

Abstract
Current FDA-approved l-asparaginases also possess significant l-glutaminase activity, which correlates with many of the toxic side effects of these drugs. Therefore, l-asparaginases with reduced l-glutaminase activity are predicted to be safer. We exploited our recently described structures of the Erwinia chrysanthemi l-asparaginase (ErA) to inform the design of mutants with diminished ability to hydrolyze l-glutamine. Structural analysis of these variants provides insight into the molecular basis for the increased l-asparagine specificity. A primary role is attributed to the E63Q mutation that acts to hinder the correct positioning of l-glutamine but not l-asparagine. The substitution of Ser-254 with either an asparagine or a glutamine increases the l-asparagine specificity but only when combined with the E63Q mutation. The A31I mutation reduces the substrate Km value; this is a key property to allow the required therapeutic l-asparagine depletion. Significantly, an ultra-low l-glutaminase ErA variant maintained its cell killing ability. By diminishing the l-glutaminase activity of these highly active l-asparaginases, our engineered ErA variants hold promise as l-asparaginases with fewer side effects.
Keywords: cancer therapy, enzyme kinetics, enzyme mutation, leukemia, structural biology, structure-function, substrate specificity
Introduction
l-Asparaginases are amidohydrolases that catalyze the hydrolysis of the amino acid l-asparagine (Asn)2 to l-aspartate (Asp) and ammonia. Currently, the Food and Drug Administration has approved the l-asparaginase from Escherichia coli (EcA) and Erwinia chrysanthemi (ErA) for treatment of select blood cancers. These enzyme drugs function by depleting Asn from the blood, thereby affecting certain blood cancers that are dependent on exogenous Asn. Sensitivity to l-asparaginase has been attributed to a reduced ability of those cancers to synthesize this amino acid de novo (1). Because the concentration of Asn in human blood is ∼50 μm (2), for an l-asparaginase to be clinically useful, its Asn Km value must be in the low micromolar range. This property is present in EcA (Km = 15 μm) and ErA (Km = 48 μm).
Patients treated with l-asparaginase often confront two types of adverse effects. The first is due to an immune response against the bacterial enzymes. To decrease the immunogenicity, the current standard of care in the United States employs a PEGylated version of EcA (3, 4). If immunogenicity still arises, ErA is used as second line treatment (5).
The second source of adverse effects of l-asparaginase treatment relates to the enzyme's ability to hydrolyze the amino acid l-glutamine (Glu). That is, in addition to catalyzing the hydrolysis of Asn (l-asparaginase activity), both EcA and ErA also catalyze the hydrolysis of Gln to l-glutamate (Glu) and ammonia (l-glutaminase activity). l-Glutamine is the most abundant amino acid in the blood with a concentration range of 400–650 μm (2). The l-glutaminase activity of these bacterial l-asparaginases is significant, ∼2% of the EcA activity and as high as 10% for ErA (3). Of note, the l-glutaminase activity of the clinically used l-asparaginases has been implicated in many of the side effects of this treatment, which include immunosuppression (2, 6, 7), hepatotoxicity (8), pancreatitis (9), and coagulation dysfunction (9, 10). Many of the side effects are believed to be due to the disruption of protein synthesis induced by the l-glutaminase activity of l-asparaginase drugs (2, 7, 8, 11, 12).
To eliminate the toxic side effects related to Gln depletion, enzymes that have retained their l-asparaginase activity but have reduced l-glutaminase activity would be beneficial. To this goal, Derst et al. (13) exploited crystal structures of the E. coli enzyme to design mutants with reduced l-glutaminase activity. These authors reported that the substitution of Asn-248 by alanine resulted in an enzyme with ∼300-fold reduced efficiency in the l-glutaminase reaction (13). However, this same mutant was also 22-fold less efficient in the l-asparaginase reaction, and most significantly, the parameter most important for Asn depletion, the Km value, was increased >6-fold (13). Therefore, whereas this variant had the desired reduction in l-glutaminase activity, a penalty was paid in its l-asparaginase activity.
ErA wild-type enzyme (ErA-WT) is characterized by a high turnover rate and a relatively low Km value for both Asn and Gln. The goal of the work presented here is to identify a mutant of ErA that maintains its low Km l-asparaginase activity concomitant with reduced l-glutaminase activity. ErA was chosen over EcA for the enzyme engineering effort for two reasons: (i) ErA has a much higher intrinsic l-glutaminase activity, so it would benefit more from a reduction in this property, and (ii) ErA is a much faster l-asparaginase, which is the property important for cell killing. We recently reported the crystal structures of ErA in complex with the products of the reactions, Asp and Glu (14). These structures revealed the molecular basis for Asn, a superior substrate over Gln for this enzyme, allowed us to predict regions that may act to control substrate selectivity. Four candidate sites that may influence the l-glutaminase/l-asparaginase ratio were identified. Of the initial 18 single and double mutants generated at these positions, the single mutant E63Q (referred to as ErA-E63Q) and the double mutant A31I/E63Q (ErA-DM1) had the best combined properties, retaining 60–90% of the wild-type l-asparaginase efficiency alongside a remarkable 95% reduction in the l-glutaminase activity. An additional double mutant, E63Q/S254N (ErA-DM2), had a >99% reduction in its l-glutaminase activity, but this was coupled with a ∼4-fold increase in the Asn Km value. To understand the molecular reasons for the increased preference for Asn versus Gln of our mutants, we solved the crystal structures of both double mutants and of the E63Q single mutant in complex with Asp. The ErA-E63Q Asp complex structure provides a structural basis for the pivotal role played by the E63Q mutation in increasing the enzyme's Asn selectivity. The structures of ErA-DM1 and ErA-DM2 in complex with Asp showcase the importance of these additional sites for substrate discrimination. Moreover, the structural analysis suggested additional variants that combine aspects from ErA-DM1 and ErA-DM2. One of these variants, the ErA-A31I/E63Q/S254Q triple mutant (ErA-TM2), proved to possess even superior discrimination between Asn and Gln. Most importantly, we could demonstrate that the ultra-low l-glutaminase property of ErA-TM2 does not hamper its ability to kill human acute lymphoblastic leukemia (ALL) cells in culture relative to ErA-WT.
Together, the ErA mutants presented here hold potential for development as replacements for the current Food and Drug Administration-approved l-asparaginase enzymes as they are predicted to maintain their cancer killing properties due to their high l-asparaginase activity but at the same time have reduced side effects due to their much-diminished l-glutaminase activity.
Results
Residue Selection for Mutagenesis
We seek ErA variants with reduced activity toward Gln but retained high activity toward Asn. Due to their role in catalysis, residues present in consensus motifs among the l-asparaginase/l-glutaminase superfamily (highlighted red in the multiple sequence alignment in Fig. 1, including 12TGGT15, 93HGTDT97, Ser-62, and Lys-168, were excluded from consideration for mutation. Instead, our strategy was to seek non-conserved residues that surround the active site and that do not necessarily interact with the substrate. Based on a multiple sequence alignment (Fig. 1) and the analysis of the crystal structure of ErA in complex with the products Asp and Glu (14), we identified Ala-31, Glu-63, Pro-123, and Ser-254 as four potential candidate sites for mutagenesis (Fig. 2). Interestingly, of these four residues only Glu-63 makes a direct interaction with the substrate.
FIGURE 1.

Sequence alignment of select l-asparaginases. Strictly and mostly conserved residues are highlighted in red and yellow, respectively. Arrows indicate the four sites selected for mutagenesis. The secondary structure shown above the primary sequence is based on the atomic structure of E. chrysanthemi (PDB ID 1O7J). The E. chrysanthemi, Erwinia carotovora and E. coli enzymes contain an N-terminal signal peptide, which is not included in the alignment. The UniProt entries used for this alignment are: E. chrysanthemi, P06608; E. carotovora, Q6Q4F4; E. coli, P00805; W. succinogenes, P50286; Helicobacter pylori, O25424; Cavia porcellus (guinea pig), H0W0T5. For the guinea pig enzyme, only residues in its N-terminal l-asparaginase domain are shown.
FIGURE 2.

Location of residues selected for mutagenesis. A, surface representation of the ErA-WT·Asp tight dimer (PDB ID 5F52) used for the design of mutants; one protomer is colored pale green, and the other protomer is dark green. The bound Asp molecule is shown in stick representation. Zoom: distances between the bound Asp and mutation sites 1, 2, 3, and 4. B, overlay of the ErA-WT·Asp (pale and dark green) and ErA-WT-Glu (PDB ID 5HW0; pale and dark gray) structures. Displayed are select conserved active site residues with their relative locations to the four mutation sites examined in this study. Site 4 originates from the neighboring protomer (dark green) as denoted by the prime in Ser-254′. Note the 0.6 Å displacement of Glu versus Asp in the direction of Glu-63. All distances are in angstroms.
Site 1, Ala-31, is part of the flexible N-terminal loop that closes and becomes ordered in the presence of ligand at the active site. When ordered, Ala-31 is located at the entrance of the substrate binding pocket (Fig. 2A). Although not interacting directly with the preferred substrate Asn, it is at a 4.1 Å distance to the Cα atom of the amino acid substrate. In the crystal structure of ErA in complex with the reaction product Glu (PDB ID 5HW0), which we used as a reference for the binding of the substrate Gln, we noticed that the extra methylene group present in this substrate (compared with Asn) twists in the direction of Ala-31. Therefore, to sterically repel the extra Gln methylene group we replaced the small alanine side chain with a bulkier side chain. Based on modeling studies, the side chains as present in valine, isoleucine, leucine, and methionine seemed the most appropriate. Additionally, as shown in the sequence alignment (Fig. 1), Ala-31 is replaced by a valine or serine in the case of the E. coli and H. pylori l-asparaginases, respectively. Compared with ErA, these enzymes have been reported to have negligible l-glutaminase activity (15, 16). For this reason, we also investigated mutating Ala-31 into either serine or threonine (the latter isosteric with valine but retaining the hydroxyl group of serine).
Site 2 for mutagenesis comprised Glu-63, a residue that directly interacts with the α-amino group of the amino acid substrate (Fig. 2B). Our structures of ErA in complex with Asp and Glu showed that to accommodate the larger Glu, the substrate shifts (by 0.6 Å) in the direction of Glu-63 relative to the binding position adopted by the smaller Asp (14). To make this required repositioning by Gln less favorable, we opted to replace the non-conserved residue Glu-63 with the isosteric glutamine. Both the glutamate and glutamine side chains contain an oxygen atom that can act as an H-bond acceptor to the α-amino group of either Asn or Gln. However, we predicted that the amide nitrogen atom (H-bond donor) present in the glutamine side chain (contrasted with a second oxygen atom in glutamate) may be less favorable for accommodating Gln. This choice of mutation was reinforced by the fact that the counterpart of Glu-63 in the low l-glutaminase H. pylori and E. coli l-asparaginases is indeed a glutamine. In addition to the E63Q mutant we also created the E63L mutation based on the recent study of the E. coli enzyme conducted by Chan et al. (17) reporting Q59L (analogues position to Glu-63 in ErA) as a highly l-asparaginase-specific variant.
The third candidate site for mutagenesis was Pro-123. This site was chosen because in two l-asparaginase homologues reported to have low l-glutaminase activity (16, 18), this residue is either a serine (in Wolinella succinogenes) or asparagine (in H. pylori; Fig. 1). Based on the location of Pro-123, we predicted that this residue may play an indirect role in increasing the preference for Asn over Gln. To test this prediction, we prepared the ErA-P123S and P123N mutants.
Residue Ser-254, site 4 for modification, is located at the entrance of the substrate binding pocket and originates from an adjacent protomer (Fig. 2A). Therefore, as rationalized above for Ala-31, we postulated that replacing this serine with a residue with a larger side chain would constrict the pocket, resulting in preferential exclusion of Gln. The multiple sequence alignment (Fig. 1) revealed the counterparts of this residue in the E. coli (low l-glutaminase activity) and guinea pig (no l-glutaminase activity) enzymes are asparagine and proline, respectively. We, therefore, created the mutants S254N and S254P to probe the effect of this site on Gln/Asn preference. The location of the residues at these four sites and their relationship to other conserved active site residues, as observed in the ErA Asp and Glu complex structures (ErA-WT·Asp and ErA-WT-Glu, respectively), is shown in Fig. 2B.
Kinetic Characterization of ErA Mutants Designed to Have Reduced l-Glutaminase Activity
In addition to ErA-WT, we initially expressed and purified the 18 ErA mutants listed in Table 1. We employed colorimetric enzyme-coupled assays to quantitate the l-asparaginase and l-glutaminase activities of the enzymes using 10 mm Gln and 2 mm Asn as substrates for the l-glutaminase and l-asparaginase assays, respectively. 17 of the 18 mutants had the desired reduction in l-glutaminase activity (Table 1). However, for the majority of mutants, this decrease coincides with a drastic decline in the l-asparaginase activity. Using the criteria of (i) ≥60% retained l-asparaginase activity (relative to ErA-WT) and (ii) ≤5% l-glutaminase:l-asparaginase ratio, we selected four mutants for further study (Table 1).
TABLE 1.
l-Glutaminase (GLNase) and l-asparaginase (ASNase) activity of ErA WT and mutant enzymes
| Enzyme | GLNase vs ErA-WT | ASNase vs ErA-WT | GLNase/ASNase within same proteina |
|---|---|---|---|
| ErA-WT | 100 | 100 | 14 |
| ErA-A31V | 72 | 81 | 13 |
| ErA-A31S | 124 | 110 | 16 |
| ErA-A31T | 18 | 28 | 9 |
| ErA-A31N | 3 | 33 | 1 |
| ErA-A31M | 7 | 26 | 3 |
| ErA-A31I | 65 | 85 | 12 |
| ErA-A31L | 6 | 12 | 4 |
| ErA-E63L | 0 | 12 | 0 |
| ErA-E63Qb | 31 | 90 | 5 |
| ErA-P123S | 2 | 3 | 11 |
| ErA-P123N | 1 | 2 | 6 |
| ErA-S254N | 34 | 40 | 12 |
| ErA-S254Pb | 0 | 120 | 0 |
| ErA-A31T/E63Q | 8 | 15 | 7 |
| ErA-A31I/E63Qb | 22 | 60 | 5 |
| ErA-S254N/E63Qb | 0 | 82 | 0 |
| ErA-S254N/A31T | 9 | 18 | 7 |
| ErA-S254N/A31I | 22 | 25 | 12 |
a Observed l-glutaminase rate measured with 10 mm Gln and 5 nm enzyme; observed l-asparaginase rate measured with 2 mm Asn and 0.5 nm enzyme.
b Mutants that were selected for determination of kcat and Km parameters.
From the initial screen, the single mutant ErA-S245P appeared to have the most desired properties. The l-glutaminase activity was below the detection sensitivity at the assay conditions, whereas the l-asparaginase activity was 120% relative to the wild-type enzyme (Table 1). Further kinetic characterization of the mutant's l-asparaginase activity confirmed the high turnover rate with Asn. However, disappointedly, it also revealed a 24-fold increase (relative to ErA-WT) in the Km value for Asn (Table 2). Because clinical use of l-asparaginase requires complete depletion of Asn from the blood, the Km of the enzyme must be in the low micromolar range to be effective. Hence, although the goal of largely eliminating the l-glutaminase activity was achieved, an unacceptable increase in the Km value of Asn for the l-asparaginase reaction eliminated the ErA-S245P mutant from the list of potential drug candidates.
TABLE 2.
l-Asparaginase kinetic parameters for ErA wild type and select mutants
| Enzyme | kcat | km | kcat/km | kobs at 50 μm | kobs at 20 μm | specific activitya |
|---|---|---|---|---|---|---|
| s−1 | μm | s−1μm−1 | s−1 | s−1 | IU/mg | |
| ErA-WT | 207.5 ± 3.6 | 47.5 ± 3.5 | 4.37 | 105.3 | 50.7 | 353.2 ± 6.1 |
| ErA-E63Q | 186.8 ± 1.7 | 50.7 ± 2.0 | 3.68 | 92.2 | 46.3 | 317.8 ± 2.9 |
| ErA-A31I/E63Q (DM1) | 123.0 ± 1.8 | 36.9 ± 2.2 | 3.33 | 69.7 | 38.8 | 209.1 ± 3.1 |
| ErA-E63Q/S254N (DM2) | 169.8 ± 1.5 | 185.3 ± 5.5 | 0.92 | 36.3 | 18.9 | 288.8 ± 2.6 |
| ErA-S254P | 250.0 ± 2.8 | 1154.2 ± 25.5 | 0.22 | 10.5 | 4.5 | 425.4 ± 4.8 |
| ErA-E63Q/S254Q (DM3) | 147.3 ± 1.0 | 179.1 ± 5.6 | 0.82 | 30.8 | 14.2 | 250.4 ± 1.7 |
| ErA-A31I/E63Q/S254N (TM1) | 109.7 ± 0.8 | 124.2 ± 4.3 | 0.89 | 29.2 | 16.4 | 186.4 ± 1.4 |
| ErA-A31I/E63Q/S254Q (TM2) | 261.2 ± 2.8 | 95.0 ± 3.5 | 2.75 | 89.1 | 40.8 | 443.5 ± 4.8 |
| EcA-WT | 44.4 ± 0.3 | 15.0 ± 0.5 | 2.96 | 41.3 | 28.3 | 76.3 ± 0.5 |
a 1 IU of enzyme is defined as the amount of enzyme used to produce 1 μmol of product in 1 min at 37 °C.
Further inspection of the properties of the mutants from the initial screen (Table 1) suggested that the next three most promising variants are the E63Q (ErA-E63Q) single mutant and the A31I/E63Q (ErA-DM1) and E63Q/S254N (ErA-DM2) double mutants. Detailed kinetic characterization of the l-asparaginase activity of the mutants revealed that the critical Km value for the substrate Asn is comparable with that of ErA-WT (Table 2). In fact, ErA-DM1 acquired an even lower Asn Km than that of ErA-WT (37 versus 48 μm). In contrast, ErA-DM2 variant exhibited a 4-fold increase in Asn Km. Together with near wild-type kcat (range 60–90% of ErA-WT), these mutants fulfill the criteria required to achieve efficient depletion of Asn from the blood.
We next determined the precise kinetic parameters for the undesired l-glutaminase activity. This work established that the reduced l-glutaminase activity of these mutants is due to a reduced kcat (3–9-fold lower) and much increased Km (8–44-fold higher) compared with ErA-WT (Table 3). In terms of efficiency, as indicated by the kcat/Km parameter, the l-glutaminase efficiency of the mutants was reduced by ∼20–400-fold. Likewise, at the physiological blood concentration of 0.5 mm, the observed l-glutaminase rate was reduced by 95–99% relative to ErA-WT. This analysis demonstrates that these three mutants largely fulfill our design criteria of being severely impaired in their l-glutaminase activity but with near ErA-WT l-asparaginase activity.
TABLE 3.
l-Glutaminase kinetic parameters for ErA wild type and select mutants
N.D., not determined.
| Enzyme | kcat | Km | kcat/Km | kobs at 1 mm | kobs at 0.5 mm | Specific activitya |
|---|---|---|---|---|---|---|
| s−1 | mm | s−1mm−1 | s−1 | s−1 | IU/mg | |
| ErA-WT | 26.84 ± 0.26 | 0.36 ± 0.02 | 74.56 | 19.22 | 15.87 | 45.68 ± 0.44 |
| ErA-E63Q | 8.33 ± 0.16 | 3.86 ± 0.23 | 3.68 | 2.16 | 0.74 | 14.17 ± 0.27 |
| ErA-A31I/E63Q (DM1) | 6.01 ± 0.13 | 2.73 ± 0.20 | 3.33 | 2.20 | 0.83 | 10.22 ± 0.22 |
| ErA-E63Q/S254N (DM2) | 2.93 ± 0.03 | 15.80 ± 0.30 | 0.19 | 0.18 | 0.11 | 4.98 ± 0.05 |
| ErA-E63Q/S254Q (DM3) | 7.17 ± 0.64 | 84.04 ± 12.15 | 0.09 | 0.19 | 0.02 | 12.17 ± 1.19 |
| ErA-A31I/E63Q/S254N (TM1) | 5.60 ± 0.20 | 28.20 ± 2.30 | 0.19 | 0.33 | 0.11 | 9.51 ± 0.34 |
| ErA-A31I/E63Q/S254Q (TM2) | 1.84 ± 0.11 | 47.46 ± 6.95 | 0.04 | N.D. | 0.01 | 3.12 ± 0.02 |
| EcA-WT | 0.89 ± 0.01 | 1.38 ± 0.09 | 0.64 | 0.36 | 0.22 | 1.53 ± 0.02 |
a 1 IU of enzyme is defined as the amount of enzyme used to produce 1 μmol of product in 1 min at 37 °C.
Toward the Molecular Understanding Behind the Improved l-Asparaginase Selectivity of the Designed Mutants
Although structural considerations were incorporated into the design of the mutants discussed above, we still sought to verify the mechanism for the increased Asn selectivity. Toward this goal we crystallized the ErA-E63Q, ErA-DM1, and ErA-DM2 enzyme variants and soaked the crystals with either Asp or Glu (the products of the l-asparaginase and l-glutaminase reaction, respectively). High resolution diffraction data were collected on both Asp- and Glu-soaked crystals. For the Asp-soaked crystals (see Table 4 for data collection and refinement statistics), inspection of the electron density at the enzymes' active site unambiguously showed the presence of the ligand (supplemental Fig. S1). However, despite several attempts under different soak conditions (e.g. increasing soak time, increasing Glu concentration), we failed to observe electron density for the soaked Glu. Instead, the active site of the Glu-soaked crystals had an electron density for HEPES buffer, which is a component of the crystallization solution. A molecule of HEPES was previously observed bound at the active site of apo ErA-WT, which was crystallized using the same conditions (14). Of note, for ErA-WT, both soaked Asp and Glu are able to displace the buffer molecule from the active site (14). In contrast, for the three ErA mutants discussed here, only Asp (but not Glu) was successful in fully displacing the bound HEPES molecule. This inability of soaked Glu to displace the buffer molecule in the ErA mutant crystals correlates with their much-increased Gln Km value relative to the wild-type enzyme (Table 3).
TABLE 4.
Data collection and refinement statistics
a.u., asymmetric unit; r.m.s.d., root mean square deviation.
| Structure |
|||
|---|---|---|---|
| ErA-E63Q+ASP | ErA-DM1+ASP | ErA-DM2+ASP | |
| PDB codes | 5I3Z | 5I48 | 5I4B |
| Data collection statistics | |||
| X-ray source and detector | LS-CAT ID-D MARCCD 300 | LS-CAT ID-F MARCCD 225 | LS-CAT ID-D MARCCD 300 |
| Wavelength (Å) | 1.008264 | 0.97872 | 1.008264 |
| Temperature (K) | 100 | 100 | 100 |
| Resolutiona (Å) | 2.05 (2.18-2.05) | 1.50 (1.59-1.50) | 1.60 (1.69-1.60) |
| Number of reflections | |||
| Observed | 437,716 (49641) | 1,278,167 (118,300) | 682,869 (105,570) |
| Unique | 74,069 (10093) | 183,230 (23,339) | 122,786 (19,113) |
| Completeness (%) | 97.2 (83.5) | 95.7 (76.2) | 98.2 (95.7) |
| Rsym (%) | 12.6 (53.7) | 5.5 (60.8) | 10.1 (61.6) |
| Average I/σ(I) | 16.88 (4.08) | 23.09 (3.01) | 14.71 (3.36) |
| Space group | P212121 | P212121 | I222 |
| Unit cell (Å): a, b, c | 77.89, 87.89, 175.99 | 77.19, 87.69, 175.11 | 76.98, 123.12, 198.44 |
| Wilson B-factors (Å2) | 19.7 | 17.1 | 13.7 |
| Refinement statistics | |||
| Refinement program | REFMAC5 | REFMAC5 | REFMAC5 |
| Rcryst (%) | 17.39 | 12.02 | 15.43 |
| Rfree (%) | 20.93 | 17.06 | 18.28 |
| Resolution range (Å) | 30.00-2.05 | 30.0-1.5 | 30.0-1.6 |
| Protein molecules per a.u. | 4 | 4 | 3 |
| Number of atoms | |||
| Protein (protA, protB, protC, protD) | 2455, 2455, 2464, 2463 | 2487, 2481, 2466, 2487 | 2521, 2499, 2503 |
| Water molecules | 976 | 1004 | 954 |
| Asp molecules | 4 | 4 | 3 |
| r.m.s.d. from ideal | |||
| Bond length (Å) | 0.0135 | 0.0226 | 0.0193 |
| Bond angles (°) | 1.5853 | 1.9611 | 1.9204 |
| Average B-factors (Å2) | 21.0 | 20.0 | 16.0 |
| Protein (ProtA, protB, protC, protD) | 22.1, 20.0, 19.9, 20.4 | 18.7, 19.9, 21.4, 19.4 | 14.9, 15.1, 16.5 |
| Water molecules | 27.4 | 30.9 | 26.7 |
| Asp molecules | 21.3, 22.6, 18.8, 22.1 | 17.5 | 16.3, 16.6, 16.4 |
| Ramachandran plot statistics (%) | |||
| Most favored regions | 96.81 | 96.67 | 97.38 |
| Additionally allowed regions | 2.88 | 3.01 | 2.29 |
| Outlier regions | 0.31 | 0.32 | 0.33 |
a High resolution shell in parenthesis.
Overall Structure of the ErA Mutants
ErA-WT builds a tetramer composed of a dimer-of-dimers, irrespective of the presence or absence of Asp/Glu at the active site (14). However, in the absence of Asp/Glu, the N-terminal region (residues 18–34) lacks clear electron density. This so-called flexible N-terminal loop was observed ordered and approaching the active site when either Asp or Glu bound to ErA-WT (14). For the three ErA mutants analyzed here (ErA-E63Q, ErA-DM1, ErA-DM2), the tetrameric organization was maintained, and the flexible N-terminal loop displayed clear electron density and adopted its closed conformation. The ErA-E63Q·Asp, ErA-DM1·Asp, and ErA-DM2·Asp structures were solved at a resolution of 2.05 Å, 1.50 Å, and 1.60 Å, respectively (Table 4), and all were very similar to the ErA-WT·Asp structure (PDB ID 5F52; root mean square deviation of 0.09–0.135 Å).
Binding of Asp to ErA-E63Q
As stated earlier, in the wild-type enzyme Glu-63 directly interacts with the α-amino group of the amino acid ligand (Asp or Glu); in the specific case of ErA-WT·Asp, that interaction distance is 2.8 Å (arrow, Fig. 3A). We postulated that replacing Glu-63 with a glutamine would result in interaction that is more distance-sensitive as, whereas Glu-63 forms a salt-bridge with the α-amino group of the substrate, Gln-63 would form an H-bond. The increase in distance sensitivity of this interaction was predicted to affect the binding of Gln but not of Asn (for more on this point, see “Discussion”). The assignment of the Gln-63 side chain orientation (carbonyl oxygen atom versus amide nitrogen atom) was done based on chemical reasoning, where the amide nitrogen atom of Gln-63 (an H-bond donor) would be the one farther away from the α-amino group of the ligand (also an H-bond donor).
FIGURE 3.

The E63Q mutation has a minor effect on Asp binding. A, overlay of the ErA-WT·Asp (a region from two of the four protomers that build the tetramer are shown in pale and dark green) and the ErA-E63Q·Asp (beige and brown) structures. The structures are basically identical, with the most significant difference being an increased distance (3.0 versus 2.8 Å) between the side chain of residue 63 (Gln-63/Glu-63) and the α-amino group of Asp (gray arrow). B, zoom on the Asp-Glu-63/Gln-63 interaction. Both oxygen atoms of the Glu-63 side chain are compatible with a close distance to the α-amino group of Asp. In contrast, for Gln-63, the side chain amide nitrogen atom (H-bond donor) would not be compatible with proximity to the α-amino group of Asp (also an H-bond donor). This is not a factor when the ligand is Asp but becomes paramount for the larger amino acid Glu (also see section “Modeling the Glu Binding Mode to the Mutant ErA Variants”).
As predicted, in the ErA-E63Q·Asp structure the interaction distance between the α-amino group of the ligand (Asp) and the carbonyl moiety of the glutamine side chain of Gln-63 has increased (to 3.0 Å; Fig. 3B). Importantly, the distance between the amide nitrogen atom of Gln-63 and the α-amino group of the Asp ligand is acceptably long at 3.9 Å (Fig. 3B). In other words, in the case of Asp (and holding true also for the substrate Asn), there is sufficient separation between these H-bond donor moieties so as not to negatively effect the l-asparaginase reaction. This explains why this mutant maintains a high l-asparaginase activity.
Binding of Asp to ErA-DM1
We next determined the crystal structure of the A31I/E63Q double mutant (ErA-DM1) in complex with Asp (ErA-DM1·Asp). The minimal effect of the E63Q mutation on ErA-DM1 was similar to that seen for ErA-E63Q. In contrast, the effect of the A31I mutation was more significant, impacting the conformation of the flexible N-terminal loop. Although still closed, this loop adopted a different conformation to the one seen in the ErA-WT·Asp complex structure (Fig. 4A). The isoleucine in position 31 that replaced the smaller alanine, prevents this section of the N-terminal flexible loop from approaching the substrate binding site (Fig. 4A, zoom). In ErA-WT·Asp, the alanine at position 31 is 4.1 and 5.3 Å away from the Cα and Cβ atoms of the bound Asp, respectively (Fig. 4B). When Ala-31 is replaced by the larger isoleucine, the position of the flexible N-terminal loop adjusts such that the tip of the isoleucine in the ErA-DM1·Asp is nearly at the same distance from the Asp (4.2 and 5.3 Å, Fig. 4B). Hence, this structure reveals how the mutant adapts to maintain its l-asparaginase activity.
FIGURE 4.

The A31I mutation affects the conformation of the flexible N-terminal loop. A, overlay of a single protomer from the ErA-WT·Asp (pale green) and ErA-DM1·Asp (blue) structures. The structures are almost identical except for the conformations of the N-terminal loop (zoom). B, stereo view showing an overlay of ErA-WT·Asp and ErA-DM1·Asp structures with an emphasis on the flexible N-terminal loop. In ErA-WT, the methyl group of Ala-31 is relatively close (4.1 Å) to the Cα-atom of Asp. To accommodate the larger isoleucine in ErA-DM1, the N-terminal loop cannot fully close, and it adopts a conformation that places the tip of the isoleucine side chain 4.2 Å from the Asp Cα-atom. Despite these adjustments in ErA-DM1, this flexible loop conformation must be compatible with hydrolysis of Asn, as indicated by the similar l-asparaginase kinetic properties between ErA-WT and ErA-DM1 (Table 2).
Binding of Asp to ErA-DM2
The single substitution of Ser-254 to asparagine results in a mutant with a comparable decrease in l-asparaginase and l-glutaminase activities (Table 1). Interestingly, combining the S254N mutation with the E63Q mutation (ErA-DM2) results in a variant that is highly selective toward Asn (Tables 2 and 3). To understand this phenomenon, we solved the crystal structure of ErA-DM2 in complex with Asp (ErA-DM2·Asp). Again, the minor effect of the E63Q mutation was similar to that previously discussed. Residue 254, the site of the second mutation, originates from a neighboring protomer in the tetrameric enzyme (denoted with a prime after the number). In ErA-WT, Ser-254′ does not interact directly with the bound Asp (distance 4.0 Å to the α-amino group of Asp) but, rather, functions to position the side chain of the conserved Asp-96 (2.7 Å distance between the Ser-254′ hydroxyl and the side chain of Asp-96) (Fig. 5). When Ser-254′ was mutated to an asparagine, the larger side chain was now closer to the bound Asp (3.5 Å to the α-amino group of Asp) and maintained the positioning interaction with Asp-96 (3.1 Å). The orientation of the Asn-254′ was dictated by Asp-96 such that the Asn-254′ side chain amide nitrogen atom (H-bond donor) faced the carboxylic acid moiety of Asp-96 (H-bond acceptor). This is an important point, as the Asn-254′ side chain amide nitrogen atom, as positioned by Asp-96, now generates repulsion to the α-amino group of Asn or Gln. This explains why the ErA-S254N single mutant has decreased activity with both Asn and Gln (Table 1).
FIGURE 5.

The S254N mutation introduced an unfavorable interaction to the α-amino group of the ligand, and this effect was largely reduced for Asn (but not Gln) when combined with the E63Q mutation. Overlay of ErA-WT·Asp (pale and dark green) and ErA-DM2·Asp (plum and cyan) reveals a very similar active site except at the mutation sites. Asn-254′ (the prime denotes residue from neighboring protomer) makes an unfavorable (H-bond donor to H-bond donor) interaction with the α-am ino group of Asp. However, the presence of the E63Q mutation allows Asp to shift slightly away, resulting in an acceptable 3.5 Å distance to the side chain of Asn-254′.
The kinetic property of the enzyme is markedly different when the S254N mutation is combined with the E63Q mutation, as is present in ErA-DM2. Similar to the previous E63Q-containing variants, the distance in the ErA-DM2·Asp structure between the side chain at position 63 and Asp increases to 3.0 Å. This increase in distance, whereas small, increased the separation between Asn-254′ and the substrate Asn to 3.5 Å (Fig. 5). This shift in the position of Asp (due to the E63Q mutation) allowed it to lessen the repulsive interaction with the side chain of Asn-254′. In other words, the E63Q mutation sufficiently displaced the ligand such as to limit the repulsive interaction between the ligand α-amino group (H-bond donor) and the Asn-254′ amide nitrogen atom (also a H-bond donor). This allowed ErA-DM2 to maintain high l-asparaginase activity.
Modeling the Glu Binding Mode to the Mutant ErA Variants
The preceding structural analysis showcases the compatibility of the mutations with the enzymatic activity with Asn. Now we wanted to understand the basis for the incompatibility of the mutations with the hydrolysis of Gln. However, a direct experimental view of how the mutants bind Gln was not possible because the Glu-soaked crystals of the mutant enzymes failed to bind the amino acid. Therefore, we resorted to overlaying the ErA-WT-Glu and mutant structures. In this type of analysis we make the postulation that Glu binds to the mutant enzymes the same as observed to ErA-WT. We are aware that this simplification is likely to be not totally accurate. Nonetheless, this analysis does provide insight into the potential Glu binding mode to the mutant variants.
The E63Q mutation must play a key role in the discrimination between Asn and Gln as it is present in all three mutants discussed so far that have high l-asparaginase/low l-glutaminase properties. The overlay of ErA-WT-Glu and the ErA-E63Q structure provides the putative Glu binding mode to ErA-E63Q, and this revealed a short distance between the Glu α-amino group and the Gln-63 amide nitrogen atom (3.5 Å) (Fig. 6A). Because both of these moieties are H-bond donors, such proximity would be detrimental for the binding of this amino acid. As discussed earlier, in the case of Asp, whose smaller size allows it to reposition away from Gln-63, the analogous H-bond:H-bond distance is 3.9 Å (Fig. 6A) and is, therefore, less detrimental. In the wild-type enzyme, instead of an amide nitrogen there is the second oxygen atom of the Glu-63 side chain carboxylate, which is an H-bond acceptor, and this explains why Gln is a relatively good substrate of ErA-WT. Why does the Glu not simply reposition, as Asp does, so as to increase the distance to the amide nitrogen atom of Gln-63? The answer lies in the fact that the Glu side chain carboxylate (and that of Asp) is anchored by Thr-95 (Fig. 6A). This threonine, in combination with Thr-15, is important for the enzymatic hydrolysis of Asn and Gln (19). Hence, the E63Q mutation discriminates against Gln by exploiting the requirement of this large amino acid to fit in the active site by shifting in the direction of Glu-63/Gln-63, which in turn would place the Gln α-amino group at an unfavorable proximity to the Gln-63 amide nitrogen atom (but is compatible with the wild-type Glu-63).
FIGURE 6.

Structural basis for selective discrimination against Gln. A, docking of Glu from the ErA-WT-Glu structure (light and dark gray) into the ErA-E63Q·Asp active site (beige). To fit in the active site, the extra methylene group of Glu (versus Asp) requires the larger amino acid to shift in the direction of Glu-63. The substitution of Glu-63 by a glutamine introduces a H-bond donor (the amide nitrogen atom) in proximity to the Glu α-amino group (also a H-bond donor). It is this proximity of the H-bond donor moieties that selects against Gln. Glu (and pertaining to Gln) cannot shift away as the opposite side of the substrate is anchored by Thr-95. For the smaller amino acid Asn, this negative constellation is mitigated by the larger distance to Gln-63. B, docking of Glu from the ErA-WT-Glu structure into the ErA-DM2·Asp active site (plum and cyan). Like the E63Q mutation, the S254N mutation introduces a H-bond donor (the amide nitrogen atom of the Asn-254′ side chain) in proximity to the substrate α-amino group. Glu (and pertaining to Gln), due to the extra methylene group, cannot avoid this negative interaction.
The A31I (as present in ErA-DM1) did not act to increase the discrimination against Gln but, rather, reduced the Km of Asn (from 51 μm in ErA-E63Q to 37 μm in ErA-DM1). This mutation also reduced the Km of Gln. The ErA-DM1·Asp structure revealed that this mutation forced the flexible N-terminal loop to adopt a different closed conformation relative to the one adopted by the wild-type enzyme. How this reduced the Asn Km value is not apparent from the structure. We speculate that this mutation influenced the dynamics of loop closure and that this ultimately influenced the substrate Km value.
The S254N mutation (as present in ErA-DM2) did increase the selectivity between Asn and Gln, and it did so in a similar manner to the E63Q mutation. Namely, this mutation introduced a H-bond donor in proximity to the Gln α-amino group (Fig. 6B). Overlay of ErA-WT-Glu and ErA-DM2·Asp revealed the likely position Glu would adopt when bound to the mutant enzyme. This superposition revealed that the larger Glu, by shifting toward Asn-254′, would result in an unfavorably short (3.2 Å) separation between H-bond groups. The reason that the smaller substrate Asn is not affected by this mutation to a significant degree (although its Km is also increased from 51 μm in ErA-E63Q to 185 in ErA-DM2) is due to the larger separation (3.5 Å) between these H-bond donor groups (Fig. 6B).
Exploiting the Structural Analysis to Develop Improved Second Generation Mutants
The previous work has identified positions 63 and 254 as “hot spots” for Asn/Gln selectivity and position 31 for reducing the substrate Km value. We first asked whether a variant with substitutions at all three hot spots would have improved properties. The triple mutant ErA-A31I/E63Q/S254N (ErA-TM1) did indeed have a lower Km for Asn (110 versus 170 μm for ErA-DM2; Table 2), but its l-glutaminase rate increased (Table 3). Together, ErA-TM1 was only marginally better than our previous best variant, ErA-DM2.
The structure of ErA-DM2·Asp suggested that glutamine at position 254 may better discriminate against Gln. Indeed, the ErA-E63Q/S254Q (ErA-DM3) variant is endowed with an even higher Gln Km compared with ErA-DM2 (83 versus 15.8 mm; Table 2). Critically, the l-asparaginase activity of ErA-DM3 was not negatively impacted by changing Ser-254 from an asparagine (as in ErA-DM2) to a glutamine (Table 2).
Because we now identified a variant with an extremely high Gln Km value (ErA-DM3), we speculated that the benefit of adding to this variant the A31A mutation would be advantageous by reducing the Asn Km value. Indeed, ErA-A31I/E63Q/S254Q (ErA-TM2) has an Asn Km value that is approximately half that of ErA-DM3 (95 versus 179 μm; Table 2). As expected, the Km value of Gln was also reduced (47.5 versus 84 mm), but despite this reduction remained 100-fold higher than the physiological blood Gln concentration. In sum to the enzyme engineering studies, the ErA triple mutant A31I/E63Q/S254Q (ErA-TM2) possessed the best combination of high l-asparaginase/low l-glutaminase properties.
The Ultra-low l-Glutaminase ErA-TM2 Variant Maintains Its Cell Killing Properties
This work designed and characterized several ErA variants that have high l-asparaginase and low l-glutaminase activity, with two variants (ErA-DM3 and ErA-TM2) having ultra-low l-glutaminase properties. To test whether variants with ultra-low l-glutaminase activity maintain the ability to kill ALL cells, we exposed the human T-cell ALL LOUCY cell line to increasing concentrations of ErA-TM2 and determined the IC50 value of this designed enzyme. We compared the killing power of this enzyme to the Food and Drug Administration-approved ErA-WT. As shown in Fig. 7, ErA-TM2 has a comparable IC50 value to that of ErA-WT.
FIGURE 7.
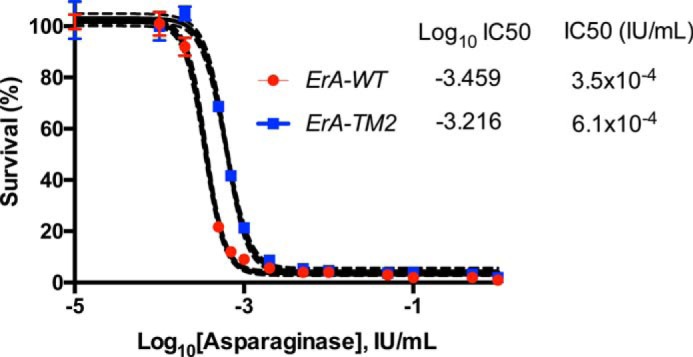
Lack of l-glutaminase activity had a negligible effect on the ability of the l-asparaginase to reduce the proliferation of human T-cell ALL LOUCY cells. Dose response of LOUCY cells to ErA-WT (high l-glutaminase) and ErA-TM2 (ultra-low l-glutaminase) is shown. The marginally higher IC50 value of ErA-TM2 relative to that of ErA-WT can be attributed to the general cell killing l-glutaminase activity of the latter.
Discussion
l-Asparaginases, although important drugs for the treatment of ALL, suffer from serious side effects. Compelling evidence (2, 8, 12, 20, 21) suggests that the thromboembolic coagulopathy (10, 22–25), pancreatitis (26, 27), and liver dysfunction (8, 28) are direct results of the l-glutaminase side activity present in the Food and Drug Administration-approved bacterial l-asparaginases and not from the cancer-killing l-asparaginase activity. Therefore, an enzyme with high l-asparaginase activity (and here a low Asn Km value is critical) and low l-glutaminase activity may be advantageous clinically. Toward this goal, we produced and tested several ErA variants that were designed to have these properties.
The biggest challenge in this enzyme engineering goal was the small structural difference between the substrates; a single methylene group differentiates Asn and Gln. Therefore, it is not surprising that many of the mutants tested had both reduced l-asparaginase and l-glutaminase activities (Table 1). Moreover, as many of the enzyme residues in the immediate surrounding of the substrate are conserved and required for activity, the possible regions for mutagenesis are limited. Despite these challenges, we were successful in identifying two hot spots that can be exploited to increase the selectivity for Asn over Gln. We also identified a hot spot that can be used to reduce the substrate's Km value. This factor is important because to achieve complete Asn depletion of the patient's blood, the enzyme drug must have a low Km for this amino acid.
A key factor for the success of this effort was the realization that the larger Gln substrate must position its side chain (the moiety hydrolyzed in this reaction) in a very similar position to that adopted by Asn, where it is sandwiched by the conserved threonines Thr-15 and Thr-95. This being the case, to accommodate the extra methylene group, Gln is limited to adjusting the position of its main-chain atoms. In the wild-type enzyme, this repositioning brings the Gln α-amino group in close proximity to Glu-63. Replacing Glu-63 with a glutamine introduces a H-bond donor that acts to repel the α-amino group, as it too is a H-bond donor. Similarly, replacing Ser-254′ (a residue originating from a neighboring protomer) by the larger asparagine (as in ErA-DM2) or glutamine (ErA-DM3) introduces an H-bond donor in the proximity of the α-amino group. These substitutions effect Gln to a larger extent compared with Asn, as the smaller size of Asn permits it to shift away from these H-bond donor moieties.
However, Asn is somewhat effected by these mutations, which increase its Km value from 47.5 μm (ErA-WT) to nearly 200 μm in ErA-DM2 and ErA-DM3 (Table 2). Fortuitously, we also identified a mutation that reduced the substrate's Km, the A31I mutation. Adding this mutation to ErA-DM3 reduced the Asn Km to a clinically relevant value of 95 μm.
So how does our best variant (ErA-TM2) compare with the Food and Drug Administration-approved enzymes? Enzymologists often use the kcat/Km ratio as a measure of an enzyme's efficiency. However, this value (presented in Tables 2 and 3 for the l-asparaginase and l-glutaminase reactions, respectively) is not the best indicator for the ability of the enzymes to affect the blood Asn and Gln levels at physiological concentrations of these amino acids. Therefore, we focused on the amino acid hydrolysis activity of the enzymes at the typical concentration of Asn (50 μm) and Gln (500 μm) present in human blood and at a lower one (20 μm for Asn, 100 μm for Gln). In the following analysis of the enzymes, we ignore additional very important properties that affect clinical efficacy, such as in vivo stability, and focus solely on the in vitro kinetic properties of the enzymes. We are aware that a multitude of parameters, with the in vitro rate only one of them, are critical to the success of these enzymes as drugs.
ErA-WT displays the highest l-asparaginase activity, as it is ∼2-fold faster than the other Food and Drug Administration-approved enzyme, EcA-WT (Table 2). That is favorable for its ErA-WT's anti-cancer effect. However, it also displays the highest l-glutaminase activity, ∼70-fold higher than EcA-WT. This would be detrimental to its safety profile. When considering our designed enzymes, ErA-TM2 largely maintained ErA-WT's l-asparaginase activity. Notably, ErA-TM2 had an ∼2-fold higher l-asparaginase activity at physiological Asn concentration relative to EcA-WT. This demonstrated that ErA-TM2 fulfilled the criteria of being a clinically relevant l-asparaginase.
The extraordinary change in the activity profile of ErA-TM2 became apparent when inspecting its l-glutaminase activity. As a derivative of ErA-WT, which is a relatively active l-glutaminase, ErA-TM2 was transformed into an enzyme with ultra-low l-glutaminase activity. Its rate at relevant Gln concentrations was reduced by >1000-fold. In fact, it was an even much worse l-glutaminase (by ∼25-fold) when compared with EcA-WT. Moreover, as patients are treated at doses that supply a set amount of l-asparaginase activity and because ErA-TM2 has nearly twice the specific activity of EcA-WT (Table 2), the l-glutaminase activity of ErA-TM2 at any desired l-asparaginase dose would be lower by an additional factor of two relative to EcA-WT. As a result, the differential in l-glutaminase power of these enzymes is ∼50-fold.
Notably, the ultra-low ErA-TM2 variant shows strong killing of human T-cell ALL LOUCY cells in vitro (Fig. 7). The marginally reduced sensitivity of the LOUCY cells to ErA-TM2 compared with ErA-WT is expected and can be explained by the high l-glutaminase activity of the latter. In other words, an enzyme with l-glutaminase activity would always be toxic to cells grown in culture. Our work demonstrates that this general cell-toxicity property is not required to kill cells that are dependent on exogenous Asn. We next plan to proceed to the evaluation of this variant in vivo, where we will compare not only its anticancer effects but also its toxicity profile. The ultra-low l-glutaminase activity of ErA-TM2 suggests that it would not deplete the blood Gln, and this in turn should mitigate toxicity.
Experimental Procedures
Gene Cloning and Mutagenesis
A codon-optimized synthetic gene corresponding to the amino acid sequence of ErA (UniProt entry P06608) lacking the first 21-amino acid signal peptide was synthesized by Genscript as described earlier (29). The synthetic gene was digested with NdeI and BamHI-HF restriction enzymes, gel-purified, and ligated into a His6-SUMO-pET14b vector (where the His6 tag is followed by the yeast protein SUMO (small ubiquitin modifier, Smt3p) using Instant Sticky End DNA ligase (New England BioLabs), generating the His6-SUMO-pET14b-ErA-WT plasmid. This plasmid was subsequently used as template to create 13 ErA single-mutant variants (A31S, A31I, A31L, A31M, A31N, A31T, A31V, E63L, E63Q, P123S, P123N, S254N, S254P). The plasmid carrying the E63Q mutation was then used as the template to create four ErA double-mutant variants (A31T/E63Q, A31I/E63Q, S254N/E63Q, and S254Q/E63Q), whereas the mutant S254N was used to generate two other double-mutants (A31I/S254N and A31T/S254N). The plasmid carrying the double mutation A31I/E63Q was used as a template to create two triple mutants A31I/E63Q/S254N and A31I/E63Q/S254Q.
Protein Expression and Purification
Protein expression and purification were performed as previously described (29). In brief, plasmids corresponding to wild-type ErA (ErA-WT) or the mutant constructs (verified by sequencing) were transformed into E. coli BL21(DE3) C41 cells for expression. A single colony was picked and grown up overnight at 37 °C in 2×YT medium. Protein expression was induced with 0.3 mm isopropyl β-d-1-thiogalactopyranoside when the culture reached an optical density (at 600 nm) of 0.6–0.8. The incubation temperature was then reduced and maintained at 18 °C for overnight. The cells were disrupted by sonication, and the debris was cleared by centrifugation at 20,000 × g at 4 °C for 30 min. The supernatant was loaded onto a 5-ml HisTrap nickel affinity column (GE Healthcare). The column was subsequently washed with buffers composed of 25 mm Tris-HCl, pH 8.5, 500 mm NaCl, and either 25, 50, or 75 mm imidazole. The bound protein was eluted with the same buffer but containing 500 mm imidazole. The N-terminal His6-SUMO tag was cleaved by His6-tagged SUMO protease, and the protein solution was loaded back onto a nickel affinity column to capture the tag and protease. The flow-through fraction containing the purified enzyme was dialyzed into 25 mm Tris, pH 8.5, 100 mm NaCl, concentrated to 20–80 mg/ml, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C.
Kinetic Assays
l-Asparaginase activity was determined using a continuous spectroscopic enzyme-coupled assay as previously described (29, 30). In brief, the assay measures the production of l-aspartate through the 1:1 oxidation of reduced NADH. The conversion of NADH to NAD+ was measured spectrophotometrically as a decrease in absorbance at 340 nm at 37 °C. All measurements were taken in triplicate. Rates were fit to the Michaelis-Menten equation using SigmaPlot software (Systat Software Inc.).
For the determination of the l-glutaminase activity, we adapted a reported method (31) with some modifications. The purified enzyme was mixed into a solution containing 50 mm Tris-HCl, pH 8.5, 1 mg/ml BSA, 7 units of glutamate dehydrogenase (Sigma G2501), 3 mm iodonitrotetrazolium chloride (Sigma I10406), 100 mm phenazine methosulfate (Sigma P9625), and 100 mm NAD+. l-Glutamine was added in varying concentrations to trigger the first reaction, which produced ammonia and l-glutamic acid. The latter, in the presence of NAD+ and under the catalysis of glutamate dehydrogenase, was broken down further into α-ketoglutarate and ammonia. NADH produced by this second reaction together with iodonitrotetrazolium chloride served as substrates for the third reaction, where the formation of iodonitrotetrazolium chloride-formazan under the catalysis of phenazine methosulfate can be followed spectrophotometrically as an increase in absorbance at 500 nm. All reactions were carried out at 37 °C.
Crystallization, X-ray Data Collection, and Refinement
Crystals of the ErA mutants were grown at 285 K using the hanging-drop vapor-diffusion method. 2 μl of enzyme at 2.5–10 mg/ml was mixed with 1–4 μl of reservoir buffer solution. The reservoir solution consisted of 0.1 m HEPES (Sigma), pH 7.5, and 24% of PEG methyl ether Mr 2000 (Fluka).
Before data collection, crystals were soaked for 5 min with 10 mm l-aspartic acid (Sigma A6683) or 5 mm l-glutamic acid (Sigma 128420) in 0.1 m HEPES, pH 7.5 and 24% of PEG methyl ether Mr 2000. Soaked crystals were then transferred to the same solutions, respectively, but supplemented with 25% of glycerol for cryoprotection.
Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) beamline 21-ID-F located at Argonne National Laboratory. Data were processed with the XDS package (32). Structures were determined by molecular replacement with MOLREP (33) using the atomic resolution structure (PDB entry 1O7J) as a search model. Refinement was conducted using REFMAC 5 (34), and model building was conducted using Coot (35). Data collection and refinement statistics are listed in Table 4. Structural figures were prepared using the PyMOL Molecular Graphics System (Version 1.6.0, Schrödinger).
Cultivation of Cells
The T-cell ALL LOUCY cell line was a generous gift from Dr. Pieter Van Vlierberghe, Ghent University, Belgium. The cell line was verified to be mycoplasma free and confirmed to match 100% to corresponding STR (Short Tandem Repeat) profile data from the Global Bioresource Center ATCC. Cells were cultivated in a humid atmosphere (5% CO2, 37 °C) using RPMI 1640 media supplemented with 10% FBS (Hyclone) and 1× penicillin-streptomycin solution (Invitrogen). l-Glutamine was added directly into cell cultures to a final concentration of 2 mm.
Cell Viability-Proliferation Assay
90-μl aliquots of cell suspension (2.5 × 105 cells per ml) were cultured in triplicate in round-bottomed 96-well microtiter plate in the presence of 10 μl of either Dulbecco's phosphate-buffered saline (Mediatech) or l-asparaginases to a final concentration ranging from 0.0001 to 1 IU/ml. After incubating the plates for 4 days at 37 °C in humidified air containing 5% CO2, Alamar Blue (Invitrogen) was added to a final concentration of 10% v/v, and the plates were incubated for an additional 4 h followed by reading of the fluorescence signal. The leukemic cell viability was calculated as the percentage of fluorescence counts in the presence of l-asparaginase versus that in the Dulbecco's phosphate-buffered saline control.
Author Contributions
H. A. N. and A. L. designed the ErA mutants. H. A. N. expressed, purified, and kinetically analyzed the mutants. H. A. N. crystallized the three variants described. H. A. N. and A. L. collected the diffraction data and solved the structure. Y. S. expressed several of the mutants and provided technical support. H. A. N. and A. L. wrote the manuscript.
Supplementary Material
Acknowledgments
We appreciate the technical support of the staff at LS-CAT during data collection. We also thank Dr. Schalk for review of the manuscript. This research used the resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817).
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 EB013685. This work was also supported by Merit Review Award I01BX001919 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service. A. L. and H. A. N. are listed as inventors on a patent application for the presented enzyme mutants. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 5I3Z, 5I48, and 5I4B) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Asn
- l-asparagine
- ALL
- acute lymphoblastic leukemia
- DM
- double mutant
- EcA
- E. coli L-asparaginase
- ErA
- E. chrysanthemi l-asparaginase
- ErA-WT
- E. chrysanthemi l-asparaginase wild type
- ErA-E63Q
- E. chrysanthemi l-asparaginase E63Q mutant
- ErA-DM1
- E. chrysanthemi l-asparaginase A31I/E63Q double mutant
- ErA-DM2
- E. chrysanthemi l-asparaginase E63Q/S254N double mutant
- ErA-DM3
- E. chrysanthemi l-asparaginase E63Q/S254Q double mutant
- ErA-TM1
- E. chrysanthemi l-asparaginase A31I/E63Q/S254N triple mutant
- ErA-TM2
- E. chrysanthemi l-asparaginase A31I/E63Q/S254Q triple mutant
- ErA-WT·Asp
- ErA-WT in complex with Asp
- ErA-WT·Glu
- ErA-WT in complex with Glu
- ErA-E63Q·Asp
- ErA-E63Q in complex with Asp
- ErA-DM1·Asp
- ErA-DM1 in complex with Asp
- ErA-DM2·Asp
- ErA-DM2 in complex with Asp
- Gln
- l-glutamine.
References
- 1. Horowitz B., Madras B. K., Meister A., Old L. J., Boyes E. A., and Stockert E. (1968) Asparagine synthetase activity of mouse leukemias. Science 160, 533–535 [DOI] [PubMed] [Google Scholar]
- 2. Ollenschläger G., Roth E., Linkesch W., Jansen S., Simmel A., and Mödder B. (1988) Asparaginase-induced derangements of glutamine metabolism: the pathogenetic basis for some drug-related side-effects. Eur. J. Clin. Invest. 18, 512–516 [DOI] [PubMed] [Google Scholar]
- 3. Narta U. K., Kanwar S. S., and Azmi W. (2007) Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. Hematol. 61, 208–221 [DOI] [PubMed] [Google Scholar]
- 4. Rizzari C., Conter V., Stary J., Colombini A., Moericke A., and Schrappe M. (2013) Optimizing asparaginase therapy for acute lymphoblastic leukemia. Curr. Opin. Oncol. 25, S1–S9 [DOI] [PubMed] [Google Scholar]
- 5. Salzer W. L., Asselin B. L., Plourde P. V., Corn T., and Hunger S. P. (2014) Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann. N.Y. Acad. Sci. 1329, 81–92 [DOI] [PubMed] [Google Scholar]
- 6. Berenbaum M. C. (1970) Immunosuppression by l-asparaginase. Nature 225, 550–552 [DOI] [PubMed] [Google Scholar]
- 7. Kafkewitz D., and Bendich A. (1983) Enzyme-induced asparagine and glutamine depletion and immune system function. Am. J. Clin. Nutr. 37, 1025–1030 [DOI] [PubMed] [Google Scholar]
- 8. Durden D. L., Salazar A. M., and Distasio J. A. (1983) Kinetic analysis of hepatotoxicity associated with antineoplastic asparaginases. Cancer Res. 43, 1602–1605 [PubMed] [Google Scholar]
- 9. Aldoss I., Douer D., Behrendt C. E., Chaudhary P., Mohrbacher A., Vrona J., and Pullarkat V. (2016) Toxicity profile of repeated doses of PEG-asparaginase incorporated into a pediatric-type regimen for adult acute lymphoblastic leukemia. Eur. J. Haematol. 96, 375–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Truelove E., Fielding A. K., and Hunt B. J. (2013) The coagulopathy and thrombotic risk associated with l-asparaginase treatment in adults with acute lymphoblastic leukaemia. Leukemia 27, 553–559 [DOI] [PubMed] [Google Scholar]
- 11. Durden D. L., and Distasio J. A. (1981) Characterization of the effects of asparaginase from Escherichia coli and a glutaminase-free asparaginase from Vibrio succinogenes on specific ell-mediated cytotoxicity. Int. J. Cancer 27, 59–65 [DOI] [PubMed] [Google Scholar]
- 12. Distasio J. A., Salazar A. M., Nadji M., and Durden D. L. (1982) Glutaminase-free asparaginase from Vibrio succinogenes: an antilymphoma enzyme lacking hepatotoxicity. Int. J. Cancer 30, 343–347 [DOI] [PubMed] [Google Scholar]
- 13. Derst C., Henseling J., and Röhm K. H. (2000) Engineering the substrate specificity of Escherichia coli asparaginase. II. Selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci. 9, 2009–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen H. A., Su Y., and Lavie A. (2016) Structural Insight into Substrate Selectivity of Erwinia chrysanthemi l-asparaginase. Biochemistry 55, 1246–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parmentier J. H., Maggi M., Tarasco E., Scotti C., Avramis V. I., and Mittelman S. D. (2015) Glutaminase activity determines cytotoxicity of l-asparaginases on most leukemia cell lines. Leuk. Res. 39, 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cappelletti D., Chiarelli L. R., Pasquetto M. V., Stivala S., Valentini G., and Scotti C. (2008) Helicobacter pylori l-asparaginase: a promising chemotherapeutic agent. Biochem. Biophys. Res. Commun. 377, 1222–1226 [DOI] [PubMed] [Google Scholar]
- 17. Chan W. K., Lorenzi P. L., Anishkin A., Purwaha P., Rogers D. M., Sukharev S., Rempe S. B., and Weinstein J. N. (2014) The glutaminase activity of l-asparaginase is not required for anticancer activity against AsnS-negative cells. Blood 123, 3596–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Distasio J. A., Niederman R. A., and Kafkewitz D. (1977) Antilymphoma activity of a glutaminase-free l-asparaginase of microbial origin. Proc. Soc. Exp. Biol. Med. 155, 528–531 [DOI] [PubMed] [Google Scholar]
- 19. Schalk A. M., Antansijevic A., Caffrey M., and Lavie A. (2016) Experimental data in support of a direct displacement mechanism for type I/II l-asparaginases. J. Biol. Chem. 291, 5088–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reinert R. B., Oberle L. M., Wek S. A., Bunpo P., Wang X. P., Mileva I., Goodwin L. O., Aldrich C. J., Durden D. L., McNurlan M. A., Wek R. C., and Anthony T. G. (2006) Role of glutamine depletion in directing tissue-specific nutrient stress responses to l-asparaginase. J. Biol. Chem. 281, 31222–31233 [DOI] [PubMed] [Google Scholar]
- 21. Ramya L. N., Doble M., Rekha V. P., and Pulicherla K. K. (2012) l-Asparaginase as potent anti-leukemic agent and its significance of having reduced glutaminase side activity for better treatment of acute lymphoblastic leukaemia. Appl. Biochem. Biotechnol. 167, 2144–2159 [DOI] [PubMed] [Google Scholar]
- 22. Bushman J. E., Palmieri D., Whinna H. C., and Church F. C. (2000) Insight into the mechanism of asparaginase-induced depletion of antithrombin III in treatment of childhood acute lymphoblastic leukemia. Leuk. Res. 24, 559–565 [DOI] [PubMed] [Google Scholar]
- 23. Hernández-Espinosa D., Miñano A., Martínez C., Pérez-Ceballos E., Heras I., Fuster J. L., Vicente V., and Corral J. (2006) l-Asparaginase-induced antithrombin type I deficiency: implications for conformational diseases. Am. J. Pathol. 169, 142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Payne J. H., and Vora A. J. (2007) Thrombosis and acute lymphoblastic leukaemia. Br. J. Haematol. 138, 430–445 [DOI] [PubMed] [Google Scholar]
- 25. Alsaid Y., Gulab S., Bayoumi M., and Baeesa S. (2013) Cerebral sinus venous thrombosis due to asparaginase therapy. Case Rep. Hematol. 2013, 841057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alvarez O. A., and Zimmerman G. (2000) Pegaspargase-induced pancreatitis. Med. Pediatr. Oncol. 34, 200–205 [DOI] [PubMed] [Google Scholar]
- 27. Raja R. A., Schmiegelow K., Albertsen B. K., Prunsild K., Zeller B., Vaitkeviciene G., Abrahamsson J., Heyman M., Taskinen M., Harila-Saari A., Kanerva J., Frandsen T. L., and Nordic Society of Paediatric Haematology and Oncology (NOPHO) group)(2014) Asparaginase-associated pancreatitis in children with acute lymphoblastic leukaemia in the NOPHO ALL2008 protocol. Br. J. Haematol. 165, 126–133 [DOI] [PubMed] [Google Scholar]
- 28. Bhojwani D., Darbandi R., Pei D., Ramsey L. B., Chemaitilly W., Sandlund J. T., Cheng C., Pui C. H., Relling M. V., Jeha S., and Metzger M. L. (2014) Severe hypertriglyceridaemia during therapy for childhood acute lymphoblastic leukaemia. Eur. J. Cancer 50, 2685–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schalk A. M., Nguyen H. A., Rigouin C., and Lavie A. (2014) Identification and structural analysis of an l-asparaginase enzyme from guinea pig with putative tumor cell killing properties. J. Biol. Chem. 289, 33175–33186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fernandez C. A., Cai X., Elozory A., Liu C., Panetta J. C., Jeha S., Molinelli A. R., and Relling M. V. (2013) High-throughput asparaginase activity assay in serum of children with leukemia. Int. J. Clin. Exp. Med. 6, 478–487 [PMC free article] [PubMed] [Google Scholar]
- 31. Gella F. J., and Pascual M. A. (1982) Assay of glutaminase activity by colorimetric determination of glutamate. Anal. Biochem. 127, 322–325 [DOI] [PubMed] [Google Scholar]
- 32. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 34. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 35. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.