

Abstract
Bacterial chemosensory signal transduction systems that regulate motility by type IV pili (T4P) can be markedly more complex than related flagellum-based chemotaxis systems. In T4P-based systems, the CheA kinase often contains numerous potential sites of phosphorylation, but the signaling mechanisms of these systems are unknown. In Pseudomonas aeruginosa, the Pil-Chp system regulates T4P-mediated twitching motility and cAMP levels, both of which play roles in pathogenesis. The Pil-Chp histidine kinase (ChpA) has eight “Xpt” domains; six are canonical histidine-containing phosphotransfer (Hpt) domains and two have a threonine (Tpt) or serine (Spt) in place of the histidine. Additionally, there are two stand-alone receiver domains (PilG and PilH) and a ChpA C-terminal receiver domain (ChpArec). Here, we demonstrate that the ChpA Xpts are functionally divided into three categories as follows: (i) those phosphorylated with ATP (Hpt4–6); (ii) those reversibly phosphorylated by ChpArec (Hpt2–6), and (iii) those with no detectable phosphorylation (Hpt1, Spt, and Tpt). There was rapid phosphotransfer from Hpt2–6 to ChpArec and from Hpt3 to PilH, whereas transfer to PilG was slower. ChpArec also had a rapid rate of autodephosphorylation. The biochemical results together with in vivo cAMP and twitching phenotypes of key ChpA phosphorylation site point mutants supported a scheme whereby ChpArec functions both as a phosphate sink and a phosphotransfer element linking Hpt4–6 to Hpt2–3. Hpt2 and Hpt3 are likely the dominant sources of phosphoryl groups for PilG and PilH, respectively. The data are synthesized in a signaling circuit that contains fundamental features of two-component phosphorelays.
Keywords: bacterial signal transduction, chemotaxis, histidine kinase, phosphoryl transfer, Pseudomonas aeruginosa (P. aeruginosa), type IV pili
Introduction
Two-component systems (TCSs)2 are a prominent method of signal transduction in bacteria and regulate diverse processes, including many that impact human health (1, 2). The canonical TCS features His → Asp phosphotransfer between a signal-sensing histidine kinase and the receiver domain of a partner response regulator, whereby the phosphorylated response regulator executes an output response. Although this canonical scheme is common, the basic TCS has evolved a variety of more complicated circuitries (3). Chemosensory-like TCSs represent a specialized subset of TCSs that often regulate cell motility (4, 5). The chemotaxis system that mediates flagellum-based swimming behavior has been extensively characterized in the Escherichia coli/Salmonella model system (6) as well as other organisms (7, 8). In the E. coli/Salmonella paradigm, the chemoreceptor-associated histidine kinase CheA transfers a phosphoryl group to the CheY response regulator (a stand-alone receiver domain), which then interacts with the flagellar motor to control swimming. Another subtype of the chemosensory system regulates bacterial cell motility via type IV pili (T4P) (5, 9). T4P are filamentous structures on the surface of many Gram-negative bacteria that function in cell adherence, biofilm formation, and DNA uptake (10, 11). T4P mediate twitching and gliding motility, types of surface movement exhibited by a variety of bacteria (12, 13). In contrast to the detailed characterization of flagellar chemotaxis, less is known about the molecular mechanisms of chemosensory systems that regulate motility by T4P.
Pseudomonas aeruginosa is an opportunistic pathogen that is pervasive in the environment and a cause of life-threatening infections for cystic fibrosis patients and other immunocompromised individuals (14). P. aeruginosa utilizes an arsenal of virulence factors for infection (15, 16), including polarly localized T4P, which function in adherence and invasion of host tissues (17). P. aeruginosa exhibits T4P-mediated twitching motility and has been used as a model system to better understand this behavior (18). Twitching motility in P. aeruginosa is regulated by the Pil-Chp chemosensory system (9) and involves repeated cycles of extrusion, surface adhesion, and retraction of T4P (10, 19, 20). In addition to regulating twitching motility, the P. aeruginosa Pil-Chp chemosensory system also regulates intracellular levels of cAMP (21), a second messenger that activates Vfr, a master transcriptional regulator of genes involved in P. aeruginosa pathogenesis (22). The relationship between Pil-Chp regulation of cAMP levels and twitching motility has yet to be fully elucidated (21, 23); however, it has been recently proposed that Pil-Chp regulation of twitching motility and cAMP synthesis is linked to mechanical tension resulting from surface attachment of T4P (24).
Proteins comprising the P. aeruginosa Pil-Chp chemosensory system are encoded by the pil-chp operon. The operon encodes homologs for all of the core flagellar chemotaxis proteins (25) but with some notable differences. As in flagellar systems, there is a transmembrane chemoreceptor (PilJ), a CheA histidine kinase (ChpA), and adaptation proteins (ChpB and PilK). The operon contains two genes encoding CheY-like response regulators (PilG and PilH) and two genes encoding CheW-like scaffolding proteins (PilI and ChpC), both present in one copy in the E. coli chemotaxis paradigm.
A striking difference in the proteins encoded by pil-chp compared with flagellar chemotaxis proteins is the domain architecture of ChpA, the histidine kinase CheA homolog. Flagellar CheAs typically contain five domains (Fig. 1A) as follows: a histidine containing phosphotransfer domain (Hpt; P1); a CheY-binding domain (P2); a dimerization domain (P3); a catalytic HATPase_c (P4) domain; and a CheW-like scaffolding domain (P5). ATP bound to the HATPase_c domain reacts with a conserved His on the Hpt domain. The phosphoryl group is subsequently transferred to a conserved aspartyl residue on CheY (a stand-alone receiver domain). In contrast, P. aeruginosa ChpA is a large protein composed of 2478 amino acids (cf. 654 in E. coli CheA) (Fig. 1A). Like CheA, ChpA contains Hpt, dimerization, HATPase_c, and CheW-like domains. However, ChpA has a string of seven additional “Xpt” domains (9), some separated from each other by long stretches of residues, together comprising the N-terminal ∼1800 amino acids of the protein. Five of the additional Xpt domains are Hpts, predicted to fold into monomeric helical bundles that contain a phosphorylatable His at the predicted position. In addition, there is an Spt domain (predicted to have an Hpt fold but contains a Ser instead of a His) and a Tpt domain, with a Thr in lieu of the His. ChpA also contains a C-terminal receiver domain (ChpArec), which is also present in some flagellar CheAs (26) but is absent in E. coli CheA.
FIGURE 1.

P. aeruginosa ChpA domain architecture and protein constructs used in these studies. Predicted domains are represented by rectangles with Xpt domains (solid gray), combined dimerization/HATPase_c/CheW domains (vertical stripe), and receiver domains (diagonal stripe). A, domain architecture of full-length ChpA with conserved His (H), Thr (T), Ser (S), and Asp (D) labeled. The domains present in E. coli CheA are indicated; in addition, CheA contains a CheY-binding (P2) domain absent from ChpA. B, protein construct designations and corresponding domain architectures used in this study. The presence of conserved His, Asp, Thr, or Ser or its alanine (A) substitution is indicated.
Phosphotransfer involving the ChpA, PilG, and PilH proteins is essential for functioning of the Chp chemosensory system. Single substitutions at predicted phosphorylation sites gave a variety of twitching phenotypes in P. aeruginosa PAO1 (27), and the ChpA catalytic domain and phosphorylation sites of PilG and PilH are required for T4P function (28). pilG deletion mutants phenocopy chpA deletions in multiple assays (21, 28), suggesting that PilG functions as an output response regulator. In contrast, pilG and pilH deletion mutants give opposite phenotypes for surface pili abundance (21, 28) and intracellular cAMP levels (21). This could reflect that PilH acts as a phosphate sink, acting to keep phosphoryl groups off of PilG (21). Alternatively, PilH could target protein(s) that have opposite function to PilG targets, e.g. it has been proposed that PilG mediates pilus extrusion and PilH mediates retraction (28). The greatly enhanced complexity of the ChpA domain architecture (with nine possible phosphorylation sites) relative to flagellar CheAs (one or two phosphorylation sites) as well as the presence of two CheY-like stand-alone receiver domains precludes the prediction of phosphoryl group flow and hence the signaling strategy of the Chp chemosensory system.
Here, we investigated the phosphotransfer reactions of the P. aeruginosa Pil-Chp system as an essential step in understanding how this system regulates twitching motility and cAMP levels. The Pil-Chp system also serves as a representative of an entire clade of T4P-based chemosensory systems with similar protein compositions and domain architectures (5). We demonstrated that of the eight Xpt domains, three (Hpt4–6) could be phosphorylated with ATP in the presence of the ChpA HATPase_c catalytic domain. These same Hpts plus two others, Hpt2 and Hpt3, could become reversibly phosphorylated by ChpArec. The remaining Xpts (Hpt1, Spt, and Tpt) displayed no detectable phosphotransfer reactivity. Phosphoryl groups transferred rapidly from all five of the phosphorylated Hpts to ChpArec and from Hpt3 to PilH, but phosphorylation of PilG was slower. Taken together, the in vitro studies demonstrated that ChpArec could act as a phosphate sink or a source of phosphoryl groups for Hpts 2 and 3. In vivo analysis of bacteria containing substitutions at the phosphorylation sites of ChpArec, Hpt2, and/or Hpt3 indicated that ChpArec operates upstream of Hpt2 and Hpt3. Furthermore, the genetic analysis suggested a model where Hpt2 and Hpt3 act as the source of phosphoryl groups for PilG and PilH, respectively. The in vitro and in vivo observations are synthesized in a signaling scheme with features of two-component phosphorelays, a subtype of TCS with multiple reversible His↔Asp phosphotransfer events (29, 30).
Results
ChpA Uses ATP to Autophosphorylate Hpt4–6
ChpA in P. aeruginosa strain PAK is a 2478-residue protein with nine potential phosphorylation sites (Fig. 1A). To characterize the phosphotransfer biochemistry of this complex system, we utilized a modular approach via a series of isolated constructs (Fig. 1B). In addition to the single Hpt domain present in E. coli CheA (designated Hpt6 in P. aeruginosa ChpA) (Fig. 1A), ChpA has five additional Hpt domains (Hpt1–5), an Spt domain (a conserved Ser in place of the His), and a Tpt domain (a conserved Thr in place of the His). In CheA, the γ-phosphoryl group of an ATP molecule bound to the HATPase_c domain reacts with the conserved His on the Hpt domain. To investigate whether ChpA could catalyze the reaction of more than one Hpt domain with ATP, we compared the abilities of various ChpA constructs containing Hpt6 and/or Hpt5 to incorporate the 32P-phosphoryl groups from [γ-32P]ATP. In some constructs, one or both His phosphorylation sites were replaced by a non-phosphorylatable Ala or a domain was deleted. Each construct was designated “ChpA” followed by three letters: the 1st and 2nd letters indicate the status of Hpt5 and Hpt6, respectively (− for deleted, H for intact His, and A for Ala substitution); the 3rd letter indicates the status of ChpArec (− for deleted, D for intact Asp, and A for Ala substitution). Under buffer conditions optimized for E. coli CheA, ChpA HH−, −H−, HA−, and AH− incorporated 32P in a time-dependent manner until a steady state was achieved (Fig. 2). ChpA AA− incorporated no detectable label, consistent with the predicted His residues being the sites of phosphorylation. Incorporation of 32P into both ChpA HA− and AH− demonstrates that both Hpt5 and Hpt6 react with ATP. The level of 32P incorporated at steady state was similar for the three constructs that had one phosphorylatable His and about twice that for Chp HH−, consistent with the phosphorylation reactions not interfering with each other and that both Hpt5 and Hpt6 can get phosphorylated on the same molecule. The enhanced kinetics for the establishment of steady state for ChpA −H− over the other constructs suggests that the physical presence of Hpt5 retarded the reaction of Hpt6. The modest kinetic enhancement of ChpA HA− over AH− could be due to differences in accessibility and/or inherent reactivities between Hpt5 and Hpt6.
FIGURE 2.

Time courses for autophosphorylation of ChpA constructs that do not contain the C-terminal receiver domain (ChpArec). The ChpA construct (1.4 μm) was incubated with [γ-32P]ATP (150 μm) in 50 mm Tris, pH 7.5, 50 mm KCl, 5 mm MgCl2, 2 mm DTT at room temperature. Aliquots were removed at designated times, and analyzed by SDS-gel electrophoresis followed by phosphorimaging analysis. Error bars reflect standard deviations of 2–4 replicates. ChpA HH− (closed diamonds); ChpA −H− (open squares); ChpA HA− (open triangles); ChpA AH− (closed triangles), and ChpA AA− (closed circles).
After establishing that at least two Xpts (Hpt5 and Hpt6) can react with ATP in the presence of the ChpA catalytic domain, we investigated whether any of the other Xpts could react with ATP. We assessed the abilities of eight isolated Xpt domains (Hpt1–6, Spt, and Tpt) to react in trans with a ChpA construct (ChpA AA−) that had an active HATPase_c domain but no phosphorylatable Hpts in cis. Precedence for reactivity of the CheA Hpt domain with the HATPase_c domain in trans has been established for both E. coli (31, 32) and Thermotoga maritima (33) CheA. The extent of incorporation of 32P into each of the eight Xpts upon incubation with ChpA AA− and 150 μm [γ-32P]ATP is shown in Fig. 3A. Hpt5 and 6 both reacted with ATP, just as they did in cis (Fig. 2), providing a positive control for the assay. Only one other Xpt (Hpt4) was phosphorylated under these conditions. There was no detectable phosphorylation of Hpt1, Hpt2, Hpt3, Spt, or Tpt under the reaction conditions used, although a duplicate Coomassie-stained gel confirmed the presence of the proteins in the reaction mix (Fig. 3B). The sensitivity of the assay is estimated to be about 10% of the Hpt6 activity. The overall ranking of inherent reactivities with ATP was Hpt4 > Hpt5 > Hpt6.
FIGURE 3.

Reactivities of the ChpA Xpts with ATP in the presence of the HATPase_c catalytic domain. ChpA AA− (1.9 μm) was incubated with each Xpt (Hpt1–Hpt6, Spt, and Tpt present at 40 μm) and [γ-32P]ATP (150 μm) for 60 min at room temperature. Reaction samples were divided and then electrophoresed on two gels for analysis by phosphorimaging (A) and Coomassie stain (B).
Phosphoryl Groups Can Transfer from Hpt5 and 6 to All Three Receivers, but ChpArec Is Kinetically Preferred over PilG and PilH
In flagellar chemotaxis, the single phosphorylated Hpt on CheA (Fig. 1A) transfers the phosphoryl group rapidly to the receiver domains of the two response regulators, CheY and CheB. There are three receiver domains encoded in the P. aeruginosa pil-chp operon as follows: (i) ChpArec, which comprises the C-terminal domain of the ChpA protein, and two stand-alone receivers (ii) PilG and (iii) PilH. ChpB, the CheB analog, has a pseudoreceiver domain (missing one of the two conserved metal-binding Asp residues and the conserved active site Ser/Thr) and thus is not predicted to be regulated by phosphorylation. To probe the relative propensities of Hpt5 and Hpt6 to transfer to the three receivers, we used the ChpA constructs containing both a HATPase_c domain and wild type versions of either Hpt5 or Hpt6 (Fig. 1B). ChpA HA− and AH− were reacted with [γ-32P]ATP in the presence of each receiver (ChpArec, PilG, or PilH) in trans (Fig. 4A). In the absence of added receiver (Fig. 4A, lanes 1 and 6), the ChpA phosphorylation intensity reflects ChpA autophosphorylation. In the presence of receiver, the ChpA signal reflects the difference between rates of ChpA autophosphorylation and phosphotransfer to the receiver. Similarly, the radioactive signal on the receiver reflects the difference between rates of phosphotransfer from ChpA and the self-catalyzed autodephosphorylation (34). For both phospho-Hpt6 (ChpA AH−) and phospho-Hpt5 (ChpA HA−), low and even substoichiometric concentrations of ChpArec were sufficient to deplete the ChpA phosphorylation signal (Fig. 4A, lanes 2, 3, 7, and 8), indicating rapid phosphotransfer from the Hpt to ChpArec. In contrast, ∼20-fold higher concentrations of PilG (Fig. 4A, lanes 4 and 9) or PilH (lanes 5 and 10) were not sufficient to deplete phosphorylated ChpA, indicating slower transfer to PilG and PilH. There were clear bands representing both phosphorylated PilG (Fig. 4A, lanes 4 and 9) and phosphorylated PilH (lanes 5 and 10) confirming that phosphotransfer occurred, albeit at a substantially slower rate than to ChpArec. Taking into account the amounts of phospho-ChpA and relative concentrations of ChpArec, PilH, and PilG, we estimate the inherent phosphotransfer reactivities for both Hpt5 and Hpt6 as ChpArec ≫ PilH > PilG. In an experiment identical to that shown in Fig. 4A, ChpA AH− and ChpA HA− were found to transfer phosphoryl groups to the E. coli CheY response regulator at a rate comparable with that of PilH (data not shown), suggesting that the rates for transfer to PilG and PilH are not what is expected for a cognate Hpt/response regulator pair.
FIGURE 4.

Reactivities of ChpArec, PilG, and PilH: phosphotransfer from Hpt5 and Hpt6 and autodephosphorylation rates. A, gel phosphorimage of reactions containing 1.4 μm ChpA AH− (left set) or ChpA HA− (right set) with 30 μm [γ-32P]ATP and either no receiver (−) or ChpArec (rec), PilG (G) or PilH (H), present in the indicated molar ratio relative to the ChpA construct. Reactions proceeded for 14 min before gel separation and phosphorimaging. B, representative time courses for single turnover phosphotransfer from purified 32P-ChpA −H− to ChpArec (open circles), PilH (closed triangles), and PilG (closed diamonds). The intensities for the phosphorylated receivers were not plotted for clarity. C, representative time courses for autophosphorylation of ChpA HHA (closed diamonds), ChpA −HA (closed squares), ChpA −HD (open squares), and ChpA HHD (open diamonds). The reaction conditions were the same as for Fig. 2 except [γ-32P]ATP was present at 30 μm. D, semi-log plot of representative time courses for autodephosphorylation rates for ChpArec (closed circles), PilH (closed triangles), and PilG (closed diamonds). E, scheme summarizing flow of phosphoryl groups based on data so far. The length of the arrow reflects the magnitude of the inherent reactivity.
Time courses that directly monitor phosphotransfer from ChpA −H− (contains only Hpt6) to the three receiver domains (ChpArec, PilG, and PilH) were also measured. ChpA −H− was reacted with [γ-32P]ATP, followed by removal of the unreacted ATP by size exclusion chromatography. The purified [32P]Chp −H− was then mixed with excess ChpArec, PilG, or PilH. The results following the loss of radioactivity from ChpA −H− over time (Fig. 4B) show that phosphotransfer from 1.4 μm 32P-labeled Chp −H− to 16 μm ChpArec was completed by the first 15-s time point, indicating a rate as rapid as typically observed with cognate histidine kinase/response regulator pairs (35). In contrast, transfer to 37 μm PilH required more than 80 s, and transfer to 37 μm PilG was still incomplete after 5 min. Thus, the results from the time courses are consistent with the steady state experiment (Fig. 4A) and indicate that the relative rates of phosphotransfer from Hpt6 to the three receivers as ChpArec ≫ PilH > PilG.
Phosphotransfer from Hpt5/6 → ChpArec → H2O Occurs Readily
Having established that Hpt5/6 → ChpArec is a facile route for phosphotransfer, we next investigated the subsequent fate of the phosphoryl group. Autophosphorylation time courses for the ChpA constructs that contain ChpArec in cis showed less phosphoryl groups incorporated at steady state relative to the corresponding ChpA construct in which the conserved Asp on ChpArec was replaced with a non-phosphorylatable Ala (Fig. 4C). This difference is consistent with phosphotransfer from Hpt5/6 to ChpArec followed by ChpArec autodephosphorylation (transfer of the phosphoryl group to water), a process theoretically requiring a fast rate of ChpArec autodephosphorylation. Subsequent measurement of the rate of ChpArec autodephosphorylation (τ½ = 80 ± 10 s; Fig. 4D) indicated that it would indeed be rapid enough to account for the decreased accumulation of 32P-phosphoryl groups seen in ChpA constructs with wild type ChpArec (Fig. 4C). The ChpArec autodephosphorylation half-life was significantly shorter than that of PilH (11 ± 0.33 min) and PilG (24 ± 4.4 min) (Fig. 4D). A scheme summarizing the phosphotransfer results so far is shown in Fig. 4E.
Reversible Phosphotransfer between ChpArec and Hpt2/Hpt3
Phosphorelays are a subset of two component systems that transfer phosphoryl groups according to Scheme 1,
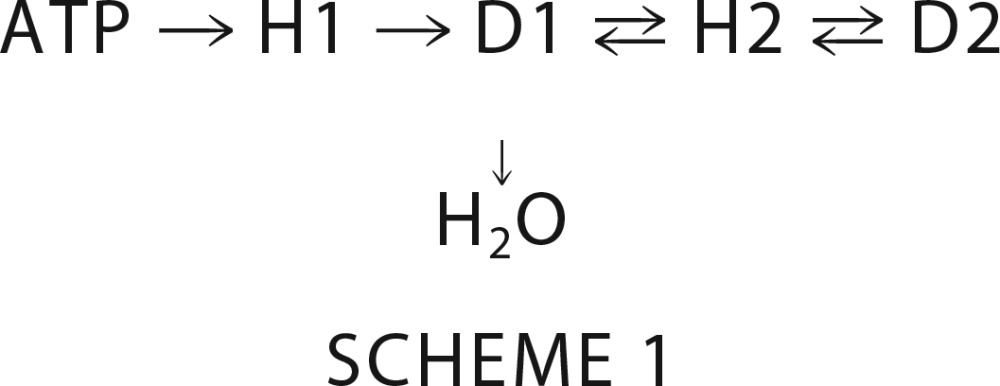 |
where H1 and H2 represent the phosphorylatable His residues of a histidine kinase DHp (dimerization and histidine phosphotransfer) and an Hpt domain, respectively, and D1 and D2 are the conserved Asp residues on two different receiver domains (3). The Hpt domain can either be a monomeric (36) or dimeric (37) 4-helix bundle. Thus, Asp to His phosphotransfer is essential for operation of phosphorelays. To determine whether any of the ChpA Xpts that did not detectably react with ATP could receive a phosphoryl group from ChpArec, a preliminary experiment entailed reaction of ChpA −HD and [γ-32P]ATP in the presence of excess Xpt for 90 min. Gel phosphorimage analysis showed no detectable label on Hpt1, Spt, and Tpt, but both Hpt2 and Hpt3 acquired label, although the great majority of the label was on the ChpA −HD (data not shown). A full time course for the reaction of ChpA −HD with Hpt2 and 30 μm ATP was bell-shaped for incorporation of 32P onto Hpt2 and ChpA −HD (Fig. 5A). In contrast, if ChpA −HA (lacking the ChpArec phosphorylation site) was used under identical reaction conditions, there was no detectable radioactivity on Hpt2, and ChpA −HA did not display a bell-shaped curve but accumulated radioactivity gradually until reaching a steady state (Fig. 5A, inset). Therefore, the data are consistent with phosphoryl group flow from ATP → Hpt6 → ChpArec → Hpt2 → ChpArec → H2O. A similar bell-shaped time course was obtained for Hpt3, except there was less label incorporated into Hpt3 relative to ChpA −HD at the peak (data not shown). Thus it appears that ChpArec transfers phosphoryl groups to both Hpt2 and Hpt3 and that the transfer is readily reversible.
FIGURE 5.
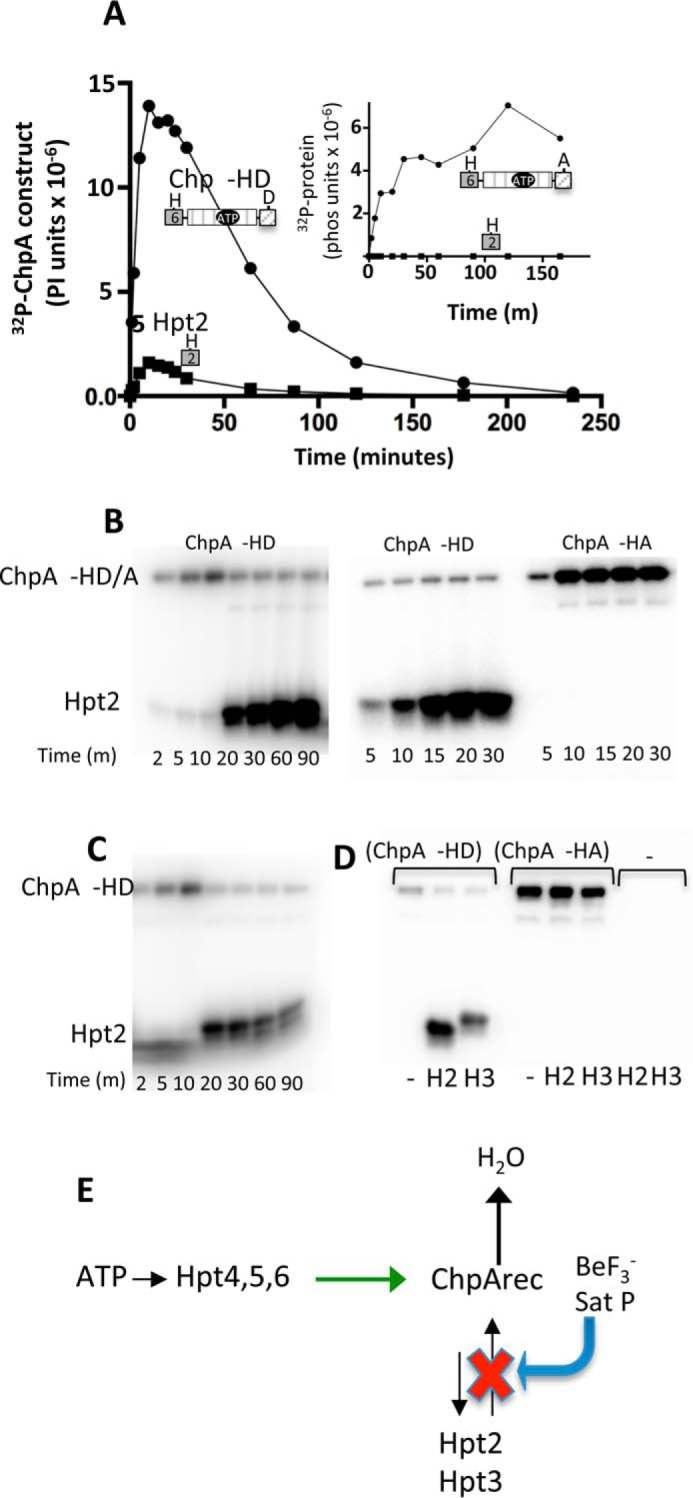
Reversible phosphotransfer between ChpArec and Hpt2/3. A, time courses for incorporation of 32P into ChpA −HD (closed circles) and His6-Hpt 2 (closed squares) in a reaction that contained 7.3 μm ChpA −HD and 34 μm Hpt2. The reaction was initiated by the addition of 30 μm [γ-32P]ATP. Aliquots were removed at the indicated times and analyzed by gel electrophoresis and phosphorimaging analysis. The inset shows results from the identical experiment except ChpA −HA was used instead of ChpA −HD. B, effect of excess BeF3− on the distribution of 32P-phosphoryl groups between ChpA −HD and Hpt2. In the left panel, the reactions were initiated as for A, and then 70 μm BeF3− (70 μm BeCl2 and 10 mm NaF) was added immediately after taking the 10-min time point. In the middle and right panels, 70 μm BeF3− was mixed with 34 μm untagged Hpt2 and 7.3 μm ChpA −HD (middle panel) or ChpA −HA (right panel). 30 μm [γ-32P]ATP was added to initiate the reactions, and the time points were removed at the indicated times. C, effect of addition of a swamp of unlabeled ATP on the distribution of 32P-phosphoryl groups between ChpA −HD and Hpt2. The reaction was carried out as in A except that 2 mm unlabeled ATP was added immediately after the 10-min time point. D, reactions contained 7.3 μm ChpA −HD, ChpA −HA, or no ChpA construct (−), 56 μm Hpt 2, Hpt3, or no Hpt (−), and 30 μm [γ-32P]ATP. After 10 min, 2 mm ATP was added and the reactions proceeded for 10 min before quenching and analysis. E, scheme summarizing reversible flow of phosphoryl groups between ChpArec and Hpt2,3. Binding BeF3− or saturating the phosphorylation sites of ChpArec inhibits Hpt2 and 3 → ChpArec phosphotransfer (red X) but continues to allow Hpt4–6 → ChpArec (green arrow) and ChpArec → Hpt2 and 3. The net result is dramatic increase in accumulation of phosphoryl groups on Hpt2 and 3.
Saturation of the ChpArec Active Site Dramatically Increased Phosphorylation of Hpt2 and Hpt3
As a further test of the ATP → Hpt6 → ChpArec → Hpt2/3 → ChpArec → H2O model, we predicted that saturating the ChpArec active site after allowing for incorporation of some 32P into Hpt2/3 would impair reverse transfer from Hpt2/3 and thus stabilize the signal on Hpt2/3. To test this prediction, the same reaction conditions were used that produced the bell-shaped curve for phosphorylation of Hpt2/3 (Fig. 5A). The reaction was allowed to proceed for 10 min (the top of the bell curve), at which time, excess BeF3−, a phosphomimic that binds tightly to the active sites of receiver domains, was added. Notably, rather than the expected stabilization of the label on Hpt2, there was a dramatic increase in the label on Hpt2 that continued for over an hour (Fig. 5B, left panel). The accumulation of label on Hpt2 also occurred if BeF3− was added before the [γ-32P]ATP (Fig. 5B, middle panel). However, no label on Hpt2 was detectable with ChpA −HA (Fig. 5B, right panel) indicating that the enhanced Hpt2 phosphorylation was entirely dependent on the presence of the ChpArec site of phosphorylation. Because the amount of label on Hpt2 was much greater than that accumulated on ChpA −HD (Fig. 5B, left and middle panels), it was concluded that saturating ChpArec with BeF3− allowed phosphoryl groups from [γ-32P]ATP to accumulate on Hpt2. An analogous experiment to that described in Fig. 5B (left panel) was carried out using a high concentration of unlabeled ATP to saturate the ChpArec active site after allowing reaction of ChpA −HD with Hpt2 and [γ-32P]ATP for 10 min. For the unlabeled ATP swamp reaction (Fig. 5C), the radioactivity that had accumulated on the ChpA −HD before the swamp was transferred onto Hpt2, vastly increasing the amount of 32P-Hpt and demonstrating that Hpt2 is receiving phosphoryl groups from the ChpA −HD protein. This result rules out the formal possibility that activation of ChpArec (with BeF3− or phosphoryl groups) somehow allows Hpt2 to react directly with ATP, as this scenario would have caused the Hpt2 signal to diminish rather than dramatically increase as a result of adding an excess of unlabeled ATP. The unlabeled ATP swamp gave similar results for Hpt2 and Hpt3 (Fig. 5D, left panel). As a control, no labeling of Hpt2 and Hpt3 occurred when Chp −HA (lacking the ChpArec phosphorylation site) was used or when either ChpA −HD or Hpt was absent (Fig. 5D, right panel).
An explanation that can account for both the BeF3− and the unlabeled ATP swamp result (summarized in Fig. 5E) is as follows. The ChpArec → Hpt2/3 → ChpArec → H2O model is valid. Both the BeF3− (tightly but reversibly bound to the ChpArec) and the saturating phosphorylation of ChpArec (the net result of phosphorylation and dephosphorylation) were effective in preventing Hpt2 → ChpArec phosphotransfer, as predicted. But, surprisingly, these same modifications continued to allow the Hpt6 → ChpArec phosphotransfer. Hpt6 transfer to ChpArec is intramolecular (versus intermolecular for Hpt2 to ChpArec) and intramolecular reactions can give very highly effective local concentrations of reactants that could drive the reaction. The unlabeled ATP swamp experiment resulted in less radioactivity on Hpt2 than the BeF3− conditions because the unlabeled ATP greatly decreased the specific activity of the phosphoryl groups that end up on Hpt2. Thus, saturation of ChpArec with phosphoryl groups or a phosphomimic appears to allow transfer from Hpt6 to ChpArec in cis but to disallow back transfer from Hpt2 to ChpArec, present here in trans, as shown in the scheme in Fig. 5E.
Next, we exploited the BeF3− and unlabeled ATP swamp strategies for sensitive measure of phosphotransfer from ChpArec to the other Xpts. We used this approach to more stringently test whether any of the other Xpts that did not react with ATP (Hpt1, Hpt3, Spt, and Tpt) were able to obtain a phosphoryl group from ChpArec. Each Xpt was incubated with Chp −HD, [γ-32P]ATP, and BeF3− and assessed for incorporation of radioactivity into the Xpt after 90 min. Only Hpt3 behaved like Hpt2 and received a phosphoryl group from ChpArec (Fig. 6A). Hpt1, Tpt, and Spt did not accumulate any detectable 32P and thus did not receive a phosphoryl group from ChpArec. Finally, we tested whether the ATP-reactive Hpts (Hpt4–6) could receive a phosphoryl group from ChpArec. Based on earlier results with Hpt2 and Hpt3 (Fig. 5, C and D), we evaluated whether label that had accumulated on ChpA −HD transferred efficiently to the Hpt present in trans when the system was swamped with nonradioactive ATP. All of the Hpts except Hpt1 received phosphoryl groups with similar kinetics (Fig. 6B). Therefore, phosphoryl groups were transferred from phosphorylated ChpArec to all of the Hpts except Hpt1.
FIGURE 6.

ChpArec phosphotransfer to Xpts under conditions of ChpArec saturation. A, single time point analysis for reactions containing BeF3− as a sensitive detection of ChpArec to Xpt phosphotransfer. The reactions contained 6 μm ChpA −HD, 40 μm Xpt, 70 μm BeF3− (70 μm BeCl2 and 10 mm NaF), and 30 μm [γ-32P]ATP and were incubated for 90 min before quenching and analysis. B, reactions contained 7.3 μm ChpA −HD, 56 μm HPt, and 30 μm [γ-32P]ATP. After 10 min, 2 mm unlabeled ATP was added; samples were removed at designated times for quenching and analysis.
Rapid Phosphotransfer from Hpt2–4 to ChpArec and from Hpt3 to PilH
In Fig. 4, we demonstrated rapid phosphotransfer from Hpt5 and 6 to ChpArec, in cis and in trans. To evaluate the reactivities of the other phosphorylatable Hpts (Hpt2–4) with various receiver domains, we used the unlabeled ATP swamp approach. The experiment entailed incubation of ChpA −HD, the Hpt, and [γ-32P]ATP for 10 min, followed by a swamp of unlabeled ATP to generate appreciable amounts of [32P]Hpt2–4. These conditions should also result in saturation of the Hpt with phosphoryl groups. We followed with addition of ChpArec, PilG, or PilH and evaluated time points within the first 2 min of reaction. This approach is only capable of measuring rapid phosphotransfer because the receiver proteins will, in short time, become saturated with unlabeled phosphoryl groups obtained from Hpt6 on ChpA −HD. The results (Fig. 7, A–C) showed that Hpt2, Hpt3, and Hpt4 all rapidly transferred to ChpArec. Hpt3 also transferred rapidly to PilH (Fig. 7B), albeit slower than to ChpArec. None of the Hpts were observed to rapidly transfer to PilG. This was surprising because PilG has been proposed to be a functional response regulator that receives phosphoryl groups from ChpA (28), and many response regulators are rapidly phosphorylated by their cognate histidine kinase (35) or Hpt (38) under similar assay conditions. The possibility that PilG bound the catalytic Mg2+ ion with relatively low affinity was considered. However, increasing the concentration of MgCl2 in the phosphotransfer reaction from 5 to 20 mm did not result in rapid phosphotransfer to PilG (data not shown). A scheme summarizing the in vitro results is shown in Fig. 8.
FIGURE 7.

Can Hpt2, Hpt3, and Hpt4 transfer rapidly to ChpArec, PilG, or PilH? Gel phosphorimages following phosphotransfer from Xpts (Hpt2–4) to PilG, PilH, and ChpArec. Reactions were initiated by addition of 30 μm [γ-32P]ATP to mixtures of 7.3 μm ChpA −HD and 58 μm Hpt2 (A), Hpt3 (B), or Hpt4 (C). After 10 min, a sample was removed and quenched (*), and 2 mm unlabeled ATP was added to the reaction. At 20 min, a second sample was removed and quenched (#). A different receiver (indicated) was then added to a final concentration of 30 μm, and time points were removed and quenched at various times. The noted times refer to the time after addition of the receiver domain. The buffer was 50 mm Tris, pH 7.5, 100 mm KCl, 5 mm MgCl2, and 4 mm DTT.
FIGURE 8.
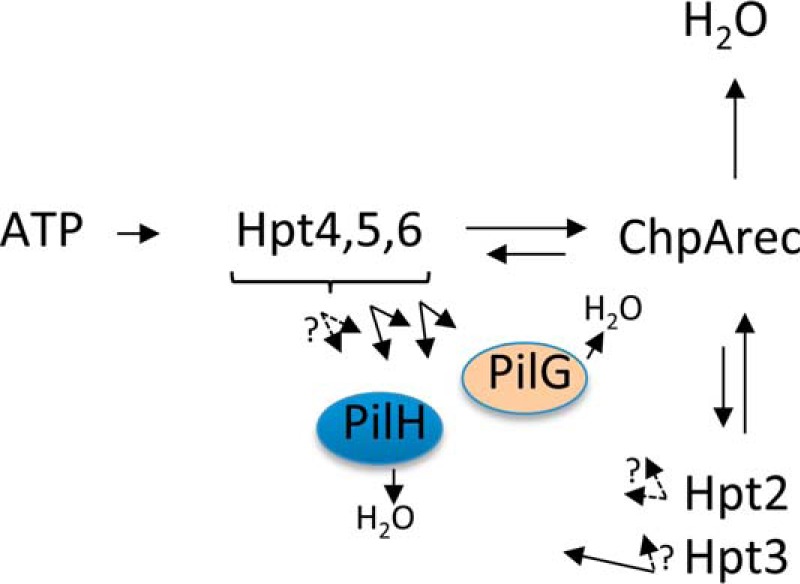
Scheme summarizing in vitro phosphotransfer reactivities between Pil-Chp domains. The length of the arrow reflects the relative reactivity. Solid black arrows reflect rates measured here. Dashed arrows indicate the absence of rapid phosphotransfer (experimental design precluded measurement of slower phosphotransfer).
ChpA Phosphorylation Site Mutant Phenotypes
Genetic analysis provides an alternative approach to probe phosphoryl group flow in the Pil-Chp system. In a previous study (27), P. aeruginosa strain PAO1 was engineered to encode ChpA single substitutions with non-phosphorylatable amino acids at seven of the nine potential phosphorylation sites (all except Tpt and Hpt6). Single site mutants of Hpt1 and Spt gave wild type levels of twitching, consistent with our inability to detect phosphorylation on these Xpt domains (Figs. 3 and 6). Similarly, phosphorylation site mutants of Hpt4 and Hpt5 displayed wild type twitching, which is consistent with the functional redundancy of Hpt4–6, as proposed here. In contrast, phosphorylation site mutants of Hpt2 and Hpt3 gave partial twitching, and a phosphorylation site mutant of ChpArec displayed a severe twitching defect. Here, we carried out further genetic analyses of mutants in strain PAK altered at the ChpA phosphorylation sites that gave non-wild type twitching phenotypes in the aforementioned study (27).
cAMP and Twitching Motility Phenotypes of Hpt2, Hpt3, and ChpArec Phosphorylation Site Mutants
Twitching motility is a relatively insensitive measure of Pil-Chp output as it involves complex interplay between pilus extension and retraction dynamics, such that gain-of-function and loss-of-function mutations both result in reduced motility (28). In contrast, gain and loss of Pil-Chp function can be distinguished based on intracellular cAMP levels (21). Specifically, chpA and pilG deletion strains both display loss-of-function (low cAMP relative to wild type), indicating phosphorylated PilG (PilGp) is necessary for cAMP production via regulation of the adenylate cyclase CyaB (21). In contrast, pilH deletion mutants display gain-of-function (elevated cAMP over wild type), indicating that PilH suppresses cAMP production (21). Thus, cAMP levels reflect the relative amount of phosphorylated PilG (PilGp) present.
We engineered strains encoding all seven possible combinations of non-phosphorylatable alanine substitutions at the phosphorylation sites of ChpArec, Hpt2, and/or Hpt3 onto the chromosome of a P. aeruginosa PAK strain lacking the constitutive low activity adenylate cyclase CyaA and carrying a lacZ reporter gene under the control of a synthetic cAMP-responsive promoter. As controls, we included isogenic chpA, pilG, and pilH deletion strains with previously reported phenotypes. The mutant strains were characterized by a combination of Western blot analysis for ChpA levels, reporter gene assay for intracellular cAMP levels, and twitching motility. All mutant strains produced ChpA at a level closely similar to wild type except the chpA deletion mutant (ΔchpA), which served as a negative control in Western blots (data not shown). As such, differences in cAMP or twitching cannot be attributed to variation in expression or stability of various ChpA mutants. Consistent with previous results (21), deletion of chpA or pilG resulted in a significant loss of intracellular cAMP-dependent reporter activity relative to the wild type strain, whereas deletion of pilH gave cAMP levels that were 3–4 times higher than wild type (Fig. 9A). Most strikingly, the Hpt3HA mutant showed gain-of-function resulting in cAMP levels that were elevated 3–4-fold over wild type and thus phenocopied the pilH deletion mutant. In contrast, cAMP levels of the Hpt2HA mutant were slightly lower than wild type levels. The Hpt2HA/Hpt3HA double mutant gave a cAMP reporter readout similar to the Hpt2HA mutant, indicating that Hpt2 is epistatic to Hpt3.
FIGURE 9.

P. aeruginosa Pil-Chp-dependent phenotypes are modulated by phosphotransfer events involving Hpt2, Hpt3, and ChpArec in vivo. Intracellular cAMP production (A) and twitching motility (B) were measured for a wild type (wt) version of P. aeruginosa strain PAK, a chpA deletion strain (ΔchpA), and chpA chromosomal substitution mutants in which the site of phosphorylation was ablated in ChpArec (rec), ChpAHpt2 (Hpt2), ChpAHpt 3 (Hpt3), individually and in all possible combinations. Previously reported deletion mutants of pilG (ΔpilG) and pilH (ΔpilH) were included for comparison. Cyclic AMP was assessed using a synthetic transcriptional reporter (lacP1ΔlacI-lacZ), where β-galactosidase activity is directly proportional to intracellular cAMP accumulation (21). The parental strain PAKΔcyaA::lacP1ΔlacI-lacZ, which lacked the minor adenylate cyaA, served as the wild type genetic background for all mutants. cAMP reporter assays were repeated a minimum of three times, and data represent average reporter activity ± S.E. Reporter activity was significantly different (p < 0.05) for all strains compared with wild type. Twitching motility was measured as the diameter of bacterial subsurface spread 43 h post-inoculation to the plastic-agar interface (21). Twitching motility data represent the average for a minimum of seven experimental replicates ± S.E. Zones of motility were significantly different (p < 0.0001) for all strains compared with wild type. C, plots of cAMP-dependent reporter activity versus twitching motility diameter using the data from A and B. The results are shown for wild type (open circle) and different mutant groupings as follows: ΔpilG, ΔchpA (closed circles); Hpt2HA, Hpt2HA/Hpt3HA (closed squares); ChpArecDA, ChpArecDA/Hpt2HA, ChpArecDA/Hpt3HA, ChpArecDA/Hpt2HA/Hpt3HA (closed triangles); ΔpilH, Hpt3HA (closed diamonds). The error bars represent ±S.E.
The ChpArecDA single mutant displayed a gain-of-function phenotype, with cAMP levels elevated about 1.5-fold over wild type (Fig. 9A). This result suggests that PilG can be phosphorylated even in the absence of ChpArec (i.e. by Hpt4–6), which is consistent with our observation of phosphotransfer (albeit slow) from Hpt5 and Hpt6 to PilG. Furthermore, the ChpArecDA phenotype suggests that like PilH, ChpArec functions to decrease cAMP. This could be due to a combination of ChpArec actions as a phosphate sink (fast autodephosphorylation) or its function as a phosphotransfer protein to Hpt3, which in turn transfers to PilH. Notably, the double mutants ChpArecDA/Hpt2HA and ChpArecDA/Hpt3HA and the triple mutant ChpArecDA/Hpt2HA/Hpt3HA all had levels of cAMP that were indistinguishable from the ChpArecDA single mutant (Fig. 9A). This result indicates that ChpArec is epistatic to Hpt2 and Hpt3; thus, Hpt2 and Hpt3 function downstream from ChpArec, which is consistent with the scheme assembled in Fig. 8 from biochemical results and forms the framework for a holistic scheme that includes conclusions based on the genetic results (Fig. 10A). In this scheme, phosphoryl groups cannot reach Hpt2 or Hpt3 in either the ChpArecDA single mutant or the Hpt2HA/Hpt3HA double mutant. The increased level of cAMP in the former mutant compared with the latter is consistent with loss of phosphoryl groups from the system by rapid autodephosphorylation of ChpArec when present.
FIGURE 10.
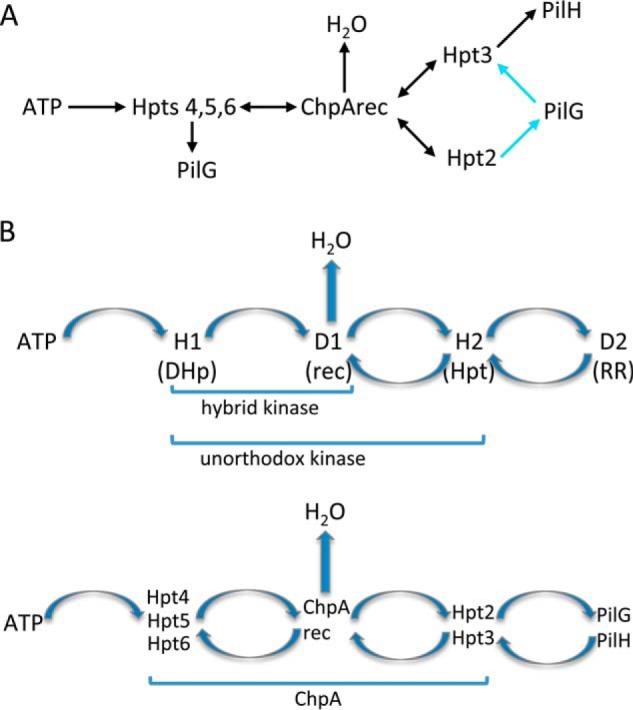
Pil-Chp system displays fundamental features of TCS phosphorelays. A, working model of phosphoryl group flow in Pil-Chp taking into account both biochemical and genetic data. The portions of the model supported by both biochemistry and genetics are colored black; reactions supported by genetics only are colored cyan. Phosphotransfer from Hpt5 and 6 to PilH (not shown) was observed in vitro, but there was no genetic test for this reaction. There was no biochemical demonstration of Hpt4 to PilG phosphotransfer. B, general schemes of phosphoryl group flow in TCS phosphorelays (upper panel) and within the Pil-Chp system (lower panel) are similar. The fusion of the various reactive domains into a single polypeptide chains is indicated by brackets. RR stands for response regulator.
The observation that the Hpt3HA mutant gave a cAMP phenotype similar to the pilH deletion mutant is consistent with our demonstration of rapid phosphoryl group transfer from Hpt3 to PilH, and thus both the biochemistry and genetics support a scheme whereby the primary path for phosphorylation of PilH is through Hpt3 (Fig. 10A). However, the observation that addition of the hpt2HA mutation to the hpt3HA mutation diminished cAMP to near the levels seen in the Hpt2HA mutant indicates that the Hpt3 gain-of-function phenotype is contingent on the presence of the Hpt2 conserved histidine. This epistasis could be explained by inclusion of phosphotransfer from Hpt2 to PilG as the preferred route for PilG phosphorylation in our model. In the absence of functional Hpt3, phosphoryl groups would be routed through Hpt2 to PilG to give the gain-of-function phenotype. Inclusion of Hpt2 to PilG phosphotransfer suggests that the Hpt2HA mutant should have a loss-of-function phenotype, which was not observed. However, also incorporating reverse phosphotransfer (a common feature of Hpt domains) from PilG to Hpt3 resulted in a model (Fig. 10A) that was able to account for all of the cAMP phenotypes. Specifically, the Hpt3HA mutant would not dephosphorylate PilG, leading to high cAMP. In the Fig. 10A model, wild type cAMP levels arise from the balance between rapid rates of PilG phosphorylation via Hpt2 and dephosphorylation via Hpt3. The Hpt2HA and Hpt2HA/Hpt3HA mutants could give near wild type levels of cAMP by being impaired in both PilG phosphorylation (no Hpt2) and dephosphorylation (Hpt3 either missing or saturated with phosphoryl groups redirected from Hpt2 and so unable to accept phosphoryl groups from PilG). Phosphotransfer from Hpt2 to PilG was not observed in our biochemical assays, but our assay was only sensitive to rapid transfer; in the case of Hpt5 and 6, there was evidence for transfer to PilG in vivo despite slow rates in vitro. Reverse phosphotransfer from PilG or PilH to any Hpt was not assessed in vitro. Thus, the genetics suggest that PilG can receive phosphoryl groups from Hpt4-6, but the preferred route of transfer may be from Hpt2, and the primary route of loss may be via Hpt3.
Correlation of Twitching and cAMP Phenotypes
The same set of mutants was evaluated for type IV pili mediated twitching motility (Fig. 9B). The chpA and pilG deletion mutants exhibited minimal twitching, as observed previously (21, 28). The Hpt3HA and ΔpilH mutants, which shared strong gain-of-function cAMP phenotypes (Fig. 9A), exhibited similar twitching defects with zone expansion rates 34–45% of the wild type strain. Likewise, all four mutants containing the ChpArecDA substitution (ChpArecDA, ChpArecDA/Hpt2HA, ChpArecDA/Hpt3HA, and ChpArecDA/Hpt2HA/Hpt3HA), which shared similar modest gain-of-function cAMP phenotypes, gave similar twitching rates of 53–64% of wild type. Thus the twitching data are also consistent with the ChpArec mutant being epistatic (upstream) to both Hpt2 and Hpt3. Finally, the Hpt2HA and Hpt2HA/Hpt3A mutants, which had cAMP phenotypes just slightly lower than wild type, also had similar twitching of 86–90% of wild type. Our twitching result for ChpArecDA (partial twitching) was inconsistent with previous results reported for P. aeruginosa strain PAO1, which was reported to have an absolute defect in twitching (27). To validate our results, we utilized a second strategy in which the chpA deletion mutant was complemented with either wild type ChpA or ChpArecDA expressed from a plasmid at wild type levels as confirmed by Western blots. Plasmid complementation yielded results for both twitching and cAMP phenotypes that were indistinguishable from chromosomal expression (data not shown).
Grouping mutants based on twitching motility gave the same groupings described above for cAMP phenotypes. Furthermore, replotting the Fig. 9, A and B, data as twitching motility versus the cAMP reporter for wild type and the four groups of mutants yields a bell-shaped curve (Fig. 9C) in which wild type displays optimal twitching, and both elevated and diminished levels of cAMP correlate with impaired twitching. Because twitching motility is largely independent of cAMP in strain PAK (21), this correlation presumably reflects underlying differences in PilGp and/or PilHp that influence cAMP and twitching independently.
Taken together, previously reported genetic data (21, 27) as well as the phenotypes of seven new mutants reported here are consistent with multiple fundamental features of the Chp/Pil signaling network deduced from our biochemical results. In particular, all genetic results are consistent with the intramolecular phosphoryl group flow within ChpA proposed here. However, some aspects of the genetic data suggest the scheme based on biochemical results alone was incomplete. In particular, the preferred routes of phosphotransfer to and from PilG suggested by genetics were not revealed by the biochemistry.
Discussion
Multiple Xpts with Differential Phosphotransfer Reactivities
Chemosensory systems specialized to mediate motility by T4P contain multiple Xpt domains within their CheA analogs (5). The current Pfam 29.0 database (pfam.xfam.org) (39) lists domain architectures for probable CheAs containing 2–8 N-terminal Xpt domains. Together, these architectures comprise 158 representative proteins, with most representing many individual sequence entries. All but one of the 158 proteins have a C-terminal receiver domain, suggesting that multiple Xpts and a C-terminal receiver are functionally related.
This work represents the first biochemical analysis, of which we are aware, of a CheA with multiple Xpts. All eight of the Xpts were stably overexpressed as isolated domains and easily purified, consistent with modeling that predicted that each Xpt would form a monomeric domain with a four-helix bundle core (9). However, only Hpt4–6 reacted with ATP in the presence of the ChpA HATPase_c domain to become phosphorylated. Once phosphorylated, Hpt4–6 transferred rapidly and reversibly to ChpArec. In contrast, Hpt2 and 3 (separated in the ChpA primary sequence from Hpt4–6 by Spt) did not react with ATP; however, Hpt2 and 3 could reversibly obtain phosphoryl groups from ChpArec. Spt, Tpt, and Hpt1 did not become phosphorylated by either ChpArec or ATP in these studies.
Accounting for Differential Xpt Phosphorylation with Sequence Variation
To what extent were the differential abilities of the Xpts to get phosphorylated consistent with known structural elements that modulate the different phosphotransfer reactions? For the reaction of the CheA Hpt with ATP, a trio of residues, Glu at position H + 22 (22 residues C-terminal to the phosphorylated His), Lys at position H + 3, and His at position H+19, form a hydrogen bonding network that orients and enhances the nucleophilicity of the attacking His and is required for optimal reactivity (32, 33, 40). Inspection of the Xpt residues at the three positions indicates that P. aeruginosa Hpt4–6, the only Xpts to give detectable phosphorylation with ATP, contain all three hydrogen bonding residues; Hpt2 and Hpt3 have the Lys and Glu, but not the His at H + 19; Hpt1 has only the Glu; Spt and Tpt have none of the requisite residues (data not shown). Thus, the differences in Xpt reactivities with ATP (Fig. 3) are consistent with the known enzymology of homologous systems.
The absence of detectable phosphotransfer from ChpArec to Hpt1 (in contrast to the easily detectable transfer to Hpt2–6, see Fig. 6B) may be due to weak binding and/or the absence of residues on Hpt1 that provide catalytic support for the phosphotransfer. Comparison of residues predicted to be at the Hpt/ChpArec interface based on the Rhodobacter sphaeroides CheA2 Hpt-CheY6 complex crystal structure Protein Data Bank code 3KYJ (41) reveals that Hpt1 does not share any of the four predicted interface residues located on the αA helix of Hpt2–6, including a conserved Phe, and only three out of the six residues on the αB helix (data not shown), consistent with the possibility that Hpt1 has diminished affinity for ChpArec. Additionally, the Lys at position H + 3 that is found in Hpt2–6 and is missing from Hpt1 is required for efficient phosphorylation of the YPD1 Hpt protein by partner and non-partner receiver domains (42). Similarly, the absence in Hpt1 of an Arg at position H − 3 that is present in Hpt2–6 may contribute to the diminished reactivity of Hpt1 (32).
Like Hpt1, Spt and Tpt also failed to be phosphorylated by ChpArec. In this case, the absence of detectable phosphotransfer from ChpArec to Spt and Tpt likely has large contributions from the diminished inherent nucleophilicities of Ser/Thr versus His. Amino acids with hydroxyl side chains, unlike His, are not in their most nucleophilic state at neutral pH. Enzymatic phosphorylation of Thr/Ser/Tyr generally requires specific catalytic machinery such as a residue to act as a general base that deprotonates the hydroxyl group to make it more nucleophilic (43). In contrast, there is no necessity for a general base (nor has there been one identified) for HATPase_c domains nor for Asp to His transfer. In addition to the absence of a general base, the Tpt and Spt domains also lack multiple residues present in Hpt2–6 that are predicted to be at the receiver domain interface (data not shown). With no positive evidence for phosphorylation, and wild type twitching motility displayed by strains containing Hpt1 and Spt phosphorylation site mutants (27), the roles of the Hpt1, Spt, and Tpt in Pil-Chp signaling have yet to be established.
cAMP Phenotypes Suggest Sources of Phosphoryl Groups for the PilG Response Regulator
Here, we saw phosphotransfer in vitro from all of the phosphorylatable Hpts (Hpt2–6) to ChpArec, even in trans, with the rapid kinetics expected for a cognate histidine kinase/response regulator. Additionally, we observed rapid transfer from Hpt3 to PilH, and this result together with the phosphorelay scheme suggested a route to phosphorylate the PilH response regulator (ATP → Hpt4–6 → ChpArec → Hpt3 → PilH), which was supported by the genetic result that the Hpt3HA mutant phenocopied the ΔpilH mutant. However, the biochemistry did not reveal an Hpt that transferred rapidly to the PilG response regulator and thus did not reveal a likely pathway for phosphoryl groups to flow to PilG. In contrast, the genetic data suggested that PilG could receive phosphoryl groups from Hpt2,4,5, and 6 in vivo. With genetic evidence consistent with Hpt2 and Hpt4–6 as sources of phosphoryl groups for PilG, it is not clear why PilG was not rapidly phosphorylated by these Hpts in our biochemical assays. It is possible that rapid transfer to PilG requires experimental conditions (e.g. buffer, ionic strength) different from ours or that our purified PilG was not optimally folded. It is also possible that rapid phosphotransfer to PilG requires another protein or other cofactor that was not present in our in vitro assays. This cofactor could potentially function to bind to PilG and/or the Hpt to enhance Hpt to PilG phosphotransfer, either by changing protein conformation or by increasing local concentration of reactants. Many flagellar CheAs have a specialized CheY-binding domain (“P2 domain”) that contributes significantly to the binding energy with CheY. However, a P2 domain is not present in ChpA. PilG was slower to phosphorylate not only with the isolated Hpts but also with the larger ChpA constructs. Thus, if a cofactor exists, it is likely a factor that is not part of the ChpA constructs used here.
P. aeruginosa ChpA Functionally Resembles an Unorthodox Histidine Kinase
Division of the phosphorylatable Hpts into those that react with ATP and those that react only with ChpArec conferred essential properties of two-component phosphorelays to the Pil-Chp signaling circuit (Fig. 10B). Phosphorelays are a common variation of the two-component system (3) that contains sequential His (H1) → Asp (D1) ↔ His (H2) ↔ Asp (D2) phosphotransfer. The second two steps are reversible, and often phosphoryl groups are lost from the system via D1 autodephosphorylation (Scheme 1) (44, 45). In Pil-Chp, the ATP-reactive ChpA Xpts (Hpt4–6) have a similar role as the phosphorelay DHp domain (H1), whereas the ATP-unreactive Xpts (Hpt2 and 3) play the role of H2. ChpArec plays the role of D1 in that it can act as both a phosphotransfer protein and a phosphate sink (removes phosphoryl groups from the system via autodephosphorylation). Our genetic analysis supported this essential scheme. Epistatic analysis based on two different phenotypes, cAMP levels and twitching motility, of the four mutants that include chpArecDA indicated that Hpt2 and Hpt3 function downstream of ChpArec. Furthermore, the genetics supported the conclusion that ChpArec had roles as both a phosphotransfer protein and as a phosphate sink. However, unlike canonical phosphorelays, it appears that Pil-Chp has multiple copies of H1- and H2-containing domains and that ChpA H1 domains (Hpt4–6) can also transfer to D2, the terminal response regulators, PilG and PilH. H1 to D2 transfer also occurs in the ArcAB phosphorelay, although at a slower rate than the H1 → D1 → H2 → D2 transfer (29). In addition, because the domains containing H1 are Hpt domains in the Pil-Chp system (in contrast to a DHp domain in phosphorelays), the H1 to D1 transfer is bidirectional in the Chp system instead of unidirectional as in phosphorelays. The chain of phosphorylation sites in basic phosphorelays endows such pathways with potentially useful signaling properties such as non-linear stimulus/response characteristics, multistate stability, and robustness to noise (46–50). The additional parallel and reversible pathways evident within ChpA presumably confer further signal transduction attributes upon proteins with multiple Hpt domains. The existence of proteins with multiple Hpt domains attests to the evolutionary value of such arrangements, but the exact contribution of the additional elements remains to be discerned.
Within phosphorelays, there is variation as to how the phosphorylatable domains are arranged on polypeptide chains. In some phosphorelays (e.g. the Spo0 system that regulates initiation of Bacillus subtilis sporulation), the functional domains carrying H1, D1, H2, and D2 are all on separate proteins, so that all phosphotransfer is intermolecular. Alternatively, another subfamily of phosphorelay systems (e.g. the S. cerevisiae osmoregulatory pathway) contains a “hybrid kinase,” a single polypeptide that contains both the DHp domain (containing H1) and a C-terminal receiver domain (D1) so that H1 to D1 transfer is an intramolecular event. In these systems, an Hpt domain (H2) and terminal response regulator (D2) are separate proteins. In the yeast osmoregulatory system, the separate Hpt (H2) domain (YPD1) is phosphorylated by a cell membrane-associated hybrid kinase (SLN1) and then shuttles between the cytoplasm and nucleus to transfer phosphoryl groups to the two terminal response regulators (51). Finally, another class of phosphorelays (represented by the well characterized ArcAB and BvgAS systems) contains an “unorthodox” kinase that contains H1, D1, and H2 domains, so all phosphotransfer between H1, D1, and H2 occurs intramolecularly. P. aeruginosa ChpA thus functionally resembles an unorthodox histidine kinase in that H1, D1, and H2 domains are on the same polypeptide chain (Fig. 10B). The arrangement of the H1 (Hpt4–6), D1 (ChpArec), and H2 (Hpt2 and 3) phosphotransfer domains in space in the context of the ChpA dimer will inevitably affect the local concentrations of the reacting domains and hence the rates of the phosphotransfer reactions.
Experimental Procedures
Plasmid Construction and Mutagenesis for Purified Proteins
DNA encoding each protein construct was obtained by PCR using P. aeruginosa strain PAK (GenBankTM accession NZ_KE137323.1) genomic DNA as template (21). In addition to the complementary region, primers for each construct also contained a restriction enzyme recognition site (NdeI for the forward primer and either HindIII or BamHI for the reverse primer) and a 6-bp random sequence overhang to allow efficient restriction enzyme cleavage. Pfu Turbo (Agilent Technologies) was used as the polymerase, and yields were sometimes optimized with inclusion of betaine (1.2 m), DMSO (8% v/v), or formamide (4% v/v) in the amplification mix. PCR products were purified from the reaction mix (Qiagen PCR Cleanup), cleaved with NdeI and either HindIII or BamHI (New England Biolabs), and gel-purified (Qiagen). The purified fragments were ligated between the NdeI and HindIII/BamHI sites in the pET28a vector (Merck Millipore), and the final construct was confirmed by sequencing. The final expression vector encoded proteins with an N-terminal thrombin-cleavable His6 tag. After thrombin cleavage, there was an additional GSH tripeptide present at the N terminus of each protein construct. Site-directed mutagenesis was carried out using QuikChange methodology (Agilent Technologies). All mutations were confirmed by sequencing the entire gene. The residue compositions of the ChpA constructs (Fig. 1B) were as follows. Constructs with N-terminal Hpt5 start at residue 1525; constructs with N-terminal Hpt6 start at residue 1674; and constructs with ChpArec absent end at 2334. The residue spans for the isolated Xpt domains are as follows: Hpt1(1–135); Tpt(425–559); Hpt2(863–994); Hpt3(1032–1171); Spt(1180–1309); Hpt4(1356–1486); Hpt5(1525–1656); and Hpt6(1674–1807). Isolated ChpArec contained ChpA residues 2352–2478.
Protein Expression and Purification
Overnight cultures of E. coli BL21(DE3) transformed with the appropriate pET28a expression plasmid were inoculated into 1 or 2 liters of LB media supplemented with kanamycin (30 μg/ml). Cultures were grown to an absorbance at 600 nm of 0.4 to 0.6 and induced with 0.5 or 1 mm isopropyl β-d-1-thiogalactopyranoside. Expression was allowed to proceed overnight at room temperature with shaking. Cells were harvested, resuspended in lysis buffer (50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 10 mm imidazole), and lysed by probe tip sonication (five times 1-min pulses on ice). The lysed cells were centrifuged (120,000 × g, 45 min) to sediment insoluble matter. The cleared lysate was applied to a 2-ml nickel-nitrilotriacetic acid affinity column (Qiagen); the column was washed (50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 20 mm imidazole), and the protein was eluted (50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 150 mm imidazole). The protein pool was incubated with human α-thrombin (Hematologic Technologies Inc.) 1:1000–10,000 (w/w thrombin to protein) overnight at 4 °C. For the higher molecular weight ChpA constructs that contained a dimerization domain (Fig. 1B), the cleaved protein was gel-filtered on a 2.6 × 95-cm Superose 12 column in TEDG buffer (50 mm Tris, pH 7.5, 0.5 mm EDTA, 5 mm DTT, and 10% (v/v) glycerol). In each case, the purified construct eluted at a position consistent with formation of a dimer of chains that varied from 72 to 105 kDa. The thrombin-cleaved pools for the smaller constructs (Hpt1–6, Tpt, Spt, ChpArec, PilG, and PilH) were gel-filtered in TEDG on a Superdex 75 1660 column and eluted as monomers. Fractions containing pure protein were pooled, concentrated, divided into aliquots, and stored at −80 °C. Protein concentrations were determined by the Bradford assay (Bio-Rad) using bovine serum albumin as a standard.
Phosphotransfer Experiments Using 32P-Phosphoryl Groups
A variety of phosphotransfer reactions were monitored by quantitative detection of 32P-labeled phosphoproteins by phosphorimaging analysis of SDS-polyacrylamide gels (52). Unless stated otherwise, the buffer was TKMD (50 mm Tris, pH 7.5, 50 mm KCl, 5 mm MgCl2, and 2 mm DTT), and reactions were carried out at room temperature. Experiments fell into two broad groups: 1) time courses where the distribution of 32P was determined at various time points for a single reaction, or 2) single time point experiments where the distribution of 32P was determined for multiple reactions on a single gel at a single time point. In all cases, aliquots from reactions were removed and immediately quenched with an equal volume of 2× SDS-PAGE sample buffer. A portion of the quenched samples (8 μl) was electrophoresed on an 18% (w/v) SDS-polyacrylamide gel; the gel was dried, and distribution of 32P on the various proteins was determined by phosphorimaging analysis (GE Storm or Typhoon) and reported as phosphorimaging units.
For ChpA autophosphorylation time courses (Figs. 2 and 4C), the ChpA construct (1.4 μm) was incubated with [γ-32P]ATP (30 μm or 150 μm final concentration) in a reaction volume of 60 μl. The [γ-32P]ATP was prepared by mixing [γ-32P]ATP (PerkinElmer Life Sciences, 6000 Ci/mmol) with unlabeled ATP. At various times after reaction initiation, aliquots (7 μl) were removed and quenched with an equal volume of 2× SDS sample buffer. An analogous procedure was used for determining time courses for distribution of 32P between ChpA −HD (7.3 μm) and Hpt2 (34 μm) upon addition of 30 μm [γ-32P]ATP (Fig. 5A). This reaction was modified by the addition of 70 μm BeF3− (formed in situ from 70 μm BeCl2 and 10 mm NaF) or 2 mm unlabeled ATP after the reaction had proceeded for 10 min.
Single time point reactions were used to characterize the reactivities of each Xpt domain (Hpt1–6, Spt, and Tpt) with ChpA AA−. ChpA AA− (1.9 μm) was incubated with each Xpt (Hpt1–Hpt6, Spt, and Tpt at 40 μm) and [γ-32P]ATP (150 μm) for 60 min at room temperature. Reaction samples were divided and then electrophoresed on two gels for analysis by Coomassie stain and phosphorimaging. To assess relative reactivities of ChpArec, PilG, and PilH, 1.4 μm ChpA HA−, or ChpA HA− was incubated with either ChpArec, PilG, or PilH (1–22 μm), and 30 μm [γ-32P]ATP. Reactions proceeded for 14 min before samples were quenched and subjected to electrophoresis and phosphorimaging.
Isolation of [32P]ChpA–H and Use for Measuring Phosphotransfer to ChpArec, PilG, and PilH
ChpA −H− (20 μm) was mixed with 100 μm [γ-32P]ATP in 50 mm Tris, pH 7.5, 7 mm MgCl2, 50 mm KCl, and 2 mm DTT in a total volume of 150 μl. After 10 min of reaction time at room temperature, 45 mg of solid ammonium sulfate was added and mixed, and the reaction was incubated on ice for 20 min to precipitate the protein. The sample was centrifuged, and the supernatant (containing the majority of the unincorporated ATP) was transferred to waste. The pellet (containing precipitated protein) was resuspended in 200 μl of TEG (50 mm Tris, pH 7.5, 0.5 mm EDTA, 10% (v/v) glycerol) and chromatographed on a G-100 column in TEG to separate the phosphorylated protein from remaining free ATP.
For measurement of phosphotransfer kinetics, purified [32P]ChpA −H− (0.34–0.44 μm) was diluted into reaction buffer (100 mm Tris, pH 7.5, 10 mm MgCl2), and an aliquot was removed and quenched with 2× sample buffer for a zero time point. The phosphotransfer reaction was initiated by addition of excess receiver (16.4 μm for ChpArec or 37 μm PilG or PilH). Aliquots were removed at desired time points, quenched with 2× sample buffer, and analyzed by SDS-gel electrophoresis and phosphorimaging as described above. The intensity of the zero time point was corrected for the differences in total reaction volume due to the addition of the receiver domain.
Receiver Domain Autodephosphorylation Rate Constants
The method was based on a previously described protocol (52). Reactions were initiated by mixing 32P-labeled ChpA −H− (0.4- 0.5 μm) with 37 μm ChpArec, PilH, or PilG in a total volume of 60 μl. For ChpArec, where phosphotransfer was complete within 15 s, aliquots (7 μl) were removed and quenched in 2× sample buffer at time points spanning 15–300 s. For PilG and PilH, aliquots were similarly removed over a span of 5–60 min. At the 5-min time point, at least 95% of the 32P had been transferred to PilG or PilH. The samples were electrophoresed and phosphorimaged as described above. The signal intensity of the phosphorylated receiver was plotted as a function of reaction time. The data were fit to a single exponential decay (Prism 6) to give the autodephosphorylation half-life. The experiment was carried out in duplicate or triplicate and the average and standard deviation reported.
Construction of Combinatorial P. aeruginosa ChpA Hpt2, Hpt3, and ChpArec Phosphorylation Site Mutants for in Vivo Studies
To construct a ChpArec phosphorylation site mutation, the receiver domain region of chpA was amplified as two separate but overlapping fragments, such that the overlapping portion of the sequences introduced an Asp to Ala codon substitution. The substitution was designed to simultaneously introduce a unique MscI endonuclease restriction site. The 5′ fragment was amplified using primers 5′fwd chpArecDA (5′ AttB1-ACCTCAGCGATCATGAAGTGCTGC) and 5′rev chpArec (5′ GCGCGGCATCTCGATGGCCAGCAGGAGGATGTC). The 3′ fragment was amplified using primers 3′fwd chpArec (5′ GACATCCTCCTGCTGGCCATCGAGATGCCGCGC) and 3′rev chpArec (5′ AttB2-GCGGACGTTCTTCACATGCCACTG). The resulting fragments were joined by splice PCR using AttB1 and AttB2 primers. The spliced fragment was inserted into plasmid pDONR201 by Gateway Cloning Technology (ThermoFisher). Correct assembly was confirmed by sequencing. The mutagenized rec domain fragment was transferred to the P. aeruginosa suicide plasmid pEXG2GW by Gateway cloning and mated with the parental P. aeruginosa strain PAKΔcyaA::lacP1ΔlacI-lacZ. This strain (denoted as wild type for this study) lacks the minor adenylate cyclase cyaA and carries a synthetic chromosomal reporter gene for indirect assessment of intracellular cAMP (21). Introduction of the ChpArec Asp to Ala substitution mutation onto the chromosomal copy of chpA was determined by PCR of the chromosomal chpArec region followed by restriction digest with MscI. Positive candidates were confirmed by sequencing.
To construct Hpt2 and Hpt3 phosphorylation site substitution mutations, a 2243-bp region of chpA, encompassing the Hpt2 and Hpt3 domains, was amplified from full-length chpA using the primers 5′ Hpt23 (5′ AttB1-AGCGACGACAACTGGACCCTTG) and 3′ Hpt23 (3′ AttB2-CGACACCATTTCCTCGTCCAGC). Primers were tailed with AttB sites for recombinational cloning. The fragment was introduced into plasmid pDONR201 by Gateway cloning. Codons for the phosphor-accepting His residues of Hpt2 and Hpt3 were mutagenized to Ala by QuikChange site-directed mutagenesis (Agilent) using primers GAGGTACGCCGTGCCTTCGCGACCCTCAAGGGCAGCGGACGG and GCCTTGCAGCGTGCCCTGGCCACCCTCAAGGGTAGCGCGC, respectively. The altered codons were designed to introduce NruI and MscI restriction endonuclease cleavage sites at the substitution positions in Hpt2 and Hpt3, respectively. The mutagenized fragments were sequence-confirmed and moved by Gateway cloning to the P. aeruginosa suicide vector pEXG2GW (21). The resulting suicide vectors were mobilized into the parental strain PAKΔcyaA::lacP1ΔlacI-lacZ with and without the ChpArec phosphorylation site mutation to generate Hpt2, Hpt3, and Hpt23 substitution mutations in the chromosomal copy of chpA. Incorporation of the mutations was determined by PCR of the chromosomal chpA Hpt23 region followed by restriction digest with NruI and MscI. Positive candidates were confirmed by sequencing.
In Vivo Assays
All phenotyping experiments utilized the seven combinations of the Hpt2, Hpt3, and ChpArec phosphorylation site mutations and previously described derivatives of P. aeruginosa strain PAK (21). Twitching motility and cAMP reporter assays were performed as described previously (21). For routine passage, E. coli and P. aeruginosa strains were maintained at 37 °C in LB broth or on LB agar. Growth was monitored by absorbance at 600 nm. For Western blottings, whole cell lysates were prepared from culture aliquots obtained at A600 = 0.5–1.0 as described previously (21). Proteins were separated on 4–15% gradient SDS-polyacrylamide gels (Bio-Rad), transferred to nitrocellulose, and simultaneously probed with rabbit anti-ChpA sera and mouse monoclonal antibody specific for the β-subunit of RNA polymerase (RNAP) (Neoclone). ChpA antiserum was generated in rabbits immunized with purified ChpA construct (Cocalico Biologicals). Anti-RNAP antibody was included as a loading and normalization control as reported previously (53). Antigen-antibody complexes were detected using goat anti-rabbit IRDye800CW and goat anti-mouse IRDye680LT (LI-COR) and visualized using a LI-COR Odyssey Classic Infrared Imaging System and Odyssey Software version 3.0. ChpA and RNAP band intensity was quantified using ImageJ (53, 54).
Author Contributions
R. E. S. and B. W. conducted the experiments. M. C. W. and N. B. F. provided reagents and helped conceive the idea for the project. R. E. S. and R. B. B. helped conceive the idea for the project. R. E. S., R. B. B., and M. C. W. wrote the paper.
Acknowledgments
We thank Robert M. Immormino, Stephani Page, and Rachel Craeger-Allen for helpful discussions and Thane Miller for computer simulations of the Chp/Pil network of phosphorylation reactions, which helped inform our thinking about this complex system. We also thank Joseph Hatch and Lauren Radlinski for assistance in generating P. aeruginosa reagents.
This work was supported by National Institutes of Health Grants R01GM050860 (to R. B. B.) and AI069116 (to M. C. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TCS
- two-component system
- Hpt
- histidine-containing phosphotransfer domain
- T4P
- type IV pili
- RNAP
- RNA polymerase.
References
- 1. Bourret R. B., and Silversmith R. E. (2010) Two-component signal transduction. Curr. Opin. Microbiol. 13, 113–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gotoh Y., Eguchi Y., Watanabe T., Okamoto S., Doi A., and Utsumi R. (2010) Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 13, 232–239 [DOI] [PubMed] [Google Scholar]
- 3. Goulian M. (2010) Two-component signaling circuit structure and properties. Curr. Opin. Microbiol. 13, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kirby J. R. (2009) Chemotaxis-like regulatory systems: unique roles in diverse bacteria. Annu. Rev. Microbiol. 63, 45–59 [DOI] [PubMed] [Google Scholar]
- 5. Wuichet K., and Zhulin I. B. (2010) Origins and diversification of a complex signal transduction system in prokaryotes. Sci. Signal. 3, ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parkinson J. S., Hazelbauer G. L., and Falke J. J. (2015) Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends Microbiol. 23, 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porter S. L., Wadhams G. H., and Armitage J. P. (2011) Signal processing in complex chemotaxis pathways. Nat. Rev. Microbiol. 9, 153–165 [DOI] [PubMed] [Google Scholar]
- 8. Lertsethtakarn P., Ottemann K. M., and Hendrixson D. R. (2011) Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 65, 389–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitchurch C. B., Leech A. J., Young M. D., Kennedy D., Sargent J. L., Bertrand J. J., Semmler A. B., Mellick A. S., Martin P. R., Alm R. A., Hobbs M., Beatson S. A., Huang B., Nguyen L., Commolli J. C., et al. (2004) Characterization of a complex chemosensory signal transduction system which controls twitching motility in Pseudomonas aeruginosa. Mol. Microbiol. 52, 873–893 [DOI] [PubMed] [Google Scholar]
- 10. Mattick J. S. (2002) type IV pili and twitching motility. Annu. Rev. Microbiol. 56, 289–314 [DOI] [PubMed] [Google Scholar]
- 11. Craig L., and Li J. (2008) type IV pili: paradoxes in form and function. Curr. Opin. Struct. Biol. 18, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zusman D. R., Scott A. E., Yang Z., and Kirby J. R. (2007) Chemosensory pathways, motility and development in Myxococcus xanthus. Nat. Rev. Microbiol. 5, 862–872 [DOI] [PubMed] [Google Scholar]
- 13. Burrows L. L. (2012) Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol. 66, 493–520 [DOI] [PubMed] [Google Scholar]
- 14. Driscoll J. A., Brody S. L., and Kollef M. H. (2007) The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368 [DOI] [PubMed] [Google Scholar]
- 15. Coggan K. A., and Wolfgang M. C. (2012) Global regulatory pathways and cross-talk control Pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr. Issues Mol. Biol. 14, 47–70 [PubMed] [Google Scholar]
- 16. Gellatly S. L., and Hancock R. E. (2013) Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173 [DOI] [PubMed] [Google Scholar]
- 17. Hahn H. P. (1997) The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa–a review. Gene 192, 99–108 [DOI] [PubMed] [Google Scholar]
- 18. Jin F., Conrad J. C., Gibiansky M. L., and Wong G. C. (2011) Bacteria use type-IV pili to slingshot on surfaces. Proc. Natl. Acad. Sci. U.S.A. 108, 12617–12622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skerker J. M., and Berg H. C. (2001) Direct observation of extension and retraction of type IV pili. Proc. Natl. Acad. Sci. U.S.A. 98, 6901–6904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Whitchurch C. B., Hobbs M., Livingston S. P., Krishnapillai V., and Mattick J. S. (1991) Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene 101, 33–44 [DOI] [PubMed] [Google Scholar]
- 21. Fulcher N. B., Holliday P. M., Klem E., Cann M. J., and Wolfgang M. C. (2010) The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol. Microbiol. 76, 889–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolfgang M. C., Lee V. T., Gilmore M. E., and Lory S. (2003) Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4, 253–263 [DOI] [PubMed] [Google Scholar]
- 23. Inclan Y. F., Huseby M. J., and Engel J. N. (2011) FimL regulates cAMP synthesis in Pseudomonas aeruginosa. PLoS ONE 6, e15867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Persat A., Inclan Y. F., Engel J. N., Stone H. A., and Gitai Z. (2015) type IV pili mechanochemically regulate virulence factors in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci., U.S.A. 112, 7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darzins A. (1994) Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and the gliding bacterium Myxococcus xanthus. Mol. Microbiol. 11, 137–153 [DOI] [PubMed] [Google Scholar]
- 26. Jiménez-Pearson M. A., Delany I., Scarlato V., and Beier D. (2005) Phosphate flow in the chemotactic response system of Helicobacter pylori. Microbiology 151, 3299–3311 [DOI] [PubMed] [Google Scholar]
- 27. Leech A. J., and Mattick J. S. (2006) Effect of site-specific mutations in different phosphotransfer domains of the chemosensory protein ChpA on Pseudomonas aeruginosa motility. J. Bacteriol. 188, 8479–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bertrand J. J., West J. T., and Engel J. N. (2010) Genetic analysis of the regulation of type IV pilus function by the Chp chemosensory system of Pseudomonas aeruginosa. J. Bacteriol. 192, 994–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Georgellis D., Lynch A. S., and Lin E. C. (1997) In vitro phosphorylation study of the Arc two-component signal transduction system of Escherichia coli. J. Bacteriol. 179, 5429–5435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burbulys D., Trach K. A., and Hoch J. A. (1991) Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell 64, 545–552 [DOI] [PubMed] [Google Scholar]
- 31. Garzón A., and Parkinson J. S. (1996) Chemotactic signaling by the P1 phosphorylation domain liberated from the CheA histidine kinase of Escherichia coli. J. Bacteriol. 178, 6752–6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishiyama S., Garzón A., and Parkinson J. S. (2014) Mutational analysis of the P1 phosphorylation domain in Escherichia coli CheA, the signaling kinase for chemotaxis. J. Bacteriol. 196, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quezada C. M., Hamel D. J., Gradinaru C., Bilwes A. M., Dahlquist F. W., Crane B. R., and Simon M. I. (2005) Structural and chemical requirements for histidine phosphorylation by the chemotaxis kinase CheA. J. Biol. Chem. 280, 30581–30585 [DOI] [PubMed] [Google Scholar]
- 34. Bourret R. B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laub M. T., Biondi E. G., and Skerker J. M. (2007) Phosphotransfer profiling: systematic mapping of two-component signal transduction pathways and phosphorelays. Methods Enzymol. 423, 531–548 [DOI] [PubMed] [Google Scholar]
- 36. Xu Q., and West A. H. (1999) Conservation of structure and function among histidine-containing phosphotransfer (HPt) domains as revealed by the crystal structure of YPD1. J. Mol. Biol. 292, 1039–1050 [DOI] [PubMed] [Google Scholar]
- 37. Varughese K. I., Madhusudan, Zhou X. Z., Whiteley J. M., and Hoch J. A. (1998) Formation of a novel four-helix bundle and molecular recognition sites by dimerization of a response regulator phosphotransferase. Mol. Cell 2, 485–493 [DOI] [PubMed] [Google Scholar]
- 38. Stewart R. C., Jahreis K., and Parkinson J. S. (2000) Rapid phosphotransfer to CheY from a CheA protein lacking the CheY-binding domain. Biochemistry 39, 13157–13165 [DOI] [PubMed] [Google Scholar]
- 39. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mourey L., Da Re S., Pédelacq J. D., Tolstykh T., Faurie C., Guillet V., Stock J. B., and Samama J. P. (2001) Crystal structure of the CheA histidine phosphotransfer domain that mediates response regulator phosphorylation in bacterial chemotaxis. J. Biol. Chem. 276, 31074–31082 [DOI] [PubMed] [Google Scholar]
- 41. Bell C. H., Porter S. L., Strawson A., Stuart D. I., and Armitage J. P. (2010) Using structural information to change the phosphotransfer specificity of a two-component chemotaxis signalling complex. PLos Biol. 8, e1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janiak-Spens F., and West A. H. (2000) Functional roles of conserved amino acid residues surrounding the phosphorylatable histidine of the yeast phosphorelay protein YPD1. Mol. Microbiol. 37, 136–144 [DOI] [PubMed] [Google Scholar]
- 43. Roskoski R., Jr. (2015) A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 100, 1–23 [DOI] [PubMed] [Google Scholar]
- 44. Georgellis D., Kwon O., De Wulf P., and Lin E. C. (1998) Signal decay through a reverse phosphorelay in the Arc two-component signal transduction system. J. Biol. Chem. 273, 32864–32869 [DOI] [PubMed] [Google Scholar]
- 45. Ansaldi M., Jourlin-Castelli C., Lepelletier M., Théraulaz L., and Méjean V. (2001) Rapid dephosphorylation of the TorR response regulator by the TorS unorthodox sensor in Escherichia coli. J. Bacteriol. 183, 2691–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Csikász-Nagy A., Cardelli L., and Soyer O. S. (2011) Response dynamics of phosphorelays suggest their potential utility in cell signalling. J. R. Soc. Interface 8, 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J. R., and Cho K. H. (2006) The multi-step phosphorelay mechanism of unorthodox two-component systems in E. coli realizes ultrasensitivity to stimuli while maintaining robustness to noises. Comput. Biol. Chem. 30, 438–444 [DOI] [PubMed] [Google Scholar]
- 48. Knudsen M., Feliu E., and Wiuf C. (2012) Exact analysis of intrinsic qualitative features of phosphorelays using mathematical models. J. Theor. Biol. 300, 7–18 [DOI] [PubMed] [Google Scholar]
- 49. Kothamachu V. B., Feliu E., Cardelli L., and Soyer O. S. (2015) Unlimited multistability and Boolean logic in microbial signalling. J. R. Soc. Interface 12, 20150234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kothamachu V. B., Feliu E., Wiuf C., Cardelli L., and Soyer O. S. (2013) Phosphorelays provide tunable signal processing capabilities for the cell. PLoS Comput. Biol. 9, e1003322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fassler J. S., and West A. H. (2013) Histidine phosphotransfer proteins in fungal two-component signal transduction pathways. Eukaryot. Cell 12, 1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bourret R. B., Thomas S. A., Page S. C., Creager-Allen R. L., Moore A. M., and Silversmith R. E. (2010) Measurement of response regulator autodephosphorylation rates spanning six orders of magnitude. Methods Enzymol. 471, 89–114 [DOI] [PubMed] [Google Scholar]
- 53. Topal H., Fulcher N. B., Bitterman J., Salazar E., Buck J., Levin L. R., Cann M. J., Wolfgang M. C., and Steegborn C. (2012) Crystal structure and regulation mechanisms of the CyaB adenylyl cyclase from the human pathogen Pseudomonas aeruginosa. J. Mol. Biol. 416, 271–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]