

Abstract
The cellular transport of the cofactor heme and its biosynthetic intermediates such as protoporphyrin IX is a complex and highly coordinated process. To investigate the molecular details of this trafficking pathway, we created a synthetic lesion in the heme biosynthetic pathway by deleting the gene HEM15 encoding the enzyme ferrochelatase in S. cerevisiae and performed a genetic suppressor screen. Cells lacking Hem15 are respiratory-defective because of an inefficient heme delivery to the mitochondria. Thus, the biogenesis of mitochondrial cytochromes is negatively affected. The suppressor screen resulted in the isolation of respiratory-competent colonies containing two distinct missense mutations in Nce102, a protein that localizes to plasma membrane invaginations designated as eisosomes. The presence of the Nce102 mutant alleles enabled formation of the mitochondrial respiratory complexes and respiratory growth in hem15Δ cells cultured in supplemental hemin. Respiratory function in hem15Δ cells can also be restored by the presence of a heterologous plasma membrane heme permease (HRG-4), but the mode of suppression mediated by the Nce102 mutant is more efficient. Attenuation of the endocytic pathway through deletion of the gene END3 impaired the Nce102-mediated rescue, suggesting that the Nce102 mutants lead to suppression through the yeast endocytic pathway.
Keywords: endocytosis, heme, membrane transport, mitochondria, mitochondrial metabolism, Nce102, eisosomes, ferrochelatase
Introduction
Heme is an essential cofactor in eukaryotes and bacteria and functions in a myriad of pathways including oxygen transport, nitric oxide and carbon monoxide sensing, oxygenase reactions, and electron transport reactions (1–3). The heme biosynthetic pathway in eukaryotes, including the yeast Saccharomyces cerevisiae, is a complex process involving eight enzymes (see Fig. 1A) (4). The process starts in the mitochondrial matrix with the condensation of glycine with succinyl-CoA to produce ∂-aminolevulinic acid (ALA)3 by the enzyme 5-aminolevulinate synthase (Hem1 is the yeast designation) (2, 4). ALA is translocated across the mitochondrial membranes to the cytosol, where the next four enzymes in the pathway convert two ALA molecules to form the intermediate pyrrole porphobilinogen and subsequent formation of coproporphyrinogen III, which is transported back into the mitochondria for conversion to protoporphyrin IX (PPIX). Ferrous iron is inserted into PPIX by ferrochelatase (Hem15), tethered to the inner membrane and facing the matrix, to form the heme cofactor. A defect in any of heme biosynthetic enzymes leads to one of the porphyric disorders in humans, each with a unique phenotype resulting from a buildup of toxic intermediates (3).
FIGURE 1.

Short circuits in the heme biosynthetic pathway in respiring cells are not rescued by the addition of exogenous hemin in heme synthesis-deficient yeast mutants. A, depiction of the heme biosynthetic pathway in the yeast S. cerevisiae highlighting the first and the last in the eight steps involving 5-aminolevulinate synthase (Hem1) and ferrochelatase (Hem15). Although both enzymes are located in the mitochondrial matrix, the intermediates of heme synthesis including ALA (the product of Hem1) are routed to the exterior of the mitochondria, but for the last three steps of synthesis, they are routed back to the mitochondria. Descriptions of all eight enzymes in the heme biosynthetic pathway are provided in the text. B, deletion of either HEM1 or HEM15 creates heme auxotrophy and requires the addition of exogenous hemin that must reach the mitochondria for proper hemylation of mitochondrial cytochromes. C, WT and hem1Δ and hem15Δ strains were serially diluted (10× dilution series) and spotted on fermentable (glucose) and non-fermentable (glycerol/lactate) medium and incubated at 30 °C. D, SDS-PAGE and immunoblot analysis of purified mitochondria isolated from strains in C. E, HPLC analysis of hemes extracted from purified mitochondria from strains in C showing the decreased levels of heme b in the hem1Δ and hem15Δ strains. Gly/Lac, glycerol/lactate; IM, inner membrane; IMS, inner membrane space; OM, outer membrane.
The mechanism of heme transport between cellular compartments remains poorly understood (4). Nevertheless, significant inroads have been recently made utilizing the heme auxotroph Caenorhabditis elegans, resulting in the identification of heme and porphyrin transporters, including the HRG-1-related heme transporters (4, 5). These transporters have mammalian homologs with similar function and specificity. Additionally, the feline leukemia virus subgroup C receptor (FLVCR1) protein was reported as a heme exporter from erythroid precursor cells (6). An additional isoform, FLVCR1b, was reported to mediate heme export from mitochondria in erythroid cell differentiation (7). Despite these advances, the overall mechanism of intracellular transport of these tetrapyrroles remains undefined. Insoluble in water, heme will partition nonspecifically to hydrophobic proteins and membranes if left without a “carrier” to mediate transit to different cellular locations.
In the interest of exploring heme trafficking processes, we utilized a strain of S. cerevisiae lacking the terminal enzyme in heme biosynthesis, ferrochelatase encoded by HEM15, which is a heme auxotroph. This mutant strain is able to propagate by fermentation with supplemental heme, which is utilized for sterol synthesis and non-mitochondrial enzymes (8). However, the mutant cells are incompetent for respiratory metabolism because of a non-functional respiratory chain.
We screened for spontaneous mutations of hem15Δ cells that were able to utilize exogenous hemin (oxidized heme) to support the mitochondrial respiration function. We report on the isolation of two suppressor strains each with a single point mutation in the plasma membrane Nce102 protein. The protein is a component of the plasma membrane invagination structures called eisosomes or membrane compartment of arginine permease Can1 (MCC) (9–12). It has been implicated in a variety of cellular functions, including endocytosis of nutrient permeases, stress response, and control of plasma membrane transport proteins (9–12). Utilizing a combination of molecular genetics and analytical techniques, we highlight the importance of the endocytic pathway in restoring heme levels in the mitochondria.
Results
The Respiratory Defect in Heme Auxotrophic Cells Is Not Rescued by Exogenous Hemin
Deletion of the ferrochelatase HEM15 in S. cerevisiae results in heme auxotrophy as depicted in Fig. 1 (A and B). The cells become fluorescent as a result of the accumulation of porphyrin. The addition of exogenous hemin (20 μm) to the culture medium enables the hem15Δ cells to ferment and propagate with glucose as a carbon source (Fig. 1C). However, when cells are forced to utilize the mitochondrial respiratory chain by using the non-fermentable carbon sources glycerol/lactate, the cells are not viable (Fig. 1C). This respiratory defect in the presence of exogenous hemin is also observed when the gene encoding for 5-aminolevulinate synthase (HEM1) (Fig. 1C) or uroporphyrinogen decarboxylase (HEM12) was deleted (data not shown), indicating that the inability to respire in these cells is a result of the lesion in the heme biosynthetic pathway, as opposed to a loss of the specific proteins.
The respiratory growth defects of hem15Δ and hem1Δ cells grown in the presence of hemin are due to the absence of the mitochondrial electron chain complexes as seen by SDS-PAGE analysis of mitochondrial lysates. Steady-state levels of subunits to complex II (Sdh2), complex III (Cob, Rip1, Cyt1), and complex IV (Cox1, Cox2) were markedly diminished (Fig. 1D). The inner membrane space heme protein cytochrome b2 (l-lactate cytochrome c reductase) was also attenuated. For comparisons to WT levels, we analyzed cells grown in the presence and absence of hemin.
In general, the impairment in steady-state abundance of mitochondrial respiratory proteins was greater in hem15Δ compared with hem1Δ cells (Fig. 1D, compare Sdh2 in hem15Δ versus hem1Δ cells). The accumulation of the PPIX porphyrin, a pro-oxidant, in hem15Δ cells, may contribute to the enhanced mitochondrial defect. However, both strains were markedly impaired in maturation of cytochrome c oxidase (CcO), whereas the defect in two other respiratory complexes succinate dehydrogenase (complex II) and cytochrome c reductase (complex III) was less apparent. A heme deficiency was validated by quantifying total mitochondrial heme in mutant cells relative to the WT control cells. Heme was extracted from purified mitochondria using an HCl-acetone extraction protocol followed by HPLC analysis. Heme levels in the mitochondria of both hem15Δ and hem1Δ cells were 3–4-fold lower compared with WT cells even when supplemented with exogenous heme (Fig. 1E). Thus, the respiratory deficiency likely arises from inefficient delivery of exogenous heme to mitochondria.
The Respiratory Defect in hem15Δ Cells Is Suppressed by Mutations in the Plasma Membrane Protein Nce102
In an attempt to gain insights into the inefficient transport of heme to mitochondria to support formation of respiratory complexes, we screened for extragenic compensatory suppressors by plating hem15Δ cells at high cell density on glycerol/lactate-containing medium supplemented with 20 μm exogenous hemin. The selective pressure allowed for spontaneous revertants to proliferate that could more efficiently utilize the supplemental heme for mitochondrial functions. Within 3–4 days of incubation at 30 °C, colonies appeared that continued to propagate on glycerol/lactate respiratory medium.
We isolated four independent suppressor colonies and designated them S5, S15, S19, and S23 for initial characterization. Each clone exhibited significant growth on glycerol-lactate medium supplemented with hemin (Fig. 2A). These suppressors retained the intense cellular fluorescence arising from PPIX accumulation, indicating that the mode of suppression was not the elimination of PPIX that could generate harmful endogenous reactive oxygen species (Fig. 2B) (13). Three suppressor strains (S5, S19, and S23) were grown for analyses of CcO activity in purified mitochondria. The three suppressors, unlike the parental hem15Δ cells, possessed appreciable levels of CcO activities (Fig. 2C).
FIGURE 2.
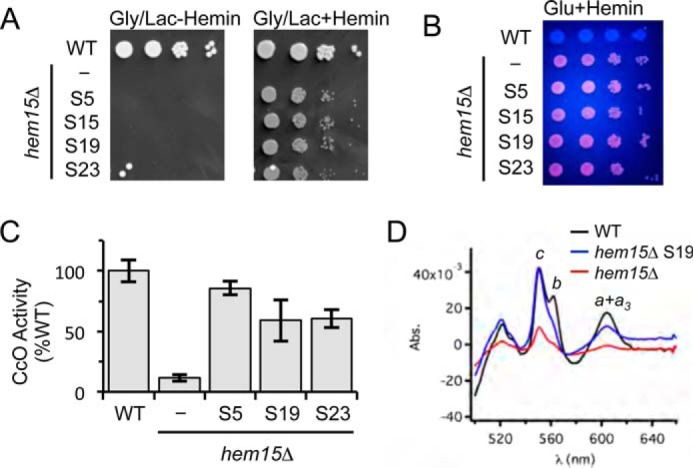
The respiratory growth of hem15Δ suppressors is mediated by restored mitochondrial respiratory complexes. A, growth test of spontaneous suppressors S5, S15, S19, and S23 relative to WT and the parent hem15Δ mutant strains on non-fermentable (glycerol/lactate) medium. B, UV-induced fluorescence of strains in A grown on glucose + hemin, indicating the presence of PPIX in the parent hem15Δ and suppressor strains. C, CcO activities from isolated mitochondria from the indicated suppressor strains. Activities are expressed as percentages of WT (N ≥ 3). D, optical spectrum of solubilized mitochondria (1 mg) from the indicated strains showing the recovery of hemes a, b, and c in the suppressor strain S19. Gly/Lac, glycerol/lactate; Glu, glucose; Abs., absorption.
Heme levels were quantified in mitochondria purified from the suppressor strains. UV-visible absorption spectroscopy was performed on detergent solubilized, purified mitochondria for the presence of cytochromes b, cytochromes c (from bc1 and soluble cytochrome c), and cytochromes a + a3 (from CcO). The difference spectra (reduced minus oxidized) of the three suppressors revealed a restoration of ∼50% of cytochromes b and a + a3 levels (Fig. 2D). Remarkably, the levels of cytochrome c were restored to WT levels. For clarity, we show the spectrum only for the S19 suppressor; the spectra for the three suppressors were similar. The higher efficiency in hemylation of cytochrome c relative to CcO is likely because that Cyc1 hemylation occurs in the inner membrane space compartment, whereas Cox1 heme-mediated synthesis and hemylation with exogenous heme requires passage through the mitochondrial inner membrane.
To identify the suppressor mutation(s), we utilized deep genomic DNA sequencing technology of genomic DNA from three of the four suppressors (S5, S19, and S23). Missense mutations in the nuclear gene NCE102 were identified. The genomic DNA sequences of the three suppressors were compared with the hem15Δ parent strain as a reference. Two of the isolates (S5 and S19) possessed mutant Nce102 with an A125E substitution, whereas the third suppressor (S23) had a mutation corresponding to a G22R substitution. Both mutations are located in putative transmembrane helices of the protein (Fig. 3A). Sequencing identified other mutations with high read confidence in the same strains possessing the Nce102 suppressor alleles, but the hits were unique to individual isolates. For example, isolate S19 possessed a missense mutation in the gene YBR139W that encodes for a putative serine-type carboxypeptidase; however, this mutation was not identified in other isolates. Additional unique mutations appearing with high confidence reads included DYN1, SOF1, and YAL064W-B.
FIGURE 3.

Point mutations in the plasma membrane protein Nce102 suppress the growth impairment in hem1Δ and hem15Δ mutants. A, topology prediction of Nce102 utilizing SOSUI with point mutations in red that are present in the isolated suppressors. B, fluorescent microscopy of Nce102-GFP expressed in WT and hem15Δ cells cultured to mid-log phase with 40 μm hemin. Nce102 is present in hem15Δ cells but is not confined to eisosomes. C, growth test of newly constructed hem15Δ nce102Sup on glycerol/lactate. D, growth test of hem15Δ cells transformed with a high copy plasmid for expressing the Nce102 or Nce102 (A125E) mutant. E, growth test of hem1Δ strain bearing the Nce102 (A125E) suppressor allele on non-fermentable (glycerol/lactate) medium. Deletion of the NCE102 suppressor allele in this background abrogates respiratory growth. F, growth of WT and nce102Δ cells on glucose and glycerol/lactate medium. Gly/Lac, glycerol/lactate; Glu, glucose; TM, transmembrane.
We focused on Nce102 because the gene was mutated in all three suppressor isolates, and the sequence reads were very high in confidence. Nce102 is a tri- or tetra-spanning membrane protein that localizes to specific furrow-like domains called eisosomes or MCC on the plasma membrane (10). We generated a strain with chromosomally encoded Nce102-GFP fusion protein and verified it localized to the plasma membrane as distinct foci consistent with a localization in MCCs (Fig. 3B). The presence of Nce102 within MCC foci was shown previously (10). In hem15Δ strains, the number of foci decreased, and in some replicates, the foci were completely absent. We attribute this to the known disruption of eisosome-associated proteins in cells with impaired mitochondrial function (14). The presence of the nce102 (A125E) suppressor allele restored the distinct foci formation likely as a result of restoration mitochondrial respiration.
We took two approaches to test whether the point mutations in Nce102 were responsible for the suppression of hem15Δ phenotype. First, we crossed each of the suppressors with WT cells and segregated spores by tetrad dissection to isolate spores bearing the nce102 suppressor allele but possessing the WT HEM15 allele (i.e. HEM15WTnce102Sup) by DNA sequencing. We isolated three spores harboring the A125E Nce102 mutant and subsequently deleted the HEM15 in those clones. The resulting nNce102, hem15Δ clones grew on glycerol/lactate medium containing heme (Fig. 3C).
We tested whether overexpression of Nce102 A125E mutant would restore respiratory growth in hem15Δ cells containing a WT Nce102 allele. Expression of the A125E Nce102 in hem15Δ cells with WT chromosomal NCE102 resulted in slight but reproducible improvement in growth on glycerol/lactate compared with the strain expressing only the WT allele (Fig. 3D). However, the growth of cells with the episomal nce102 suppressor was less robust than hem15Δ cells with the chromosomal nce102 mutant. Thus, the Nce102 A125E mutant is only partially dominant.
To address whether the Nce102 mutants were effective in restoring respiratory growth to other heme biosynthetic mutants, we used a haploid strain with the NCE102 suppressor allele (A125E mutant) and the WT heme biosynthetic pathway to delete HEM1. The resulting strain was respiratory-competent, unlike hem1Δ cells that contained WT NCE102 (Fig. 3E). The respiratory growth of the nce102Sup hem1Δ cells was abrogated upon deletion of the nce102 allele (Fig. 3E). These nce102Δ,hem1Δ cells were then transformed with either vector-borne WT NCE102 or the NCE102 suppressor allele. The presence of Nce102 A125E enabled respiratory growth unlike the WT Nce102 (Fig. 3E). Cells lacking only Nce102 remain respiratory-competent (Fig. 3F). The nce102 suppressor alleles are not linked to the specific loss of HEM15; rather, the suppressor mutation appears to enhance delivery of heme to the mitochondrial compartment. From here on, we describe the biochemical characterization of the chromosomal Nce102 A125E mutant.
The Nce102 Suppressor Enhances Heme Availability to Mitochondria
The marked improvements in growth and restoration of mitochondrial heme b and a pools in the suppressor strains strongly suggested that the mitochondrial respiratory complexes are partially restored. To verify this, we used purified mitochondria from the strains to quantify steady-state levels of subunits of complexes II, III, and IV by SDS-PAGE and immunoblotting. Cells expressing Nce102 A125E showed near WT restoration of subunit abundance in all three respiratory complexes (Fig. 4A). CcO subunits Cox1 and Cox2 were significantly restored in the suppressor relative to the parent hem15Δ cells in which the subunits were undetectable. The robust levels of Cox1 imply that the exogenous heme entered the mitochondrial matrix, because translation of Cox1 is dependent on the heme-activated Mss51 translational activator (15). The steady-state abundance of the intermembrane space protein Cyb2 was also restored in hem15Δ cells containing the Nce102 suppressor.
FIGURE 4.

Nce102 A125E enhances delivery of exogenous heme into the mitochondrial matrix. A, SDS-PAGE and immunoblot analysis of purified mitochondria isolated from the indicated strains showing the restoration of subunits to Complexes II, III, and IV when hem15Δ cells possess the nce102 suppressor allele. B, SDS-PAGE and immunoblot analysis of purified mitochondria from hem1Δ cells harboring matrix-targeted myoglobin (Mb) on a multicopy plasmid grown either on ALA or Tween-ergosterol-methionine to demonstrate the instability of the myoglobin in the absence of heme in the mitochondrial matrix. C, SDS-PAGE and immunoblot analysis of purified mitochondria isolated from the indicated strains harboring matrix-targeted myoglobin on a multicopy plasmid. D, growth test of hem15Δ strains relative to exogenous hemin concentrations on raffinose carbon source showing better growth when cells possess the Nce102 suppressor. Glu, glucose; TEM, Tween-ergosterol-methionine; Raff, raffinose.
To test whether heme in the mitochondrial matrix was limiting in hem15Δ Nce102 A125E cells, we expressed a matrix-targeted heterologous heme-binding protein in which the protein abundance is directly correlated with heme occupancy. Human myoglobin was expressed in yeast with a Sod2 mitochondrial targeting sequence. Expression of Sod2-myoglobin in WT cells revealed specific targeting to the mitochondrial matrix (data not shown). To demonstrate the hemes dependence in protein stability, we expressed the myoglobin chimera in hem1Δ cells that were depleted in heme but supplied with heme-dependent biosynthetic products by growing the cells in the presence of Tween (source of unsaturated fatty acids), ergosterol, and methionine (collectively referred to as Tween-ergosterol-methionine (8) and verified that myoglobin targeted to the mitochondrial matrix was indeed unstable using Western blot analysis of steady-state levels of myoglobin (Fig. 4B). The addition of ∂-aminolevulinic acid (ALA) to bypass the hem1Δ defect restored heme levels and stabilized mitochondrial myoglobin (Fig. 4B).
The myoglobin chimera was then expressed in hem15Δ cells cultured in exogenous hemin, but only minimal levels of myoglobin were apparent in mitochondria from these cells (Fig. 4C). In contrast, the presence of the suppressor allele in hem15Δ cells restored matrix myoglobin to levels seen in WT cells. This suggests that heme levels in the mitochondrial matrix of hem15Δ cells grown in the presence of exogenous hemin is limiting. A heme deficiency in the matrix would impair synthesis of the Cox1 subunit of CcO, because its translation is mediated by the heme-activated Mss51 translational activator (15). These data with matrix myoglobin indicate that cells containing Nce102 A125E are able to uniformly restore heme levels and, consequently, levels of heme-dependent proteins in all mitochondrial subcompartments.
The Nce102 Suppressor Mutant Promotes Better Growth at Low Hemin Concentrations
The biochemical phenotype of the Nce102 suppressor mutant presented thus far suggests that the steady-state heme level in the mitochondria is increased relative to the parental hem15Δ cells. We tested whether the increase in heme levels is due to an increase in efficiency of heme uptake (i.e. higher uptake rate at lower concentrations of extracellular hemin). We plated hem15Δ and hem15Δ nce102 suppressor cells on medium supplemented with limiting levels of hemin. With glucose as the sole carbon source and the resulting glucose attenuation of mitochondrial function, growth was similar between hem15Δ and hem15Δ cells expressing the Nce102 A125E suppressor with hemin concentrations between 0 and 0.8 μm (Fig. 4D). In contrast, when cells were grown on medium with raffinose as the carbon source leading to derepression of mitochondrial genes, the hem15Δ cells bearing the Nce102 mutant exhibited enhanced growth. These data suggest that the Nce102 mutant is advantageous when the heme requirement is high.
The Heme Transporter CeHRG-4 Also Rescues hem1Δ and hem15Δ Cell Respiratory Growth
The C. elegans HRG-4 protein localizes to the plasma membrane and functions to import heme into worm cells (5). Heterologous expression of this permease in yeast was shown to result in similar plasma membrane localization and import of exogenous heme into the cytosol (16). We tested whether the expression of HRG-4 in hem1Δ and hem15Δ cells would restore respiratory growth to these cells when cultured on medium containing supplemental heme. Indeed, expression of HRG-4 enabled both hem1Δ and hem15Δ cells to propagate on glycerol/lactate medium (Fig. 5A). Thus, respiratory growth in the heme biosynthetic mutants can be achieved either by expression of a bona fide heme permease or the eisosomal Nce102 suppressor.
FIGURE 5.

The heme transporter from C. elegans (CeHRG-4) also suppresses the respiratory phenotype in hem15Δ and hem1Δ cells; however, the mechanism of suppression could be different from that mediated by the Nce102 suppressor. A, growth test of hem1Δ and hem15Δ cells expressing the CeHRG-4 protein on a multicopy plasmid showing the rescue mediated by the heme transporter. B, β-galactosidase assay of hem1Δ cells bearing either the nce102 suppressor allele or expressing the CeHRG-4 plasmid that were grown in the presence of the indicated concentrations of hemin. C, growth test in the presence of the toxic heme analog gallium PPIX of WT yeast cells transformed with a plasmid bearing CeHRG-4 or cells possessing the Nce102 A125E mutation. D, CcO activities from purified mitochondria of hem15Δ cells bearing either the nce102 suppressor allele or expressing the CeHRG-4 plasmid. E, SDS-PAGE analysis of purified mitochondria showing the restoration of Cox2 subunit of CcO relative to hem15Δ nce102 suppressor allele or hem15Δ cells harboring the CeHRG-4 plasmid. Gly/Lac, glycerol/lactate; Raff, raffinose; GaPPIX, gallium PPIX.
To compare the effectiveness of the two import pathways, we utilized the heme-responsive Hap1 transcriptional response using the well established pCYC1-lacZ promoter-reporter fusion (17, 18). Cells containing this Hap1-reporter construct reveal that β-galactosidase activity correlates with intracelluar heme levels. As expected, the β-galactosidase activity was very low in hem1Δ cells grown in the presence of either 5 or 40 μm exogenous hemin. This suggests that the low level of mitochondrial heme detected in hem1Δ cells (Fig. 1E) may arise from inefficient heme import into the cytosol. When the hem1Δ cells contain Nce102 A125E, the level of β-galactosidase activity was significantly enhanced (Fig. 5B). In comparison, hem1Δ cells expressing the HRG-4 showed markedly higher β-galactosidase activities when compared with the cells containing Nce102 A125E, with levels approaching that of WT cells.
As a second test to assess whether the Nce102 suppressor enhances heme uptake, we performed a toxicity assay with the heme analog gallium PPIX in WT cells expressing either HRG-4 or Nce102 A125E and propagated on raffinose medium. Fig. 5C shows that at 10 μm concentration of gallium PPIX, cells expressing either HRG-4 or Nce102 A125E showed diminished growth on plates relative to WT cells, suggesting increased toxicity of the gallium PPIX in Nce102 A125E cells, as well as HRG-4-containing cells.
We quantified CcO levels in hem15Δ cells expressing either HRG-4 or Nce102 A125E (Fig. 5D). The presence of HRG-4 in hem15Δ cells led to a marked increase in CcO activity; however, the magnitude of the increase was lower compared with hem15Δ cells bearing Nce102 A125E. The CcO activity in hem15Δ cells with HRG-4 plasmid was ∼50% of WT levels, whereas hem15Δ cells bearing the Nce102 mutant was ∼70%.
This pattern with CcO enzymatic activity paralleled the protein expression pattern of the Cox2 subunit of CcO (Fig. 5E). In fact, Cox2 levels in hem15Δ cells bearing the suppressor allele was comparable with WT levels, unlike in hem15Δ cells with HRG-4. The difference in CcO activities and Cox2 steady-state levels between cells with Nce102 A125E or HRG-4 suggests that the molecular mechanism of suppression between Nce102 A125E and HRG-4 is different, and that the Nce102 mechanism involves more than simply increasing heme flux into the cells.
Depletion of the Endocytic End3 Protein Nullifies the Nce102 Mutant Rescue
Nce102 has been implicated to affect a variety of cellular process, primarily because of its role in directly regulating the activities of two membrane-associated protein kinases: Pkh1 and Pkh2. These paralogous kinases are implicated in endocytosis (11), maintenance of yeast cell wall integrity (19), and eisosome organization (20). The candidate role of Nce102 in endocytosis may relate to the mechanism of uptake of exogenous heme. To address this putative link, we tested whether deletion of END3, important for cellular endocytosis (21, 22), would affect the Nce102 suppressor activity. The respiratory growth observed in hem15Δ cells harboring the Nce102 suppressor allele was impaired in the absence of End3 (Fig. 6A). However, respiratory growth persisted in hem15Δ cells containing the HRG-4 heme permease in the absence of End3. Thus, we conclude that the mode of suppression between the Nce102-mediated and HRG-4-mediated rescue of hem15Δ is different.
FIGURE 6.

Deletion of END3 nullifies Nce102-mediated suppression, but not HRG-4 mediated suppression A, growth test of hem15Δ end3Δ cells harboring either the nce102 suppressor allele or CeHRG-4 on a multicopy plasmid. B, fluorescence microscopy of WT and hem1Δ yeast expressing Fur4-GFP. The cells were cultured in 40 μm hemin. The fluorescence microscopy pictures are inverted, and thus black indicates the localization of GFP as described previously (23). Gly/Lac, glycerol/lactate; Glu, glucose.
A variety of nutrient transporters associate with eisosomes in yeast. The arginine/proton symporter Can1, the uracil/proton symporter Fur4, and tryptophan permease Tat2 localize with eisosomes in WT cells (14). The abundance of Fur4 at the plasma membrane is modulated by its substrate availability (23) and the plasma membrane potential (14). Dissipation of the membrane potential with an uncoupler or impairment to the respiratory capacity of mitochondria results in rapid movement of the symporters out of eisomes, whereas the Sur7 component of eisosomes remains in the membrane foci (14). The movement of Fur4 out of eisosomes leads to internalization and degradation. We tested whether the eisosomal localization of Fur4 was altered in cells harboring the Nce102 suppressor mutants. Fur4 was expressed as a Fur4-GFP fusion protein in hem1Δ cells containing either WT Nce102 or the Nce102 A125E suppressor (Fig. 6B). Whereas Fur4 is seen in plasma membrane foci in WT cells, it is internalized in hem1Δ cells propagated with exogenous hemin. Fur4 was also internalized in hem1Δ cells harboring the Nce102 suppressor. Thus, despite the suppressor cells exhibiting oxidative growth, the mutant Nce102 fails to retain Fur4 within eisosomes. The Nce102 suppressor enables heme uptake to the mitochondrial compartment yet fails to maintain eisosomal retention of nutrient transporters.
Discussion
The heme biosynthetic pathway in eukaryotes is coupled to the trafficking of its biosynthetic intermediates. This process is coordinately complex, involving no less than eight enzymes, the mitochondria, the cytosol, and the necessary transmembrane transport mechanisms of heme intermediates, and finally the distribution of the product (heme) to their targets (4). We discovered that a synthetic lesion in this biosynthetic pathway, created by a deletion of the genes for either the enzymes ferrochelatase (Hem15) or ALA synthase (Hem1) in yeast, results in a respiratory growth defect when the cells are grown in the presence of exogenous hemin. This respiratory defect appears to arise from a deficiency in the delivery of exogenous hemin to the mitochondria for use in assembly of respiratory complexes. This inefficiency is consistent with the fact that, unlike bona fide heme auxotrophs such as C. elegans, S. cerevisiae lacks an identified heme import mechanism.
The cellular heme deficiency in hem1Δ and hem15Δ cells cultured in exogenous heme was demonstrated by quantifying mitochondrial heme and by measuring Hap1 reporter β-galactosidase activity, with the latter a readout for heme levels in the cytosol (18, 24). Increasing cytosolic heme levels by expression of the heterologous HRG-4 heme permease restored respiratory growth, suggesting that mitochondrial function can be maintained by increasing, even by artificial means, cytosolic heme levels.
We demonstrate presently that mitochondrial function can also be restored in hem15Δ or hem1Δ cells in the presence of Nce102 mutations containing either A125E or G22R substitutions in transmembrane helices. Unlike the rescue mediated by the heterologous HRG-4, we presume that Nce102-mediated suppression exploits specific endogenous trafficking pathway(s) to bypass the heme limitation defect. Cells with the Nce102 suppressor allele possessed higher CcO activity compared with cells with HRG-4, even though the latter cells contained higher steady-state levels of hemin in the cytosol.
Nce102 was originally defined as being involved in NCE (nonclassical export) pathway for protein secretion (25) but more recently as a component of plasma membrane eisosome structures (10). The mutant Nce102-mediated restoration of mitochondrial function in hem15Δ or hem1Δ cells cultured in exogenous heme is suggestive of an endocytic pathway of heme uptake, similar to the mechanism proposed for the internalization of ferric siderophore in S. cerevisiae (26, 27). We showed that depletion of End3, which is required for an internalization step in endocytosis, abolished suppression in the hem15Δ suppressor mutant. Furthermore, deletion of END3 had less of an effect in hem15Δ cells containing the HRG-4 permease. Endocytosis is a significant mechanism for the uptake of small molecules such as vitamin B12 from the extracellular environment, so heme uptake may follow a similar path. However, the distribution of internalized heme within the cell is unknown. The enhanced efficiency of heme restoration of mitochondrial function when acquired via Nce102 suppressor cells relative to HRG-4-containing cells may suggest a direct transfer of endocytic heme to mitochondria rather than being routed as free heme through the cytoplasmic compartment.
Nce102 is a component of the yeast plasma membrane compartment designated as eisosomes or MCC (10). This compartment has structural components Pil1, Lsp1, and Sur7 and associates with numerous nutrient permeases including Can1, Fur4, Tat2, and Hup1, as well as regulatory components (10–12, 28). The mutations in Nce102 did not alter its eisosomal localization. However, the localization of wild-type Nce102 in cells lacking Hem1 or Hem15 is unclear. The abundance of Nce102 is attenuated in hem15Δ cells, and eisosomal function is impaired. The colocalization of nutrient permeases with eisosomes is impaired in cells with dysfunctional mitochondria (11). Although the mutant Nce102 resides within eisosome structures, it fails to retain Fur4-GFP within eisosomes on the plasma membrane. We postulate that another unidentified transporter mediating heme uptake also translocates out of eisosome furrows like Fur4 and is endocytosed with heme as cargo leading to heme internalization.
Heme is synthesized on the inner membrane ferrochelatase (2), and as such, heme is trafficked out of mitochondria by poorly understood pathways. We show that heme import into the mitochondria can also occur. Unlike yeast, however, heme-auxotrophs such as worms are dependent on this process of mitochondrial heme import to support respiration. The HRG-4 permease is responsible for bringing heme into cells in worms, but nothing is known regarding how that heme is imported into the mitochondria. Likewise, human cells have a HRG-4 ortholog HRG-1 that resides within endocytic vesicles and facilitates heme import into the cytoplasm (16, 29). HRG-1 is known to mediate heme transport from phagolysosomes of macrophages to the cytoplasm compartment during erythrophagocytosis in a process of iron recycling (30). Because HRG-1 is ubiquitously expressed, it remains unresolved whether heme imported via endosomal HRG-1 in other cells is available as a cofactor in the cytoplasm or mitochondria.
It remains unclear whether labile heme pools or a heme reservoir exists for ready mobilization. Heme trafficking to the mitochondria may be significant in certain cell physiological conditions. Endosomal routing of heme may occur in a direct interorganellar transfer from endosomes to mitochondria as has been described for iron (31). Mitochondrial import would necessitate passage across the ion impervious inner membrane. That may be accomplished by a promiscuous transporter or carrier. Future research is needed to address the transport process.
Experimental Procedures
Yeast Strains and Vectors
All S. cerevisiae strains used in this study were W303 (Mat a or Matα ura3-1 ade2-1 trp1-1 his3-11,15 leu2-3,112). Deletion strains hem1Δ and hem15Δ were constructed by homologous recombination using either HIS3MX6 or the Candida albicans URA3 disruption cassettes; end3Δ and nce102Δ using HIS3MX6 (32). Strains bearing the nce102 point mutations in HEM15WT background were isolated by crossing hem15Δ nce102 suppressor strains with WT and isolating spores with the nce102 suppressor alleles and WT HEM15. Construction of the hem1Δ strain bearing the nce102 suppressor allele was generated by deleting the HEM1 in the nce102 suppressor background. The Nce102 (A125E)-GFP strain was constructed by amplifying the 3′ region of NCE102 that had been fused to a GFP tag containing a HIS3MX6 marker (a kind gift from Tobias Walther). We designed the primers to anneal adjacent to the suppression mutation (NCE102_inter 379, 5′-CTCTACTTCTCTTGTGCCATC-3′) and 140 bp of downstream of NCE102 ORF (NCE102_dn 140, 5′-CCGAAAGACGGAAAGGGATG-3′). The resulting PCR product was transformed into hem15Δ and hem15Δ suppressor to generate hem15Δ NCE102-GFP and hem15Δ NCE102 (A125E)-GFP, respectively. All deletion and tagged strains were confirmed by PCR analysis of the locus.
The NCE102 ORF of WT and mutant allele were amplified by PCR and incorporated into pRS424 vector containing MET25 promoter and CYC1 terminator to generate pNCE102 WT and pNCE102 A125E, respectively. The plasmid pCeHRG-4 was a kind gift from Dr. Iqbal Hamza. The pCYC1-lacZ was a kind gift from Dr. L. Zhang. All strains were grown either in yeast peptone medium or synthetic complete medium lacking the amino acid(s) necessary to maintain plasmid selection with either 2% galactose or 2% glycerol/lactate as the carbon source. For heme auxotrophic strains, 20 μm hemin was added to the medium or to a prescribed concentration. Yeast strains were transformed using lithium acetate with DMSO (33).
Mitochondria Isolation
Intact mitochondria were isolated using previously described methods of Glick and Pon (34) and Diekert et al. (35). For experiments requiring HPLC quantification of heme in the mitochondria, we ensured removal of any exogenous hemin that were adventitiously bound to the cell surface (to prevent carry over to mitochondria) by several washes of cells using the 100 mm Tris buffer, pH 9.4, with 10 mm DTT. For some experiments, isolated mitochondria were further purified using ultracentrifugation through a Histodenz (Sigma-Aldrich) step gradient (14 and 22%). Total mitochondrial protein was quantified using either the Bradford (36) or the bicinchoninic acid assays (37).
Immunoblotting
Steady-state levels of mitochondrial proteins were analyzed using the NuPAGE Bis-Tris Gel system (Thermo Fisher) using MES as the buffer system. Proteins were subsequently transferred to nitrocellulose membrane and probed using the indicated primary antibodies and visualized using ECL reagents with horseradish peroxidase-conjugated secondary antibodies. Primary antibodies were obtained from the following: anti-HA was from Rockland, and anti-Cox2, anti-Cox1, and anti-Porin were from Mitoscience. Anti-Rip1, anti-Cob, and anti-Cyt1 were provided by Dr. Vincenzo Zara, and anti-Cyb2 was from Dr. Carla Koehler.
β-Galactosidase Heme Reporter Assay
A heme-responsive β-galactosidase reporter plasmid that is under the control of the CYC1 promoter (pCYC1-lacZ fusion) was obtained from Dr. L. Zhang. The cells were transformed with pCYC1-lacZ plasmid or cotransformed with pCYC1-lacZ + pCeHRG-4 plasmids. Cells were grown to early to mid-log phase in the appropriate synthetic complete auxotrophic medium supplemented with 2% raffinose. β-Galactosidase was assayed as described (18, 24).
Microscopy
For fluorescence microscopy, Nce102-GFP tagged yeast cells were grown overnight to early to mid-log phase (A600 nm = 0.6–1) and mounted on polylysine-coated slides and analyzed on an Olympus BX51 microscope. For confocal microscopy, yeast cells were grown to early to mid-log phase and mounted on chambered microscope slides. Fluorescent images were obtained on a Nikon A1 at the University of Utah Core Facility. Fur4-GFP fluorescence microscopy was conducted on deconvolution microscope from Delta Vision (Applied Precision, Issaquah, WA).
Respiratory Complex Assays
Cytochrome c oxidase activity was determined by measuring the oxidation of ferrous cytochrome c by purified mitochondria (5–10 μg of protein) in 20 mm K2HPO4 + 0.5% Tween 20 (38). The oxidation of ferrous cytochrome c was measured spectrophotometrically at 550 nm.
Heme Analysis
Mitochondrial heme was quantified in two ways. To quantify respiratory complex hemes b, c, and a, 0.75–1 mg of purified mitochondria were pelleted at 17,000 × g and solubilized in 400 μl of 10 mm Tris, pH 7.2, containing either 1% digitonin or 5% Triton X-100. Unsolubilized material was pelleted at 17,000 × g, and the supernatant was analyzed on a HP-8453 spectrophotometer.
Second, total mitochondrial hemes b and PPIX were analyzed using a modified version of the acid-acetone extraction of hemes and subsequent analysis using C18 reverse phase HPLC (39). Briefly, 0.75–1 mg of purified mitochondria were pelleted as above and resuspended in 100–200 μl of acetone, 2.5% HCl solution. The volume of the solution was minimized to ∼10–20 μl using a vacuum centrifuge and resuspended in 200 μl of HPLC starting buffer A (25% acetonitrile + 0.1% TFA). Insoluble material was clarified using a second centrifugation, and the supernatant was applied to a SunFire C18 column (4.6 mm × 150 mm) and eluted at 1 ml/min using a 25–50% gradient for the first 4 ml and 50–75% for the 22 ml. Elution of heme and PPIX were monitored at 400 nm.
Deep Sequencing
To identify mutations in the hem15Δ suppressors, we submitted genomic DNA for Illumina high throughput sequencing (HiSeq, single read) to the High Throughput Genomics Core Facility at the University of Utah. Cell pellet (from 50-ml cultures at A600 nm = ∼1) was pretreated with lyticase in the resuspension buffer (20 mm Tris, pH 8.0, 0.1 m EDTA, 0.5 m β-mercaptoethanol) for 30 min at 37 °C. Genomic DNA from yeast spheroplast was purified using a Qiagen DNeasy Blood & Tissue kit. (Further purification, DNA library generation, tagging, and sequencing were performed at the sequencing core facility.)
Author Contributions
H. J. K. and M.-Y. J. conducted the yeast studies. T. J. P. assisted in the bioinformatics identification of the NCE102 suppression mutants. M. B. performed the fluorescence microscopy of Fur4-GFP. J. D. P. contributed valuable discussions on the project, and D. R. W. supervised the entire project.
Acknowledgments
We acknowledge Tobias Walther, Iqbal Hamza, Jerry Kaplan, Diane Ward, and the University of Utah Microscopy Core for reagents and helpful discussions.
This work was supported by National Institutes of Health Grant GM110755 (to D. R. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ALA
- aminolevulinic acid
- CcO
- cytochrome c oxidase
- MCC
- membrane compartment of Can1, the arginine permease
- PPIX
- protoporphyrin IX.
References
- 1. Storbeck S., Rolfes S., Raux-Deery E., Warren M. J., Jahn D., and Layer G. (2010) A novel pathway for the biosynthesis of heme in Archaea: genome-based bioinformatic predictions and experimental evidence. Archaea 2010, 175050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamza I., and Dailey H. A. (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 1823, 1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ajioka R. S., Phillips J. D., and Kushner J. P. (2006) Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763, 723–736 [DOI] [PubMed] [Google Scholar]
- 4. Schultz I. J., Chen C., Paw B. H., and Hamza I. (2010) Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 285, 26753–26759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rajagopal A., Rao A. U., Amigo J., Tian M., Upadhyay S. K., Hall C., Uhm S., Mathew M. K., Fleming M. D., Paw B. H., Krause M., and Hamza I. (2008) Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quigley J. G., Yang Z., Worthington M. T., Phillips J. D., Sabo K. M., Sabath D. E., Berg C. L., Sassa S., Wood B. L., and Abkowitz J. L. (2004) Identification of a human heme exporter that is essential for erythropoiesis. Cell 118, 757–766 [DOI] [PubMed] [Google Scholar]
- 7. Chiabrando D., Marro S., Mercurio S., Giorgi C., Petrillo S., Vinchi F., Fiorito V., Fagoonee S., Camporeale A., Turco E., Merlo G. R., Silengo L., Altruda F., Pinton P., and Tolosano E. (2012) The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122, 4569–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gollub E. G., Liu K. P., Dayan J., Adlersberg M., and Sprinson D. B. (1977) Yeast mutants deficient in heme biosynthesis and a heme mutant additionally blocked in cyclization of 2,3-oxidosqualene. J. Biol. Chem. 252, 2846–2854 [PubMed] [Google Scholar]
- 9. Desmyter L., Verstraelen J., Dewaele S., Libert C., Contreras R., and Chen C. (2007) Nonclassical export pathway: overexpression of NCE102 reduces protein and DNA damage and prolongs lifespan in an SGS1 deficient Saccharomyces cerevisiae. Biogerontology 8, 527–535 [DOI] [PubMed] [Google Scholar]
- 10. Fröhlich F., Moreira K., Aguilar P. S., Hubner N. C., Mann M., Walter P., and Walther T. C. (2009) A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J. Cell Biol. 185, 1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grossmann G., Malinsky J., Stahlschmidt W., Loibl M., Weig-Meckl I., Frommer W. B., Opekarová M., and Tanner W. (2008) Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 183, 1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Douglas L. M., Wang H. X., Li L., and Konopka J. B. (2011) Membrane compartment occupied by Can1 (MCC) and eisosome subdomains of the fungal plasma membrane. Membranes 1, 394–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu H., Sun Y., Zhang Y., Wang W., Dan J., Yao J., Chen H., Tian F., Sun X., Guo S., Tian Z., and Tian Y. (2014) Protoporphyrin IX induces a necrotic cell death in human THP-1 macrophages through activation of reactive oxygen species/c-Jun N-terminal protein kinase pathway and opening of mitochondrial permeability transition pore. Cell Physiol. Biochem. 34, 1835–1848 [DOI] [PubMed] [Google Scholar]
- 14. Grossmann G., Opekarová M., Malinsky J., Weig-Meckl I., and Tanner W. (2007) Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J. 26, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soto I. C., Fontanesi F., Myers R. S., Hamel P., and Barrientos A. (2012) A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab. 16, 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan X., Protchenko O., Philpott C. C., and Hamza I. (2012) Topologically conserved residues direct heme transport in HRG-1-related proteins. J. Biol. Chem. 287, 4914–4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guarente L., and Mason T. (1983) Heme regulates transcription of the CYC1 gene of S. cerevisiae via an upstream activation site. Cell 32, 1279–1286 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L., and Guarente L. (1995) Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roelants F. M., Torrance P. D., Bezman N., and Thorner J. (2002) Pkh1 and Pkh2 differentially phosphorylate and activate Ypk1 and Ykr2 and define protein kinase modules required for maintenance of cell wall integrity. Mol. Biol. Cell 13, 3005–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo G., Gruhler A., Liu Y., Jensen O. N., and Dickson R. C. (2008) The sphingolipid long-chain base-Pkh1/2-Ypk1/2 signaling pathway regulates eisosome assembly and turnover. J. Biol. Chem. 283, 10433–10444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bénédetti H., Raths S., Crausaz F., and Riezman H. (1994) The END3 gene encodes a protein that is required for the internalization step of endocytosis and for actin cytoskeleton organization in yeast. Mol. Biol. Cell 5, 1023–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raths S., Rohrer J., Crausaz F., and Riezman H. (1993) end3 and end4: two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae. J. Cell Biol. 120, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keener J. M., and Babst M. (2013) Quality control and substrate-dependent downregulation of the nutrient transporter Fur4. Traffic 14, 412–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thorsness M., Schafer W., D'Ari L., and Rine J. (1989) Positive and negative transcriptional control by heme of genes encoding 3-hydroxy-3-methylglutaryl coenzyme A reductase in Saccharomyces cerevisiae. Mol. Cell. Biol. 9, 5702–5712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cleves A. E., Cooper D. N., Barondes S. H., and Kelly R. B. (1996) A new pathway for protein export in Saccharomyces cerevisiae. J. Cell Biol. 133, 1017–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim Y., Yun C. W., and Philpott C. C. (2002) Ferrichrome induces endosome to plasma membrane cycling of the ferrichrome transporter, Arn1p, in Saccharomyces cerevisiae. EMBO J. 21, 3632–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim Y., Lampert S. M., and Philpott C. C. (2005) A receptor domain controls the intracellular sorting of the ferrichrome transporter, ARN1. EMBO J. 24, 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Douglas L. M., Wang H. X., and Konopka J. B. (2013) The MARVEL domain protein Nce102 regulates actin organization and invasive growth of Candida albicans. MBio 4, e00723–00713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Callaghan K. M., Ayllon V., O'Keeffe J., Wang Y., Cox O. T., Loughran G., Forgac M., and O'Connor R. (2010) Heme-binding protein HRG-1 is induced by insulin-like growth factor I and associates with the vacuolar H+-ATPase to control endosomal pH and receptor trafficking. J. Biol. Chem. 285, 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. White C., Yuan X., Schmidt P. J., Bresciani E., Samuel T. K., Campagna D., Hall C., Bishop K., Calicchio M. L., Lapierre A., Ward D. M., Liu P., Fleming M. D., and Hamza I. (2013) HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 17, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheftel A. D., Zhang A. S., Brown C., Shirihai O. S., and Ponka P. (2007) Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110, 125–132 [DOI] [PubMed] [Google Scholar]
- 32. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 33. Hill J., Donald K. A., and Griffiths D. E. (1991) DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 19, 5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glick B. S., and Pon L. A. (1995) Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 260, 213–223 [DOI] [PubMed] [Google Scholar]
- 35. Diekert K., de Kroon A. I., Kispal G., and Lill R. (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65, 37–51 [DOI] [PubMed] [Google Scholar]
- 36. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 37. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., and Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 38. Capaldi R. A., Marusich M. F., and Taanman J. W. (1995) Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 260, 117–132 [DOI] [PubMed] [Google Scholar]
- 39. Barros M. H., Carlson C. G., Glerum D. M., and Tzagoloff A. (2001) Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 492, 133–138 [DOI] [PubMed] [Google Scholar]