

Abstract
Activation of Group I metabotropic glutamate receptors (mGluRs) activates signaling cascades, resulting in calcium release from intracellular stores, ERK1/2 activation, and long term changes in synaptic activity that are implicated in learning, memory, and neurodegenerative diseases. As such, elucidating the molecular mechanisms underlying Group I mGluR signaling is important for understanding physiological responses initiated by the activation of these receptors. In the current study, we identify the multifunctional scaffolding protein spinophilin as a novel Group I mGluR-interacting protein. We demonstrate that spinophilin interacts with the C-terminal tail and second intracellular loop of Group I mGluRs. Furthermore, we show that interaction of spinophilin with Group I mGluRs attenuates receptor endocytosis and phosphorylation of ERK1/2, an effect that is dependent upon the interaction of spinophilin with the C-terminal PDZ binding motif encoded by Group I mGluRs. Spinophilin knock-out results in enhanced mGluR5 endocytosis as well as increased ERK1/2, AKT, and Ca2+ signaling in primary cortical neurons. In addition, the loss of spinophilin expression results in impaired mGluR5-stimulated LTD. Our results indicate that spinophilin plays an important role in regulating the activity of Group I mGluRs as well as their influence on synaptic activity.
Keywords: calcium imaging, endocytosis, extracellular-signal-regulated kinase (ERK), glutamate receptor, receptor regulation
Introduction
Glutamate is the main excitatory neurotransmitter in the central nervous system. The actions of glutamate are mediated via two types of receptors: ionotropic glutamate receptors, which are ligand-gated cation channels, and metabotropic glutamate receptors, which are G protein-coupled receptors (GPCRs)2 (1, 2). Activation of mGluRs produces long term changes in synaptic plasticity, including long term potentiation and long term depression (LTD) (3, 4). Consequently, mGluRs have been implicated in both memory and learning as well as neurodegenerative disorders, such as Alzheimer and Parkinson diseases (5).
The mechanism by which Group I mGluR signaling contributes to neurodegenerative disease remains unclear. However, genetic deletion of mGluR5 has been shown to improve cognitive performance and reduce Alzheimer disease-like pathology in an APPswe/PS1ΔE9 mouse model of Alzheimer disease (6). Multiple protein partners have been shown to interact with Group I mGluRs to alter their signaling and trafficking (7, 8). Recently, via a tandem affinity purification proteomic screen with the mGluR1a C-terminal tail, our laboratory has discovered a novel Group I mGluR-interacting protein, protein phosphatase 1, regulatory subunit 9B (spinophilin/neurabin II). Spinophilin is a multifunctional synaptic scaffolding protein that is compartmentalized to the heads of dendritic spines and interacts with protein phosphatases (PP1α and -γ) and F-actin (9–11). It also encodes three putative Src homology 3 domains, a GPCR-binding domain, a PDZ (PSD95/Disc Large/zona occludens) domain, three coiled-coiled domains, and a potential leucine/isoleucine zipper motif (12).
Spinophilin is recruited to the synapse in response to N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) activation to maintain LTD (11, 13–15). Thus, in addition to regulating AMPAR trafficking (16), the interaction of PP1 and spinophilin with other components of the synapse may be extremely important for controlling synaptic strength. Spinophilin has been shown to interact with the third intracellular loop (IL3) domain of several GPCRs (17–21). Spinophilin competes with G protein-coupled receptor kinase 2 (GRK2) for binding to the α2-adrenergic receptor (α2AR) following agonist stimulation and prevents β-arrestin recruitment, blocking endocytosis of α2AR and regulating Ca2+ signaling by scaffolding RGS2 with the α2AR (22). In contrast, spinophilin has been demonstrated to increase the internalization of the μ-opioid receptor without affecting δ-opioid receptor-mediated ERK1/2 phosphorylation (20, 23). Spinophilin also regulates synaptic transmission by targeting PP1 to ionotropic glutamate receptors (15). In the present study, we have identified spinophilin as a protein that interacts with the C-terminal PDZ binding motifs of Group I mGluRs and functions to regulate their endocytosis and signaling and the expression of mGluR5-dependent activity.
Results
Identification of Spinophilin as a Novel Group I mGluR-interacting Protein
To identify novel Group I mGluR-interacting proteins, we performed a proteomic screen in human embryonic kidney (HEK) 293 cells transfected with a FLAG-tagged plasmid construct encoding the mGluR1a C-terminal tail (amino acids 841–1199), which encodes both a PDZ binding motif and a PP1γ binding site. We found that a number of novel and known Group I mGluR-interacting proteins were identified (Table 1). The previously identified mGluR1/5-interacting proteins included Homer1–3, PP1γ, PP2A, and GOPC (Golgi-associated PDZ and coiled-coil motif-containing) (24–27). The novel Group I mGluR C-terminal tail-interacting proteins identified in the screen included spinophilin, insulin substrate 4, reticulocalbin, and NRAGE (Table 1). Because the mGluR1a and mGluR5a C-terminal tails encode PDZ binding motifs and spinophilin is the regulatory subunit for PP1γ, a known mGluR5a-interacting protein (25, 28), we tested whether spinophilin could be co-immunoprecipitated with mGluR1a, mGluR1b, and mGluR5a.
TABLE 1.
Proteins co-precipitated with FLAG-mGluR1-CT from HEK 293 cells
| Gene ID | Protein name | Unique peptides | Total peptides | Percentage of coverage |
|---|---|---|---|---|
| % | ||||
| 2911 | Metabotropic glutamate receptor 1a | 14 | 90 | 6.1 |
| 9454 | Homer 3 | 11 | 14 | 46 |
| 9456 | Homer 1 | 9 | 9 | 41.1 |
| 9455 | Homer 2 | 5 | 9 | 35 |
| 84687 | Protein phosphatase 1, regulatory subunit 9B | 10 | 10 | 16 |
| 8471 | Insulin receptor substrate 4 | 8 | 11 | 8 |
| 5931 | Retinoblastoma-binding protein 7 | 7 | 9 | 28 |
| 57120 | Golgi-associated PDZ and coiled-coil motif-containing | 8 | 8 | 26 |
| 5518 | Protein phosphatase 2, regulatory subunit A, α isoform | 6 | 6 | 16 |
| 5955 | Reticulocalbin 2, EF-hand calcium binding domain | 4 | 5 | 22.1 |
| 5501 | Protein phosphatase 1, catalytic subunit, γ isoform | 4 | 4 | 14.2 |
| 9500 | Melanoma antigen family D, 1 (NRAGE) | 4 | 4 | 6.7 |
| 5515 | Protein phosphatase 2, catalytic subunit, α isoform | 3 | 3 | 18.4 |
Co-immunoprecipitation of Spinophilin with Group I mGluRs
To validate the interaction of spinophilin with Group I mGluRs, HEK 293 cells were transfected with either FLAG epitope-tagged mGluR1a or mGluR5a along with either empty GFP vector or GFP-spinophilin. We found that spinophilin was co-immunoprecipitated with both FLAG-mGluR1a and FLAG-mGluR5a (Fig. 1, A and B), confirming that spinophilin was able to interact with both full-length Group I mGluRs. mGluR1a and mGluR5a both encode a functional PDZ binding motif (STL) and PP1γ binding domain (KSCSW) that could mediate either direct or indirect interactions of spinophilin with the receptors (26). Therefore, we either deleted the mGluR1a PDZ binding motif or mutated the mGluR1a PP1γ binding motif (KSVTW) to alanine residues to assess whether this prevented spinophilin interactions with the resulting receptor mutants. When tested, we found that spinophilin could still be co-immunoprecipitated with either mutant construct (Fig. 1C). To further evaluate spinophilin interactions with Group I mGluRs, we tested whether spinophilin could be co-immunoprecipitated with mGluR1b, an alternatively spliced variant in which the extended C-terminal tail is replaced by a 20-amino acid segment (29). However, spinophilin was found to co-immunoprecipitate with FLAG-mGluR1b, indicating that spinophilin must interact with Group I mGluRs via an additional binding site that was not encoded by the C-terminal tail (Fig. 1D). We previously demonstrated that binding of several Group I interacting proteins was mediated via interactions with both the mGluR second intracellular loop domain (IL2) and the C-terminal tail (29–32). Therefore, we tested whether GFP-spinophilin could be co-precipitated in a GST pull-down assay with purified GST-IL2. We found that co-incubation of GFP-spinophilin-expressing HEK 293 cell lysates with GST-IL2 resulted in the co-precipitation of GFP-spinophilin (Fig. 1E). To further delineate Group I mGluR motifs and residues required for spinophilin interactions, we assessed whether spinophilin could be co-immunoprecipitated with FLAG-mGluR1b-K691A, FLAG-mGluR1b-K692b, or a FLAG-tagged mGluR1a construct lacking both the PDZ and PP1γ binding site (FLAG-mGluR1a-PP1-ΔPDZ). We found that GFP-spinophilin could be co-immunoprecipitated with both FLAG-mGluR1b-K692A and FLAG-mGluR1a-PP1Ala-ΔPDZ, but not FLAG-mGluR1b-K691A, which not only lacks a PDZ binding motif but was previously shown to be defective in GRK2 binding (33) (Fig. 1F). Thus, spinophilin interactions with Group I mGluRs appeared to be mediated by multiple domains in a manner similar to what we previously characterized for GRK2.
FIGURE 1.

Spinophilin co-immunoprecipitates with Group I mGluRs. Shown is a representative immunoblot (IB) showing GFP-spinophilin co-immunoprecipitated (IP) with either FLAG-mGluR1a (A) or FLAG-mGluR5a (B) from HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding either FLAG-mGluR1a or FLAG-mGluR5a and 2 μg of either pEGFP or GFP-spinophilin as indicated. C, representative immunoblots showing Myc-spinophilin co-immunoprecipitation with FLAG-mGluR1a mutants either lacking the PP1γ (FL-PP1-Ala) or PDZ (FL-ΔPDZ) binding sites from HEK 293 cells transfected with 2 μg of plasmid cDNA encoding mGluR1a receptor mutants and either pEGFP or Myc-spinophilin as indicated. A line denotes where the blot was cut to remove other co-immunoprecipitations not included in the figure. D, representative immunoblot showing Myc-spinophilin co-immunoprecipitated with FLAG-mGluR1b in HEK 293 cells transiently transfected with 2 μg of FLAG-mGluR1b and 2 μg of either pEGFP or Myc-spinophilin as indicated. Data are representative of 4–5 independent experiments. E, GST and GST-IL2 fusion proteins were purified from E. coli using glutathione-Sepharose and 1 μg of each fusion protein was incubated for 1 h with 500 μg of HEK 293 cell lysates transfected with GFP-spinophilin. Shown is a representative immunoblot demonstrating the co-precipitation of GFP-spinophilin with GST-IL2. Data are representative of four independent experiments. F, representative immunoblot showing GFP-spinophilin co-immunoprecipitated with wild-type and mutant FLAG-mGluR1a and FLAG-mGluR1b constructs in HEK 293 cells transiently transfected with 2 μg of cDNA encoding receptor and 2 μg of either pEGFP or GFP-spinophilin as indicated. Data are representative of 4–5 independent experiments.
Effect of Spinophilin on Group I mGluR Endocytosis
Spinophilin was previously shown to interact with the agonist-activated α2AR-Gβγ complex and was proposed to tether the receptor to the plasma membrane, thereby blocking receptor endocytosis (34, 35). Therefore, we examined whether the overexpression of spinophilin would similarly affect agonist-stimulated endocytosis of both FLAG-mGluR1a and FLAG-mGluR5a. We found that overexpression of GFP-spinophilin significantly reduced FLAG-mGluR1a internalization in response to stimulation with 30 μm quisqualate at 5 min and showed a trend to antagonize FLAG-mGluR1a endocytosis following 15 min of agonist activation (Fig. 2A). In contrast, FLAG-mGluR5a internalization was not significantly reduced following 5 min of agonist stimulation following GFP-spinophilin overexpression but was significantly attenuated following 15 min of agonist treatment (Fig. 2B). Because the overexpression of spinophilin in HEK 293 cells attenuated endocytosis of both mGluR1a and mGluR5a, we examined whether the internalization of endogenous mGluR5a was modulated by endogenous spinophilin in cortical neurons. To test this, cortical neurons derived from E18 spinophilin knock-out mice and littermate controls were cultured until 12–14 days in vitro (DIV) and assessed the intracellular redistribution of cell surface biotinylated mGluR5a in response to agonist stimulation (10 μm dihydroxyphenylglycine (DHPG)) for 5 and 10 min.
FIGURE 2.

Effect of spinophilin expression on of Group I mGluR endocytosis. A, determination of internalized biotin-labeled FLAG-mGluR1a in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding FLAG-mGluR1a along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin (GFP-Sp) following treatment with 30 μm quisqualate for 0, 5, and 15 min. B, determination of internalized biotin-labeled FLAG-mGluR5a in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding FLAG-mGluR5a along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin following treatment with 30 μm quisqualate for 0, 5, and 15 min. C, determination of internalized biotin-labeled FLAG-mGluR1b in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding FLAG-ΔPDZ along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin following treatment with 30 μm quisqualate for 0, 5, and 15 min. D, determination of internalized biotin-labeled FLAG-mGluR1a lacking the PPIγ binding site (FLAG-PP1Ala) in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding FLAG-PP1Ala along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin following treatment with 30 μm quisqualate for 0, 5, and 15 min. E, determination of internalized biotin-labeled FLAG-mGluR1a lacking the PDZ binding motif (FLAG-ΔPDZ) in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding FLAG-ΔPDZ along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin following treatment with 30 μm quisqualate for 0, 5, and 15 min. The bar graphs represent the densitometric analysis of internalized biotin-labeled mGluR1a protein normalized to total cell surface mGluR1a biotinylation. Data represent the mean ± S.D. (error bars) of 4–6 independent experiments. *, p < 0.05 versus GFP-transfected cells.
To further assess domains that may be required for spinophilin-mediated attenuation of Group I mGluR endocytosis, we assessed the ability of spinophilin to prevent the internalization of FLAG-mGluR1b, which lacks an extended C-tail, and FLAG-mGluR1a mutants lacking either the PP1γ domain (FLAG-mGluR1a-PP1Ala) or the PDZ binding motif (FLAG-mGluR1a-ΔPDZ). When tested, spinophilin overexpression did not alter the extent of FLAG-mGluR1b internalization (Fig. 2C). In addition, the loss of the FLAG-mGluR1a PP1γ binding site did not alter the capacity of GFP-spinophilin to antagonize FLAG-mGluR1a endocytosis (Fig. 2D), but deletion of the PDZ binding motif completely abrogated the ability of GFP-spinophilin to attenuate FLAG-mGluR1a endocytosis (Fig. 2E). Thus, these data indicate that a functional association of spinophilin with the C-terminal PDZ binding motif of Group I mGluRs was essential for the regulation of their endocytosis.
We found that genetic deletion of endogenous spinophilin resulted in a significantly large increase in mGluR5a internalization at both 5 and 10 min in response to DHPG treatment (Fig. 3, A and B). Thus, spinophilin expression plays a significant role in the regulation of endogenous mGluR5a.
FIGURE 3.
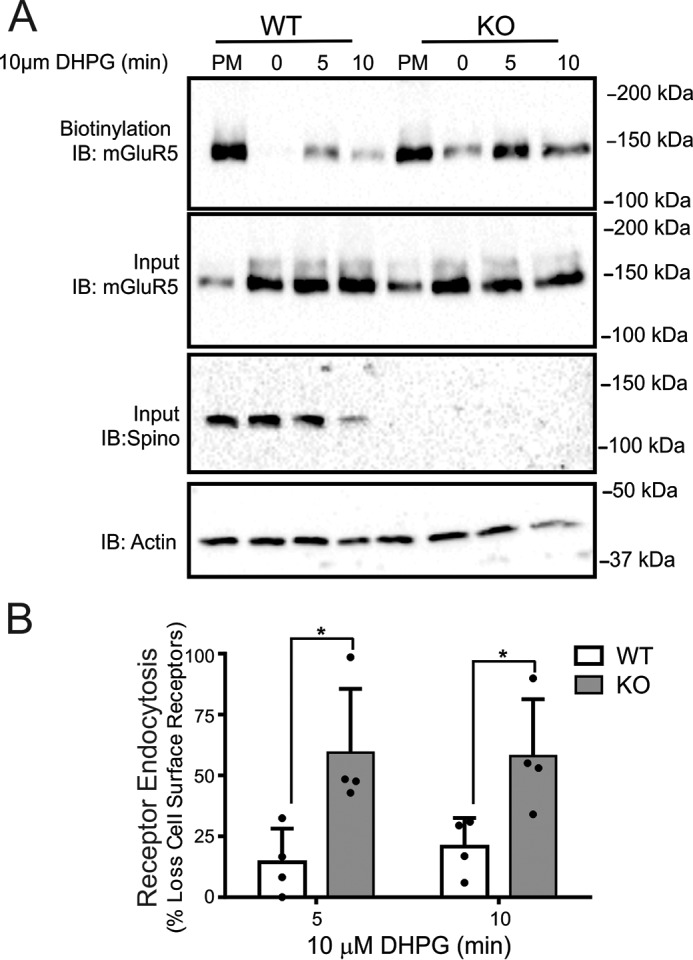
Regulation of Group I mGluR endocytosis in cortical neurons derived from wild-type and spinophilin knock-out mice. A, the top blot shows a representative immunoblot (IB) for internalized biotin-labeled endogenous mGluR5 in primary cortical neurons (12–14 DIV) derived from E18 wild-type and spinophilin knock-out embryos in response to 10 μm DHPG treatment for 0, 5, and 10 min. The bottom blots show cell lysates (50 μg) for endogenous mGluR5 and spinophilin protein expression. B, the bar graph shows the densitometric analysis of internalized biotin-labeled mGluR5 protein normalized to total cell surface mGluR5 biotinylation and normalized for receptor and actin loading controls. Data represent the mean ± S.D. (error bars) of four independent experiments. *, p < 0.05 versus untreated control.
Effect of Spinophilin on Group I mGluR-mediated Inositol Phosphate Formation and Ca2+ Signaling
Because spinophilin interacts with and modulates internalization of Group I mGluRs, we examined whether overexpression of spinophilin would alter the extent of Group I mGluR-stimulated inositol phosphate (IP) formation. We assessed IP formation using wild-type FLAG-mGluR1a and FLAG-mGluR1b constructs as well as a mutant FLAG-mGluR5a cDNA construct that has a single amino acid mutation (A154V) in the glutamate binding region. This residue was analogous to Ala-168 in mGluR1a, which when mutated to valine causes low basal IP formation so that agonist-stimulated responses could be measured (36). Agonist stimulation of either FLAG-mGluR1a, FLAG-mGluR1b, or FLAG-mGluR5a-A154V resulted in a dose-dependent increase in IP formation over 30 min that was not altered by the expression of GFP-spinophilin (Fig. 4, A–C). We also found that FLAG-mGluR1a-PP1Ala- and FLAG-mGluR1a-ΔPDZ-stimulated IP formation was not significantly different (123 ± 27 and 88% ± 16%, respectively) from FLAG-mGluR1-mediated IP formation. In addition, we tested whether overexpression of GFP-spinophilin affected either FLAG-mGluR1a-, FLAG-mGluR1b-, or FLAG-mGluR5a-mediated Ca2+ release. Treatment of HEK 293 cells with 30 μm quisqualate significantly increased Fura-2/AM fluorescence, but the overexpression of GFP-spinophilin did not alter either FLAG-mGluR1a-, FLAG-mGluR1b-, or FLAG-mGluR5a-mediated Ca2+ release, quantified by area under the curve (Fig. 5, A–C). However, when we measured Ca2+ release from primary cortical neurons, we found that 10 μm DHPG treatment resulted in significantly increased Ca2+ release from neurons derived from E18 spinophilin knock-out mice as compared with wild-type cortical neurons (Fig. 5D). Thus, although spinophilin overexpression in HEK 293 cells does not influence Group I mGluR-dependent Ca2+ signaling, endogenous spinophilin contributes to the regulation of Group I mGluR signaling in neurons.
FIGURE 4.
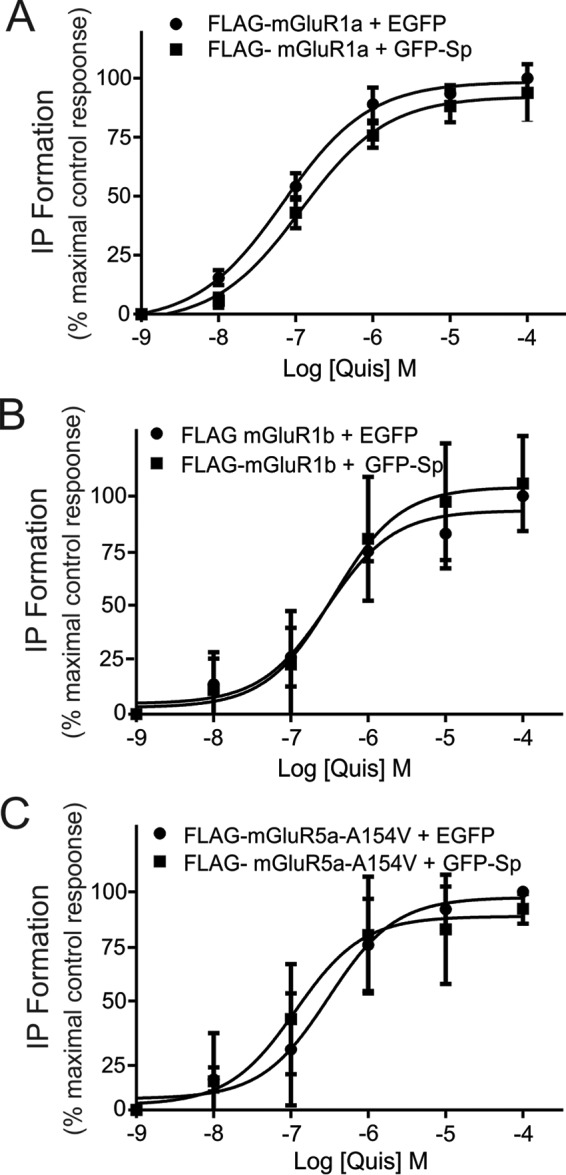
Effect of spinophilin on agonist-stimulated Group I mGluR-stimulated inositol phosphate formation. Dose response for quisqualate-stimulated (30 min, 0–30 μm) inositol formation (IP) in HEK 293 cells transfected with 2 μg of pcDNA3.1 plasmid cDNA encoding FLAG-mGluR1a (A), FLAG-mGluR1b (B), or FLAG-mGluR5a-A154V (C) along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin. The data represent the mean ± S.D. (error bars) for 4–6 independent experiments.
FIGURE 5.
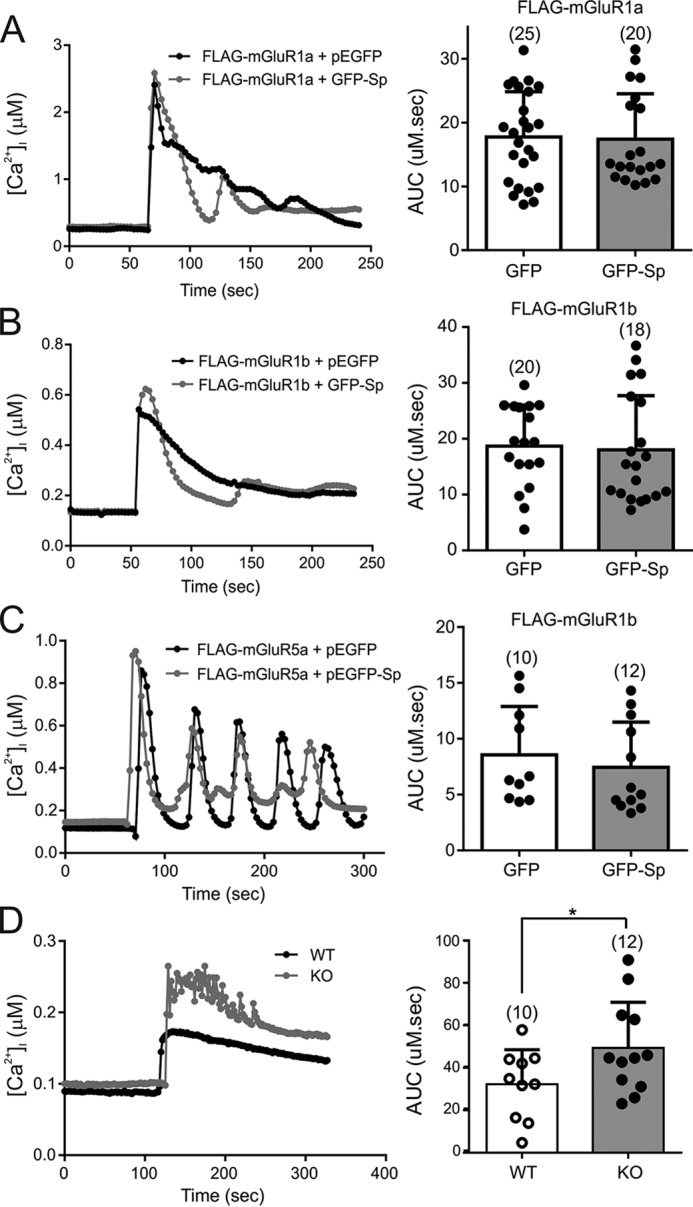
Effect of spinophilin on Group I mGluR-mediated Ca2+ release. Shown are representative traces and quantification, represented as area under the curve (AUC), of intracellular Ca2+ release in response to treatment with 30 μm quisqualate in HEK 293 cells transfected with 2 μg of pcDNA3.1 plasmid cDNA encoding FLAG-mGluR1a (A), FLAG-mGluR1b (B), or FLAG-mGluR5a (C) along with 2 μg of either GFP or GFP-spinophilin. The number of cells tested is shown in parentheses. D, representative traces and quantification, represented as area under the curve, of intracellular Ca2+ release in response to treatment with 10 μm DHPG in primary cortical neurons (12–14 DIV) derived from E18 wild-type and spinophilin knock-out embryos. The area under the curve is representative of the number of cells tested, shown in parentheses. The data represent the mean ± S.D. (error bars) of the number of cells tested as indicated. *, p < 0.05 compared with wild-type neurons.
Effect of Spinophilin on Group I mGluR-stimulated ERK1/2 Phosphorylation
Spinophilin was previously shown to modulate α2AAR-stimulated ERK1/2 phosphorylation (22). Similar to other GPCRs, Group I mGluRs activate ERK1/2 phosphorylation by a number of different molecular mechanisms (19, 31, 37). Therefore, we examined whether spinophilin overexpression in HEK 293 cells altered ERK1/2 phosphorylation in response to the activation of either FLAG-mGluR1a, FLAG-mGluR1b, or FLAG-mGluR5a with 30 μm quisqualate. We found that agonist stimulation for 2, 5, and 15 min resulted in a significant increase in ERK1/2 phosphorylation (Fig. 6, A–C). However, in cells expressing either FLAG-mGluR1a or FLAG-mGluR5a, spinophilin overexpression significantly attenuated ERK1/2 phosphorylation when compared with control cells (Fig. 6, A and B). In contrast, overexpression of spinophilin did not alter FLAG-mGluR1b-stimulated ERK1/2 phosphorylation in HEK 293 cells (Fig. 7A). The deletion of the PP1γ binding motif in the C-terminal tail of mGluR1a did not prevent spinophilin overexpression-dependent attenuation of FLAG-mGluR1a-stimulated ERK1/2 phosphorylation (Fig. 7B). However, the spinophilin-mediated antagonism of FLAG-mGluR1a-stimulated ERK1/2 phosphorylation was mitigated by the deletion of the C-terminal PDZ binding motif (Fig. 7C). Consequently, similar to what we observed for the regulation of Group I mGluR-mediated endocytosis, functional spinophilin-dependent blockade of ERK1/2 phosphorylation required PDZ domain interactions. To assess whether endogenous spinophilin contributed to the regulation of ERK1/2 phosphorylation by endogenous Group I mGluRs expressed in primary cortical neurons, we examined DHPG-stimulated ERK1/2 phosphorylation in primary cortical neurons (12–14 DIV) derived from either E18 wild-type littermate or spinophilin knock-out embryos. We found that in the absence of agonist stimulation, basal ERK1/2 phosphorylation was increased in neurons derived from spinophilin knock-out mice when compared with wild-type neurons (Fig. 8, A and B). Moreover, agonist-stimulated ERK1/2 responses were increased in primary neurons derived from spinophilin knockouts, and DHPG treatment for 10 min induced a significant increase in ERK1/2 phosphorylation over basal (Fig. 8B). Similar to what we observed for ERK1/2 phosphorylation, basal AKT phosphorylation was also increased in spinophilin knock-out cultures when compared with littermate control cultures (Fig. 8, A and C).
FIGURE 6.

Effect of spinophilin on mGluR1a and mGluR5 ERK1/2 phosphorylation. Shown are representative immunoblots of pERK1/2 activity and total ERK1/2 expression in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding either FLAG-mGluR1a (A) or FLAG-mGluR5 (B) along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin and treated with 30 μm quisqualate for 0, 2, 5, and 15 min. Also shown are cell lysates (50 μg) for mGluR1a, mGluR5a, and GFP-spinophilin expression. Bar graphs in each panel show the densitometric analysis of ERK1/2 phosphorylation normalized to both basal activity and total ERK1/2 protein expression. Data represent the mean ± S.D. (error bars) of four independent experiments. *, p < 0.05 versus untreated cells lacking GFP-spinophilin.
FIGURE 7.

Functional regulation of Group I mGluR ERK1/2 phosphorylation is regulated by the C-terminal tail PDZ binding motif. A, representative immunoblot for pERK1/2 activity and total ERK1/2 expression in HEK 293 cells transfected with 2 μg of pcDNA3.1 of FLAG-mGluR1b along with 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin and treated with 30 μm quisqualate for 0, 2, 5, and 15 min. Also shown are cell lysates (50 μg) for mGluR1b and GFP-spinophilin expression. The bar graph shows the densitometric analysis of ERK1/2 phosphorylation normalized to both basal activity and total ERK1/2 protein expression. Data represent the mean ± S.D. (error bars) of five independent experiments. Shown are representative immunoblots for pERK1/2 activity and total ERK1/2 expression in HEK 293 cells transfected with 2 μg of pcDNA3.1 encoding either FLAG-PP1Ala (B) or FLAG-ΔPDZ (C) along with either 2 μg of plasmid cDNA encoding either GFP or GFP-spinophilin and treated with 30 μm quisqualate for 0, 5, and 15 min. Also shown are cell lysates (50 μg) for FLAG-PP1Ala, FLAG-ΔPDZ, and GFP-spinophilin expression. The bar graphs in each panel show the densitometric analysis of ERK1/2 phosphorylation normalized to both basal activity and total ERK1/2 protein expression. Data represent the mean ± S.D. of 5–7 independent experiments. *, p < 0.05 versus untreated cells lacking GFP-spinophilin.
FIGURE 8.

Effect of spinophilin knock-out on Group I mGluR-mediated ERK1/2 and AKT phosphorylation in cortical neurons. A, representative immunoblots for both pERK1/2 and pAKT along with total ERK and AKT in primary cortical neurons (12–14 DIV) derived from E18 wild-type and spinophilin knock-out embryos treated for 0, 5, and 10 min with 10 μm DHPG. Also shown are cell lysates (50 μg) for endogenous mGluR1, mGluR5, and spinophilin expression. Bar graphs show the densitometric analysis of ERK1/2 (B) and AKT (C) phosphorylation normalized to both basal activity and total ERK1/2 or AKT protein expression. Data represent the mean ± S.D. (error bars) of 5–6 independent experiments. *, p < 0.05 versus wild-type neurons.
Effect of Spinophilin on mGluR5-stimulated LTD
To assess the role of spinophilin in mGluR5-mediated synaptic plasticity, LTD was induced by perfusing hippocampal slices for 10 min with the mGluR1/5 agonist DHPG (50 μm) in the presence of the mGluR1 antagonist LY367385 (10 μm). A repeated measurement analysis of variance, with genotype as between subject and drug treatment (control/acute/long term) as within subject measurements, revealed a significant effect of drug on fEPSP amplitudes (F(2,24) = 63.774, p < 0.0001), a significant effect of genotype (F(1,25) = 14.855, p = 0.001), and a significant interaction between genotype and drug (F(2,24) = 3.52, p = 0.037). The application of DHPG in the presence of LY367385 initially reduced the fEPSP amplitudes in both wild-type (to 61 ± 3%, p < 0.0001, n = 17) and spinophilin knock-out brain slices (to 72 ± 5%, p < 0.0001, n = 10 animals; Fig. 9, A and B). In wild-type slices, the depression of fEPSP persisted 1 h after DHPG perfusion (to 83 ± 3%, p = 0.0018), whereas fEPSP amplitudes fully recovered in slices derived from spinophilin knock-out mice and were not different from control fEPSPs before drug perfusion (100 ± 5%, p = 0.81; Fig. 9C). There was also a significant effect on paired pulse ratio (PPR) by drug treatment (F(2,24) = 15.430, p < 0.0001), a significant effect of genotype (F(1,25) = 5.428, p = 0.028), and a significant interaction with genotype (F(2,24) = 4.047, p = 0.023). The PPR was also significantly lower in knock-out slices (1.33 ± 0.3) when compared with wild-type slices (1.54 ± 0.05, p = 0.029; Fig. 9D). The PPR did not significantly change during acute or chronic administration of DHPG in knock-out slices (p = 0.74 and p = 0.15, respectively), whereas in slices derived from littermate controls, it was unchanged during acute DHPG application (p = 0.63) but significantly increased during LTD to 1.66 ± 0.08 (p = 0.0003; Fig. 9, D and E). In summary, acute DHPG inhibited fEPSPs in both wild-type and spinophilin knock-out mice without affecting the PPR, but it induced LTD only in wild-type animals, accompanied by an increase of the PPR. DHPG failed to induce either LTD or changes in PPR in knock-out animals.
FIGURE 9.
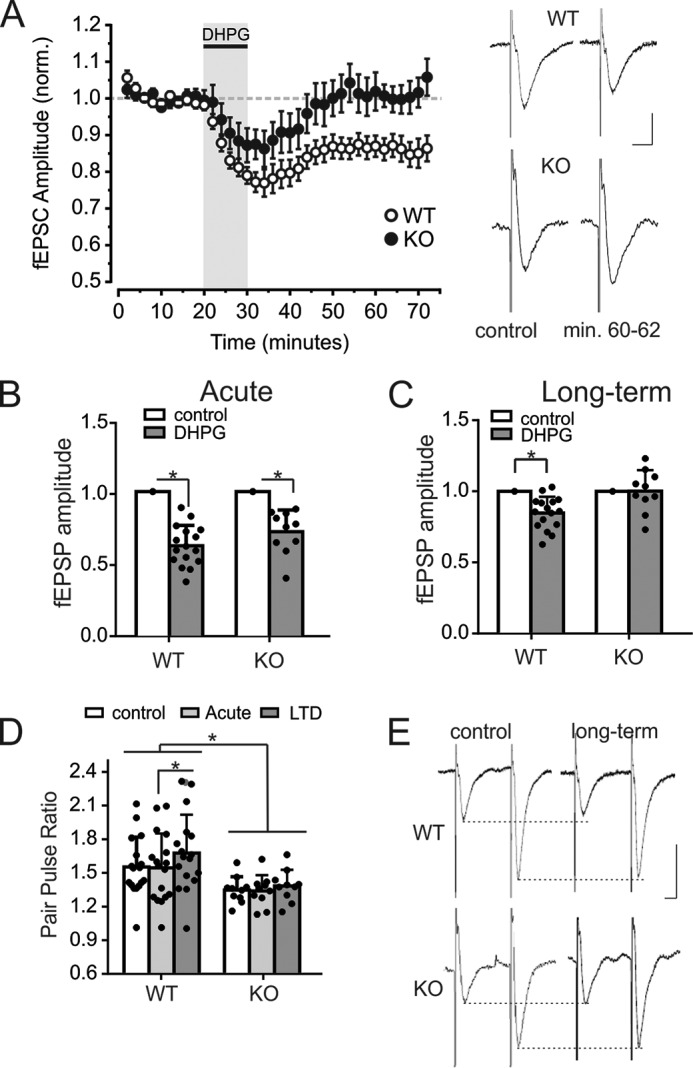
The mGluR5 agonist DHPG failed to induce LTD in knock-out mice. A, fEPSP amplitudes in the CA1 of wild-type and knock-out animals over 70 min. of stimulation (0.05 Hz). fEPSPs of each animal were normalized to the average amplitude before perfusion of DHPG. The shaded area indicates the slice perfusion with DHPG (50 μm) and LY367385 (10 μm). Normalized amplitudes of 17 wild-type and 10 knock-out mice were averaged, and error bars indicate S.D. On the right-hand side, exemplary fEPSPs are displayed for each genotype under control conditions (before perfusion with DHPG) and 30 min after DHPG perfusion. Vertical scale bar, 0.5 mV; horizontal bar, 10 ms. B, average fEPSP amplitudes in the CA1 during control measurements and acute DHPG perfusion (in the presence of LY367385). 10 fEPSPs of control measurements just before drug perfusion and at the end of a 10-min perfusion period were averaged per animal. Amplitudes are significantly reduced in both wild-type and knock-out animals. C, average fEPSP amplitudes in the CA1 during control and 30 min after DHPG perfusion (in the presence of LY367385). 10 fEPSPs during control measurements just before drug perfusion and from recording min 60–62 were averaged per animal. Amplitudes are significantly reduced only in wild-type and not in knock-out animals, indicating that DHPG failed to induce LTD in knock-out animals. D, PPR of fEPSPs with an interstimulus interval of 50 ms in wild-type and knock-out animals under control conditions, during DHPG perfusion, and 30 min after DHPG perfusion. The PPR was significantly lower in knock-out animals than in wild-type animals. Also, the PPR was significantly increased during LTD in wild-type animals but did not change during acute DHPG administration or in knock-out animals. E, exemplary fEPSP of a wild-type and a knock-out mouse during control measurements and 30 min after DHPG perfusion. Scale bars, 0.5 mV (vertical) and 10 ms (horizontal).
Discussion
In the present study, we have screened for novel proteins that may interact with the C-terminal tail of mGluR1a and identified spinophilin as a novel Group I mGluR-interacting protein, in addition to a number of previously known interacting proteins. Spinophilin is a multifunctional scaffolding protein that is the regulatory subunit for PP1γ and encodes both GPCR-binding and PDZ domains (9, 12). Both Group I mGluR subtypes, mGluR1a and mGluR5a, encode a PP1γ binding site in the proximal region of their C-terminal tails and a Class I PDZ binding motif at the distal end of their intracellular terminal tails. Therefore, we have assessed the potential role for spinophilin in the regulation of Group I mGluR activity. We find that overexpression of spinophilin in HEK 293 cells antagonized both agonist-stimulated endocytosis and ERK1/2 signaling mediated by either mGluR1a or mGluR5a. Consistent with this observation, the internalization of endogenous mGluR5 in cortical neurons derived from spinophilin knock-out mice is enhanced when compared with wild-type littermate control neurons. Similarly, basal ERK1/2 and AKT phosphorylation is increased in spinophilin knock-out neurons, and agonist-stimulated ERK1/2 signaling is significantly enhanced by the loss of spinophilin expression. The increase in ERK1/2 signaling in spinophilin knock-out neurons is accompanied by increased DHPG-stimulated intracellular Ca2+ release. Spinophilin was also required for the induction of chemically induced LTD mediated by the activation of mGluR5 because LTD in response to DHPG treatment is lost in spinophilin knock-out mice. The mechanism by which spinophilin interacts with Group I mGluRs appears to require the functional PDZ motif interactions as opposed to an indirect interaction with PP1γ, which is also scaffolded on Group I mGluRs (25, 26). Taken together, spinophilin appears to play a generalized role in antagonizing Group I mGluR activity in neurons.
Spinophilin was previously shown to interact with a number of other GPCRs, including the α1AR, α2AR, cholecystokinin A (CCKA) and CCKB, μ- and δ-opioid, D2 dopamine receptor (D2R), and M3 muscarinic receptors (m3AChR) (17–20, 22, 35). The interaction of spinophilin with each of these receptors was shown to be mediated by an interaction with their IL3. In the case of the α2AR, spinophilin binding to IL3 blocked GRK2 interactions with the receptor-Gβγ complex, thereby antagonizing β-arrestin recruitment to the receptor, resulting in impaired endocytosis and reduced ERK1/2 activation (22). Spinophilin interactions with the α1AR, α2AR, D2R, CCKA, CCKB, and m3AChR also negatively regulated Ca2+ signaling mediated by each of these receptors (19, 38). In contrast, spinophilin overexpression increased μ-opioid endocytosis but negatively regulated both μ- and δ-opioid-mediated ERK1/2 phosphorylation (20, 23). In the striatum, spinophilin knock-out increased μ-opioid receptor-mediated ERK1/2 phosphorylation while contributing to a loss of Gαi-mediated inhibition of cAMP signaling (20). In the present study, we showed that spinophilin overexpression did not affect either mGluR1a-, mGluR1b-, or mGluR5a-mediated IP formation and the subsequent release of intracellular Ca2+ calcium stores in HEK 293 cells but that spinophilin knock-out resulted in increased Ca2+ release in cortical neurons in response to Group I mGluR activation. This suggested that there are, as can be expected, distinct differences in how intracellular Ca2+ release is regulated in HEK 293 cells and neurons. Moreover, similar to what was observed for the α1AR, α2AR, D2R, CCKA, CCKB, and m3AChR, spinophilin binding to Group I mGluRs functioned to antagonize their internalization. Although we observed differences in G protein signaling in HEK 293 cells versus primary cortical neurons, spinophilin overexpression in HEK 293 cells antagonized both mGluR1a internalization and ERK1/2 phosphorylation, whereas agonist-stimulated internalization and agonist-stimulated ERK1/2 phosphorylation mediated by endogenous mGluR5 in cortical neurons was enhanced. The observation that spinophilin overexpression did not result in the attenuation of IP formation in HEK 293 cells could be due to the G protein coupling not being rate-limiting in these cells as a consequence of mGluR1a overexpression or because the column chromatography method utilized in the measurement of IP formation of an extended period of time lacked sensitivity. In contrast, agonist-stimulated signaling via endogenous mGluR5 was measured by Ca2+ release over a shorter time period, which had greater sensitivity for changes in second messenger levels.
We identified spinophilin as a protein that binds to the C-terminal tail of Group I mGluRs. However, we also found that, similar to what was previously observed for other GPCRs, spinophilin bound to the mGluR1/5 IL2 domain and was also co-immunoprecipitated with the alternative mGluR1 splice variant (mGluR1b), which lacked the extended mGluR1a C-terminal tail but retained the PP1γ binding domain (26, 39, 40). This raised the possibility that spinophilin was scaffolded on the receptor as a consequence of its interactions with IL2 and/or its indirect association with the receptor by virtue of its potential ability to form a functional complex with PP1γ (9). However, we found that an intact C-terminal PDZ binding motif was absolutely required for spinophilin-dependent antagonism of Group I mGluR endocytosis and ERK1/2 phosphorylation. Thus, the mechanism underlying the binding of spinophilin to Group I mGluRs was distinct from that of other GPCRs because it required intact PDZ domain interactions with the receptors, as opposed to the association of receptor IL domains with the spinophilin GPCR binding domain. Thus, despite the observation that the overall functional consequence of spinophilin binding to Group I mGluRs was similar to what was observed for other GPCRs, the mechanism by which spinophilin bound to Group I mGluRs to regulate their activity was uniquely mediated by PDZ interactions.
Spinophilin-mediated attenuation of Group I mGluR endocytosis and ERK1/2 signaling may also be the consequence of impaired β-arrestin recruitment. However, there is limited evidence to suggest that β-arrestins interact with mGluRs to regulate either their endocytosis or signaling, with the majority of studies suggesting that β-arrestins play no role in regulating the activity of mGluRs (28). Nevertheless, spinophilin has been demonstrated to bind directly to GRK2 and Gβ5, and GRK2 plays a central role in regulating Group I mGluR desensitization and internalization (34, 41). Thus, it is possible that spinophilin interactions with Group I mGluRs may regulate GRK2 interactions with Group I mGluRs, thereby contributing to the regulation of their endocytosis and signaling. The observation that spinophilin binds to IL2 of mGluR1/5 may be similar to the secondary interactions observed for spinophilin with the C-terminal tails of the μ- and δ-opioid receptors (23). Moreover, a number of mGluR1/5 IL2-interacting proteins exhibit secondary interactions with the C-terminal tail of the receptors, including GRK2, Pyk2, Arf6, Ral, phospholipase D1, and CAMKII (30–33). Thus, the effects of spinophilin overexpression may result as a consequence of spinophilin competing with these other mGluR-interacting proteins that have previously been shown to contribute to the mGluR1/5 endocytosis and ERK1/2 signaling. Similarly, spinophilin knock-out may enhance the ability of Pyk2 to bind and couple the receptor to the activation of ERK1/2 phosphorylation and/or facilitate GRK2-dependent endocytosis of the receptor (31, 42).
Interestingly, we find that PPRs are different between wild-type and knock-out mice. This probably occurs as a consequence of the fact that DHPG-induced LTD in the Schaffer collateral/CA1 synapse is induced by activation of postsynaptic mGluR5 receptors but has been shown also by others to be associated with an increase of paired pulse ratio, indicating a decrease in transmitter release probability during long-term depression (41). Accordingly, we observed an increase of PPR in wild-type mice after DHPG during LTD expression but not in knock-out mice after DHPG application. Interestingly, PPR in knock-out mice also showed a lower PPR at baseline recordings, indicating a lack of any preexisting depression and therefore a higher release probability when animals were sacrificed.
Spinophilin functions as the regulatory subunit for PP1γ and is structurally similar to neurabin I (9). These structurally similar cytoskeletal proteins not only regulate the structure of dendritic spines but also function as scaffolding proteins for protein phosphatases and are involved in the regulation of synaptic strength at excitatory synapses (9, 11, 15, 43). Studies using peptides to inhibit the interaction of PP1 with spinophilin and neurabin I result in the inhibition of NMDAR-dependent LTD but not LTD triggered by the activation of Group I mGluRs (15). However, these studies do not distinguish the specific role of different PP1 regulatory subunits. Similarly, LTD induced by low frequency stimulation is not observed in spinophilin knock-out mice (11). The previous observation that disruption of PP1 interactions with spinophilin does not affect LTD indicates that a loss of PP1 targeting does not contribute to the observed loss of mGluR5-induced LTD in spinophilin knock-out mice observed in the present study. Studies have also demonstrated that long term potentiation is deficient in neurabin I but not spinophilin knock-out mice (43). In contrast, LTD is lost in spinophilin but not neurabin I knock out mice and can be rescued by D2R activation (43). We also show that chemical LTD induced by the selective activation of mGluR5 is absent in spinophilin knock-out mice. Concomitant with this loss of mGluR5-mediated LTD, we observe that mGluR5 endocytosis is significantly enhanced in spinophilin knock-out mice when compared with littermate controls. Group mGluRs undergo both constitutive and agonist-stimulated endocytosis (28). Group I mGluR-mediated LTD is considered to be associated with increased post-synaptic AMPAR endocytosis (4). It is likely that the accelerated endocytosis of Group I mGluRs under basal conditions is the underlying factor for the observation that mGluR5-dependent LTD is not induced in spinophilin knock-out mice. Because LTD is an indicator of long-term changes in synaptic activity, we propose that spinophilin-dependent regulation of Group I mGluR activity may play a key role in long-term neuroplasticity associated with learning and memory.
Experimental Procedures
Materials
HEK 293 cells were from American Type Culture Collection (Manassas, VA). Cell culture reagents were from Invitrogen (Burlington, Canada): minimal essential medium, DMEM, neurobasal medium, FBS, and 0.25% trypsin-EDTA. l-Quisqualic acid (quisqualate), DHPG, and LY-367385 were from Tocris Bioscience (Minneapolis, MN). Biotinylation reagents EZ-Link sulfo-NHS-SS-biotin and NeutrAvidin agarose resin were purchased from Thermo Scientific (Rockford, IL). myo-[3H]Inositol was purchased from PerkinElmer Life Sciences. Protein G-Sepharose beads were from GE Healthcare (Oakville, Canada). The DC protein assay kit, 0.45-mm nitrocellulose, Clarity Western ECL substrate was purchased from Bio-Rad (Mississauga, Canada). Fura-2/AM was purchased from Thermo Fisher Scientific. Spinophilin, phospho-p44/42 MAPK, p44/42 MAPK, phospho-AKT, and AKT antibodies were purchased from Cell Signaling Technology (Danvers, MA). Rabbit mGluR1a, mGluR1b, and mGluR5a antibodies and mouse Myc tag antibody were purchased from Millipore (Billerica, MA), and GFP antibody was purchased from Invitrogen (Mississauga, Canada). Actin antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). GST antibody and secondary mouse and rabbit antibodies were purchased from GE Healthcare and Bio-Rad, respectively. All other biochemical reagents were purchased from Sigma-Aldrich.
Cell Culture
HEK 293 cells were maintained in minimal essential medium supplemented with 8% (v/v) heat-inactivated FBS (Invitrogen). Cells were seeded in 100-mm dishes and were transfected using a modified calcium phosphate method (44). 18 h following transfection, cells were washed, and medium was replaced. Cells were then allowed to recover for 24 h for co-immunoprecipitation experiments or were reseeded into 6- or 24-well plates or confocal dishes for all other experiments.
Mass Spectroscopy Identification of mGluR1-interacting Proteins
Ten 100-mm dishes of HEK 293 cells were transfected with FLAG-tagged mGluR1a C-terminal tail construct (amino acids 841–1199). Subsequently, HEK 293 cells were washed three times with PBS and solubilized in 1% Triton X-100 lysis buffer (25 mm HEPES, pH 7.5, 300 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, and 1% Triton X-100) containing protease inhibitors (1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) and 20 μg/ml of both leupeptin and aprotinin). Cellular debris was precipitated by centrifugation at 13,000 × g for 30 min at 4 °C. Cellular lysates were precleared by incubation with a 20-μl volume of Protein A-agarose beads for 6 h. Lysates were then incubated with a 40-μl volume of FLAG M2 resin for 4 h and washed three times with lysis buffer and three times with 50 mm NH4HCO3, pH 7.8. Subsequently, co-immunoprecipitates were eluted with 500 mm NH4OH at pH 11 in three 100-μl volumes and lyophilized to remove NH4OH. The samples were then resuspended in a 100-μl volume of H2O and subjected to a second round of lyophilization. Subsequently, samples were resuspended in a 50-μl volume of NH4HCO3, pH 8.0, and directly digested with sequencing grade trypsin (Promega). The resulting peptide mixture was then analyzed by liquid chromatography-tandem mass spectrometry using an LTQ-XL linear ion trap mass spectrometer (Thermo Scientific). The acquired tandem mass spectra were searched against a FASTA file containing the human NCBI sequences using a normalized implementation of SEQUEST running on the Sorcerer platform (Sage-N Research). The resulting peptide identifications returned by SEQUEST were filtered and assembled into protein identifications using PeptideProphet and ProteinProphet (Institute of Systems Biology, Seattle, WA) as described previously (45).
Mouse Model
Spinophilin knock-out mice were generously provided by Dr. Paul Greengard (Rockefeller University, New York) and bred to a C57BL/6 background. Mice were housed in an animal care facility at 23 °C on a 12-h/12-h light/dark cycle with food and water available ad libitum. Animal care was in accordance with the Animal Care and Use Committee at the University of Western Ontario and the Canadian Council on Animal Care.
Neuronal Primary Cultures
Neuronal cultures were prepared from the cortical and hippocampal regions of embryonic day 18 spinophilin knock-out and wild-type mouse brains. Following dissection, tissue underwent trypsin digestion followed by cell dissociation with an extra long 1250-μl filter tip pipette. Cells were then counted on a hemocytometer and plated on poly-l-ornithine-coated dishes with neurobasal medium supplemented with N2 and B27 supplements, 2 mm GlutaMAX, 50 μg/ml penicillin, 50 μg/ml streptomycin. Cells were incubated at 37 °C and 5% CO2 in a humidified incubator and grown for 12–14 DIV. Every 4 days, medium was replenished.
Plasmid Constructs
FLAG-mGluR1a, FLAG-mGluR1b, and FLAG-mGluR5a were described previously (46). Spinophilin cDNA was a generous gift from Dr. Qin Wang (University of Alabama). FLAG-mGluR1a lacking a PDZ binding motif (FLAG-mGlurR1a-ΔPDZ), lacking a PP1γ binding site (FLAG-mGluR1a-PP1Ala), or lacking both the PDZ and PP1γ binding site (FLAG-mGluR1a-PP1-ΔPDZ) was generated by QuikChangeTM site-directed mutagenesis (Stratagene, La Jolla, CA). To obtain FLAG-mGluR1a-ΔPDZ, we introduced a stop before the C-terminal PDZ motif at position 1192 (1192STL1194). The PP1γ binding site, composed of amino acids 891–895 (891KSVSW895), was mutated to alanine, thereby generating FLAG-mGluR1a-PP1Ala.
Co-immunoprecipitation
HEK 293 cells were transiently transfected with the cDNAs described in the figure legends. Cells were then incubated in HEPES balanced salt solution (HBSS) with or without 30 μm quisqualate for 15 min. Cells were placed on ice, washed twice with cold PBS, and lysed with cold lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, and 0.1% Triton X-100) containing protease inhibitors (1 mm AEBSF, 10 μg/ml leupeptin, and 5 μg/ml aprotinin) for 20 min. Cells were scraped and transferred to a 1.5-ml Eppendorf tube and centrifuged at 14,000 × g for 15 min at 4 °C to pellet insoluble material. Protein concentration was determined using a Bradford assay. 200–250 μg of lysate was then incubated with 50 μl of FLAG M2-affinity beads for 2 h at 4 °C with rotation to immunoprecipitate mGluR1a, mGluR1b, or mGluR5a. Following incubation, beads were washed twice with cold 0.1% Triton X-100 lysis buffer and once with cold PBS. Proteins were solubilized in 3× SDS sample buffer containing 2-mercaptoethanol.
Samples then were separated by SDS-PAGE, transferred to 0.45-mm nitrocellulose membrane, and immunoblotted to identify co-immunoprecipitated GFP- or Myc-tagged spinophilin protein using a primary rabbit antibody against GFP (1:10,000 dilution) or a primary mouse antibody against Myc (1:1000). This was followed by an HRP-conjugated secondary anti-rabbit (1:5000 dilution) or anti-mouse (1:2500) antibody. Receptor and spinophilin protein expression was determined by immunoblotting 20 μg of protein from each cell lysate used for immunoprecipitation. Proteins were detected by chemiluminescence.
GST Pull-down
GST-mGluR1a intracellular loop 2 (IL2) was cloned into a pGEX4T1 vector and transformed into Escherichia coli recombinant bacteria. Bacteria were grown at 37 °C with shaking until A600 = 0.6–1.0. Cultures were then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 23 °C for 3 h. Cells were pelleted by centrifugation and lysed in lysis buffer (500 mm NaCl, 0.5% Nonidet P-40, 50 mm Tris, pH 7.6, 5 mm EDTA, 5 mm EGTA) containing protease inhibitors (2 mm AEBSF, 50 mg/ml aprotinin, 20 mg/ml leupeptin) and sonicated (three times for 10 s each) at 4 °C. Insoluble material was pelleted by centrifugation at 14,000 × g for 15 min at 4 °C. To purify GST fusion constructs, 1 ml of solubilized protein was incubated overnight with 50 μl of glutathione-Sepharose bead slurry. Beads were then washed three times in cold PBS, and 500 μl of HEK 293 cell lysate overexpressing GFP-spinophilin was added to the GST fusion peptide and rotated for 1 h at 4 °C. Glutathione-Sepharose beads were then washed five times in PBS and eluted with 3× SDS loading buffer containing 2-mercaptoethanol. Samples were subjected to SDS-PAGE, and membranes were immunoblotted with GFP to determine whether GFP-spinophilin was pulled down with the GST-mGluR1a-IL2 as described previously (33).
Internalization Biotinylation
For both neurons and HEK 293 cells, cells were placed on ice, washed one time with cold HBSS, and allowed to cool on ice for 10 min to prevent recycling events. Cells were biotinylated on ice with 1 μg/ml sulfo-NHS-SS-biotin for 1 h. Biotin was quenched by washing twice and incubating on ice with cold 100 mm glycine. Cells were washed with cold HBSS and then stimulated with or without 30 μm quisqualate (HEK 293 cells) or 10 μm DHPG (neurons) for the amount of time specified in the figure legends to induce internalization of Group I mGluRs. Cell surface biotin was then stripped using 50 mm sodium-2-mercaptoethane sulfate in 0.5 m Tris-EDTA (TE) buffer plus 0.2% BSA. Cells were then washed three times with cold HBSS and lysed using 1% lysis buffer containing protease inhibitors (1 mm AEBSF, 10 μg/ml leupeptin, and 5 μg/ml aprotinin). Lysates were rocked for 15 min at 4 °C and centrifuged at 14,000 × g for 15 min. 200 μg of protein was incubated with 50 μl NeutrAvidin-agarose resin for 90 min with rotation at 4 °C. Beads were then washed twice with 1% lysis buffer and one time with PBS, and proteins were solubilized in 3× SDS sample buffer with 2-mercaptoethanol. Samples were then separated by SDS-PAGE, transferred to 0.45 μm nitrocellulose, and incubated in primary anti-FLAG (1:1000) or anti-mGluR5 antibody (1:1000), followed by HRP-conjugated secondary antibody (1:10,000). Total levels of mGluRs and spinophilin were determined by immunoblotting 20 μg of protein from cell lysates used for biotinylation. Proteins were detected by chemiluminescence. Internalization is expressed as a percentage of total cell surface receptor normalized to receptor and actin loading.
Inositol Phosphate Formation
48 h following transfection, cells were incubated overnight with 100 μCi/ml in myo-[3H]inositol containing glutamine-free DMEM. The following morning, cells were washed twice with warm HBSS and incubated for 1 h at 37 °C. Cells were then incubated for 10 min with 10 mm LiCl, followed by incubation with increasing concentrations of quisqualate for 30 min. Cells were then placed on ice, and the reaction was stopped with 500 μl of 0.8 m perchloric acid for 30 min. Perchloric acid was neutralized with 400 μl of 0.72 m KOH, 0.6 m KHCO3. Incorporation of [3H]inositol was determined by counting the radioactivity in 50 μl of total cell lysate. Total inositol phosphate was purified by anion exchange chromatography using Dowex 1-X8 (formate form) 200–400-mesh anion exchange resin. [3H]Inositol phosphate formation was determined by using liquid scintillation and a Beckman LS 6500 scintillation counter.
ERK Activation
HEK 293 Cells
Cells were starved overnight at 37 °C in serum- and glutamine-free DMEM. Cells were then washed twice and incubated in warm HBSS for 1 h and then stimulated with 30 μm quisqualate for the amount of time indicated in the figure legends. Cells were then placed on ice, washed twice with cold PBS, and lysed with 1% lysis buffer containing protease inhibitors and phosphatase inhibitors (sodium orthovanadate and sodium fluoride). 20 μg of cell lysate were solubilized in 3× SDS sample buffer and 2-mercaptoethanol, separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted for phosphorylated and total ERK levels, followed by HRP-conjugated secondary antibodies. Receptor and spinophilin levels were also detected by chemiluminescence.
Neurons
Primary neuronal cultures from spinophilin knock-out mice and wild-type littermates were washed twice and incubated at 37 °C in warm HBSS for 30 min. Neurons were then stimulated with 10 μm DHPG for 0, 5, or 10 min. Neurons were then placed on ice, washed twice with cold PBS, and lysed in radioimmune precipitation buffer containing protease and phosphatase inhibitors. 20 μg of protein from the cell lysate was solubilized in 3× SDS buffer, and Western blotting was performed as described above.
Calcium Imaging
HEK 293 Cells
HEK 293 cells were transfected and reseeded into 35-mm confocal dishes. Cells were washed twice with KRH buffer (125 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2.6 mm MgSO4, 5 mm HEPES, pH 7.2). FLAG-mGluR1a, FLAG-mGluR1b, or FLAG-mGluR5a was then labeled with rabbit anti-FLAG-conjugated Zenon Alexa Fluor 555 antibody for 20 min. Intracellular Ca2+ was labeled with 5 μm Fura-2/AM in KRH buffer for 30 min at 37 °C. Cells were washed twice with KRH buffer before loading onto a PTI DeltaRam microscope. Cells expressing either FLAG-mGluR1a, FLAG-mGluR1b, or FLAG-mGluR5a and GFP-spinophilin were selected using ImageMaster software. Fluorescence intensity was examined by illuminating the cells with 340 and 380 nm, and the intensity values at each excitation were recorded. After obtaining a 1-min baseline recording, cells were stimulated with 30 μm quisqualate for 5 min. Calcium concentration was obtained by using the formula, [Ca2+] = Kd × (R − Rmin)/(Rmax − R) × Fmax/Fmin, where R is the Fura-2/AM 340/380 ratio, Kd = 0.761 μm, Rmin = 0.196, Rmax = 6.907, and Fmax/Fmin = 9.558. The area under the curve was calculated using GraphPad Prism software.
Neurons
Neuronal primary cultures from spinophilin knock-out and wild-type mice were seeded onto 35-mm confocal dishes and grown in culture for 21 days with twice weekly feeding. Neurons were washed twice with warm HBSS, and intracellular Ca2+ was labeled with 1 μm Fura-2/AM in HBSS for 20 min at 37 °C. Neurons were washed twice and loaded onto the PTI DeltaRam microscope. A 3-min baseline was recorded, and cells were then stimulated with 10 μm DHPG for 5 min. Recording and analysis were conducted as described for HEK 293 cells.
Electrophysiology
LTD was measured in acute brain slices (400 μm) of 4–6-week-old wild-type and knock-out littermates. Mice were briefly anesthetized with isoflurane and decapitated, and the brain was quickly removed and cut with a vibratome (Microm, Walldorf, Germany) in 4 °C artificial cerebrospinal fluid, containing 119 mm NaCl, 4 mm KCl, 1.5 mm MgSO4, 2.5 mm CaCl2, 26.2 mm NaHCO3, 1 mm NaHPO4, 11 mm glucose. The slices were stored in oxygenated artificial cerebrospinal fluid at room temperature. For recordings, a slice was transferred into the superfused chamber on a fixed stage microscope (Zeiss Axioscope, Oberkochen, Germany) and constantly perfused at 1 ml/min with oxygenated 28 °C warm artificial cerebrospinal fluid. A concentric bipolar stimulation electrode was placed in the stratum radiatum of the CA1 region, and extracellular field potentials were recorded using a glass pipette filled with artificial cerebrospinal fluid (∼4-megaohm resistance) placed around 0.7–1 mm medial to the stimulus electrode. Slices were equilibrated for 1 h in the recording chamber before recordings were started. Stimulus intensity was adjusted to produce a response of 1–1.5-mV amplitude, with an initial slope of 0.5 mV/ms. Paired pulse synaptic stimulation was administered at 0.05 Hz with an interstimulus interval of 50 ms between paired pulses. To induce mGluR5-dependent LTD, the slice was perfused with the mGluR1/5 agonist DHPG (50 μm) in the presence of the mGluR1 antagonist LY367385 (10 μm) for 10 min. For LTD analysis, fEPSP amplitudes of each cell were normalized to the average amplitude of the first 10 min of baseline stimulation. For assessing the acute effect of drug, the last 10 trials during drug perfusion were averaged per cell. For assessing LTD, the last 10 trials of each cell were averaged. Paired pulse ratio was calculated as fEPSP2/fEPSP1 amplitude.
Statistical Analysis
Immunoblots were quantified using Image Lab software. GraphPad Prism software was used to analyze data for statistical significance as well as to analyze and fit dose-response curves. Statistical significance was determined by either an unpaired two-tailed t test or two-way analysis of variance followed by a Holm-Sidak multiple-comparison test with a significance level of p < 0.05. For electrophysiology experiments, statistical analyses were performed using mixed analysis of variance in SPSS and Bonferroni adaptions for post hoc comparisons.
Author Contributions
A. R. D. helped to design the experiments and performed most of the experiments, analyzed the data, and wrote the manuscript. S. F., H. A. D., C. W., and F. M. R. performed experiments and analyzed data. S. P. C. provided intellectual input and help with experimental design. S. A. performed the proteomic screen. S. S. performed the electrophysiology experiments and analyzed the data. S. S. G. F. conceived the study, supervised the project, and wrote and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Canadian Institutes of Health Research Grant MOP-119437 (to S. S. G. F.). The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptors
- α1AR and α2AR
- α1- and α2-adrenergic receptor, respectively
- NMDA
- N-methyl-d-aspartate
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- GRK2
- G protein-coupled receptor kinase 2
- IL2 and IL3
- intracellular loop domain 2 and 3, respectively
- IP
- inositol phosphate
- LTD
- long-term depression
- mGluR
- metabotropic glutamate receptor
- quisqualate
- l-quisqualic acid
- fEPSP
- field excitatory postsynaptic potential
- DIV
- days in vitro
- PPR
- paired pulse ratio
- DHPG
- dihydroxyphenylglycine
- CCKA and CCKB
- cholecystokinin A and B, respectively
- D2R
- D2 dopamine receptor
- m3AChR
- M3 muscarinic receptor(s)
- AEBSF
- 4-(2-aminoethyl)benzenesulfonyl fluoride
- HBSS
- HEPES balanced salt solution.
References
- 1. Dingledine R., Borges K., Bowie D., and Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 2. Ribeiro F. M., Paquet M., Cregan S. P., and Ferguson S. S. (2010) Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol. Disord. Drug Targets 9, 574–595 [DOI] [PubMed] [Google Scholar]
- 3. Ayala J. E., Chen Y., Banko J. L., Sheffler D. J., Williams R., Telk A. N., Watson N. L., Xiang Z., Zhang Y., Jones P. J., Lindsley C. W., Olive M. F., and Conn P. J. (2009) mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collingridge G. L., Peineau S., Howland J. G., and Wang Y. T. (2010) Long-term depression in the CNS Nat. Neurosci. Rev. 11, 459–473 [DOI] [PubMed] [Google Scholar]
- 5. Hamilton A., Zamponi G. W., and Ferguson S. S. G. (2015) Glutamate receptors as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol. Brain 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamilton A., Esseltine J. L., DeVries R. A., Cregan S. P., and Ferguson S. S. G. (2014) Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer's disease. Mol. Brain 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mao L. M., Liu X. Y., Zhang G. C., Chu X. P., Fibuch E. E., Wang L. S., Liu Z., and Wang J. Q. (2008) Phosphorylation of group I metabotropic glutamate receptors (mGluR1/5) in vitro and in vivo. Neuropharmacology 55, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magalhaes A. C., Dunn H., and Ferguson S. S. G. (2012) Regulation of G protein-coupled receptor activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 165, 1717–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen P. B., Ouimet C. C., and Greengard P. (1997) Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 94, 9956–9961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hsieh-Wilson L. C., Allen P. B., Watanabe T., Nairn A. C., and Greengard P. (1999) Characterization of the neuronal targeting protein spinophilin and its interactions with protein phosphatase-1. Biochemistry 38, 4365–4373 [DOI] [PubMed] [Google Scholar]
- 11. Feng J., Yan Z., Ferreira A., Tomizawa K., Liauw J. A., Zhuo M., Allen P. B., Ouimet C. C., and Greengard P. (2000) Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 97, 9287–9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarrouilhe D., di Tommaso A., Métayé T., and Ladeveze V. (2006) Spinophilin: from partners to functions. Biochimie 88, 1099–1113 [DOI] [PubMed] [Google Scholar]
- 13. Wang L. Y., Orser B. A., Brautigan D. L., and MacDonald J. F. (1994) Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature 369, 230–232 [DOI] [PubMed] [Google Scholar]
- 14. Yan Z., Hsieh-Wilson L., Feng J., Tomizawa K., Allen P. B., Fienberg A. A., Nairn A. C., and Greengard P. (1999) Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat. Neurosci. 2, 13–17 [DOI] [PubMed] [Google Scholar]
- 15. Morishita W., Connor J. H., Xia H., Quinlan E. M., Shenolikar S., and Malenka R. C. (2001) Regulation of synaptic strength by protein phosphatase 1. Neuron 32, 1133–1148 [DOI] [PubMed] [Google Scholar]
- 16. Hu X. D., Huang Q., Yang X., and Xia H. (2007) Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J. Neurosci. 27, 4674–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith F. D., Oxford G. S., and Milgram S. L. (1999) Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J. Biol. Chem. 274, 19894–19900 [DOI] [PubMed] [Google Scholar]
- 18. Richman J. G., Brady A. E., Wang Q., Hensel J. L., Colbran R. J., and Limbird L. E. (2001) Agonist-regulated interaction between α2-adrenergic receptors and spinophilin. J. Biol. Chem. 276, 15003–15008 [DOI] [PubMed] [Google Scholar]
- 19. Wang X., Zeng W., Kim M. S., Allen P. B., Greengard P., and Muallem S. (2007) Spinophilin/neurabin reciprocally regulate signaling intensity by G protein-coupled receptors. EMBO J. 26, 2768–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Charlton J. J., Allen P. B., Psifogeorgou K., Chakravarty S., Gomes I., Neve R. L., Devi L. A., Greengard P., Nestler E. J., and Zachariou V. (2008) Multiple actions of spinophilin regulate μ opioid receptor function. Neuron 58, 238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruiz de Azua I., Nakajima K., Rossi M., Cui Y., Jou W., Gavrilova O., and Wess J. (2012) Spinophilin as a novel regulator of M3 muscarinic receptor-mediated insulin release in vitro and in vivo. FASEB J. 26, 4275–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Q., Zhao J., Brady A. E., Feng J., Allen P. B., Lefkowitz R. J., Greengard P., and Limbird L. E. (2004) Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science 304, 1940–1944 [DOI] [PubMed] [Google Scholar]
- 23. Fourla D. D., Papakonstantinou M. P., Vrana S. M., and Georgoussi Z. (2012) Selective interactions of spinophilin with the C-terminal domains of the δ- and μ-opioid receptors and G proteins differentially modulate opioid receptor signaling. Cell. Signal. 24, 2315–2328 [DOI] [PubMed] [Google Scholar]
- 24. Kato A., Ozawa F., Saitoh Y., Fukazawa Y., Sugiyama H., and Inokuchi K. (1998) Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J. Biol. Chem. 273, 23969–23975 [DOI] [PubMed] [Google Scholar]
- 25. Croci C., Sticht H., Brandstätter J. H., and Enz R. (2003) Group I metabotropic glutamate receptors bind to protein phosphatase 1C: mapping and modeling of interacting sequences. J. Biol. Chem. 278, 50682–50690 [DOI] [PubMed] [Google Scholar]
- 26. Mao L., Yang L., Arora A., Choe E. S., Zhang G., Liu Z., Fibuch E. E., and Wang J. Q. (2005) Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J. Biol. Chem. 280, 12602–12610 [DOI] [PubMed] [Google Scholar]
- 27. Cheng S., Zhang J., Zhu P., Ma Y., Xiong Y., Sun L., Xu J., Zhang H., and He J. (2010) The PDZ domain protein CAL interacts with mGluR5a and modulates receptor expression. J. Neurochem. 112, 588–598 [DOI] [PubMed] [Google Scholar]
- 28. Dhami G. K., and Ferguson S. S. (2006) Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol. Ther. 111, 260–271 [DOI] [PubMed] [Google Scholar]
- 29. Dhami G. K., Anborgh P. H., Dale L. B., Sterne-Marr R., and Ferguson S. S. G. (2002) Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J. Biol. Chem. 277, 25266–25272 [DOI] [PubMed] [Google Scholar]
- 30. Bhattacharya M., Babwah A. V., Godin C., Anborgh P. H., Dale L. B., Poulter M. O., and Ferguson S. S. (2004) Ral and phospholipase D2-dependent pathway for constitutive metabotropic glutamate receptor endocytosis. J. Neurosci. 24, 8752–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicodemo A. A., Pampillo M., Ferreira L. T., Dale L. B., Cregan T., Ribeiro F. M., and Ferguson S. S. (2010) Pyk2 uncouples metabotropic glutamate receptor G protein signaling but facilitates ERK1/2 activation. Mol. Brain 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raka F., Di Sebastiano A. R., Kulhawy S. C., Ribeiro F. M., Godin C. M., Caetano F. A., Angers S., and Ferguson S. S. G. (2015) Ca2+/calmodulin-dependent protein kinase II interacts with group I metabotropic glutamate and facilitates receptor endocytosis and ERK1/2 signaling: role of β-amyloid. Mol. Brain 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dhami G. K., Babwah A. V., Sterne-Marr R., and Ferguson S. S. (2005) Phosphorylation-independent regulation of metabotropic glutamate receptor 1 signaling requires G protein-coupled receptor kinase 2 binding to the second intracellular loop. J. Biol. Chem. 280, 24420–24427 [DOI] [PubMed] [Google Scholar]
- 34. Brady A. E., Wang Q., Allen P. B., Rizzo M., Greengard P., and Limbird L. E. (2005) α2-Adrenergic agonist enrichment of spinophilin at the cell surface involves β γ subunits of Gi proteins and is preferentially induced by the α2A-subtype. Mol. Pharmacol. 67, 1690–1696 [DOI] [PubMed] [Google Scholar]
- 35. Brady A. E., Wang Q., Colbran R. J., Allen P. B., Greengard P., and Limbird L. E. (2003) Spinophilin stabilizes cell surface expression of α2B-adrenergic receptors. J. Biol. Chem. 278, 32405–32412 [DOI] [PubMed] [Google Scholar]
- 36. Esseltine J. L., Willard M. D., Wulur I. H., Lajiness M. E., Barber T. D., and Ferguson S. S. (2013) Somatic mutations in GRM1 in cancer alter metabotropic glutamate receptor 1 intracellular localization and signaling. Mol. Pharmacol. 83, 770–780 [DOI] [PubMed] [Google Scholar]
- 37. Iacovelli L., Salvatore L., Capobianco L., Picascia A., Barletta E., Storto M., Mariggiò S., Sallese M., Porcellini A., Nicoletti F., and De Blasi A. (2003) Role of G protein-coupled receptor kinase 4 and β-arrestin 1 in agonist-stimulated metabotropic glutamate receptor 1 internalization and activation of mitogen-activated protein kinases. J. Biol. Chem. 278, 12433–12442 [DOI] [PubMed] [Google Scholar]
- 38. Wang X., Zeng W., Soyombo A. A., Tang W., Ross E. M., Barnes A. P., Milgram S. L., Penninger J. M., Allen P. B., Greengard P., and Muallem S. (2005) Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat. Cell Biol. 7, 405–411 [DOI] [PubMed] [Google Scholar]
- 39. Pin J. P., Waeber C., Prezeau L., Bockaert J., and Heinemann S. F. (1992) Alternative splicing generates metabotropic glutamate receptors inducing different patterns of calcium release in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 89, 10331–10335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pickering D. S., Thomsen C., Suzdak P. D., Fletcher E. J., Robitaille R., Salter M. W., MacDonald J. F., Huang X. P., and Hampson D. R. (1993) A comparison of two alternatively spliced forms of a metabotropic glutamate receptor coupled to phosphoinositide turnover. J. Neurochem. 61, 85–92 [DOI] [PubMed] [Google Scholar]
- 41. Pignatelli M., Piccinin S., Molinaro G., Di Menna L., Riozzi B., Cannella M., Motolese M., Vetere G., Catania M. V., Battaglia G., Nicoletti F., Nisticò R., and Bruno V. (2014) Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling angelman syndrome. J. Neurosci. 34, 4558–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ribeiro F. M., Ferreira L. T., Paquet M., Cregan T., Ding Q., Gros R., and Ferguson S. S. (2009) Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J. Biol. Chem. 284, 23444–23453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allen P. B., Zachariou V., Svenningsson P., Lepore A. C., Centonze D., Costa C., Rossi S., Bender G., Chen G., Feng J., Snyder G. L., Bernardi G., Nestler E. J., Yan Z., Calabresi P., and Greengard P. (2006) Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience 140, 897–911 [DOI] [PubMed] [Google Scholar]
- 44. Ferguson S. S., and Caron M. G. (2004) Green fluorescent protein-tagged β-arrestin translocation as a measure of G protein-coupled receptor activation. Methods Mol. Biol. 237, 121–126 [DOI] [PubMed] [Google Scholar]
- 45. Ahmed S. M., Daulat A. M., Meunier A., and Angers S. (2010) G protein βγ subunits regulate cell adhesion through Rap1a and its effector Radil. J. Biol. Chem. 285, 6538–6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dale L. B., Bhattacharya M., Anborgh P. H., Murdoch B., Bhatia M., Nakanishi S., and Ferguson S. S. (2000) G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J. Biol. Chem. 275, 38213–38220 [DOI] [PubMed] [Google Scholar]