

Abstract
The yeast vacuole requires four SNAREs to trigger membrane fusion including the soluble Qc-SNARE Vam7. The N-terminal PX domain of Vam7 binds to the lipid phosphatidylinositol 3-phosphate (PI3P) and the tethering complex HOPS (homotypic fusion and vacuole protein sorting complex), whereas the C-terminal SNARE motif forms SNARE complexes. Vam7 also contains an uncharacterized middle domain that is predicted to be a coiled-coil domain with multiple helices. One helix contains a polybasic region (PBR) composed of Arg-164, Arg-168, Lys-172, Lys-175, Arg-179, and Lys-186. Polybasic regions are often associated with nonspecific binding to acidic phospholipids including phosphoinositides. Although the PX (phox homology) domain alone binds PI3P, we theorized that the Vam7 PBR could bind to additional acidic phospholipids enriched at fusion sites. Mutating each of the basic residues in the PBR to an alanine (Vam7-6A) led to attenuated vacuole fusion. The defective fusion of Vam7-6A was due in part to inefficient association with its cognate SNAREs and HOPS, yet the overall vacuole association of Vam7-6A was similar to wild type. Experiments testing the binding of Vam7 to specific signaling lipids showed that mutating the PBR to alanines augmented binding to PI3P. The increased binding to PI3P by Vam7-6A likely contributed to the observed wild type levels of vacuole association, whereas protein-protein interactions were diminished. PI3P binding was inhibited when the PX domain mutant Y42A was introduced into Vam7-6A to make Vam7-7A. Thus the Vam7 PBR affects PI3P binding by the PX domain and in turn affects binding to SNAREs and HOPS to support efficient fusion.
Keywords: calcium transport, diacylglycerol, membrane fusion, phosphatidic acid, phosphatidylinositol, SNARE proteins, HOPS, ycf1
Introduction
The study of membrane lipid composition is vital to gaining a complete understanding of membrane trafficking and fusion. Membranes can contain ordered microdomain platforms that concentrate and organize lipids and proteins required for specific functions such as fusion (1–7). Membrane microdomains are usually enriched in signaling lipids including phosphoinositides, sterols, and sphingolipids as well as their modified derivatives. Fusion events are essential for cellular homeostasis, and understanding the regulation and mechanics of membrane fusion can shed light on understanding diseases that subvert these pathways.
We use lysosomal vacuoles from the yeast Saccharomyces cerevisiae as a model system to test the role of signaling lipids in membrane fusion. The fusion pathway is initiated when the AAA+ protein Sec18/NSF3 (N-ethylmaleimide-sensitive factor) associates with Sec17/α-SNAP (soluble NSF adaptor protein) bound to inactive cis-SNARE (SNAP receptor) complexes (8). The ATPase activity of Sec18 leads to conformational changes that are transferred to α-SNAP, which then mechanically disrupts the cis-SNARE bundle into individual proteins in a process defined as priming. Although most SNAREs are anchored to membranes, the vacuolar Qc-SNARE Vam7 is soluble and is released from the membrane upon priming (9). Vam7 re-associates with the organelle during the docking stage through the interaction of its PX domain with the lipid phosphatidylinositol 3-phosphate (PI3P) and subsequently enters into trans-SNARE complexes via its SNARE motif (10–13). The formation of trans-SNARE complexes between partnered vesicles triggers the release of luminal Ca2+ stores (14). During the tethering and docking stages, vacuoles become tightly apposed, forming a flattened domain termed the boundary membrane. The edge of the boundary, where membranes come into contact, becomes enriched in the proteins and lipids that promote fusion (2, 6, 7). This membrane microdomain is termed the vertex ring and is the site of fusion. Fusion can occur directly through the formation of a fusion pore or through an intermediate hemifusion state where the outer leaflets of membrane mix without fusing the inner leaflets, thus preventing content mixing (15–18). Full fusion occurs when both leaflets fuse and luminal contents are mixed.
Each stage of vacuole fusion is affected by signaling lipids through distinct roles. Phosphatidic acid (PA) is converted to diacylglycerol (DAG) at the priming stage by the PA phosphatase Pah1 to facilitate the transfer of Sec18 from PA-enriched membrane domains to cis-SNARE complexes4 (19, 20). Ergosterol and PI(4,5)P2 function during priming by unknown mechanisms as well as late in the pathway where they affect actin remodeling (21–23). DAG functions after trans-SNARE pairing where it is thought to destabilize membrane bilayers to promote fusion (2, 12, 24). PI3P is made by Vps34 during the fusion cascade where it recruits Vam7 as well as the Ypt7 GEF Mon1-Ccz1 (11, 25). Phosphoinositides also aid in binding HOPS to the membrane and affect actin enrichment at the vertex microdomain (21, 26). In addition to these effects, signaling lipids are essential for the organization of the proteins and lipids at the vertex ring in an interdependent manner (2).
Vam7 is a unique SNARE in that it lacks a membrane anchor be it proteinaceous or lipidic (10). Instead, it associates with the membrane through its N-terminal PX domain that binds both PI3P and the HOPS complex (11, 26, 27). Binding to PI3P and fusion are inhibited by the Y42A mutation (11, 12). Vam7 also associates with the vacuole through the interactions of its C-terminal SNARE motif that binds its cognate SNAREs as well as the HOPS subunit Vps33 (28). Vam7-triggered fusion is blocked by the Q283R mutation in the ionic zero layer of the SNARE motif, which stalls the fusion pathway in a hemifusion state (18, 29). Vam7 is also recruited to vacuoles through a mechanism dependent on the ABC transporter Ycf1 through an unknown mechanism (30). Vam7 contains a putative α helical middle domain that is uncharacterized. In further examining the properties of the middle domain, we identified a polybasic region (PBR) containing six Arg and Lys in the third α-helix near the SNARE domain. Mutagenesis of the region was performed converting all of the basic residues to alanine (Vam7-6A). We report that the PBR affects PI3P binding and protein interactions with SNAREs and HOPS needed for efficient vacuole fusion.
Results
The Polybasic Region of the Vam7 Middle Domain Is Required for Efficient Homotypic Fusion
In previous studies we and others have examined how the PX and SNARE domains of the soluble Qc-SNARE Vam7 interact with the vacuole to stimulate fusion. Here we examined the role of the region between the PX and SNAREs domains that we refer to as the middle domain, which is predicted to be a α-helical structure. The α-helix closest to the SNARE domain has a polybasic face with six Arg and Lys in close proximity (Fig. 1A). We mutated each of these basic residues to alanine to construct Vam7-6A. To test the ability of Vam7-6A to stimulate fusion, we employed a well characterized bypass assay where priming is inhibited by anti-Sec17 IgG (12, 14, 29, 31). Recombinant GST-Vam7 can be added to these blocked in vitro reactions to stimulate fusion. Soluble Vam7 can interact with free Vam3, Vti1, and Nvy1 to form trans-SNARE pairs at the docking stage. We found that wild type Vam7 robustly stimulated fusion as observed previously, whereas Vam7-6A was impaired (Fig. 1B). Although Vam7-6A could support fusion, it required far higher concentrations relative to wild type, resulting in a significant right-shift at the half-maximal efficacy values. The EC50 of wild type Vam7 was 42.2 ± 14.8 nm, whereas the EC50 for Vam7-6A was 153.1 ± 46.1 nm. The biphasic curve seen with Vam7 is characteristic of the anti-Sec17 bypass. The inhibitory effect at high concentrations is thought to be due to competition for HOPS and PI3P on the membrane (29). We next examined Vam7-6A in a standard fusion assay in the absence of any inhibitor. Wild type Vam7 has a slight stimulatory effect on fusion as previously observed. On the other hand, Vam7-6A lacked the stimulatory effect and, rather, inhibited fusion at concentrations above 300 nm (Fig. 1C).
FIGURE 1.

Vam7-6A requires higher concentrations to reach Vam7 fusion levels. A, schematic representation of Vam7 and its polybasic region in the middle domain (MD). B, vacuole fusion reactions were performed using vacuoles from BJ3505 and DKY6281. Fusion reactions were incubated with 60 μg/ml α-Sec17 IgG for 10 min on ice followed by the addition of recombinant wild type Vam7 or Vam7-6A at the indicated concentrations for 5 min on ice. Fusion reactions were incubated at 27 °C for 90 min and assayed for Pho8 activity. C, standard fusion reactions (no inhibitors) were incubated with Vam7 or Vam7-6A at the indicated concentrations and incubated at 27 °C for 90 min to test fusion. D, differential scanning fluorimetry. Vam7 and Vam7-6A were incubated with SYPRO orange. Samples were equilibrated for 30 min before starting the melting curve between 20 °C and 95 °C. SYPRO orange fluorescence was measured at each temperature (λex = 490 nm, λem = 560 nm). Shown are the first derivatives of the fluorescence data to depict Tm for each sample. E, lipid Mixing (hemifusion) assays were performed using WT vacuoles inhibited with anti-Sec17 IgG at 4 °C. Reactions were transferred from ice to 27 °C and incubated for 5 min before the addition of ATP regenerating system. After an additional 10-min incubation wild type Vam7 or Vam7-6A was added to the indicated reactions. An increase in fluorescence occurred when the outer leaflet of vacuoles mixed during hemifusion. Shown is a representative of three trials. F, the average lipid dequenching at 35 min after the addition of ATP. Error bars indicate S.E. (n = 3).
The inhibitory effect of Vam7-6A led us to ask if the protein was structurally unstable. To examine if mutating the PBR affected protein stability, we used differential scanning fluorimetry. Fig. 1D shows the first derivative of thermal melt curves for wild type Vam7 and Vam7-6A. The Tm for both proteins was 55 °C, indicating that mutating the PBR did not have a deleterious effect on protein folding.
Vam7-6A Bypass of Anti-Sec17 IgG-blocked Fusion Promotes Lipid Mixing
Others have shown that fusion can occur rapidly by making a direct fusion pore or through a slower pathway that goes through a hemifusion intermediate (15, 17). During hemifusion, the outer leaflets of docked vesicles fuse, leaving the inner leaflets intact to prevent the mixing of luminal content. Vacuole homotypic fusion can also go through a hemifusion intermediate, and mutations in SNAREs can stall the pathway at this stage (16, 18). For instance, Vam7Q283R can form SNARE complexes but cannot trigger the full fusion of vacuoles blocked with anti-Sec17 antibody. However, Vam7Q283R could trigger lipid mixing of the outer leaflet as efficiently as wild type Vam7, indicating that the mutant SNARE could only promote hemifusion and not full bilayer mixing. In this study we saw that Vam7-6A was attenuated in the bypass of anti-Sec17 IgG inhibited priming. To determine if Vam7-6A-containing reactions were stalled before or after a hemifusion stage, we employed the previously described lipid-mixing assay. Here, a population of vacuoles was labeled with Rh-PE and mixed with an 8-fold excess of unlabeled vacuoles. Rh-PE is limited to the outer leaflet and self-quenches at elevated concentrations. Rh-PE fluorescence de-quenches when the outer leaflets of membranes fuse to dilute the fluorophore. The kinetics of lipid mixing and content mixing are separated by up to 60 min (32). Using vacuoles treated with anti-Sec17 IgG, we found that both 100 nm Vam7 and Vam7-6A promoted Rh-PE fluorescence de-quenching (Fig. 1, E and F). However, the effectiveness of Vam7-6A to promote lipid mixing was reduced by 10% relative to wild type. Importantly, content mixing was reduced by 50% when comparing 100 nm Vam7-6A to wild type Vam7. These data suggest that Vam7-6A can trigger outer leaflet mixing with wild type efficiency while having diminished content mixing. Thus, it appears that there is a delay between a hemifusion stage and full bilayer mixing.
Expression of Vam7-6A in Vivo
To test the effects of mutating the Vam7 PBR in vivo, we expressed plasmid encoded wild type Vam7 and Vam7-6A in vam7Δ cells and examined vacuole morphology. Fig. 2A shows cells incubated with the vital dye FM4-64. Wild type cells showed the characteristic vacuole staining, whereas vam7Δ cells lacked intact vacuoles and the dye-labeled scattered puncta throughout the cell. In addition, vam7Δ cells appeared to have an endocytic defect, as a significant portion of FM4-64 remained on the plasma membrane. The vacuole morphology defect was rescued with plasmid-encoded Vam7. Similarly, Vam7-6A expression restored vacuole structure. Although Vam7-6A restored vacuole morphology, it was not sufficient to restore defective vacuole fusion in vitro. Because Vam7 is soluble and associates with membrane proteins and lipids, we next examined Vam7 partitioning between the vacuole and cytosol. Defects in membrane binding would be evident by a shift to the cytosolic fraction as seen previously with Vam7Y42A (11) or in the absence of the ABC protein Ycf1 (30). Our fractionation experiments showed there was no difference in the distribution of Vam7 and Vam7-6A.
FIGURE 2.

In vivo expression of Vam7-6A. A, yeast cells (wild type, vam7Δ, vam7Δ + pVAM7, and vam7Δ + pVAM7−6A) were incubated with 5 μm FM4-64 to label vacuoles. B, distribution of proteins in cytosol versus vacuoles. Vacuoles and cytosol were collected from wild type cells or vam7Δ cells expressing plasmid encoded Vam7 or Vam7-6A. Immunoblots were performed to examine the distribution of Vam7 and Vam7-6A. Ypt7 was used as marker for vacuole enrichment.
Vam7-6A Is Inhibited in the Bypass of Multiple Fusion Blocks
In the first figure we showed that Vam7-6A was unable to fully support fusion when SNARE priming was blocked with anti-Sec17 IgG. We next asked if Vam7-6A could bypass other inhibitors relative to wild type Vam7. First, we tested a second inhibitor of priming. Previously we found that inhibiting PA phosphatase activity with propranolol blocked priming (20). In that study we showed that Vam7 was able to bypass the propranolol fusion block. Here we tested the ability of wild type Vam7 and Vam7-6A in rescuing the propranolol block. Fusion rescue was compared with uninhibited fusion that was normalized to 100%. We found that Vam7-6A was inhibited in its ability to support fusion relative to wild type (Fig. 3A). The apparent EC50 of wild type Vam7 was 150 nm, whereas 375 nm Vam7-6A was required for the same level of fusion. Because of the fusogenicity of DAG, it is possible that a reduction in its production from PA increased the energy threshold needed for fusion to be triggered by Vam7 and that Vam7-6A function did not generate sufficient energy to do so. It is also possible that the PBR affects the interaction with the other SNAREs, resulting in the absence of fusion. Perhaps the PBR interacts electrostatically with the acidic lipids of the membrane and/or acidic protein surfaces. Thus, the Vam7-6A would abolish electrostatic interactions and reduce its ability to support maximal fusion.
FIGURE 3.

Vam7-6A is unable to rescue to Vam7 levels. Fusion reactions were carried out using WT (A–D) or ycf1Δ vacuoles (E). Fusion reactions were incubated with 2 mm propranolol (A), 100 μg/ml PX (B), 347 μg/ml SigD (C), or 95 μg/ml C1b and 150 μm chlorpromazine (D). Fusion reactions were incubated with inhibitor for 10 min on ice followed by the addition of wild type Vam7 or Vam7-6A at the indicated concentrations for 5 min on ice. Fusion reactions were then incubated at 27 °C for 90 min and assayed for Pho8 activity. Error bars represent S.E. (n ≥ 3). In each panel the fusion values were normalized to untreated “control” reactions in the absence of Vam7. The control values were set at 100%, and Vam7 rescue data are expressed relative to the control.
One of the ways that Vam7 interacts with the vacuole is through the binding of PI3P by its N-terminal PX domain (11). The PI3P binding property of the PX domain can be inhibited by the Y42A mutation, which also severely attenuates the ability of Vam7 to bypass an anti-Sec17 IgG block (12). The PX domain alone binds to the HOPS complex and can block fusion when added to fusion reactions containing endogenous levels of full-length Vam7 (9, 26). The addition of exogenous Vam7 can partially overcome the inhibitory effect of the PX domain (14, 33). Here we found that Vam7-6A was unable to rescue the PX block compared with the effect of the wild type SNARE (Fig. 3B). This could be due to conformational changes in Vam7-6A that prevent displacing of bound PX domain.
Thus far we tested the ability of Vam7 to bypass blocks that target early steps in the reaction pathway. We next tested if Vam7-6A can bypass late blocks as effectively as its wild type parent. A key signaling lipid involved in fusion is PI(4,5)P2, which is made during the fusion reaction and is required for the assembly of the vertex ring at the docking stage and for regulating actin dynamics (21, 23). PI(4,5)P2 serves additional unknown functions during priming and after trans-SNARE pair formation (22, 24). Converting PI(4,5)P2 to PI4P with the PI 5-phosphatase SigD, blocks trans-SNARE pairing (34). The addition of recombinant Vam7 had previously been shown to bypass the effects of SigD (18). Here we found that Vam7-6A was able to bypass the SigD block, albeit with reduced efficacy compared with wild type Vam7 (Fig. 3C). This suggests that the Vam7 PBR plays a more important role in early stages of the fusion pathway.
To test another late-acting lipid, we examined the bypass of blocking fusion with the DAG ligand C1b. We previously found that Vam7 could rescue C1b-blocked fusion in the presence of chlorpromazine, an amphipathic cation that increases membrane fluidity and induces negative membrane curvature (18, 35). DAG accumulates at the boundary membrane and vertex microdomain, where it is thought to destabilize the bilayer to facilitate fusion after docking (2). The DAG ligand C1b disrupts the vertex ring, which would disperse the bilayer-destabilizing effects of DAG. Chlorpromazine reduces membrane tension and is theorized to lower the fusion energy barrier threshold to allow Vam7 to stimulate fusion in the presence of C1b. Here we compared the ability of wild type and Vam7-6A to bypass the C1b block in the presence chlorpromazine. Similar to what we found with the SigD bypass, we found that both wild type Vam7 and Vam7-6A restored fusion of C1b-blocked reactions. There was only a modest shift in the effective dose of Vam7-6A required to support fusion at the same level as seen with the wild type protein (Fig. 3D). This is consistent with the notion that the Vam7 PBR is more important at earlier stages of the fusion pathway.
Vam7-6A Does Not Rescue the Defective Fusion of ycf1Δ Vacuoles
Until recently, the recruitment of Vam7 to vacuoles was thought to only depend on its interactions with PI3P, HOPS, and its cognate SNAREs. However, we found that the class C ABC transporter Ycf1 was also needed for Vam7 recruitment. The deletion of YCF1 led to a reduction in vacuolar Vam7 that was linked to inhibited fusion (30). Importantly, the fusion defect observed with ycf1Δ vacuoles was rescued by the addition of recombinant Vam7 during the fusion reaction. Here, we tested if the Vam7 PBR was required for the rescue of attenuated ycf1Δ fusion. For the purpose of this paper we normalized fusion to the levels of untreated ycf1Δ vacuoles, which show a 40% reduction in fusion when compared with wild type vacuoles. We found that Vam7-6A was unable to increase ycf1Δ fusion, whereas wild type Vam7 enhanced the fusion of ycf1Δ vacuoles to wild type levels as seen previously (Fig. 3E). These data suggest that additional interactions are between Vam7 and vacuolar constituents occur that require an intact PBR.
Vam7-6A Shows Diminished Interaction with SNAREs and HOPS
In the previous experiments we showed under various conditions that Vam7-6A was unable to promote robust fusion, suggesting that the PBR plays an important role in Vam7 function. We next tested if the PBR domain would affect the formation of SNARE complexes or the interactions between SNAREs and HOPS. To this aim we performed large scale anti-Sec17 IgG bypass reactions for the isolation of protein complexes (12, 29). SNARE priming was blocked by the addition of anti-Sec17 IgG to reactions. After incubating for 15 min, secondary inhibitors were added followed by an additional 5 min of incubation. Next, 150 nm GST-Vam7 or GST-Vam7-6A was added to bypass the anti-Sec17 IgG block. Fusion reactions were incubated for a total of 90 min after which membranes were processed for GST-Vam7 isolation as described under “Experimental Procedures.” The secondary inhibitors Gyp1–46 and propranolol were used to inhibit Ypt7 and Pah1 function, respectively. The Western blots show that wild type Vam7 was able to form protein complexes with the syntaxin homologue Vam3, the R-SNARE Nyv1, and the HOPS subunit Vps16 (Fig. 4, A and B). The formation of these complexes was inhibited by both Gyp1–46 and propranolol. It should be noted that although Vam7 can bypass fusion blocked by propranolol alone, it is unable to do so when used in combination with anti-Sec17 IgG. Vam7-6A showed a significant reduction in binding to the SNAREs Vam3 and Nyv1 as well as the HOPS complex (Fig. 4, A and B). The reduced amount of Vam7-6A protein complexes was further reduced by Gyp1–46 and propranolol as seen with wild type Vam7. These data indicate the Vam7 PBR is required for the efficient formation of protein complexes.
FIGURE 4.

Vam7-6A shows a decrease in protein-protein interactions. A, to examine the interactions with other proteins in the SNARE complexes, we monitored the binding of Vam3, Nyv1, and Vps16 to isolated GST-Vam7 complexes. WT vacuoles were incubated at 27 °C for the indicated times in the presence of 150 nm Vam7 (WT or 6A). After incubation, reactions were centrifuged to isolate the membrane fraction. Membranes were solubilized, and GST-Vam7 complexes were isolated with glutathione-Sepharose. Complexes were probed by immunoblotting for GST-Vam7, Vam3, Nyv1, and Vps16. The experiment shown is representative of three trials. B, quantitation of Vam3 and Nyv1 bound to GST-Vam7. Values represent concentrations relative to time WT Vam7 at time = 0 min. Error bars represent S.E. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, calcium efflux assays were performed using WT BJ3505 vacuoles and contained 150 nm Fluo-4 dextran. Reactions were transferred to 27 °C for incubation, and after 1 min ATP or PIPES-sorbitol buffer was added. After 15 min, Vam7 or Vam7-6A was added at the indicated concentrations to reactions. Fluorescence was measured for 30 min. A representative experiment of eight repeats is shown. AU, absorbance units. D, shown is the average efflux at 25 min. Error bars indicate S.E. (n = 6). **, p < 0.01.
The formation of SNARE complexes is linked to the release of luminal Ca2+ stores, and changes in the homeostasis of Ca2+ transport has been associated to fusion efficiency (14, 36). In this study we have seen that the Vam7 PBR is essential for the optimal function. Vam7-6A is unable to fully bypass fusion blocks that wild type Vam7 supports. This difference is due in part to the reduced ability of Vam7-6A to form protein complexes with its partner SNAREs. We next asked if the reduced interactions with SNAREs and HOPS directly affected Ca2+ release. Fusion reactions were treated with buffer or anti-Sec17 IgG. After 15 min of incubation, the reactions blocked for priming were supplemented with buffer, wild type Vam7, or Vam7-6A. Ca2+ transport was detected by changes in Fluo-4 fluorescence. In all conditions we first observed the uptake of Ca2+ from the media as detected by the reduction in Fluo-4 fluorescence. The untreated control began to efflux Ca2+ at 10 min and plateaued after 17–18 min. The reactions treated with anti-Sec17 continued to uptake extraluminal Ca2+ until the addition of Vam7. These data showed that both wild type Vam7 and Vam7-6A triggered the release of Ca2+ from the vacuole lumen (Fig. 4, C and D). Vam7-6A appeared to release less Ca2+ compared with wild type. Fig. 4B shows the average of three experiments with two concentrations of Vam7. Importantly, we observed that the initial rate of Ca2+ release was slower with Vam7-6A. When the rates of Ca2+ release were fitted to kinetic curves, we found that wild type Vam7 stimulated half-maximal release of 1.03 nm Ca2+ s−1. Vam7-6A data showed half-maximal release of 0.48 nm Ca2+ s−1. Together, this suggests that wild type Vam7 is fully engaged in forming trans-SNARE complexes that trigger Ca2+ efflux. The reduced rate of release seen with Vam7-6A is indicative of this SNARE interacting with another ligand apart from trans-SNARE complexes. Because Vam7 binds to the membrane through its PX domain, it is possible that wild type Vam7 sequentially binds to PI3P and SNAREs, whereas Vam7-6A may remain associated to the membrane while forming SNARE complexes. Thus, it is important to test for differential membrane binding by Vam7 and Vam-6A.
Mutating the Vam7 PBR Does Not Alter Net Vacuole Association
We next examined if the Vam7 PBR was required for its association with vacuoles. To determine the efficacy of vacuole binding we added a curve of wild type Vam7 or Vam7-6A to vacuoles treated with anti-Sec17 IgG. After incubating for 10 min at 30 °C, the vacuoles were re-isolated by centrifugation and separated from the supernatant fraction. Bound GST-Vam7 was detected by Western blotting. In Fig. 5 it is shown that both wild type and Vam7-6A associated with vacuoles equally well. This is in contrast to Fig. 3 where we found that Vam7-6A was attenuated in its interaction with other SNAREs and HOPS. Together this suggests that the wild type levels of overall vacuole association by Vam7-6A were due to other interactions, such as those with membrane lipids.
FIGURE 5.

Vam7-6A shows wild type levels of vacuole association. A, to examine the recruitment of wild type Vam7 and Vam7-6A, we measured binding to isolated vacuoles. Vacuoles were incubated with the indicated concentrations of either WT GST-Vam7 or GST-Vam7-6A for 10 min. After incubation, vacuoles were centrifuged to isolate bound GST-Vam7. Complexes were probed by immunoblotting for Vam3 and Vam7. The experiment shown is representative of three trials. B, quantitation of Vam7 bound to vacuoles. Values were normalized to wild type Vam7 binding at 500 nm. Error bars represent S.E. (n = 3).
The Vam7 PBR Affects Binding to PI3P by the PX Domain
Polybasic regions of various proteins have been shown to interact with anionic lipids. To examine if the Vam7 PBR affects binding to lipids, we used a liposome binding assay. In these assays we reconstituted liposomes composed of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) alone or in the presence of a signaling lipid. Recombinant Vam7 was incubated with liposomes after which the membranes were isolated by flotation. Bound Vam7 floated with liposomes if there was a direct interaction. In Fig. 6A we show that wild type Vam7 bound well to PI3P. This was expected, as the PX domain preferentially interacts with PI3P (11). There were only trace amounts of Vam7 that associated with PC/PE liposomes. The lower panel of Fig. 6A showed quantitation of three experiments where binding was normalized to PC/PE liposomes and wild type Vam7. In addition to PI3P, we also included liposomes that contained PA, DAG, or phosphatidylserine (PS). Vam7 did not interact with either DAG or PS. PA liposomes showed limited binding to Vam7. When Vam7-6A was tested, we found that binding to PI3P was increased by nearly 3-fold. The increase in PI3P binding was not accompanied by changes to binding other lipids, indicating that the interaction was specific. The interaction of Vam7-6A with PA, DAG, and PS was similar to binding by wild type Vam7. This was curious, as we expected the removal of a multiple basic residues to reduce lipid binding. This led us to hypothesize that the Vam7 PBR affects the ability of the PX to bind PI3P.
FIGURE 6.

Vam7-6A shows increased binding to PI3P liposomes. To examine the recruitment of wild type Vam7 or Vam7-6A to specific lipids, we examined binding to liposomes or varying composition. Liposomes were incubated with 150 nm GST-Vam7 (WT or 6A) for 10 min. After incubation, liposomes were centrifuged to isolate bound Vam7 by flotation. Liposomes were probed by Western blotting for Vam7. The experiment shown is representative of three trials. A, liposomes composed of PC and PE alone or also containing PI3P, DAG, PA, or PS. B, liposomes composed of PC/PE alone or also containing PI, PI4P, or PI5P. Quantitation of Vam7 bound to liposomes is shown as bar graphs in each panel. Values represent concentration compared with Vam7 WT bound to PC/PE liposomes. Error bars represent S.E. (n = 3). *, p < 0.05.
To determine if mutating the Vam7 PBR affected interactions with other lipids, we performed similar flotation assays with other phosphoinositides. In Fig. 6B we tested Vam7 binding to PI alone as well as PI4P and PI5P. We found that both wild type Vam7 and Vam7-6A floated with PI5P-containing liposomes. The binding to PI5P is not unexpected as Cheever et al. (11) showed that the PX alone could bind to PI5P by lipid overlay assays. The role of PI5P in membrane trafficking is not well understood. That said, Boal et al. (37) showed that the endosomal protein TOM1 interacts with PI5P to regulate endosomal maturation. Thus it is possible that Vam7 interactions with other phosphoinositides could be part of its function. We also detected equivalent low levels of binding of Vam7 and Vam7-6A to PI and PI4P. These interactions were near the level of baseline interactions to PC/PE liposomes. To complete the liposome binding experiments, we tested polyphosphorylated phosphoinositides. Liposomes were constructed containing PC/PE and either PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, or PI(3,4,5)P3. These experiments showed that neither Vam7 nor Vam7-6A interacted with PI(3,5)P2, PI(4,5)P2, or PI(3,4,5)P3 (data not shown). Interestingly, we did find that both Vam7 and Vam7-6A interacted modestly with PI(3,4)P2 and mutating the PBR increased binding to the lipid, but the difference was not statistically significant. PI(3,4)P2 is not well characterized in its role in membrane trafficking. This unusual lipid is made by Class II PI 3-kinases that convert PI4P to PI(3,4)P2 (38, 39). PI(3,4)P2 is also made by the dephosphorylation of PI(3,4,5)P3 (40). Because there is no evidence for either lipid in yeast, we conclude that the binding of Vam7 constructs to PI(3,4)P2 is not biologically relevant.
Because Vam7-6A binds to PI3P more efficiently relative to wild type, we theorized that the PBR affects PI3P binding by the PX domain. Previous studies have shown that mutating the PX critical Tyr-42 inhibited binding to PI3P and severely blocked vacuole fusion (11, 12). Computer structure predictions indicate that the surface of the Vam7 PX domain contains two acidic patches that could in theory interact with the basic charges of the PBR. To test this notion we made the Y42A mutation in Vam7-6A to produce Vam7-7A. We compared PI3P binding by Vam7-7A with that of wild type Vam7, Vam7-6A, and Vam7Y42A. Our data show that Vam7Y42A had reduced binding to PI3P (Fig. 7A). Importantly, Vam7-7A binding to PI3P was below the level of Vam7Y42A. This is consistent with the idea that the Vam7 PBR does not directly bind lipids but, rather, affects the lipid binding ability of a separate domain. We now postulate that the Vam7 PBR affects PI3P binding by the PX domain.
FIGURE 7.
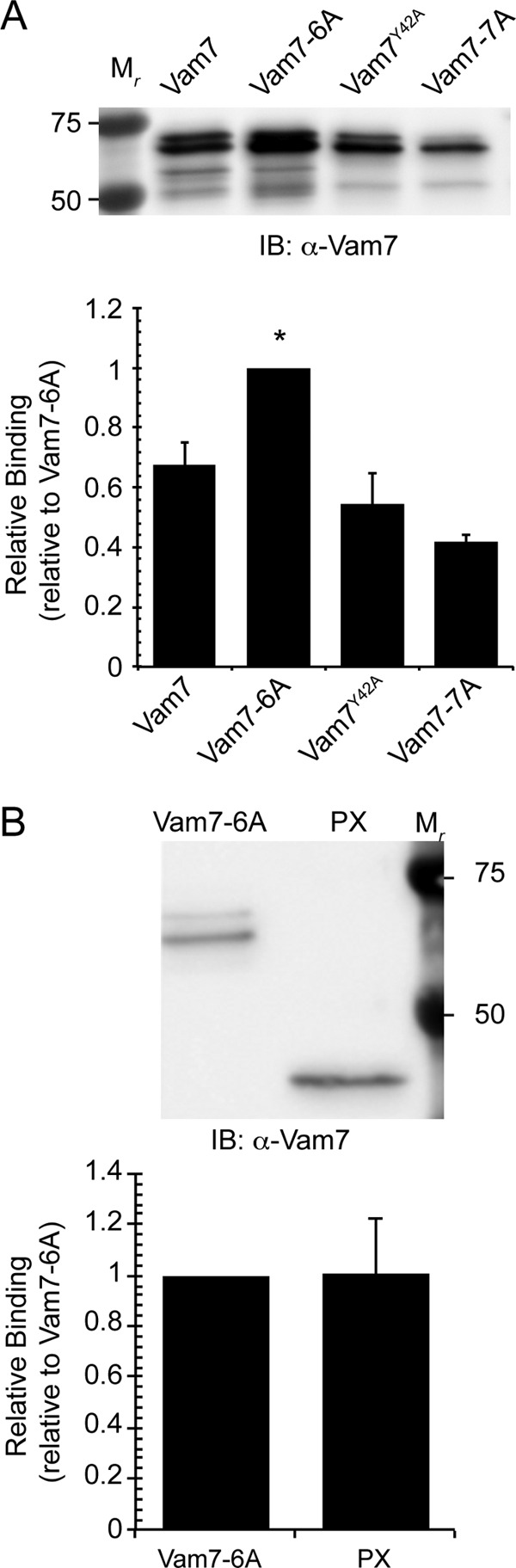
Vam7-6A affects PI3P binding by the PX domain. A, liposomes with PI3P were incubated with wild type Vam7, Vam7-6A, Vam7Y42A, or Vam7-7A. Protein binding was detected as described above and compared with Vam7-6A bound to PI3P. IB, immunoblot. B, Vam7-6A and PX were incubated with PI3P liposomes. The bar graph indicates average PI3P binding normalized to Vam7-6A. Error bars represent S.E. (n = 3). *, p < 0.05.
Due to the increased PI3P binding caused by mutating the PBR, we next tested how Vam7-6A lipid binding compared with the PX domain alone. Our data showed that the enhanced PI3P binding of Vam7-6A was equivalent to that seen with the PX domain alone (Fig. 7B). This is consistent with the notion that other domains in Vam7 regulate lipid binding by the PX domain. At this point we cannot distinguish whether the differences in PI3P binding were due to changes in binding affinity or undefined steric effects. Nevertheless, we can conclude that PI3P binding by Vam7 is auto-regulated in a manner dependent on the PBR.
Discussion
Homotypic vacuole fusion requires the R-SNARE Nyv1 and the Q-SNAREs Vam7, Vam3, and Vti1. Vam7 is a unique SNARE in that it lacks a transmembrane helix or lipid moiety to anchor it to the membrane. Instead, Vam7 associates to the membrane via an N-terminal PX domain that interacts with the lipid PI3P and the HOPS complex. Previous studies have focused on either the PX domain or the C-terminal SNARE motif. Vam7 also has a middle domain that had not been characterized. One of the predicted features of the Vam7 middle domain is an α-helix with a polybasic face containing six Lys and Arg in close proximity that we refer to as the polybasic region, or PBR. In this study we examined the role of the Vam7 PBR in supporting vacuole homotypic fusion.
Many proteins across diverse pathways contain PBRs that are associated with binding anionic lipids including PS, PA, and PIs. Vam7 is not the only SNARE that contains a PBR. The Q-SNARE Spo20 contains an amphipathic helix with a polybasic face that binds PA on prospores and the plasma membrane as a positive regulator of the protein (42). Its binding to PA depends on the presence of the acidic lipids PS and PI(4,5)P2 (43). In mammalian cells syntaxin1 contains a PBR that binds to various PIs on lipid overlay experiments, whereas liposome binding shows that it preferentially binds to PA (44). The syntaxin1 PBR regulates fusion pore dynamics, and mutating basic residues to Ala alters secretion. We originally hypothesized that the Vam7 PBR would contribute to membrane binding by directly interacting with vacuolar acidic lipids. However, contrary to what we expected, we found that mutating the basic residues of the PBR to alanines augmented the binding Vam7-6A to PI3P. Thus we were correct in predicting that the PBR would affect lipid binding, but incorrect in that the Vam7-6A mutant would exhibit reduced binding due to disrupting the interactions between membrane lipids and the PBR domain.
How does the Vam7 PBR affect fusion? To reconcile the increased binding of Vam7-6A to PI3P with the reduced ability to support fusion, we must consider two things. First, Vam7-6A showed a marked decrease in protein binding to its cognate SNAREs and HOPS. Second, the overall association of Vam7-6A with vacuoles was indistinguishable from wild type Vam7. Together this suggests that the increased binding of Vam7-6A to PI3P and decreased binding to SNAREs and HOPS cancel each other out to show an over all “wild type” vacuole association. Another question is how does this translate to the attenuated fusion phenotype? In addition to SNARE complex formation, vacuole fusion requires the downstream release of luminal Ca2+ stores. Our data showed that Vam7-6A triggered reduced Ca2+ efflux with slower initial rate relative to wild type Vam7. The fast initial Ca2+ efflux rate triggered by wild type Vam7 suggests that it operates at “saturation,” meaning that it is fully participating in SNARE complexes. The slower rate of Ca2+ release triggered by Vam7-6A suggests that it is at sub-saturation in the forming SNARE complexes and is likely binding a second ligand. Because Vam7-6A bound PI3P more than the wild type protein, we theorize that it remains bound to PI3P while forming SNARE complexes. This leads us to postulate that wild type Vam7 first binds PI3P and subsequently releases the lipid as it enters into SNARE complexes. Retention of Vam7-6A by PI3P could possible reduce its ability to be transferred to nascent trans-SNARE complexes. The reduced number of SNARE bundles would generate and transfer less energy to the membrane resulting in the stall at hemifusion.
In our model the Vam7 PBR affects PI3P binding by the PX domain. We propose that this occurs through a direct interaction between the positive charges of the PBR and the negatively charged surface of the PX domain. Our homology modeling with the PX domain of p40phox indicates that there are two acidic patches on the Vam7 PX domain where the PBR domain could electrostatically interact. This interaction could potentially reduce the affinity or access of the PX to PI3P to facilitate SNARE complex formation. Therefore, mutating the basic PBR residues to Ala would disrupt the electrostatic interaction with the PX domain, leading to increased PI3P binding at the sacrifice of optimal SNARE interactions. This model is supported by our data showing that mutating the critical PX Tyr (Y42A) in Vam7-7A abolished the augmented PI3P binding of Vam7-6A.
In addition to the PX domain there are other acidic surfaces on the vacuole that could potentially interact with the Vam7 PBR. For instance, the surface of SNARE four-helical bundles is acidic. Thus, it is possible that the Vam7 PBR interacts with the SNARE bundle to stabilize the complex and promote the transfer of energy to the membrane. The lack of interactions of the PBR with the acidic surface of SNARE bundle could reduce the energy transferred to the membrane, resulting in a stall between a hemifusion intermediate and full content mixing. In turn, conformational changes could affect PI3P binding by the PX domain.
Experimental Procedures
Reagents
Soluble reagents were dissolved in PIPES-sorbitol buffer (20 mm PIPES-KOH, pH 6.8, 200 mm sorbitol) with 125 mm KCl unless indicated otherwise. Anti-Sec17 IgG (8), GST-Vam7 (29), GST-PX (12), GST-FYVE (41), His6-SigD (2), His6-Gyp1–46 (7), GST-C1b (45), and Pbi2 (46) were prepared as described previously. PC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidycholine), PE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyethanolamine), PS (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine), DAG (1-palmitoyl-2-oleoyl-sn-glycerol), phosphatidic acid (POPA; 1,2-dipalmitoyl-sn-glycero-3-phosphate), were purchased from Avanti Polar Lipids (Alabaster, AL). (PI; 1,2-dipalmitoyl phosphatidylinositol), PI 3-phosphate (PI3P; 1,2-dipalmitoyl phosphatidylinositol 3-phosphate), PI4P, PI5P, PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3 were purchased from Echelon Biosciences Inc. (Salt Lake City, UT). Chlorpromazine (Sigma) was stored as a 4.5 mm stock solution in dimethyl sulfoxide. Propranolol (Sigma) was dissolved in PIPES-sorbitol buffer.
Recombinant GST-Vam7 Constructs
The previously described pET42a constructs encoding Vam7 and Vam7Y42A were used for expressing recombinant GST fusions and to generate new mutants (12, 29). Site-directed mutagenesis was carried out using pET42a-Vam7 to change Arg-164, Arg-168, Lys-172, Lys-175, Lys-179, and Lys-186 to alanines to generate Vam7-6A using the primers listed in Table 1. In addition, the Y42A mutation was made in Vam7-6A to produce Vam7-7A. All GST-Vam7 constructs were recombinantly expressed as described previously (12, 29).
TABLE 1.
Primers for site-directed mutagenesis
F, forward primer; R, reverse primer. To make single mutations, the forward (F) primer would be combined with a reverse single (RS) primer. The primers were synthesized by Integrated DNA Technologies. The overlapping primer sequences for making point mutations are in italics. Mutations are underlined, and the targeted mutations are in bold.
| Oligonucleotide | Sequence |
|---|---|
| R164AF | 5′-CAGCGTGGCTGGAGCACTAAGGGCACGTACGAAGCTC-3′ |
| R164AR | 5′-GCTCCAGCCACGCTGGGTGTGCCATCACTCTCCTTATC-3′ |
| R168AF | 5′-CACTAGCGGCACGTACGAAGCTCCACAAGTTAC-3′ |
| R168AR | 5′-ACGTGCCGCTAGTGCTCCAGCCACGCTGGG-3′ |
| R168ARS | 5′-ACGTGCCGCTAGTGCTCCAGCCACGCTGG-3′ |
| K172AF | 5′-CGTACGGCGCTCCACAAGTTACGAGAGCGACTAG-3′ |
| K172AR | 5′-GGAGCGCCGTACGTGCCGCTAGTGCTCCAC-3′ |
| K172ARS | 5′-GGAGCGCCGTACGTGCAGGTAGTGCTCCAC-3′ |
| K175AF | 5′-TCCACGCGTTACGAGAGCGACTAGAACAGGAT-3′ |
| K175AR | 5′-CGTAACGCGTGGAGCGCCGTACGTGCC-3′ |
| K175ARS | 5′-CGTAACGCGTGGAGCTTCGTACGTGCCCA-3′ |
| R179AF | 5′-CGAGAGGCACTAGAACAGGATGTGCAAAAG-3′ |
| R179AR | 5′-CTAGTGCCTCTCGTAACGCGTGGAGCGC-3′ |
| R179ARS | 5′-CTAGTGCCTCTCGTAACTTGTGGAGCTTCGTA-3′ |
| K186AF | 5′-GTGCAAGCGAAGTCTCTTCCAAGCACGGAAG-3′ |
| K186AR | 5′-GACTTCGCTTGCACATCCTGTTCTAGTGCCTC-3′ |
| K186ARS | 5′-GACTTCGCTTGCACATCCTGTTCTAGTCGCTC-3′ |
| SOEing 1 | 5′-GCGGGATCCCCCACACCTTTAAGTAACCGTCACC-3′ |
| SOEing 2 | 5′-GCTGCCATCAATATCAATTATCAACCCTTATATGAC-3′ |
| SOEing 3 | 5′-GATGGCAGCTAATTCTGTAGGGAAAATGAG-3′ |
| SOEing 4 | 5′-CTCATTAATTCAAGCACTGTTGTTAAAATGTCTAGCC-3′ |
| SOEing 5 | 5′-GTGCTTGAATTAATGAGTTACTATCCGGG-3′ |
| SOEing 6 | 5′-GCGGAGCTCGCTACAATAGTGTTATGGATCTCCGTCTCG-3′ |
| SOEing 7 | 5′-CGCGAATTCGCTACAATAGTGTTATGGATCTCCGTCTCG-3′ |
In Vivo Complementation
Vam7 was amplified from BJ3505 genomic DNA beginning 308 bases upstream and ending 308 bases downstream using SOEing primer 1 and SOEing primer 7. The DNA was purified and digested with BamH1-HF and EcoR1-HF. After purification, the cut DNA was ligated with pRS413 that was digested with BamH1-HF and EcoR1-HF and calf intestinal phosphatase-treated. Purified ligated DNA was transformed into NEB Turbo competent Escherichia coli cells and selected for on Luria broth with ampicillin. Plasmid was isolated from transformants, and fidelity was confirmed by sequencing using M13 forward and reverse primers. Plasmid was transformed into DKY6281 vam7Δ yeast and selected for on complete synthetic media lacking tryptophan and histidine to make RFY70.
Vam7-6A was amplified in three pieces. The 5′-untranslated region was amplified from BJ3505 genomic DNA using SOEing primer 1 and SOEing primer 2. The 3′-untranslated region was amplified from BJ3505 genomic DNA using SOEing primer 5 and SOEing primer 7. The open reading frame of Vam7-6A was amplified from pET42a-VAM7−6A plasmid using SOEing primer 3 and SOEing primer 4. PCR products were purified and then combined via SOEing reaction (62 °C annealing; 72 °C extension for 45 s; 15 cycles) followed by the addition of SOEing primer 1 and SOEing primer 7 (62 °C annealing; 72 °C extension, 1 min; 29 cycles). The band at proper size was gel-extracted and then followed the above procedure for cloning Vam7 in to pRS413 to make RFY71.
Vacuole Isolation and in Vitro Fusion
Vacuoles were isolated from the S. cerevisiae strains BJ3505 and DKY6281 as described (47). Vacuoles deleted in YCF1 were isolated from RFY32 and RFY33 (30). In vitro content mixing fusion reactions (30 μl) contained 3 μg each of vacuoles from BJ3505 (PHO8 pep4Δ) and DKY6281 (pho8Δ PEP4) backgrounds, fusion reaction buffer (20 mm PIPES-KOH, pH 6.8, 200 mm sorbitol, 125 mm KCl, 5 mm MgCl2), ATP regenerating system (1 mm ATP, 0.1 mg/ml creatine kinase, 29 mm creatine phosphate), 10 μm coenzyme A, and 283 nm Pbi2. Fusion was determined by the activation of pro-alkaline phosphatase (proPho8) by the protease Pep4. Fusion reaction mixtures were incubated at 27 °C for 90 min, after which the vacuoles were lysed in 250 mm Tris-Cl, pH 8.5, 0.4% Triton X-100, 10 mm MgCl2, and 1 mm p-nitrophenyl phosphate. Mature Pho8 activity was assayed through the dephosphorylation of p-nitrophenyl phosphate to generate p-nitrophenolate. Fusion units were measured by determining the p-nitrophenolate produced, which was measured at 400 nm. Yeast cytosol was prepared as described previously (48). Cells expressing Vam7 or Vam7-6A were disrupted by vortexing with glass beads in the presence of protease inhibitors. Lysates were centrifuged to remove large debris, supernatants were transferred to ultracentrifuge tubes, and membranes were pelleted by centrifugation (100,000 × g, 1 h, 4 °C). The supernatants were collected as the cytosol fractions.
GST-Vam7 Pulldown
GST-Vam7 protein complex isolation was performed as described (12, 29). Large-scale 6× fusion reactions (180 μl) were incubated with 85 μg/ml anti-Sec17 IgG to block priming. After 15 min, 0.5 μm Gyp1–46 or 2 mm propranolol was added to selected reactions and incubated for an additional 5 min, adding buffer or 150 nm GST-Vam7. After a total of 90 min, reactions were placed on ice for 5 min, and 30-μl aliquots were removed to measure fusion activity. The remaining vacuoles were re-isolated by centrifugation (11,000 × g, 10 min, 4 °C), and the supernatants were decanted before extracting vacuoles with solubilization buffer (20 mm HEPES-KOH, pH 7.4, 100 mm NaCl, 2 mm EDTA, 20% glycerol, 0.5% Triton X-100, 1 mm DTT) with protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 10 μm Pefabloc-SC, 5 μm pepstatin A, and 1 μm leupeptin). Vacuole pellets were gently resuspended with 200 μl of solubilization buffer and incubated for 20 min on ice. Insoluble debris was sedimented by centrifugation (16,000 × g, 10 min, 4 °C), and 176 μl of supernatants were transferred to new chilled tubes. Next, 16 μl was removed from each reaction as 10% total samples, mixed with SDS loading buffer (2X Laemmli sample buffer, 120 mm Tris-Cl pH 6.8, 4% SDS, 20% glycerol), and heated (95 °C, 5 min). The remaining extracts were incubated with 30 μl of equilibrated glutathione-Sepharose 4B beads (GE Healthcare) (12 h, 4 °C, nutating). Beads were sedimented (735 × g, 2 min, 4 °C) and washed with 1 ml of solubilization buffer 5 times, and bound material was eluted with SDS loading buffer. Protein complexes were examined by Western blotting.
Calcium Efflux
Vacuole lumen Ca2+ efflux was measured as described (36). Fusion reactions (2×) contained 20 μg of vacuoles isolated from BJ3505, fusion reaction buffer with 10 μm CoA, and 283 nm Pbi2, and the fluorescent Ca2+ probe Fluo-4 dextran at 150 nm (Invitrogen). Reaction mixtures were transferred to a black, half-volume 96-well flat-bottom plate with nonbinding surface (Corning). ATP regenerating system or buffer was added, and reactions were incubated at 27 °C while monitoring Fluo-4 fluorescence. Samples were analyzed in a fluorescence plate reader at 27 °C with the excitation filter at 485 nm and emission filter at 520 nm. The reactions were initiated with the addition of ATP regenerating system when the start of measurement subsequently followed. Calibration was done using buffered Ca2+ standards (Invitrogen).
Liposome Preparation and Co-floatation Assay
Small unilamellar liposomes containing specific lipid compositions were produced by sonication (49). Stock lipids dissolved in chloroform were mixed to produce a mixture with the desired mole percentages of 2.6 μmol of total phospholipids. Samples were then placed under a vacuum in a desiccator for an additional 14 h. PBS (2.6 ml) was added to the dried lipids, and the tubes were covered with Parafilm and incubated at room temperature for 1 h. The lipids were suspended with vortexing and disrupted in a water bath sonicator for 30 min. To measure protein binding to the liposomes we used a floatation assay as described (50). Liposome binding was conducted by adding 150 nm Vam7 to prepared liposomes (150 μl). Samples were incubated for 10 min at 30 °C, and 630 μl of 1.65 m sucrose (PBS) was added. Samples were loaded into the bottom of a centrifuge tube and overlaid with 630 μl of 0.75 m sucrose (PBS) and 1× PBS to the top of the tube. Samples were centrifuged (200,000 × g, 90 min, 4 °C), and 200 μl of floated liposomes were recovered from the top of the 0.75 m sucrose layer. The bottom 100-μl fraction was recovered, and SDS sample buffer was added to detect unbound protein levels. Liposomes were diluted in 2 ml of 1× PBS and isolated by centrifugation (16,000 × g, 20 min, 4 °C). SDS sample buffer was added to the final liposome pellet, and bound proteins were resolved by SDS-PAGE. The proteins were transferred to nitrocellulose and probed by Western blotting. Images were acquired using a ChemiDoc MP Imaging System (Bio-Rad).
Lipid Mixing
Lipid mixing assays were conducted using rhodamine B DHPE (Rh-PE; Thermo Fisher) as described (48). BJ3505 vacuoles (300 μg) were isolated and then incubated in 400 μl of PIPES-sorbitol buffer containing 150 μm Rh-PE (10 min, 4 °C, nutating). Next, 800 μl of 15% Ficoll was added and then transferred to an 11 × 60-mm ultracentrifuge tube overlaid with 1.2 ml of 8 and 4% and 0.5 ml of PIPES-sorbitol buffer. Labeled vacuoles were isolated by centrifugation (105,200 × g, 25 min, 4 °C, SW-60 Ti rotor) and recovered from the 0–4% Ficoll interface. Lipid mixing assays (90 μl) contained 2 μg of labeled vacuoles and 16 μg of unlabeled vacuoles in fusion buffer. Reaction mixtures were transferred to a black, half-volume 96-well flat-bottom microtiter plate on ice. The plate was transferred to a fluorescence plate reader at 27 °C to start the reactions. Measurements were taken every 60 s for 50 min, yielding fluorescence values at the onset (F0) and during the reaction (Ft). After 40 min 0.45% (v/v) Triton X-100 was added, and the final 10 measurements were averaged to give the value of fluorescence after infinite dilution (FTX100). The relative fluorescence change ΔFt/FTX100 = (Ft − F0)/FTX100 − F0 was calculated.
Differential Scanning Fluorimetry
Wild type Vam7 and Vam7-6A were diluted in PIPES-sorbitol buffer with 125 mm KCl and 10% glycerol to a final concentration of 0.43 mg/ml. SYPRO orange dye (6×) was added to the reactions, and 25 μl of mix was added to a white Bio-Rad hard-shell 96-well PCR plate and covered with optical Microseal “b” Adhesive seal. Samples were equilibrated for 30 min before starting the melting curve. This was run on a Bio-Rad CFX connect Real-Timer PCR detection system. The PCR protocol was 20 °C for 1 min followed by a 20 °C to 95 °C temperature gradient with 10-s dwell times at each temperature. Fluorescence was measured at each temperature (λex = 490 nm, λem = 560 nm). The first derivatives of the fluorescence data were used to determine the Tm for each sample.
Microscopy
Vacuole morphology was detected by incubating cells with 5 μm FM4-64 in yeast extract/peptone/dextrose broth (Invitrogen) as described previously (36). Images were acquired with a Zeiss Axio Observer Z1 inverted microscope with an X-Cite 120LX light source, a Plan Apochromat 63× oil objective (1.4 N.A.), and AxioCam CCD camera. FM4-64 images were acquired using a 43 HE Cy3 shift-free filter set. Z-stacks of images were taken, and images were deconvolved using AxioVision 3D software.
Statistical Analysis
All statistical analysis was calculated using one-way analysis of variance. p values of ≤ 0.05 were considered significant. Ca2+ efflux rates were calculated using Origin software.
Author Contributions
G. E. M., M. L. S., L. R. H., and R. P. S. designed and performed the experiments, analyzed the data, and prepared the figures. M. P. made genetic constructs and purified proteins. R. A. F. designed the experiments, analyzed the data, and prepared the figures. All authors wrote the manuscript and contributed to preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. William Wickner of antisera.
This work was supported, in whole or in part, by National Institutes of Health Grant GM101132 (to R. A. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Starr, M. L., Hurst, L. R., and Fratti, R. A. (2016) Traffic, 10.1111/tra.12409.
- NSF
- N-ethylmaleimide-sensitive factor
- DAG
- diacylglycerol
- SNAP
- soluble NSF adaptor protein
- SNARE
- soluble N-ethylmaleimide-sensitive factor attachment protein receptors
- HOPS
- homotypic fusion and vacuole protein sorting complex
- PA
- phosphatidic acid
- PBR
- polybasic region
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PS
- phosphatidylserine
- PI
- phosphatidylinositol
- PX
- phox homology
- Rh-PE
- rhodamine phosphatidylethanolamine.
References
- 1. Guo W., Tamanoi F., and Novick P. (2001) Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3, 353–360 [DOI] [PubMed] [Google Scholar]
- 2. Fratti R. A., Jun Y., Merz A. J., Margolis N., and Wickner W. (2004) Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J. Cell Biol. 167, 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lang T., Bruns D., Wenzel D., Riedel D., Holroyd P., Thiele C., and Jahn R. (2001) SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20, 2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts R. L., Barbieri M. A., Pryse K. M., Chua M., Morisaki J. H., and Stahl P. D. (1999) Endosome fusion in living cells overexpressing GFP-rab5. J. Cell Sci. 112, 3667–3675 [DOI] [PubMed] [Google Scholar]
- 5. TerBush D. R., Maurice T., Roth D., and Novick P. (1996) The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15, 6483–6494 [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L., Seeley E. S., Wickner W., and Merz A. J. (2002) Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell 108, 357–369 [DOI] [PubMed] [Google Scholar]
- 7. Wang L., Merz A. J., Collins K. M., and Wickner W. (2003) Hierarchy of protein assembly at the vertex ring domain for yeast vacuole docking and fusion. J. Cell Biol. 160, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mayer A., Wickner W., and Haas A. (1996) Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85, 83–94 [DOI] [PubMed] [Google Scholar]
- 9. Boeddinghaus C., Merz A. J., Laage R., and Ungermann C. (2002) A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J. Cell Biol. 157, 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ungermann C., and Wickner W. (1998) Vam7p, a vacuolar SNAP-25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO J. 17, 3269–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheever M. L., Sato T. K., de Beer T., Kutateladze T. G., Emr S. D., and Overduin M. (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 3, 613–618 [DOI] [PubMed] [Google Scholar]
- 12. Fratti R. A., and Wickner W. (2007) Distinct targeting and fusion functions of the PX and SNARE domains of yeast vacuolar Vam7p. J. Biol. Chem. 282, 13133–13138 [DOI] [PubMed] [Google Scholar]
- 13. Xu H., and Wickner W. T. (2012) N-terminal domain of vacuolar SNARE Vam7p promotes trans-SNARE complex assembly. Proc. Natl. Acad. Sci. U.S.A. 109, 17936–17941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merz A. J., and Wickner W. (2004) Trans-SNARE interactions elicit Ca2+ efflux from the yeast vacuole lumen. J. Cell Biol. 164, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giraudo C. G., Hu C., You D., Slovic A. M., Mosharov E. V., Sulzer D., Melia T. J., and Rothman J. E. (2005) SNAREs can promote complete fusion and hemifusion as alternative outcomes. J. Cell Biol. 170, 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reese C., and Mayer A. (2005) Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J. Cell Biol. 171, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Diao J., Grob P., Cipriano D. J., Kyoung M., Zhang Y., Shah S., Nguyen A., Padolina M., Srivastava A., Vrljic M., Shah A., Nogales E., Chu S., and Brunger A. T. (2012) Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. elife 1, e00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karunakaran S., and Fratti R. (2013) The lipid composition and physical properties of the yeast vacuole affect the hemifusion-fusion transition. Traffic 14, 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han G. S., Wu W. I., and Carman G. M. (2006) The Saccharomyces cerevisiae lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 281, 9210–9218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sasser T., Qiu Q. S., Karunakaran S., Padolina M., Reyes A., Flood B., Smith S., Gonzales C., and Fratti R. A. (2012) Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J. Biol. Chem. 287, 2221–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karunakaran S., Sasser T., Rajalekshmi S., and Fratti R. A. (2012) SNAREs, HOPS, and regulatory lipids control the dynamics of vacuolar actin during homotypic fusion. J. Cell Sci. 125, 1683–1692 [DOI] [PubMed] [Google Scholar]
- 22. Kato M., and Wickner W. (2001) Ergosterol is required for the Sec18/ATP-dependent priming step of homotypic vacuole fusion. EMBO J. 20, 4035–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayer A., Scheglmann D., Dove S., Glatz A., Wickner W., and Haas A. (2000) Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol. Biol. Cell 11, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collins K. M., Thorngren N. L., Fratti R. A., and Wickner W. T. (2005) Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 24, 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lawrence G., Brown C. C., Flood B. A., Karunakaran S., Cabrera M., Nordmann M., Ungermann C., and Fratti R. A. (2014) Dynamic association of the PI3P-interacting Mon1-Ccz1 GEF with vacuoles is controlled through its phosphorylation by the type-1 casein kinase Yck3. Mol. Biol. Cell 25, 1608–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stroupe C., Collins K. M., Fratti R. A., and Wickner W. (2006) Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 25, 1579–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krämer L., and Ungermann C. (2011) HOPS drives vacuole fusion by binding the vacuolar SNARE complex and the Vam7 PX domain via two distinct sites. Mol. Biol. Cell 22, 2601–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lobingier B. T., and Merz A. J. (2012) Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Mol. Biol. Cell 23, 4611–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fratti R. A., Collins K. M., Hickey C. M., and Wickner W. (2007) Stringent 3Q: 1R composition of the SNARE 0-layer can be bypassed for fusion by compensatory SNARE mutation or by lipid bilayer modification. J. Biol. Chem. 282, 14861–14867 [DOI] [PubMed] [Google Scholar]
- 30. Sasser T. L., Lawrence G., Karunakaran S., Brown C., and Fratti R. A. (2013) The yeast ABC transporter Ycf1p enhances the recruitment of the soluble SNARE Vam7p to vacuoles for efficient membrane fusion. J. Biol. Chem. 288, 18300–18310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thorngren N., Collins K. M., Fratti R. A., Wickner W., and Merz A. J. (2004) A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 23, 2765–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jun Y., and Wickner W. (2007) Assays of vacuole fusion resolve the stages of docking, lipid mixing, and content mixing. Proc. Natl. Acad. Sci. U.S.A. 104, 13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jun Y., Thorngren N., Starai V. J., Fratti R. A., Collins K., and Wickner W. (2006) Reversible, cooperative reactions of yeast vacuole docking. EMBO J. 25, 5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collins K. M., and Wickner W. T. (2007) Trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 104, 8755–8760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fang J., and Iwasa K. H. (2007) Effects of chlorpromazine and trinitrophenol on the membrane motor of outer hair cells. Biophys. J. 93, 1809–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sasser T. L., Padolina M., and Fratti R. A. (2012) The yeast vacuolar ABC transporter Ybt1p regulates membrane fusion through Ca2+ transport modulation. Biochem. J. 448, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boal F., Mansour R., Gayral M., Saland E., Chicanne G., Xuereb J. M., Marcellin M., Burlet-Schiltz O., Sansonetti P. J., Payrastre B., and Tronchère H. (2015) TOM1 is a PI5P effector involved in the regulation of endosomal maturation. J. Cell Sci. 128, 815–827 [DOI] [PubMed] [Google Scholar]
- 38. Nigorikawa K., Hazeki K., Guo Y., and Hazeki O. (2014) Involvement of class II phosphoinositide 3-kinase α-isoform in antigen-induced degranulation in RBL-2H3 cells. PLoS ONE 9, e111698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Posor Y., Eichhorn-Gruenig M., Puchkov D., Schöneberg J., Ullrich A., Lampe A., Müller R., Zarbakhsh S., Gulluni F., Hirsch E., Krauss M., Schultz C., Schmoranzer J., Noé F., and Haucke V. (2013) Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499, 233–237 [DOI] [PubMed] [Google Scholar]
- 40. Hawkins P. T., and Stephens L. R. (2016) Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 44, 307–314 [DOI] [PubMed] [Google Scholar]
- 41. Gillooly D. J., Morrow I. C., Lindsay M., Gould R., Bryant N. J., Gaullier J. M., Parton R. G., and Stenmark H. (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakanishi H., de los Santos P., and Neiman A. M. (2004) Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol. Biol. Cell 15, 1802–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horchani H., de Saint-Jean M., Barelli H., and Antonny B. (2014) Interaction of the Spo20 membrane-sensor motif with phosphatidic acid and other anionic lipids, and influence of the membrane environment. PLoS ONE 9, e113484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lam A. D., Tryoen-Toth P., Tsai B., Vitale N., and Stuenkel E. L. (2008) SNARE-catalyzed fusion events are regulated by Syntaxin1A-lipid interactions. Mol. Biol. Cell 19, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson J. E., Giorgione J., and Newton A. C. (2000) The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry 39, 11360–11369 [DOI] [PubMed] [Google Scholar]
- 46. Slusarewicz P., Xu Z., Seefeld K., Haas A., and Wickner W. T. (1997) I2B is a small cytosolic protein that participates in vacuole fusion. Proc. Natl. Acad. Sci. U.S.A. 94, 5582–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haas A., Conradt B., and Wickner W. (1994) G-protein ligands inhibit in vitro reactions of vacuole inheritance. J. Cell Biol. 126, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qiu Q. S., and Fratti R. A. (2010) The Na+/H+ exchanger Nhx1p regulates the initiation of Saccharomyces cerevisiae vacuole fusion. J. Cell Sci. 123, 3266–3275 [DOI] [PubMed] [Google Scholar]
- 49. Mima J., and Wickner W. (2009) Complex lipid requirements for SNARE- and SNARE chaperone-dependent membrane fusion. J. Biol. Chem. 284, 27114–27122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsuoka K., Morimitsu Y., Uchida K., and Schekman R. (1998) Coat assembly directs v-SNARE concentration into synthetic COPII vesicles. Mol. Cell 2, 703–708 [DOI] [PubMed] [Google Scholar]