 μs and spin-lock time of 80 ms with MLEV17 spin-lock field of 10 kHz were employed (38). A spectral width of 6 kHz was used in both dimensions with 2048 and 256 complex data points in the direct and indirect dimensions, respectively. A total of 40 transients were accumulated with a repetition delay of 2 s.
31P NMR spectra were recorded using a phase-cycled Hahn echo pulse sequence with gated broadband proton continuous wave decoupling at a field strength of 20 kHz (39). A 90° pulse length of 13 μs was used with interpulse delays of 33 μs. Typically 1024 scans were acquired with a recycle delay of 5 s. The acquisition time was set at 10 ms with a 5-μs dwell time. 2H NMR spectra were obtained using a solid-echo pulse sequence (40) with a 90° pulse length of 2.8 μs, interpulse and repetition delays of 20 μs and 0.5 s, respectively. At least 5000 data points were obtained, and typically 2048 scans were acquired with an acquisition time of 5 ms and 1-μs dwell time.
Electrophysiology Methods
We chose to use Xenopus oocytes for our electrophysiological recordings as this system enables high level expression of almost all channels, including mutant channels that result in expression defects in mammalian systems (41).
Molecular Biology
The cDNA of Kv11.1 (a gift from Dr. Gail Robertson, University of Wisconsin) was subcloned into a pBluescript vector, which contains the 5′-untranslated region (UTR) and 3′ UTR of the Xenopus laevis β-globin gene (a gift from Dr. Robert Vandenberg, University of Sydney). Site-directed mutagenesis of Kv11.1 cDNA was performed using the QuikChange method (Agilent Technologies, Mulgrave, Victoria, Australia), and mutations were confirmed by Sanger DNA sequencing. WT and mutant channel cDNAs were linearized with BamHI-HF (New England Biolabs, Ipswich, MA), and cRNA was transcribed with T7 RNA polymerase using the mMessage mMachine kit (Ambion, Austin, TX).
Oocyte Preparation
Female X. laevis frogs were purchased from Nasco (Fort Atkinson, WI). The Garvan/St Vincent's Animal Ethics Committee granted approval (ID 14/30) for all animal experiments described in this study. Frogs were anesthetized by immersion in 0.17% w/v Tricaine. One ovarian lobe was mobilized, and ∼5 ml were removed. The follicular cell layer was digested by using 1 mg/ml collagenase A (Roche Applied Science) in Ca2+-free ND96 solution containing (in mm): NaCl 96, KCl 2, MgCl2 1.0 and Hepes 5 (pH adjusted to 7.5 with 5 m NaOH) for ∼2 h. After rinsing with ND96 (as above, plus 1.8 mm CaCl2) to remove collagenase A, stage V and VI oocytes were isolated and stored at 18 °C in ND96 supplemented with 2.5 mm pyruvic acid sodium salt, 0.5 mm theophylline, and 50 μg/ml gentamicin. X. laevis oocytes were injected with cRNA and incubated at 18 °C for 24–48 h prior to electrophysiological recordings.
Electrophysiology
Two-electrode voltage-clamp experiments were performed at room temperature (∼21 °C) using a Geneclamp 500B amplifier (Molecular Devices Corp., Sunnyvale, CA). We used glass microelectrodes with tip resistances of 0.3–1.0 megohms, when filled with 3 m KCl. Oocytes were perfused with ND96 solution (see above) during all experiments. A 50-ms 20-mV depolarization step from the holding potential of −90 mV was applied at the start of each sweep to enable off-line leak-current subtraction. We assumed that the current leakage was linear in the voltage range −150 to +40 mV. Data acquisition and analysis were performed using pCLAMP software (Version 10.2, Molecular Devices, Sunnyvale, CA), Excel software (Microsoft, Seattle), and Prism 6 (GraphPad Software Inc., La Jolla, CA). All parameter values are reported as means ± S.E. of the mean (S.E.) for n experiments, where n denotes the number of different oocytes studied for each construct.
Data Analysis
To measure the voltage dependence of activation, cells were depolarized from −70 to +60 mV for 3 s (exact voltage range depending on the mutant tested) followed by a step to −70 mV to measure tail current amplitude. Tail current amplitudes were normalized to the maximum tail current value and fitted with a Boltzmann function as shown in Equation 1,
where I/Imax is the relative tail current amplitude; V0.5 is the voltage at which 50% of channels are deactivated; Vt is the test potential, and k is the slope factor. To measure rates of activation, an envelope of tail protocol was used to obtain the rates of activation at 20 mV (42). The peak tail current amplitudes recorded at −70 mV using different activation duration at 20 mV were fitted with a single exponential function to obtain a time constant for activation.
Rates of deactivation were measured from the hooked tail current traces recorded at voltages in the range −70 to −150 mV, following a 1-s depolarization step to 40 mV from a holding potential of −90 mV. The decaying phase of the current traces was fitted with a double exponential component to derive both components of channel deactivation. However, only the fast component is reported in this study. To measure the voltage dependence of deactivation, cells were depolarized to +40 mV for 1 s to ensure channels were fully activated and then stepped to voltages between +60 and −100 mV (exact voltage range depending on the mutant tested) for 3 s, followed by a step to −70 mV to measure tail current amplitude (23, 24, 43). Tail current amplitudes were normalized to the maximum tail current value and fitted with a Boltzmann function using Equation 1. In experiments in which the free energies associated with voltage-dependent deactivation were examined, the same datasets were fitted with the thermodynamic form of the Boltzmann distribution as shown in Equation 2,
where ΔG0 is the work done at 0 mV; zg is the effective number of gating charges moving across the membrane electric field (E); F is the Faraday constant; R is the universal gas constant; and T is the absolute temperature. From Equation 2 we can also calculate the effect of a mutation on ΔG0 as shown in Equation 3,
Double Mutant Cycle Analysis
To investigate whether two residues interact to stabilize the open and/or closed states we used thermodynamic mutant cycle analysis. The interaction energy, ΔΔG0int, between two mutants was calculated as the difference in free energy of the double mutant compared with the sum of the free energy differences of the individual mutants shown in Equation 4,
Absolute ΔΔG0int values of greater than 4.2 kJ mol−1 (i.e. 1 kcal mol−1) were considered significant (25–30).
Statistical Analysis
We used one-way ANOVA with Bonferroni post-test analysis to compare rates of deactivation and V0.5 of deactivation or an unpaired t test when comparing the ΔΔG0 values of double mutants (ΔΔG0Mut1 + Mut2) compared with the sum of ΔΔG0 values of individual mutants (ΔΔG0Mut1 +ΔΔG0Mut2) in double mutant cycle analysis experiments. p values <0.05 were considered significant.
Homology Modeling
The crystal structure of a Kv1.2/2.1 channel chimera (19) was used to generate the homology model of Kv11.1 using Swiss PdbViewer (44), and the final structure was optimized by using SWISS-MODEL Workspace (45, 46).
Author Contributions
J. I. V. conceived and coordinated the study. C. A. N., A. E. G., I. M., and J. I. V. wrote the paper. C. A. N. designed, performed, and analyzed the experiments shown in Figs. 1 and 3–7. A. E. G. and I. M. designed, performed, and analyzed the experiments shown in Fig. 2. M. D. P. and A. A. A. assisted with the design of experiments and acquisition of data. All authors contributed to the preparation of the figures and approved the final version of the manuscript.
This work was supported in part by National Health and Medical Research Council of Australia Program Grant App1074386 and Fellowship 1019693 (to J. I. V.). The authors declare that they have no conflicts of interest with the contents of this article.
- VSD
- voltage sensor domain
- WT
- wild-type
- NMR
- nuclear magnetic resonance
- DMPC
- dimyristoylphosphatidylcholine
- DPC
- dodecylphosphocholine
- MLV
- multilamellar vesicle
- TOCSY
- total correlation spectroscopy
- ANOVA
- analysis of variance.
References
- 1. Klabunde R. E. (2012) Cardiovascular Physiology Concepts, 2nd Ed., pp. 9–40, Lippincott Williams & Wilkins/Wolters Kluwer, Baltimore [Google Scholar]
- 2. Grant A. O. (2009) Cardiac ion channels. Circulation 2, 185–194 [DOI] [PubMed] [Google Scholar]
- 3. Warmke J. W., and Ganetzky B. (1994) A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trudeau M. C., Warmke J. W., Ganetzky B., and Robertson G. A. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95 [DOI] [PubMed] [Google Scholar]
- 5. Sanguinetti M. C., Jiang C., Curran M. E., and Keating M. T. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 [DOI] [PubMed] [Google Scholar]
- 6. Smith P. L., Baukrowitz T., and Yellen G. (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836 [DOI] [PubMed] [Google Scholar]
- 7. Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., and Hill A. P. (2012) hERG K+ channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 [DOI] [PubMed] [Google Scholar]
- 8. Lu Y., Mahaut-Smith M. P., Varghese A., Huang C. L., Kemp P. R., and Vandenberg J. I. (2001) Effects of premature stimulation on HERG K+ channels. J. Physiol. 537, 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piper D. R., Varghese A., Sanguinetti M. C., and Tristani-Firouzi M. (2003) Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 10534–10539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith P. L., and Yellen G. (2002) Fast and slow voltage sensor movements in HERG potassium channels. J. Gen. Physiol. 119, 275–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferrer T., Rupp J., Piper D. R., and Tristani-Firouzi M. (2006) The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-a-go-go-related gene (hERG) K+ channel. J. Biol. Chem. 281, 12858–12864 [DOI] [PubMed] [Google Scholar]
- 12. Ng C. A., Perry M. D., Tan P. S., Hill A. P., Kuchel P. W., and Vandenberg J. I. (2012) The S4-S5 linker acts as a signal integrator for hERG K+ channel activation and deactivation gating. PLoS ONE 7, e31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de la Peña P., Alonso-Ron C., Machín A., Fernández-Trillo J., Carretero L., Domínguez P., and Barros F. (2011) Demonstration of physical proximity between the N terminus and the S4-S5 linker of the human ether-a-go-go-related gene (hERG) potassium channel. J. Biol. Chem. 286, 19065–19075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Slyke A. C., Rezazadeh S., Snopkowski M., Shi P., Allard C. R., and Claydon T. W. (2010) Mutations within the S4-S5 linker alter voltage sensor constraints in hERG K+ channels. Biophys. J. 99, 2841–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alonso-Ron C., de la Peña P., Miranda P., Domínguez P., and Barros F. (2008) Thermodynamic and kinetic properties of amino-terminal and S4–S5 loop HERG channel mutants under steady-state conditions. Biophys. J. 94, 3893–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tristani-Firouzi M., Chen J., and Sanguinetti M. C. (2002) Interactions between S4–S5 linker and S6 transmembrane domain modulate gating on HERG K+ channels. J. Biol. Chem. 277, 18994–19000 [DOI] [PubMed] [Google Scholar]
- 17. Sanguinetti M. C., and Xu Q. P. (1999) Mutations of the S4–S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J. Physiol. 514, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lörinczi É., Gómez-Posada J. C., de la Peña P., Tomczak A. P., Fernández-Trillo J., Leipscher U., Stühmer W., Barros F., and Pardo L. A. (2015) Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat. Commun. 6, 6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Long S. B., Tao X., Campbell E. B., and MacKinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382 [DOI] [PubMed] [Google Scholar]
- 20. Gravel A. E., Arnold A. A., Dufourc E. J., and Marcotte I. (2013) An NMR investigation of the structure, function and role of the hERG channel selectivity filter in the long QT syndrome. Biochim. Biophys. Acta 1828, 1494–1502 [DOI] [PubMed] [Google Scholar]
- 21. Seelig J., and Seelig A. (1980) Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 13, 19–61 [DOI] [PubMed] [Google Scholar]
- 22. Ju P., Pages G., Riek R. P., Chen P. C., Torres A. M., Bansal P. S., Kuyucak S., Kuchel P. W., and Vandenberg J. I. (2009) The pore domain outer helix contributes to both activation and inactivation of the HERG K+ channel. J. Biol. Chem. 284, 1000–1008 [DOI] [PubMed] [Google Scholar]
- 23. Tan P. S., Perry M. D., Ng C. A., Vandenberg J. I., and Hill A. P. (2012) Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. J. Gen. Physiol. 140, 293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ng C. A., Ke Y., Perry M. D., Tan P. S., Hill A. P., and Vandenberg J. I. (2013) C-terminal β9-strand of the cyclic nucleotide-binding homology domain stabilizes activated states of Kv11.1 channels. PLoS ONE 8, e77032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Serrano L., Bycroft M., and Fersht A. R. (1991) Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J. Mol. Biol. 218, 465–475 [DOI] [PubMed] [Google Scholar]
- 26. Horovitz A., Serrano L., Avron B., Bycroft M., and Fersht A. R. (1990) Strength and co-operativity of contributions of surface salt bridges to protein stability. J. Mol. Biol. 216, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 27. Waldburger C. D., Schildbach J. F., and Sauer R. T. (1995) Are buried salt bridges important for protein stability and conformational specificity? Nat. Struct. Biol. 2, 122–128 [DOI] [PubMed] [Google Scholar]
- 28. Marqusee S., and Sauer R. T. (1994) Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in λ repressor. Protein Sci. 3, 2217–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tissot A. C., Vuilleumier S., and Fersht A. R. (1996) Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry 35, 6786–6794 [DOI] [PubMed] [Google Scholar]
- 30. Loewenthal R., Sancho J., and Fersht A. R. (1992) Histidine-aromatic interactions in barnase. Elevation of histidine pKa and contribution to protein stability. J. Mol. Biol. 224, 759–770 [DOI] [PubMed] [Google Scholar]
- 31. Zubcevic L., Herzik M. A. Jr., Chung B. C., Liu Z., Lander G. C., and Lee S. Y. (2016) Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 23, 180–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Long S. B., Campbell E. B., and Mackinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family β channel. Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 33. Cao E., Liao M., Cheng Y., and Julius D. (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zalk R., Clarke O. B., des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., and Marks A. R. (2015) Structure of a mammalian ryanodine receptor. Nature 517, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., and Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Payandeh J., Scheuer T., Zheng N., and Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nolandt O. V., Walther T. H., Grage S. L., and Ulrich A. S. (2012) Magnetically oriented dodecylphosphocholine bicelles for solid-state NMR structure analysis. Biochim. Biophys. Acta 1818, 1142–1147 [DOI] [PubMed] [Google Scholar]
- 38. Bax A., and Davis D. G. (1985) MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65, 355–360 [Google Scholar]
- 39. Rance M., and Byrd R. A. (1983) Obtaining high-fidelity spin-1/2 powder spectra in anisotropic media: phase-cycled Hahn echo spectroscopy. J. Magn. Reson. 52, 221–240 [Google Scholar]
- 40. Davis J. H., Jeffrey K. R., Bloom M., Valic M. I., and Higgs T. P. (1976) Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 42, 390–394 [Google Scholar]
- 41. Perry M. D., Ng C. A., Phan K., David E., Steer K., Hunter M. J., Mann S. A., Imtiaz M., Hill A. P., Ke Y., and Vandenberg J. I. (2016) Rescue of protein expression defects may not be enough to abolish the pro-arrhythmic phenotype of long QT type 2 mutations. J. Physiol. 10.1113/JP271805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu S., Rasmusson R. L., Campbell D. L., Wang S., and Strauss H. C. (1996) Activation and inactivation kinetics of an E-4031-sensitive current from single ferret atrial myocytes. Biophys. J. 70, 2704–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ng C. A., Phan K., Hill A. P., Vandenberg J. I., and Perry M. D. (2014) Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J. Biol. Chem. 289, 25822–25832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 45. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., and Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 46. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]