

Abstract
Lymphocyte-specific protein-tyrosine kinase (Lck) plays an essential role in T cell receptor (TCR) signaling and T cell development, but its activation mechanism is not fully understood. To explore the possibility that plasma membrane (PM) lipids control TCR signaling activities of Lck, we measured the membrane binding properties of its regulatory Src homology 2 (SH2) and Src homology 3 domains. The Lck SH2 domain binds anionic PM lipids with high affinity but with low specificity. Electrostatic potential calculation, NMR analysis, and mutational studies identified the lipid-binding site of the Lck SH2 domain that includes surface-exposed basic, aromatic, and hydrophobic residues but not the phospho-Tyr binding pocket. Mutation of lipid binding residues greatly reduced the interaction of Lck with the ζ chain in the activated TCR signaling complex and its overall TCR signaling activities. These results suggest that PM lipids, including phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate, modulate interaction of Lck with its binding partners in the TCR signaling complex and its TCR signaling activities in a spatiotemporally specific manner via its SH2 domain.
Keywords: cell signaling, phosphoinositide, plasma membrane, Src homology 2 domain (SH2 domain), T-cell receptor (TCR), Lck
Introduction
Lymphocyte-specific protein-tyrosine kinase (Lck) is a 56-kDa Src family kinase that plays an essential role in T cell receptor (TCR)4 signaling and T cell development (1, 2). In response to TCR stimulation, Lck phosphorylates immunoreceptor tyrosine-based activation motifs (ITAMs) in the CD3 and the ζ chain of the TCR-CD3 complex, thereby contributing to the initiation of TCR signaling (1, 2). This leads to ITAM recruitment and activation of ZAP-70 that subsequently phosphorylates the linker for the activation of T cells and the Src homology 2 (SH2) domain-containing leukocyte protein of 76 kDa (3). Phosphorylated linker for activation of T cells nucleates the T cell signalosome, recruiting and activating further downstream enzymes, including phospholipase Cγ1 (PLCγ1) (4, 5).
The enzymatic activity of Lck is regulated by phosphorylation and dephosphorylation of two Tyr residues, Tyr-394 and Tyr-505 (1, 2, 4). It has been generally thought that Lck has a similar activation mechanism to those of other Src family kinases (6). In the resting state, Tyr-505 near the C terminus is phosphorylated by C-terminal Src kinase, and the resulting phospho-Tyr-505 (Tyr(P)-505) interacts with the SH2 domain, leading to intramolecular tethering of its kinase domain, which is further augmented by the Src homology 3 (SH3) domain-kinase domain interaction. Upon TCR activation, CD45-mediated dephosphorylation of Tyr(P)-505 unleashes the kinase domain and primes Lck for full activation, which is thought to require autophosphorylation of Tyr-394 within the activation loop (7). More recent studies have revealed, however, that the activation mechanism of Lck is more complex than originally thought. For example, it has been reported that Lck can also exist as an enzymatically active form with Tyr-394 and Tyr-505 both phosphorylated (Tyr(P)-394/Tyr(P)-505) and that Lck-dependent tyrosine phosphorylation of TCR ITAMs does not require enzyme activation of Lck but involves other mechanisms, such as the membrane redistribution of active Lck or ligand-mediated conformational changes of Lck (8–10). It was also reported that Tyr-505 could be autophosphorylated by Lck (10). These results suggest the possibility that other factors are involved in Lck regulation under physiological conditions.
Lck is constitutively localized via N-terminal acylation to the plasma membrane (PM) where the TCR-CD3 complex is located (1, 2). Thus, PM lipids might be involved in interaction of Lck with the TCR-CD3 complex and its phosphorylation of ITAMs. It was reported that CD3 chains bind anionic lipids in the PM in resting T cells, thereby limiting the access of ITAMs to Lck (11, 12). However, a more recent study reported that ITAM phosphorylation by Lck was enhanced by an anionic lipid, phosphatidylserine (10). Overall, the mechanism and the physiological significance of Lck-lipid interaction in regulation of Lck activity remain unknown.
The SH2 domain is a prototypical modular protein interaction domain that has been identified in diverse cell-signaling proteins, including kinases, adaptors, and phosphatases (13). As a major reader of phosphotyrosine (Tyr(P)) signaling information, SH2 domain-containing proteins play crucial roles in linking various protein-tyrosine kinases to downstream molecules, thereby controlling the specificity of Tyr(P) signaling (14). Structural analysis of a wide range of SH2 domains and their complexes with Tyr(P)-peptides has revealed that SH2 domains have a common architecture made of two α-helices (αA and αB) flanking antiparallel β strands (βA to βG) (13, 15). They specifically recognize Tyr(P) and a few residues immediately C-terminal to Tyr(P) using a Tyr(P) binding pocket and a secondary binding site, respectively (13).
It was reported earlier that a small number of SH2 domains could bind lipids, which either inhibited (16) or promoted (17) the activity of their host proteins, but the lipid-binding site and the lipid specificity of these SH2 domains as well as the physiological significance of these findings remain controversial (18). More recently, genome-wide screening of PDZ (PSD95, Dlg1, Zo-1) (19–21) and SH2 domains (22) revealed that a large majority of these protein interaction domains bind PM lipids with high affinity and that their lipid binding is crucial for the cellular function of their host proteins. In this study, we explored the possibility that lipids bind the Lck SH2 domain (Lck-SH2) and control the cellular activity of Lck through this interaction.
Results
Membrane Binding of Lck SH2 Domain
To quantitatively assess the lipid binding activity and specificity of Lck-SH2, we measured binding of Lck-SH2 to lipid vesicles with varying compositions by SPR analysis. Because Lck-SH2 was not stably expressed in Escherichia coli, we expressed it as a C-terminal EGFP fusion protein, which generally improves the expression yield of modular domains without interfering with their membrane binding properties (23). Our control experiments showed that the C-terminal EGFP tag did not affect the membrane binding properties of Lck-SH2 (data not shown). Because Lck is associated with the cytofacial leaflet of the PM, we first used for binding measurements the vesicles whose lipid composition recapitulates that of the cytofacial PM (i.e. PM mimetic vesicles) (24).
Quantitative determination of the affinity of Lck-SH2 for the PM mimetic vesicles (Fig. 1, A and B, and Table 1) showed that it had a Kd of 160 ± 15 nm for the PM mimetic vesicles, which is comparable with that of other lipid-binding proteins (24), including phospholipase Cδ pleckstrin homology domain (PLCδ-PH) (Table 1). This high affinity of Lck-SH2 for PM mimetic vesicles is not due to nonspecific membrane adsorption because it was determined from the signal differences between the two channels containing PM mimetic vesicles and the neutral phosphatidylcholine vesicles, respectively. Also, the Lck SH3 domain showed no detectable affinity for the PM mimetic vesicles under the same conditions (data not shown). Finally, Lck-SH2 and the full-length Lck had comparable affinity for PM mimetic vesicles (Table 1), indicating that lipid binding activity of Lck lies within its SH2 domain and that the lipid-binding site of Lck-SH2 is fully exposed in the intact protein.
FIGURE 1.

Membrane binding properties of the Lck SH2 domain. A and B, determination of Kd for Lck-SH2-PM mimetic vesicle binding by SPR analysis. A, Lck-SH2 was injected at 5 μl/min at varying concentrations (0, 25, 50, 80, 100, 120, 150, 180, 200, and 250 nm from bottom to top) over the L1 chip coated with PM mimetic vesicles, and Req values were measured. B, binding isotherm was generated from the Req (average of triplicate measurements) versus the concentration (Po) of Lck-SH2 plot. A solid line represents a theoretical curve constructed from Rmax (= 470 ± 22) and Kd (= 160 ± 15 nm) values determined by nonlinear least squares analysis of the isotherm using the following equation: Req = Rmax/(1 + Kd/Po). Data represent means ± S.D. from triplicate measurements. C, PtdInsP selectivity of Lck-SH2. Sensorgrams are shown for PIP3 > PI4,5P2 > PI3,4P2 ≈ PI3,5P2 > PI5P > PI4P > PI3P (from top to bottom). D, effect of mutating cationic residues in the Tyr(P) pocket on membrane binding of Lck-SH2. E, effect of mutating cationic residues in ACP on membrane binding of Lck-SH2. F, PtdInsP selectivity of PLCδ-PH. Sensorgrams are shown for PI4,5P2 > PIP3 > PI3,5P2 ≈ PI3P ≈ PI3,4P2 ≈ PI4P > PI5P (from top to bottom). SPR measurements were performed in 20 mm Tris-HCl, pH 7.4, containing 0.16 m KCl. POPC vesicles were used for the control surface. PM mimetic (A and B), POPC/POPS/PtdInsP (77:20:3) (C and F), and POPC/POPS/PIP3 (77:20:3) (D and E) vesicles were used as active surfaces. SPR measurements were performed in 20 mm Tris-HCl, pH 7.4, containing 0.16 m KCl. PM mimetic vesicles contain POPC/POPE/POPS/PI/cholesterol/PI4,5P2 (12:35:22:8:22:1). The Lck-SH2 domain concentration in C–E was 150 nm. Typically, dissociation from the sensor chip was too slow to reach completion within 10 min of elution. Thus, only the association phases of the sensorgrams are shown here. All sensorgrams shown herein are representative of triplicate measurements that show essentially the same results.
TABLE 1.
Membrane binding properties of Lck, Lck-SH2, and mutants
| Proteins | Kda |
|---|---|
| nm | |
| Lck-SH2 WTb | 160 ± 15 |
| Lck-SH2 K182A/R184Ab | 430 ± 10 |
| Lck-SH2 A160Kb | 120 ± 10 |
| Lck FLc WT | 220 ± 80 |
| PLCδ-PH WT | 240 ± 60 |
| PLCδ-PH R56A/R60A | 480 ± 100 |
a Means ± S.D. were taken from triplicate equilibrium SPR measurements using PM mimetic vesicles (POPC/POPE/POPS/cholesterol/PI/PI45P2 = 12:35:22:22:8:1).
b These are EGFP fusion proteins.
c FL means full-length proteins.
We then determined the selectivity of Lck-SH2 for phosphoinositides (PtdInsP), which play key roles in cell signaling (25). When compared with canonical lipid binding domains with a well defined lipid pocket, such as PLCδ-PH (Fig. 1F), Lck-SH2 showed a low degree of PtdInsP selectivity, modestly preferring the most anionic PIP3 to other PtdInsPs (Fig. 1C). As expected from its high affinity for PM mimetic vesicles that contain PI4,5P2, Lck-SH2 bound PI4,5P2 slightly better than PI3,4P2 and PI3,5P2. Overall, it would seem that PtdInsP binding of Lck-SH2 is driven primarily not by specific PtdInsP headgroup recognition but by nonspecific electrostatic interactions.
To prove that PIP3 and PI4,5P2 play an important role in PM localization of Lck-SH2, we measured the PM localization of EGFP-tagged Lck-SH2 in response to PI4,5P2 and PIP3 depletion (26, 27). Before treatment, Lck-SH2 showed PM localization (Fig. 2A), albeit to a lesser degree than PLCδ-PH (Fig. 2B). When PI4,5P2 depletion was triggered by a rapamycin analog, Lck-SH2 (Fig. 2A) was rapidly removed from the PM, as was the case with a prototypical PI4,5P2-selective protein, PLCδ-PH (Fig. 2B). Also, PIP3 depletion by a PI3K inhibitor, LY294002, greatly reduced the PM localization of Lck-SH2 (Fig. 2A). These results support the physiological significance of PIP3 and PI4,5P2 binding of Lck-SH2.
FIGURE 2.

Effect of PI4,5P2 and PIP3 manipulation on the subcellular localization of C-terminal EGFP-tagged Lck-SH2. A, PI4,5P2 and PIP3 depletion greatly reduced PM localization of Lck-SH2. B, PI4,5P2 depletion greatly inhibited PM localization of PI4,5P2-selective EGFP-PLCδ-PH. C, K182A/R184A showed dramatically reduced PM localization, which was not affected by PI4,5P2 depletion. D, R154A behaved similarly to WT. The PI4,5P2 depletion was triggered by 2.5 μm A/C heterodimerizer that recruits FKBP-Inp to PM-anchored Lyn-FRB. All images for PI4,5P2 depletion were taken before and 1 min after heterodimerizer treatment. PIP3 depletion was achieved by pretreating the cells with 50 μm LY294002 for 1 h. HeLa cells were used for all measurements. Scale bars, 10 μm. Expression levels of EGFP-Lck-SH2 and EGFP-PLCδ-PH were kept as low as possible to minimize the possibility of non-physiological PM localization of proteins due to overexpression.
Membrane-binding Site of Lck SH2 Domain
Electrostatic potential computation of Lck-SH2 shows that it has a highly cationic Tyr(P) binding pocket (red arrow in Fig. 3A) and an alternative cationic patch (ACP) (green arrow in Fig. 3A) that is not as electropositive as the Tyr(P) pocket. Previous biochemical (16), structural (28), and computational (29) studies of some SH2 domains suggested that their Tyr(P) pockets could bind lipids. However, we found that mutation of the invariant Arg (Arg-154) in the Tyr(P) pocket of Lck-SH2 to Ala did not cause a significant decrease in binding to PM mimetic vesicles (Fig. 1D). In contrast, mutation of ACP residues (Lys-182 and Arg-184) reduced affinity for PM mimetic vesicles to a much greater extent (Fig. 1E and Table 1). About a 3-fold decrease in PM affinity of Lck-SH2 by the K182A/R184A mutation was comparable with that caused by the R56A/R60A mutation of PLCδ-PH (Table 1). These two residues are located on the membrane binding surface of PLCδ-PH that is involved in its nonspecific electrostatic interaction with anionic lipids (30). These results suggest that not the Tyr(P) pocket but the ACP serves as the primary lipid-binding site for Lck-SH2. This notion is supported by the subcellular localization patterns of Lck-SH2 mutants in HeLa cells, i.e. mutation of Lys-182 and Arg-184 abrogated its PM localization (Fig. 2C), whereas the mutation of Arg-154 had little effect on its PM recruitment (Fig. 2D).
FIGURE 3.

Membrane-binding site of Lck-SH2. A, surface electrostatic potential of Lck-SH2. The Tyr(P) binding pocket and the most prominent ACPs are indicated by red and green arrows, respectively. The minimal and maximal electrostatic potentials are −5 kT/e (red) and +5 kT/e (blue), respectively. B, location and morphology of the lipid-binding site of Lck-SH2. The structure of Lck-SH2 is shown with the Tyr(P) binding pocket (red arrow) pointing upward. Key cationic residues involved in lipid binding are shown in space-filling representation and labeled. C, model structure of Lck-SH2-Tyr(P)-IP4 ternary complex. Lck-SH2 domain (in the same orientation as in B) is shown in surface representation, and the peptide (red arrows) and IP4 (green arrows) are in stick representation. Notice that there is no steric overlap between the peptide and IP4. D, NMR CSPs for backbone amide signals of 15N-labeled Lck-SH2 observed in the presence of IP4. E, Lck-SH2 structure (Protein Data Bank code 1LCK) in ribbon representation. The residues with the highest CSPs are shown in light to dark red (top 15%), and the residues whose NMR signals are missing after IP4 titration are colored in green. Notice that all residues with the largest CSP are on the same molecular surface with Lys-182 and Arg-184. A much smaller CSP was observed with the invariant Arg (R154) and neighboring residues in the Tyr(P) binding pocket.
The ACP of Lck-SH2 does not overlap with any known protein-binding sites of Lck-SH2, including the secondary binding site to the Tyr(P) motif (13). Indeed, molecular modeling of Lck-SH2 suggests that it can bind a lipid headgroup and a Tyr(P) motif simultaneously and independently (Fig. 3, B and C). This notion was verified by its identical Tyr(P) peptide binding with and without PM mimetic vesicles and unaltered vesicle binding in the presence of a Tyr(P) peptide (Fig. 4).
FIGURE 4.
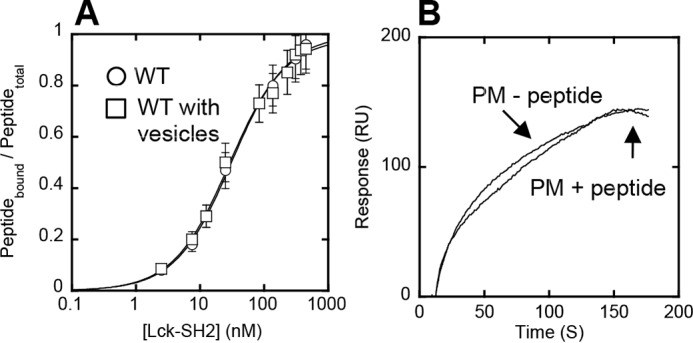
Orthogonal functionality of lipid and peptide binding sites of Lck-SH2. A, binding of Lck-SH2 to the Tyr(P) peptide (pYEEI) monitored by fluorescence anisotropy in the presence (□) and absence (○) of 40 μm PM mimetic vesicles. The peptide concentration was 50 nm. Notice that vesicles have no effect on peptide binding of Lck-SH2. Data represent means ± S.D. from triplicate measurements. B, effect of the Tyr(P) peptide on membrane binding of Lck-SH2. The SH2 domain was allowed to interact with the PM mimetic vesicles before and after a 30-min incubation with 100 μm peptide. The Lck-SH2 domain concentration was 50 nm. The peptide had a negligible effect on the membrane binding of Lck-SH2, showing the orthogonality of lipid and peptide binding. Sensorgrams are a representative from triplicate measurements that show essentially the same results.
To further investigate how Lck-SH2 interacts with membrane lipids, we performed NMR analysis of Lck-SH2-lipid binding using 15N-labeled Lck-SH2. Although Lck-SH2 showed low PtdInsP selectivity, it still has the highest affinity for PIP3 (Fig. 1C). We thus used for the solution NMR analysis a soluble headgroup analog of PIP3, IP4, which has been extensively used for both NMR and crystallographic analyses. IP4 was selected over a short-chain PIP3 for NMR titration because its high water solubility allowed cleaner NMR signals and more straightforward data analysis. Upon binding IP4, major chemical shift perturbations (CSP) were detected primarily on the residues located on the molecular surface containing the ACP (Fig. 3D). Importantly, large CSPs were observed not only with cationic residues (Arg-134 and Lys-182) but also with aromatic and hydrophobic residues (e.g. Ala-160, Phe-163, and Ile-183), suggesting that these residues also contribute to lipid binding of Lck-SH2. Although IP4 lacks the hydrophobic tail, CSPs were also observed in these residues presumably because their backbone conformational changes, which 15N NMR CSP mainly monitors, are linked to those of neighboring cationic residues during lipid binding. In contrast, CSPs were insignificant for Tyr(P) pocket residues. When Ala-160 was mutated to Lys to enhance electrostatic binding to anionic PM vesicles, the A160K mutant had ≈30% higher affinity than WT, supporting the notion that Ala-160 constitutes the membrane-binding surface (Table 1). In contrast, mutation of surface residues remote from the putative membrane-binding site (e.g. K130A and R207A) had little effect on the affinity of Lck-SH2 to PM vesicles (data not shown). Thus, lipid binding of Lck-SH2 appears to be driven by the combination of electrostatic and hydrophobic interactions, as reported for other lipid binding domains (24).
Role of Lipid Binding of Lck SH2 Domain in T Cell Signaling
To investigate the physiological significance of the lipid binding activity of Lck-SH2, we measured the effects of altering lipid binding of Lck on its TCR signaling activities. Because Lck-SH2 has high affinity for PM mimetic vesicles (Table 1) and modestly prefers PIP3 and PI4,5P2 to other PtdInsPs (Fig. 1C), both PI4,5P2 that is constitutively present in the PM and PIP3 whose production in the PM is induced by cell stimuli might contribute to its PM binding. For functional studies of Lck, we prepared a lipid-binding loss-of-function (LOF) mutant (K182A/R184A) and a gain-of-function (GOF) mutant (A160K) (Table 1). Neither mutation altered Tyr(P) binding of Lck-SH2 (Fig. 5).
FIGURE 5.

Tyr(P) binding affinity of Lck-SH2 WT and mutants. Affinity of Lck-SH2 WT (○), R154A (Tyr(P) site mutant) (□), K182A/R184A (LOF lipid site mutant) (Δ), and A160K (GOF lipid site mutant) (■) for three different Tyr(P)-containing peptides, pYEEI (A), pYSDPE (B), and pYQPQP (C), that have been reported to specifically bind the Lck-SH2 was measured by fluorescence anisotropy analysis as described in Fig. 4. For all three peptides, Lck-SH2 WT (Kd = 33 ± 7 nm for pYEEI), K182A/K184A (Kd = 40 ± 8 nm for pYEEI), and A160K (Kd = 32 ± 3 nm for pYEEI) showed comparable affinity, whereas R154A (Kd = 570 ± 200 nm for pYEEI) exhibited a ≈20-fold decrease in affinity. The peptide concentrations used varied from 5 to 500 nm depending on the Kd values. Data represent means ± S.D. from triplicate measurements.
We then compared the effects of introducing WT and mutants of Lck to Lck-deficient (JCaM1.6) Jurkat cells on TCR signaling activities. We first monitored the increase in calcium flux in JCaM1.6 cells (Fig. 6A). When stimulated with an anti-CD3 antibody, OKT3, JCaM1.6 cells showed no calcium flux, which was greatly increased by stable expression of Lck WT. Under the same conditions, K182A/R184A had ≈40% of the Lck WT activity (Fig. 6A), whereas the GOF mutant A160K was ≈30% more active than the WT (Fig. 6B). Furthermore, K182A/R184A was much less effective than WT in inducing OKT3-stimulated Tyr phosphorylation of downstream proteins, ZAP70, PLCγ, and ERK1/2 (Fig. 6C). In contrast, A160K showed ≈30% higher activity than WT in the ERK Tyr phosphorylation assay (Fig. 6D). It should be noted that for accurate comparison of signaling activities of WT and A160K, it was critical to use stable cell lines with similar expression levels for our activity assays. To compensate for the differences in expression levels of WT and the GOF mutant, we thus gated cells expressing similar levels of GFP-tagged proteins by flow cytometry and used them for quantitative analysis. Finally, we measured the signaling activity of a Tyr(P) binding-deficient mutant of Lck (R154A) as controls. As expected, it showed much reduced binding for Tyr(P) peptides (Fig. 5), and no detectable activity in all assays (Fig. 6, A and C). Collectively, strong correlation between in vitro membrane binding affinity of Lck WT and lipid-binding site mutants with their cellular signaling activities supports the notion that lipid binding of its SH2 domain is important for the cellular regulation and function of Lck.
FIGURE 6.

Lipid binding activity of Lck-SH2 domain is crucial for T cell signaling activities of Lck. A, calcium mobilization; C, Tyr phosphorylation of TCR signaling proteins in JCaM1.6 cells stably transduced with EGFP-tagged Lck WT and mutants after TCR stimulation. For calcium flux assay, cells were labeled with 4 μg/ml Indo-1 AM, and the calcium flux was monitored for 8 min after OKT3 (1 μg/ml) treatment by flow cytometry. For Tyr(P) monitoring, cells were stimulated with OKT3 (10 μg/ml for 5 min), and cell lysates were immunoblotted with indicated antibodies. B, calcium mobilization in JCaM1.6 cells stably transduced with EGFP-tagged Lck WT and A160K, respectively, after TCR stimulation with 0.1 μg/ml OKT3. D, JCaM1.6 cells stably transduced with EGFP-tagged Lck WT and A160K were stimulated with indicated doses of OKT3 for 5 min, and the levels of pERK1/2 were measured by intracellular staining and flow cytometry. To compensate for the differences in expression levels of WT and GOF mutants, cells expressing similar levels of EGFP-tagged proteins were gated and used for quantitative analysis. All data are from triplicate measurements that show essentially the same results. D, data represent means ± S.D. ***, p < 0.001 from Student's t test.
Mechanisms by Which Lipids Regulate Lck Function
Lck has been reported to interact with various proteins involved in TCR signaling (1–3, 31). To better understand how lipid-binding site mutations of Lck caused observed cellular phenotypes, we determined how lipids modulate the interaction of Lck with the TCR ζ chain whose ITAMs serve as a substrate for Lck in the activated TCR-CD3 complex (1–3). We employed two-color single molecule tracking for this study, because it can quantitatively and sensitively monitor dynamic and transient interaction among signaling proteins, which cannot be detected by conventional biochemical methods, such as coimmunoprecipitation (20, 32). We first transfected the SNAP-tagged ζ chain into JCaM1.6 cells stably expressing EGFP-Lck WT, labeled the SNAP tag with tetramethylrhodamine (TMR), and tracked two proteins simultaneously. Because TCR-ζ is a transmembrane protein and Lck is membrane-anchored by N-terminal myristoylation and palmitoylation, both proteins show typical lateral diffusion in the PM, but their trajectories exhibited a low degree of overlap in resting cells, showing lack of colocalization (Fig. 7A).
FIGURE 7.

Single molecule tracking of Lck and TCR-ζ in the TCR complex. A, representative images of Lck-GFP (WT and K182A/R184A) and SNAP-TMR-labeled TCR-ζ in a JCaM1.6 cell before and after OKT3 stimulation are shown. Green, red, and yellow lines are trajectories of Lck, TCR-ζ, and colocalized molecules, respectively. Arrows indicate the starting points of tracking. The scale bar indicates 1 μm. B and C, time courses of dynamic colocalization of SNAP-TMR-TCR-ζ and Lck-GFP (WT and K182A/R184A). The histograms display percentages of TCR-ζ molecules spending a given colocalization time (>0.2 s) with Lck on the PM of JCaM1.6 cells. D and E, time courses of the half-life of colocalization for Lck-GFP proteins and SNAP-TMR-TCR-ζ. The half-life of colocalization was calculated from the dissociation rate constant (k), which was determined by non-linear least squares analysis of histograms using a single-exponential decay equation (i.e. P = Po e−kt). The same size of PM surface was analyzed for each histogram. Error bars indicate S.D. (n = 50–100).
When cells were stimulated with OKT3, which should trigger the release of the Lck SH2 domain from intramolecular tethering (Fig. 8) (1, 2), dynamic Lck-ζ colocalization was significantly enhanced within 5 min, which lasted for ≥10 min (Fig. 7, A and B, and a black line in D). However, dynamic colocalization of the Lck LOF mutant (K182A/R184A) with TCR-ζ was only slightly and briefly increased after OKT3 stimulation (Fig. 7, A and C, and a red line in D). In contrast, the GOF mutant A160K exhibited more extensive OKT3-induced colocalization with TCR-ζ than WT (blue line in Fig. 7D). Intriguingly, pretreatment of JCaM1.6 cells with a PI3K inhibitor, LY294002, did not have a detectable effect on initial Lck-TCR-ζ colocalization up to 5 min but significantly reduced the colocalization thereafter (green line in Fig. 7D). This implies that the Lck-PIP3 interaction takes maximal effect ≈5 min after OKT stimulation under our experimental conditions and that it is important for sustaining interaction between Lck and TCR-ζ. Finally, we monitored the OKT3-induced dynamic colocalization of the Tyr(P) binding-deficient mutant, R154A, with TCR-ζ, and it consistently showed much lower colocalization with TCR-ζ than WT (Fig. 7E). This result indicates that Lck interacts with TCR-ζ via its SH2 domain. More importantly, our results indicate that binding of PM lipids via its SH2 domain is essential for the dynamic interaction of Lck with TCR-ζ upon TCR activation, although both proteins are prelocalized at the PM. Also, binding of Lck-SH2 to PIP3 produced by PI3K activation might be important for sustained interaction between Lck and TCR-ζ and sustained activation of Lck.
FIGURE 8.

Hypothetical model for spatiotemporal coordination of protein interaction and signaling activity of Lck by PM lipids during T cell activation. Lck is PM-localized by N-terminal lipidation (orange lines) but remains inactive due to intramolecular tethering of its catalytic domain (Kin) by SH2 and SH3 domains. Upon TCR activation, dephosphorylation of Tyr-505 by CD45 unleashes Lck-SH2 from the catalytic domain and allows its interaction with PM lipids, including PI4,5P2 (green hexagon) and autophosphorylation on Tyr-394 for full enzymatic activation. Lipid-Lck-SH2 binding is crucial for spatiotemporally specific interaction of Lck with proteins in the activated TCR-CD3 complex, including the ζ chain, and for preventing inactivation of another active species, Tyr(P)-394/Tyr(P)-505, by Tyr(P)-505-Lck-SH2 intramolecular tethering. Both Lck-Tyr(P)-394 and Lck-Tyr(P)-394/Tyr(P)-505 can effectively phosphorylate ITAMs in the CD3 and ζ chains to initiate the TCR signaling. PIP3 (red hexagon), which is subsequently produced by PI3K and accumulated near the TCR-CD3 complex, allows sustained activation of Lck via its tight interaction with Lck-SH2.
Discussion
This study provides new mechanistic insight into Lck activation in response to TCR stimulation by demonstrating that its SH2 domain mediates interaction of Lck with its binding partners in the activated TCR complex in a lipid-dependent manner. Our SPR analysis clearly shows that Lck-SH2 can bind PM lipids with high affinity, albeit with low lipid headgroup specificity. Consistent with its membrane binding properties, electrostatic potential calculation, mutational analysis, and NMR analysis of Lck-SH2 indicate that it neither uses its Tyr(P) binding pocket for lipid binding nor has a well defined lipid binding pocket. Instead, Lck-SH2 employs the ACP and surrounding aromatic and hydrophobic residues for membrane binding. This type of relatively flat membrane-binding sites containing basic, hydrophobic, and aromatic residues has been reported for other lipid binding domains with low lipid headgroup specificity, such as AP180 N-terminal homology domains (33, 34). As is the case with AP180 N-terminal homology domains (33), Lck-SH2 effectively binds PtdInsPs, most notably PIP3 and PI4,5P2.
Our recent study showed that the molecular location and morphology of lipid-binding sites in SH2 domains are highly variable even among related proteins, which allows SH2 domains to adopt different modes of lipid and Tyr(P) binding that ideally suit their different cellular functions (22). For each SH2 domain, however, the conservation of lipid-binding residues among species is extremely high (22). This is true for Lck-SH2 as multiple sequence alignment demonstrates that all known orthologs of Lck-SH2 have basic residues at positions 182 and 184 (supplemental Fig. S1).
Many TCR signaling activities of Lck, including Tyr phosphorylation of ZAP70, PLCγ, and ERK1/2, as well as the intracellular calcium flux mediated by PLCγ, were all significantly compromised by the LOF lipid-binding mutation of Lck while enhanced by the GOF mutation. Excellent correlation between their in vitro lipid binding properties with their cellular activities, enhanced cellular activities of the GOF mutant in particular, shows that the lipid binding activity of Lck-SH2 is essential for the TCR signaling activity of Lck and also makes it unlikely that the observed cellular phenotypes derive from lipid-independent effects, such as altered tertiary structures or post-translational modification of Lck caused by mutations.
Lck is prelocalized in the PM via N-terminal acylation, and lipids should thus play more complex roles than simple membrane recruitment of Lck. Our single molecule imaging analysis shows that lipid binding of the SH2 domain regulates spatiotemporally specific interaction of Lck with TCR-ζ in the TCR signaling complex. Although Lck has been reported to interact with various proteins involved in TCR signaling (1–3, 31), it is not fully understood whether and how Lck-SH2 is involved in specific interaction with proteins in the TCR-CD3 complex. Because the ITAM in TCR-ζ is a substrate for Lck, however, Lck should directly interact with TCR-ζ (35). Single molecule tracking of the Tyr(P) binding-deficient mutant, R154A, confirms that Lck-TCR-ζ interaction is indeed mediated by Lck-SH2. In this regard, it is interesting to find that Lck WT and LOF/GOF lipid-binding mutants, which have essentially the same affinity for Tyr(P)-containing peptides (see Fig. 5), show distinctly different dynamic colocalization with TCR-ζ in the cell in accordance with their different in vitro lipid binding activity. Clearly, Lck and TCR-ζ cannot effectively interact with each other without Lck-SH2 lipid binding, although both proteins are anchored to the PM. The question arises then as to how exactly PM lipids control interaction of Lck with TCR-ζ (and other proteins) in the TCR-CD3 complex.
On the basis of reported properties of Lck and our data presented herein, one can speculate at least two different mechanisms by which PM lipids regulate interaction of Lck with other proteins, including TCR-ζ, in the TCR-CD3 complex and its TCR signaling activities. Membrane lipids are known to regulate interactions among membrane-associated proteins by modulating their conformations and membrane-bound orientations (36). It is thus possible that lipid binding of Lck-SH2 induces conformational changes of Lck in such a way to align Lck-SH2 for optimal interaction with the Tyr(P) motif in the target proteins while preventing its intramolecular interaction with Tyr(P)-505 that would lead to Lck inactivation (see Fig. 8). It was reported that a conformational change takes place when enzymatically active Tyr(P)-394/Tyr(P)-505-Lck, which constitutes 25% of Lck population in T cells, is activated for TCR signaling (8, 9). It is possible that lipid-mediated conformational changes represent the mechanism to keep this species active (see Fig. 8). Also, PIP3 might control signaling activities of Lck and its interaction with protein partners in a spatially and temporally specific manner. Because of high affinity of Lck-SH2 for PIP3, PIP3 produced via PI3K activation, which is downstream of Lck activation (4, 5), might serve as a positive feedback mechanism to control the duration of Lck activation. Furthermore, PIP3 has been reported to be enriched in the region of the activated TCR complex (37), and thus PIP3 might allow Lck to be dynamically co-localized with the TCR-CD3 complex. It has been reported that c-Src catalyzes the multisite phosphorylation of its substrates in a processive manner via its SH2 domain that binds to Tyr(P) of partially phosphorylated substrates (38). Although it is not known whether Lck catalyzes the multisite phosphorylation processively under physiological conditions (10), PIP3-mediated sustained interaction of Lck with TCR-ζ and other proteins should contribute to its processive catalysis. Undoubtedly, further studies are needed to fully answer these complex mechanistic questions, and our results provide a new conceptual framework for further mechanistic studies of Lck activation and TCR signaling.
Experimental Procedures
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), cholesterol, and liver phosphoinositol (PI) were purchased from Avanti Polar Lipids. 1,2-Dipalmitoyl derivatives of phosphatidylinositol 4,5-bisphosphate (PI4,5P2), phosphatidylinositol 3,4,5-trisphosphate (PIP3), phosphatidylinositol 3,4-bisphosphate (PI3,4P2), phosphatidylinositol 3,5-bisphosphate (PI3,5P2), phosphatidylinositol 3-monophosphate (PI3P), phosphatidylinositol 4-monophosphate (PI4P), and phosphatidylinositol 5-monophosphate (PI5P) were from Cayman Chemical Co. d-Myo-inositol-(1,3,4,5)-tetrakisphosphate (IP4) was also from Cayman. The human anti-CD3 antibody, OKT3, was purchased from Bio X Cell, and anti-GFP antibody and anti-actin antibody were from Santa Cruz Biotechnology and Bethyl Laboratories, respectively. All other antibodies were from Cell Signaling Technologies. Phosphoinositide 3-kinase (PI3K) inhibitor, LY294002, was from Sigma.
DNA Constructs
cDNAs of full-length human Lck and TCR-ζ (CD247) were subcloned to the pSNAPf vector (New England Biolabs) using KpnI/XhoI and EcoRI/EcoRV sites, respectively.
Protein Expression and Purification
All EGFP-tagged Lck-SH2 and full-length Lck proteins were expressed as His6-tagged proteins in Escherichia coli BL21 (DE3) pLysS (Novagen). Cells were cultured to an A600 of ∼0.6, and protein expression was induced by adding 0.5–1 mm isopropyl-β-d-1-thiogalactopyranoside at 16 °C for 12 h. The transformed cells were harvested by centrifugation at 4 °C, and cell pellets were resuspended in the lysis buffer (50 mm Tris-HCl, pH 7.9, 0.3 m NaCl, 10 mm imidazole, 10% glycerol, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride) and lysed by sonication. The His-tagged proteins were purified using the nickel-nitrilotriacetic acid-agarose resin (Qiagen). Eluted proteins were further treated on a PD-10 desalting column (GE Healthcare) equilibrated with 20 mm Tris-HCl, pH 7.4, 0.16 m NaCl. Proteins were quantified by the Bradford assay (Bio-Rad).
Lipid Vesicles Preparation and SPR Analysis
PM mimetic vesicles were prepared by mixing POPC, POPE, POPS, cholesterol, PI, and PI4,5P2 in a molar ratio of 12:35:22:22:8:1 (39). All SPR measurements were performed at 23 °C using a lipid-coated L1 chip in the BIAcore X and T100 systems as described (40, 41). 20 mm Tris-HCl, pH 7.4, containing 0.16 m NaCl was used as the running buffer, whereas PM mimetic vesicles and POPC vesicles were coated on the active surface and the control surface, respectively. Equilibrium measurements were performed at a flow rate of 10 μl/min. At least five different protein concentrations were injected to collect a set of Req values that were plotted against the protein concentrations (Po). An apparent dissociation constant (Kd) was then determined by nonlinear least squares analysis of the binding isotherm using the equation, Req = Rmax/(1 + Kd/Po), where Rmax indicates the maximal Req value (42). For kinetic measurements, the flow rate was maintained at 20–30 μl/min.
PI4,5P2 and PIP3 Depletion
The PI4,5P2 depletion was performed in HeLa cells according to a reported procedure (26, 27). The method uses the Lyn-based PM-anchored FKBP12-rapamycin binding (FRB) domain of the mechanistic target of rapamycin (Lyn-FRB) and the FK506-binding protein-12-yeast inositol polyphosphate 5-phosphatase domain fusion protein (FKBP-Inp). To monitor PM translocation of EGFP-tagged Lck-SH2 WT and mutants and PLCδ-PH with this system, Lyn-FRB and FKBP-Inp were used without fluorescence protein tag (43). 2.5 μm rapamycin analog, A/C heterodimerizer (Clontech), was used to trigger the PI4,5P2 depletion. PIP3 depletion was achieved by pretreating the cells with 50 μm LY294002 for 1 h.
Stable Cell Line Preparation
A Jurkat cell line derivative, JCaM1.6 (CRL-2063), was purchased from ATCC and cultured in RPMI 1640 medium containing 5% FBS. Wild type (WT) and mutant Lck-GFP were stably expressed in JCaM1.6 by retroviral gene transduction. A retroviral construct, pMSCVneo-hLck-GFP, was cloned by PCR, and point mutations were introduced by site-directed mutagenesis. Retrovirus preparation and transduction were performed as described previously (44).
Cell Signaling Activity Assays
Changes in the intracellular calcium levels were assessed by flow cytometry. Cells were loaded with 4 μg/ml Indo-1 AM (Life Technologies, Inc.) in RPMI 1640 media containing 5% fetal bovine serum for 30 min at 37 °C, washed, and resuspended in RPMI 1640 medium supplemented with 25 mm HEPES and 2% fetal bovine serum at a cell density of 2 × 106/ml. The changes in the fluorescence emission ratio of the calcium-bound versus -unbound form of Indo-1 were measured using LSR Fortessa (BD Biosciences). Baseline measurements were conducted for 1 min, and calcium flux was monitored for an additional 8 min after the addition of OKT3. The data were analyzed with FlowJo software (TriStar).
Tyr Phosphorylation Assay
Cells expressing either WT or mutant Lck-GFP were stimulated with OKT3 at 37 °C for the indicated time. Cells were then immediately lysed by addition of ice-cold radioimmunoprecipitation assay buffer supplemented with a protease inhibitor mixture and a protein phosphatase inhibitor mixture. The proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with the indicated antibodies. The chemiluminescence was detected with ImageQuant LAS 4000 (GE Healthcare).
pERK1/2 Intracellular Staining
After TCR stimulation, cells were fixed with 3.7% formaldehyde for 10 min, washed with FACS buffer (phosphate buffer saline containing 2% bovine serum albumin and 0.1% NaN3), and permeabilized with methanol for 30 min on ice. After washing with FACS buffer, the fixed cells were incubated with mouse anti-pERK1/2 for 45 min at room temperature, washed, and then incubated with Alexa Fluor 647-conjugated goat anti-mouse IgG antibody for 30 min at room temperature. The levels of pERK1/2 were measured by flow cytometry.
SH2-Peptide Binding Assay by Fluorescence Anisotropy
Fluorescein-6-aminohexanoyl (F-Ahx)-labeled peptides used for binding studies are F-Ahx-pYEEI, F-Ahx-pYSDPE, and F-Ahx-pYQPQP. The peptide was dissolved in 20 mm Tris buffer, pH 7.4, containing 160 mm NaCl, and 5% dimethyl sulfoxide. To each well of a 96-flat bottom black polystyrol plate was added 100 μl of solution containing each peptide (5 nm) and SH2 domain (2.5 nm to 25 μm) with or without 150 μm PM vesicles. After 30 min of incubation, the plate was inserted into BioTek Synergy Neo microplate reader, and the fluorescence anisotropy (r) was measured with excitation and emission wavelengths set at 485 and 535 nm, respectively. Because under our conditions, Po ≫ Pepo, the Kd value for the SH2 domain-peptide binding was determined by the non-linear least squares analysis of the binding isotherm using Equation 1,
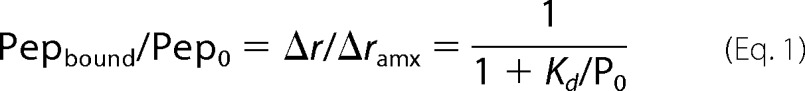 |
where Pepbound, Pepo, and Po indicate the concentration of bound peptide, total peptide, and total SH2 domain, respectively, and Δr and Δrmax are the anisotropy change for each Po and the maximal Δr, respectively.
Single Molecule Tracking by Total Internal Reflection Fluorescence Microscopy
Simultaneous single molecule imaging of two proteins was performed in Jurkat cells using a total internal reflection fluorescence microscope as described previously (20). JCaM1.6 Jurkat cells stably expressing Lck-GFP WT (or mutants) were transiently transfected with SNAP®-tagged TCR-ζ chain, which was subsequently labeled with SNAP-Cell® TMR-Star® (New England Biolabs). Labeled cells were washed, attached to a poly-l-lysine-coated glass plate for imaging, and stimulated with 10 μg/ml OKT3, and Lck and ζ molecules were simultaneously tracked. To see the effect of PIP3 depletion on interaction between Lck WT and TCR-ζ, cells were pretreated with 50 μm LY294002 for 1 h before OKT3 stimulation. All particle tracking, data analysis, and image processing were carried out with in-house programs written in MATLAB. Colocalization analysis of two molecules was performed with a fixed threshold criterion (i.e. <400 nm) for colocalization (32). The same size of PM surface was analyzed for each data. The percentage of TCR-ζ molecules spending a given colocalization time (>0.2 s) with Lck on the PM was calculated from the total number of TCR-ζ molecules and displayed as a histogram. Data were fit into a single exponential decay equation (i.e. P = Po e−kt) to determine the dissociation rate constant (k) values by non-linear least squares analysis, and the half-life values of colocalization were calculated as ln2/k. 50–100 images were analyzed for each data point.
Electrostatic Analysis
The Protein Data Bank files were prepared for further calculations using PDB2PQR interface (45, 46). We used PROPKA (47, 48) to predict protonation states of protein residues. DelPhi (49) was used for assigning electrostatic potentials to the atoms of the structures. Only linear DelPhi iterations were run. Both PROPKA and Delphi used CHARMM force field (50). The calculated potentials were loaded into GRASP2 (51) for subsequent visualization.
Docking
Docking of the lipids to the structure of SH2 domains was performed with the DOCK program of the package DOCK 6 (52, 53). The spheres representing the binding site were selected within 7 Å from the side chains of the residues shown to be important for the lipid binding. The ligand was allowed to be flexible during the orientation step. The topmost pose generated by DOCK run was rescored using Amber score program, which allows both the ligand and the active site of the receptor to be flexible (54).
Multiple Sequence Alignments
Multiple sequence alignments were generated with Clustal 2.0.10 (55).
NMR Data Acquisition and Processing
The 1H,15N-HSQC experiments were recorded at 600- and 800-MHz on Bruker Avance spectrometers at 20 °C. The samples contained 0.1, 0.5, or 1 mm 15N-labeled Lck-SH2 in 25 mm potassium phosphate, 2 mm dithiothreitol, and 5% deuterium oxide, pH 7.2. IP4 was titrated to a final concentration of 1 mm. All NMR data were processed with NMRpipe (56) and analyzed with CARA. The chemical shift perturbations, Δδ, upon addition of IP4 were calculated from 1H and 15N chemical shifts using a weighted averaging scheme previous described by Grzesiek et al. (57). Missing shifts are indicative of either unavailable experimental data, because of peak overlap, or a residue position occupied by a proline.
Author Contributions
R. S., H. K., I. S., E. K., M. J. P., K. B., and S. R. designed and performed biochemical studies, and Z. G. W. and Y. X. performed imaging work. A. S. and B. H. performed computational work, and P. T. R., S. S., and J. L. carried out NMR analysis. D. J. J. and Y. M. K. handled all cell studies. W. C. and Y. M. K. conceived the work and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Diana Murray for helpful discussions on nonspecific electrostatic interactions and Dr. Takanari Inoue for the generous gift of the PI4,5P2 and PIP3 manipulation systems.
This work was supported in part by National Institutes of Health Grants GM68849 and GM110128 (to W. C.) and GM030518, GM094597, and CA121852 (to B. H.), National Research Foundation of Korea Grant NRF-2013R1A1A2074573, and World Class University Program R31-2008-000-10105-0 by the Korean Government. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1.
- TCR
- T cell receptor
- PM
- plasma membrane
- SH
- Src homology
- ITAM
- immunoreceptor tyrosine-based activation motif
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine
- PI
- phosphoinositol
- PtdInsP
- phosphoinositide
- PI4,5P2
- phosphatidylinositol 4,5-bisphosphate
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- PI3,4P2
- phosphatidylinositol 3,4-bisphosphate
- PI3,5P2
- phosphatidylinositol 3,5-bisphosphate
- PI3P
- phosphatidylinositol 3-monophosphate
- PI4P
- phosphatidylinositol 4-monophosphate
- PI5P
- phosphatidylinositol 5-monophosphate
- IP4
- d-myo-inositol 1,3,4,5-tetrakisphosphate
- CSP
- chemical shift perturbation
- EGFP
- enhanced green fluorescence protein
- FRB
- FKBP12-rapamycin binding
- F-Ahx
- fluorescein-6-aminohexanoyl
- SPR
- surface plasmon resonance
- PLCδ-PH
- phospholipase Cδ pleckstrin homology domain
- ACP
- alternative cationic patch
- TMR
- tetramethylrhodamine
- LOF
- loss-of-function
- GOF
- gain-of-function.
References
- 1. Palacios E. H., and Weiss A. (2004) Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000 [DOI] [PubMed] [Google Scholar]
- 2. Salmond R. J., Filby A., Qureshi I., Caserta S., and Zamoyska R. (2009) T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev. 228, 9–22 [DOI] [PubMed] [Google Scholar]
- 3. Wang H., Kadlecek T. A., Au-Yeung B. B., Goodfellow H. E., Hsu L. Y., Freedman T. S., and Weiss A. (2010) ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol. 2, a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Merwe P. A., and Dushek O. (2011) Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 [DOI] [PubMed] [Google Scholar]
- 5. Pollizzi K. N., and Powell J. D. (2014) Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin G. S. (2001) The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2, 467–475 [DOI] [PubMed] [Google Scholar]
- 7. Hardwick J. S., and Sefton B. M. (1995) Activation of the Lck tyrosine protein kinase by hydrogen peroxide requires the phosphorylation of Tyr-394. Proc. Natl. Acad. Sci. U.S.A. 92, 4527–4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nika K., Soldani C., Salek M., Paster W., Gray A., Etzensperger R., Fugger L., Polzella P., Cerundolo V., Dushek O., Höfer T., Viola A., and Acuto O. (2010) Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stirnweiss A., Hartig R., Gieseler S., Lindquist J. A., Reichardt P., Philipsen L., Simeoni L., Poltorak M., Merten C., Zuschratter W., Prokazov Y., Paster W., Stockinger H., Harder T., Gunzer M., and Schraven B. (2013) T cell activation results in conformational changes in the Src family kinase Lck to induce its activation. Sci. Signal. 6, ra13. [DOI] [PubMed] [Google Scholar]
- 10. Hui E., and Vale R. D. (2014) In vitro membrane reconstitution of the T-cell receptor proximal signaling network. Nat. Struct. Mol. Biol. 21, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu C., Gagnon E., Call M. E., Schnell J. R., Schwieters C. D., Carman C. V., Chou J. J., and Wucherpfennig K. W. (2008) Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi X., Bi Y., Yang W., Guo X., Jiang Y., Wan C., Li L., Bai Y., Guo J., Wang Y., Chen X., Wu B., Sun H., Liu W., Wang J., and Xu C. (2013) Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 493, 111–115 [DOI] [PubMed] [Google Scholar]
- 13. Pawson T. (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 [DOI] [PubMed] [Google Scholar]
- 14. Lim W. A., and Pawson T. (2010) Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waksman G., Shoelson S. E., Pant N., Cowburn D., and Kuriyan J. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72, 779–790 [DOI] [PubMed] [Google Scholar]
- 16. Rameh L. E., Chen C. S., and Cantley L. C. (1995) Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell 83, 821–830 [DOI] [PubMed] [Google Scholar]
- 17. Bae Y. S., Cantley L. G., Chen C. S., Kim S. R., Kwon K. S., and Rhee S. G. (1998) Activation of phospholipase C-γ by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 4465–4469 [DOI] [PubMed] [Google Scholar]
- 18. Surdo P. L., Bottomley M. J., Arcaro A., Siegal G., Panayotou G., Sankar A., Gaffney P. R., Riley A. M., Potter B. V., Waterfield M. D., and Driscoll P. C. (1999) Structural and biochemical evaluation of the interaction of the phosphatidylinositol 3-kinase p85α Src homology 2 domains with phosphoinositides and inositol polyphosphates. J. Biol. Chem. 274, 15678–15685 [DOI] [PubMed] [Google Scholar]
- 19. Chen Y., Sheng R., Källberg M., Silkov A., Tun M. P., Bhardwaj N., Kurilova S., Hall R. A., Honig B., Lu H., and Cho W. (2012) Genome-wide functional annotation of dual-specificity protein- and lipid-binding modules that regulate protein interactions. Mol. Cell 46, 226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheng R., Chen Y., Yung Gee H., Stec E., Melowic H. R., Blatner N. R., Tun M. P., Kim Y., Källberg M., Fujiwara T. K., Hye Hong J., Pyo Kim K., Lu H., Kusumi A., Goo Lee M., and Cho W. (2012) Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat. Commun. 3, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheng R., Kim H., Lee H., Xin Y., Chen Y., Tian W., Cui Y., Choi J. C., Doh J., Han J. K., and Cho W. (2014) Cholesterol selectively activates canonical Wnt signalling over non-canonical Wnt signalling. Nat. Commun. 5, 4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park M. J., Sheng R., Silkov A., Jung D. J., Wang Z. G., Xin Y., Kim H., Thiagarajan-Rosenkranz P., Song S., Yoon Y., Nam W., Kim I., Kim E., Lee D. G., Chen Y., et al. (2016) SH2 domains serve as lipid-binding modules for Tyr(P)-signaling proteins. Mol. Cell 62, 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim H., Afsari H. S., and Cho W. (2013) High-throughput fluorescence assay for membrane-protein interaction. J. Lipid Res. 54, 3531–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho W., and Stahelin R. V. (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151 [DOI] [PubMed] [Google Scholar]
- 25. Di Paolo G., and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 26. Inoue T., Heo W. D., Grimley J. S., Wandless T. J., and Meyer T. (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Varnai P., Thyagarajan B., Rohacs T., and Balla T. (2006) Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 175, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tokonzaba E., Capelluto D. G., Kutateladze T. G., and Overduin M. (2006) Phosphoinositide, phosphopeptide and pyridone interactions of the Abl SH2 domain. Chem. Biol. Drug Des. 67, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hong Y., Chalkia D., Ko K. D., Bhardwaj G., Chang G. S., van Rossum D. B., and Patterson R. L. (2009) Phylogenetic profiles reveal structural and functional determinants of lipid-binding. J. Proteomics Bioinform. 2, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferguson K. M., Lemmon M. A., Schlessinger J., and Sigler P. B. (1995) Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 31. Pelosi M., Di Bartolo V., Mounier V., Mège D., Pascussi J. M., Dufour E., Blondel A., and Acuto O. (1999) Tyrosine 319 in the interdomain B of ZAP-70 is a binding site for the Src homology 2 domain of Lck. J. Biol. Chem. 274, 14229–14237 [DOI] [PubMed] [Google Scholar]
- 32. Koyama-Honda I., Ritchie K., Fujiwara T., Iino R., Murakoshi H., Kasai R. S., and Kusumi A. (2005) Fluorescence imaging for monitoring the colocalization of two single molecules in living cells. Biophys. J. 88, 2126–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stahelin R. V., Long F., Peter B. J., Murray D., De Camilli P., McMahon H. T., and Cho W. (2003) Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J. Biol. Chem. 278, 28993–28999 [DOI] [PubMed] [Google Scholar]
- 34. Silkov A., Yoon Y., Lee H., Gokhale N., Adu-Gyamfi E., Stahelin R. V., Cho W., and Murray D. (2011) Genome-wide structural analysis reveals novel membrane binding properties of AP180 N-terminal homology (ANTH) domains. J. Biol. Chem. 286, 34155–34163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stephen T. L., Wilson B. S., and Laufer T. M. (2012) Subcellular distribution of Lck during CD4 T-cell maturation in the thymic medulla regulates the T-cell activation threshold. Proc. Natl. Acad. Sci. U.S.A. 109, 7415–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cho W. (2006) Building signaling complexes at the membrane. Sci. STKE 2006, pe7. [DOI] [PubMed] [Google Scholar]
- 37. Gamper C. J., and Powell J. D. (2012) All PI3Kinase signaling is not mTOR: dissecting mTOR-dependent and independent signaling pathways in T cells. Front. Immunol. 3, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scott M. P., and Miller W. T. (2000) A peptide model system for processive phosphorylation by Src family kinases. Biochemistry 39, 14531–14537 [DOI] [PubMed] [Google Scholar]
- 39. Stahelin R. V., Rafter J. D., Das S., and Cho W. (2003) The molecular basis of differential subcellular localization of C2 domains of protein kinase C-α and group IVa cytosolic phospholipase A2. J. Biol. Chem. 278, 12452–12460 [DOI] [PubMed] [Google Scholar]
- 40. Stahelin R. V., and Cho W. (2001) Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry 40, 4672–4678 [DOI] [PubMed] [Google Scholar]
- 41. Stahelin R. V., and Cho W. (2001) Roles of calcium ions in the membrane binding of C2 domains. Biochem. J. 359, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melowic H. R., Stahelin R. V., Blatner N. R., Tian W., Hayashi K., Altman A., and Cho W. (2007) Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cθ. J. Biol. Chem. 282, 21467–21476 [DOI] [PubMed] [Google Scholar]
- 43. Yoon Y., Lee P. J., Kurilova S., and Cho W. (2011) In situ quantitative imaging of cellular lipids using molecular sensors. Nat. Chem. 3, 868–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim J., Huh J., Hwang M., Kwon E. H., Jung D. J., Brinkmann M. M., Jang M. H., Ploegh H. L., and Kim Y. M. (2013) Acidic amino acid residues in the juxtamembrane region of the nucleotide-sensing TLRs are important for UNC93B1 binding and signaling. J. Immunol. 190, 5287–5295 [DOI] [PubMed] [Google Scholar]
- 45. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., and Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Robertson A. D., and Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 48. Rostkowski M., Olsson M. H., Søndergaard C. R., and Jensen J. H. (2011) Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rocchia W., Alexov E., and Honig B. (2001) Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 105, 6507–6514 [Google Scholar]
- 50. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 51. Petrey D., and Honig B. (2003) GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences. Methods Enzymol. 374, 492–509 [DOI] [PubMed] [Google Scholar]
- 52. Lorber D. M., and Shoichet B. K. (1998) Flexible ligand docking using conformational ensembles. Protein Sci. 7, 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wei B. Q., Baase W. A., Weaver L. H., Matthews B. W., and Shoichet B. K. (2002) A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 322, 339–355 [DOI] [PubMed] [Google Scholar]
- 54. Graves A. P., Shivakumar D. M., Boyce S. E., Jacobson M. P., Case D. A., and Shoichet B. K. (2008) Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J. Mol. Biol. 377, 914–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 56. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 57. Grzesiek S., Stahl S. J., Wingfield P. T., and Bax A. (1996) The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 35, 10256–10261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.