

Abstract
The pancreas secretes digestive proenzymes typically in their monomeric form. A notable exception is the ternary complex formed by proproteinase E, chymotrypsinogen C, and procarboxypeptidase A (proCPA) in cattle and other ruminants. In the human and pig pancreas binary complexes of proCPA with proelastases were found. To characterize complex formation among human pancreatic protease zymogens in a systematic manner, we performed binding experiments using recombinant proelastases CELA2A, CELA3A, and CELA3B; chymotrypsinogens CTRB1, CTRB2, CTRC, and CTRL1; and procarboxypeptidases CPA1, CPA2, and CPB1. We found that proCELA3B bound not only to proCPA1 (KD 43 nm) but even more tightly to proCPA2 (KD 18 nm), whereas proCELA2A bound weakly to proCPA1 only (KD 152 nm). Surprisingly, proCELA3A, which shares 92% identity with proCELA3B, did not form stable complexes due to the evolutionary replacement of Ala241 with Gly. The polymorphic nature of position 241 in both CELA3A (∼4% Ala241 alleles) and CELA3B (∼2% Gly241 alleles) points to individual variations in complex formation. The functional effect of complex formation was delayed procarboxypeptidase activation due to increased affinity of the inhibitory activation peptide, whereas proelastase activation was unchanged. We conclude that complex formation among human pancreatic protease zymogens is limited to a subset of proelastases and procarboxypeptidases. Complex formation stabilizes the inhibitory activation peptide of procarboxypeptidases and thereby increases zymogen stability and controls activation.
Keywords: complex, metalloprotease, pancreas, proteolytic enzyme, serine protease, elastase, pancreatitis, zymogen activation
Introduction
The exocrine pancreas produces digestive enzymes including serine- and metalloproteases secreted as inactive precursors (zymogens) that attain their active form in the duodenum. Physiological activation of protease zymogens is initiated by enteropeptidase-mediated activation of trypsinogen to trypsin followed by trypsin-mediated activation of chymotrypsinogens, proelastases, and procarboxypeptidases (1). Full activation of human procarboxypeptidases A1 (proCPA1)2 and A2 (proCPA2) requires additional cleavages by chymotrypsin C (CTRC) to facilitate dissociation of the inhibitory activation peptide (2). Renewed interest in digestive protease physiology was spurred by the recognition that mutations in cationic trypsinogen (PRSS1), the pancreatic secretory trypsin inhibitor (SPINK1), CTRC and CPA1 are strongly associated with chronic pancreatitis in humans (3–6). At the same time, information obtained from genome and pancreatic transcriptome sequencing allowed correct annotation of the full complement of digestive protease isoforms and opened up avenues for investigations using genetically-defined recombinant enzymes. These studies, in turn, led to novel insight into digestive protease function such as the central role of CTRC in the regulation of human trypsinogen (7–9) and procarboxypeptidase activation (2), the role of mesotrypsin (PRSS3) in degrading trypsin inhibitors (10, 11), and the significance of mutation-induced digestive protease misfolding in acinar cell stress and chronic pancreatitis (6, 12, 13).
A long known yet still unsolved enigma in digestive protease biochemistry is the physiological function of complex formation. Although most zymogens are secreted in their monomeric forms, ternary and binary complexes have also been identified (for reviews, see Refs. 14–17). The best studied example is the bovine ternary complex formed by proCPA (subunit I), chymotrypsinogen C (subunit II), and proproteinase E (subunit III), the ortholog of human chymotrypsin-like proelastases 3A and 3B (proCELA3A and proCELA3B) (18–23). Similar ternary complexes were found in other ruminants such as goat and sheep (24). Bovine ternary complexes isolated in earlier studies all contained an autolyzed, inactive form of proproteinase E, cleaved at the Val30-Asn31 peptide bond (25). This truncated proelastase was later shown to be a purification artifact due to the catalytic action of trace amounts of active elastase (26–28). In addition to the ternary complex, the bovine pancreas also contains lower levels of binary complexes of proCPA with chymotrypsinogen C (29) or proproteinase E (30). In contrast, the pig and human pancreas secretes binary complexes only, formed by proCPA and proproteinase E in the pig (31–33), and proCPA1 and proCELA3B or proCELA2A in humans (34–36). Interestingly, a relatively poorly characterized complex between proCPA2 and chymotrypsinogen B (CTRB) was also described in the rat pancreas (37). Finally, proCPA of the sei whale was found in complex with an unidentified protein, possibly chymotrypsinogen C (38).
The bovine ternary complex has been crystallized and its structure has been solved (39, 40). In this complex chymotrypsinogen C and proproteinase E are bound to different surfaces of the centrally located proCPA. The activation peptide of proCPA participates in most of the binding interactions, suggesting that the rate of proCPA activation might be affected by complex formation and that the complex should dissociate upon proCPA activation (41). In contrast, the activation peptides of chymotrypsinogen C and proproteinase E are not involved in binding contacts; therefore, activation of these zymogens should not be affected in the complex. Earlier functional studies using the chemically dissociated pig binary complex demonstrated small differences in the rate of trypsin-mediated proCPA activation, which was inconsistent with experimental observations using components of the bovine ternary complex (23, 32, 33). This discrepancy has not been resolved and the exact effect of complex formation on proCPA activation has remained uncertain.
Even though the physiological role of ternary and binary complexes formed by pancreatic protease zymogens has remained perplexing, investigations into this problem dwindled before the advent of the genomic era. Therefore, the objective of the present study was to perform a comprehensive analysis of complex formation among human pancreatic protease zymogens using recombinant proenzymes and to characterize the binding affinities and the functional significance of the complexes.
Results
Protease Zymogens Studied
There are three human pancreatic procarboxypeptidases encoded by the CPA1, CPA2, and CPB1 genes. Four genes code for chymotrypsinogens (CTRB1, CTRB2, CTRC, and CTRL1) and five genes for chymotrypsin-like proelastases (CELA1, CELA2A, CELA2B, CELA3A, and CELA3B). The CELA1 gene, which is the ortholog of the classic pig elastase, is not expressed in the human pancreas due to evolutionary mutations in its promoter region (42, 43); therefore, this isoform was not studied here. As their gene symbols suggest, both CELA2A-CELA2B and CELA3A-CELA3B gene pairs represent gene duplications in humans. CELA2B was also excluded from the present study as this protein has never been demonstrated in pancreatic secretions and the recombinantly expressed enzyme is inactive due to multiple evolutionary mutations (44). In contrast, both CELA3A and CELA3B are expressed in the human pancreas and their concentrations appear comparable at the mRNA and protein levels (45, 46). Finally, the three human trypsinogens encoded by the PRSS1, PRSS2, and PRSS3 genes were not included as trypsinogens studied so far have been always found monomeric (1).
Autolysis of ProCELA3A and ProCELA3B
Subunit III of the bovine ternary complex was originally identified as an inactive elastase cleaved at the Val30-Asn31 peptide bond (Ref. 25 and references therein). A similar defective elastase was detected in binary complexes with proCPA1 from human pancreas (34). Subsequent studies revealed that subunit III was a purification artifact resulting from the autolytic cleavage of proproteinase E by small amounts of active proteinase E (26–28). We found that despite every precaution to prevent trypsin contamination, our freshly made proCELA3B preparations were already partially autolyzed. As judged by N-terminal Edman degradation, typically about 60% of the fresh proCELA3B preparations were intact, and the rest showed cleavages at the Ser26-Ser27 (20%) and Val30-Asn31 (20%) peptide bonds (Fig. 1). In contrast, fresh proCELA3A preparations were uncleaved. Autolysis occurred during the purification procedure as proCELA3B in the conditioned medium of transfected cells was found intact. During storage at 4 °C, autolysis proceeded slowly with about 80% of proCELA3B cleaved in 6 weeks, with the predominant cleavage (55–60%) observed at the Val30-Asn31 peptide bond. In the same time period, preparations of proCELA3A also became partially cleaved at the Ser25-Ser26 (10%), Ser27-Arg28 (10%), and Val30-His31 (3%) peptide bonds. Surprisingly, when the tryptic activation site Arg28 was mutated in proCELA3B to His (R28H mutant), the fresh zymogen preparations were even more autolyzed (55% cleaved) with enhanced cleavage observed at the Ser26-Ser27 peptide bond (40%). The behavior of the R28H mutant indicated that autolytic cleavage was more likely to occur as a result of the intrinsic zymogen activity of proCELA3B rather than through inadvertent proelastase activation by trypsin contamination. As expected, mutation of the catalytic Ser in proCELA3B (S217A mutant) yielded zymogen preparations with intact N termini. Therefore, for the quantitative binding experiments discussed below we used the S217A mutant versions of proCELA3A and proCELA3B constructs.
FIGURE 1.
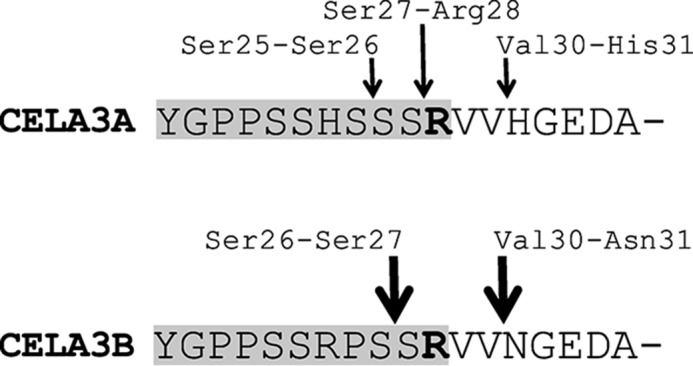
Autolytic cleavage sites in the activation peptide and N-terminal region of human proCELA3A and proCELA3B. The activation peptides (Tyr18-Arg28) are highlighted in gray. The activation site Arg28 is emboldened. See text for details.
Binding of Human Proelastases and Chymotrypsinogens to Procarboxypeptidases
To develop a simple screening assay for binding among various pancreatic protease zymogens, we took advantage of the availability of His-tagged and non-tagged expression constructs in our laboratory. We expressed all proenzymes in HEK 293T cells via transient transfection and mixed the conditioned media in different combinations of His-tagged and non-tagged zymogens. After a brief incubation to allow for binding, the mixture was passed through a nickel column; the column was washed with 20 mm imidazole and bound proteins were eluted with 250 mm imidazole. Four 0.5-ml fractions were collected and analyzed by SDS-PAGE and Coomassie Blue staining (Fig. 2). In the absence of binding, only the His-tagged zymogen was eluted, whereas complex formation resulted in co-elution of the non-tagged binding partner as well. We tested for potential binary complexes between non-tagged procarboxypeptidases (proCPA1, proCPA2, and proCPB1) and His-tagged proelastases (proCELA2A, proCELA3A, and proCELA3B) and chymotrypsinogens (CTRB1, CTRB2, CTRC, and CTRL1). As shown in Fig. 2, binding was detected between proCELA2A and proCPA1, proCELA3B and proCPA1, and proCELA3B and proCPA2, whereas other zymogen combinations did not result in measurable complex formation (data not shown for CTRB1, CTRB2, and CTRL1). The observation that proCPA2 formed a complex with human proCELA3B was novel, whereas complexes of proCELA3B and proCELA2A with proCPA1 were previously reported (34–36). The inability of proCELA3A to bind to procarboxypeptidases was surprising as this isoform is 92% identical to proCELA3B at the amino acid level. We also explored whether the CTRC zymogen might participate in ternary complexes with proCELA3B and proCPA1 or proCPA2. Non-tagged CTRC proenzyme was mixed with His-tagged proCELA3B and non-tagged proCPA1 or proCPA2. Because CTRC and proCELA3B migrate similarly on SDS-PAGE, the nickel column eluate was analyzed for the presence of CTRC by Western blots. No detectable CTRC zymogen was co-eluted with the proCELA3B-proCPA1 or proCELA3B-proCPA2 complexes (not shown).
FIGURE 2.

Binary complex formation of human pancreatic protease zymogens. Qualitative binding assays using His-tagged proelastases (proCELA, including proCELA2A, proCELA3A, and proCELA3B), His-tagged chymotrypsinogens (CTRB1, CTRB2, CTRC, and CTRL1), and non-tagged procarboxypeptidases (proCP, including proCPA1, proCPA2, and proCPB1) were performed as described under ”Experimental Procedures.“ The numbers above the lanes indicate the fractions collected after elution from the nickel column. None of the chymotrypsinogens formed complexes; representative negative results are shown for the CTRC zymogen only. Representative gel pictures of two binding experiments are shown.
Quantitative Binding Experiments with Purified Proelastases and Procarboxypeptidases
To characterize binding affinity between various human procarboxypeptidases and proelastases, we developed an equilibrium binding assay in which a fixed concentration of non-tagged proCPA1 (250 nm) and proCPA2 (100 nm) was incubated with increasing concentrations of His-tagged proelastases (0–1500 nm) in the presence of nickel resin. After 15 min incubation, His-tagged proelastases together with bound procarboxypeptidases were removed by centrifugation. The amount of the free (unbound) proCPA1 and proCPA2 remaining in the supernatant was determined by enzyme activity measurements and free proCPA1 and proCPA2 concentrations were plotted as a function of total proelastase concentrations. The equilibrium dissociation constants (KD) were determined by fitting the data points to the simple reversible binding equation, as described under “Experimental Procedures.” As shown in Fig. 3, proCELA3B bound to both proCPA1 and proCPA2 with relatively high affinity, as judged by the KD values of 43 ± 2 and 18 ± 1 nm, respectively. The slightly stronger binding observed with proCPA2 was surprising as a proCELA3B-proCPA2 complex has not been identified before in human pancreatic secretions. In contrast, only minimal binding of proCELA3A to proCPA1 and proCPA2 was detected with KD values estimated at 10.9 ± 1 and 7.1 ± 0.2 μm, respectively (Fig. 4). Thus, proCELA3A and proCELA3B exhibit at least 250- and 400-fold differences in binding affinity to proCPA1 and proCPA2, respectively. In agreement with a report of a proCELA2A-proCPA1 complex found in human pancreatic juice, we measured binding of proCELA2A to proCPA1 with a KD value of 152 ± 22 nm; which was about 4-fold weaker than binding of proCELA3B (Fig. 4). In contrast, proCELA2A bound to proCPA2A at least 33-fold weaker, with an estimated KD of 4.2 ± 0.4 μm.
FIGURE 3.
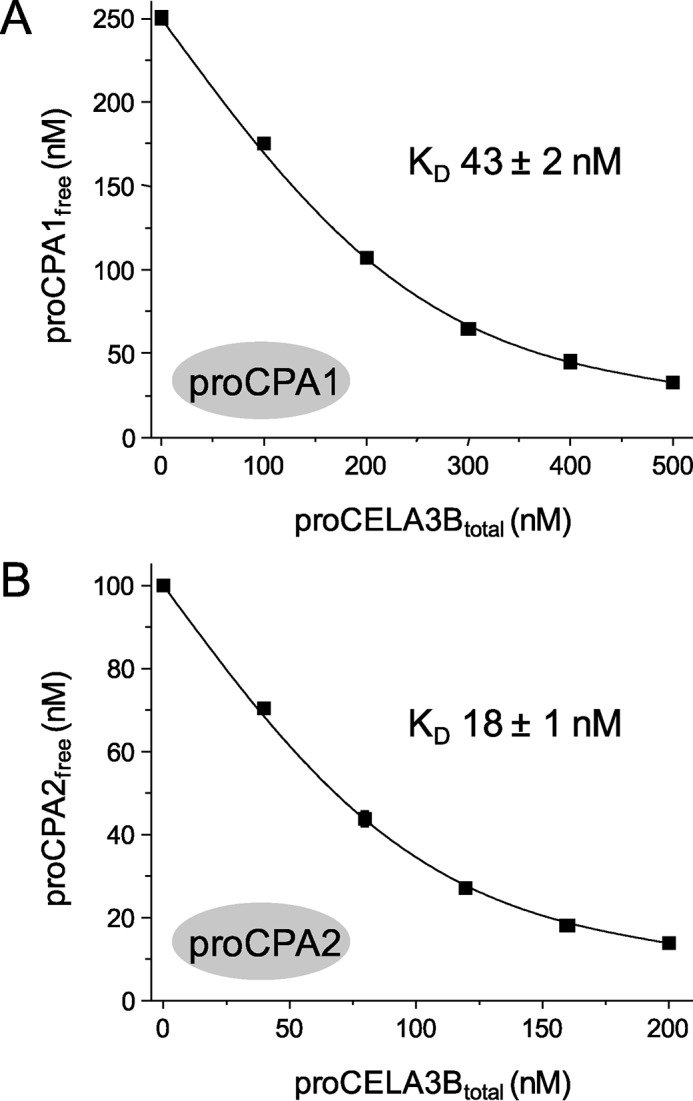
Binding of proCELA3B to proCPA1 (A) and proCPA2 (B). Equilibrium binding assays using purified proenzymes were carried out as described under ”Experimental Procedures.“ The free (unbound) proCPA concentrations were plotted as a function of the total proCELA3B concentration. Data points represent the average of three replicates ± S.E. Error bars may be smaller than symbol sizes. Data were fitted globally, the KD values and the error of the fits are indicated. The catalytically inactive S217A mutant of proCELA3B was used.
FIGURE 4.
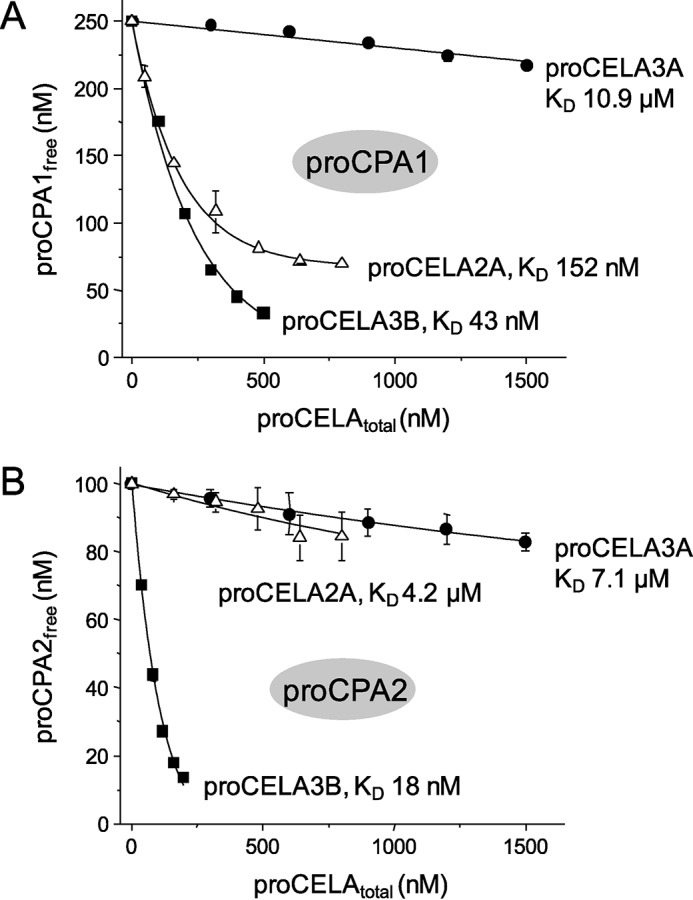
Binding of proCELA2A and proCELA3A to proCPA1 (A) and proCPA2 (B). Equilibrium binding assays using purified proenzymes were carried out as described in the legend to Fig. 3 and under ”Experimental Procedures.“ See text for the error of the fits. For comparison, binding data for wild-type proCELA3B from Fig. 3 are also indicated. Note that KD values in the micromolar range should be considered estimates. The catalytically inactive S217A mutant of proCELA3A was used.
Evolutionary Replacement of Ala241 with Gly in ProCELA3A Caused Loss of Binding to Procarboxypeptidases
Proelastases CELA3A and CELA3B exhibit 92% identity at the amino acid level yet only proCELA3B formed tight complexes with procarboxypeptidases. We mapped the differences between the two proleastases to the binding interface between bovine subunit III (proproteinase E) and proCPA in the published structure of the ternary complex (Fig. 5A). We found five amino acid differences between proCELA3A and proCELA3B at positions 77, 78, 79, 197, and 241, which could potentially affect binding. To test the significance of these evolutionary changes, we replaced these amino acids in proCELA3B with the corresponding residues from proCELA3A resulting in triple mutant S77R,S78D,W79L and single mutants S197T and A241G. Qualitative binding experiments indicated that mutation A241G had the most drastic effect as it essentially abolished binding, whereas slightly decreased binding was observed with the triple mutant and essentially unchanged binding with mutant S197T (Fig. 5B). When binding of proCELA3B mutant A241G was measured in a quantitative manner, KD values of 2.2 ± 0.1 and 1.0 ± 0.03 μm were determined against proCPA1 and proCPA2, respectively, indicating a more than 50-fold decreased affinity (Fig. 6). Still, compared with proCELA3A, binding of proCELA3B mutant A241G to proCPA1 and proCPA2 was at least 5- and 7-fold stronger, respectively, indicating that additional changes in proCELA3A contribute to the overall loss of affinity. Therefore, we measured binding of the S77R,S78D,W79L triple mutant to proCPA1 and proCPA2 and found KD values of 81 ± 5 and 130 ± 7 nm, respectively, indicating ∼2- and 7-fold reduced affinity, respectively, relative to wild-type proCELA3B (reviewed but not shown). Taken together, we can conclude that the major determinant for the defective binding of proCELA3A to procarboxypeptidases is the evolutionary change of Ala241 to Gly. This conclusion was further supported by qualitative binding experiments using the G241A proCELA3A mutant, which exhibited improved binding, particularly to proCPA1, relative to wild-type proCELA3A (Fig. 5C). Quantitative binding experiments confirmed the higher affinity of the proCELA3A G241A mutant toward proCPA1 (KD 270 ± 13 nm) and also to proCPA2 (KD 2.2 ± 0.1 μm), indicating 40- and 3.2-fold increased binding, respectively (Fig. 6).
FIGURE 5.
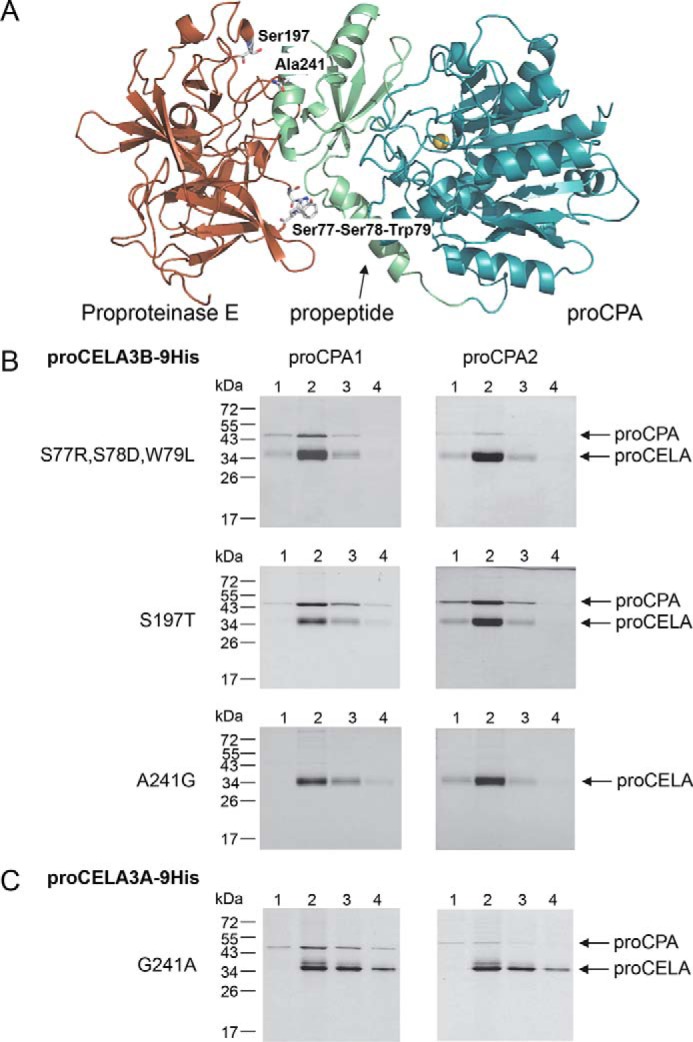
Structural determinants of defective proCELA3A binding to proCPA1 and proCPA2. A, the positions of amino acid differences between proCELA3A and proCELA3B were mapped to the binding interface between bovine proCPA and proproteinase E. For clarity, chymotrypsinogen C was omitted from the ternary complex (Protein Data Bank file 1PYT). Amino acid residues at or near the interface are indicated. B, binding of proCELA3B triple mutant S77R,S78D,W79L and single mutants S197T and A241G to proCPA1 and proCPA2. C, binding of proCELA3A mutant G241A to proCPA1 and proCPA2. Qualitative binding experiments using conditioned media with His-tagged proelastases and non-tagged procarboxypeptidases were carried out as described under ”Experimental Procedures.“ The numbers above the lanes indicate the fractions eluted from the nickel column. Representative gel pictures of two experiments are shown.
FIGURE 6.
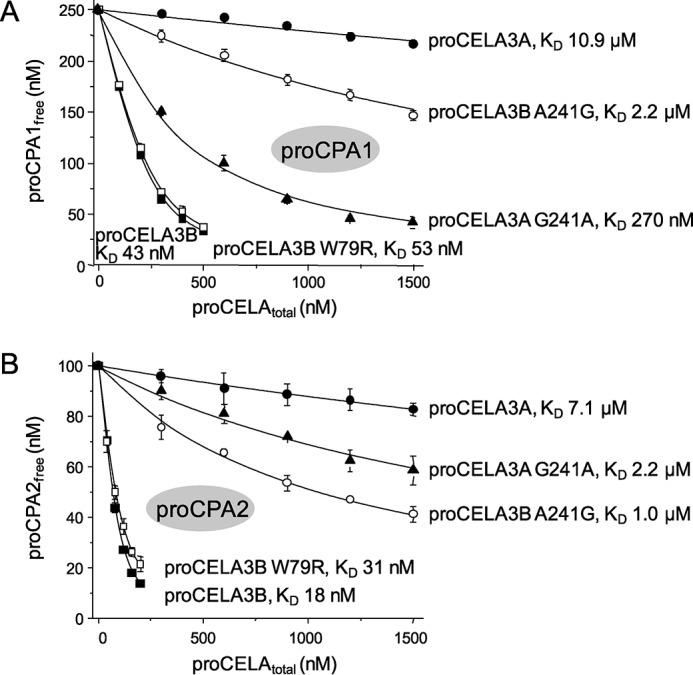
Binding of proCELA3A variant G241A and proCELA3B variants W79R and A241G to proCPA1 (A) and proCPA2 (B). Equilibrium binding assays using purified proenzymes were carried out as described in the legend to Fig. 3 and under ”Experimental Procedures.“ For comparison, binding data for wild-type proCELA3A and proCELA3B from Figs. 3 and 4 are also indicated. Data points represent the average of three replicates ± S.E. Error bars may be smaller than symbol sizes. See text for the error of the fits. Note that KD values in the micromolar range should be considered estimates. The proCELA3A and proCELA3B constructs used contained the S217A catalytic mutation.
Complex Formation of Natural CELA3A and CELA3B Variants with Procarboxypeptidases
To identify relatively common CELA3A and CELA3B variants in the general population, we queried the Exome Variant Server database of the NHLBI Exome Sequencing Project. Table 1 lists the coding sequence (exonic) variants that were found to occur with an allele frequency higher than 1%. When the variants were mapped on the structure of the bovine ternary complex, only CELA3B variants W79R and A241G and CELA3A variant G241A were found at or near the binding interface with proCPA. As described above, variant A241G diminished binding of proCELA3B to proCPA1 and proCPA2 by more than 50-fold, and, conversely, variant G214A increased binding of proCELA3A to proCPA1 by 40-fold and to proCPA2 by 3.2-fold (see Fig. 6). Because position 241 is a major determinant of proelastase binding to proCPA, the polymorphic nature of this amino acid points to individual variability of complex formation in human pancreatic secretions. On the other hand, variant W79R had a minimal effect on proCELA3B binding to proCPA1 or proCPA2; KD values of 53 ± 2 and 31 ± 1 nm were measured, respectively (Fig. 6).
TABLE 1.
Genetic variants of CELA3A and CELA3B in the population
Data were taken from the Exome Variant Server of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project. Only variants with a combined allele frequency exceeding 1% were listed. The number of European-American (EA) and African-American (AA) alleles sequenced were 8600 and 4394 for CELA3A and 8600 and 4406 for CELA3B, respectively. Note that the genomic reference sequence (chromosome 1 primary assembly) for CELA3A contains the G241A variant and the reference sequence for CELA3B contains the W79R variant. Here we used the common alleles as reference (corresponding to the chromosome 1 alternate assembly) and indicated the minor alleles as changes at these positions.
| cDNA change | Amino acid change | EA alleles | AA alleles | All alleles |
|---|---|---|---|---|
| CELA3A | ||||
| c.722G>C | G241A | 2.4% | 7.9% | 4.3% |
| CELA3B | ||||
| c.235T>C | W79R | 0.8% | 14.3% | 5.4% |
| c.401A>T | Q134L | 0.1% | 4.0% | 1.4% |
| c.625A>G | I209V | 3.2% | 0.5% | 2.3% |
| c.629G>A | R210H | 1.6% | 5.8% | 3.0% |
| c.722C>G | A241G | 1.7% | 1.8% | 1.7% |
Complex Formation Reduces the Rate of Procarboxypeptidase Activation
Because the activation peptide of procarboxypeptidases is involved in binding to proCELA3B, activation kinetics of proCPA1 and proCPA2 in complex are expected to be delayed relative to their monomeric forms. Activation of human proCPA1 and proCPA2 requires an initial cleavage at the C terminus of the activation peptide by trypsin, which results in the appearance of 10–15% of the full carboxypeptidase activity as the inhibitory activation peptide remains attached (2). Subsequently, CTRC cleaves the severed activation peptide at multiple sites and facilitates its dissociation resulting in full carboxypeptidase activity (2). We found that both activation steps were affected by complex formation. When activation of 200 nm proCPA1 and proCPA2 was initiated with 5 nm trypsin, only low levels of carboxypeptidase activity developed, which was reduced by about 50% as a result of complex formation with 2 μm proCELA3B (Fig. 7). Because trypsin also activates proCELA3B and elastase can partially activate proCPA1 and proCPA2, in these experiments we used the catalytically inactive S217A proCELA3B mutant, thereby excluding this confounding factor. Addition of 5 nm CTRC to the trypsin-cleaved proCPA1 or proCPA2 resulted in rapid development of carboxypeptidase activity and the presence of 2 μm proCELA3B reduced rates of activation by ∼2- and 5-fold, respectively (Fig. 7). The stronger effect observed with proCPA2 is consistent with its somewhat tighter binding to proCELA3B (see Fig. 3). Importantly, the low-affinity proCELA3B mutant A241G had no effect whatsoever on proCPA1 activation and had only a small effect on proCPA2 activation, confirming that the observed differences in activation rates were related to complex formation between the protease zymogens.
FIGURE 7.
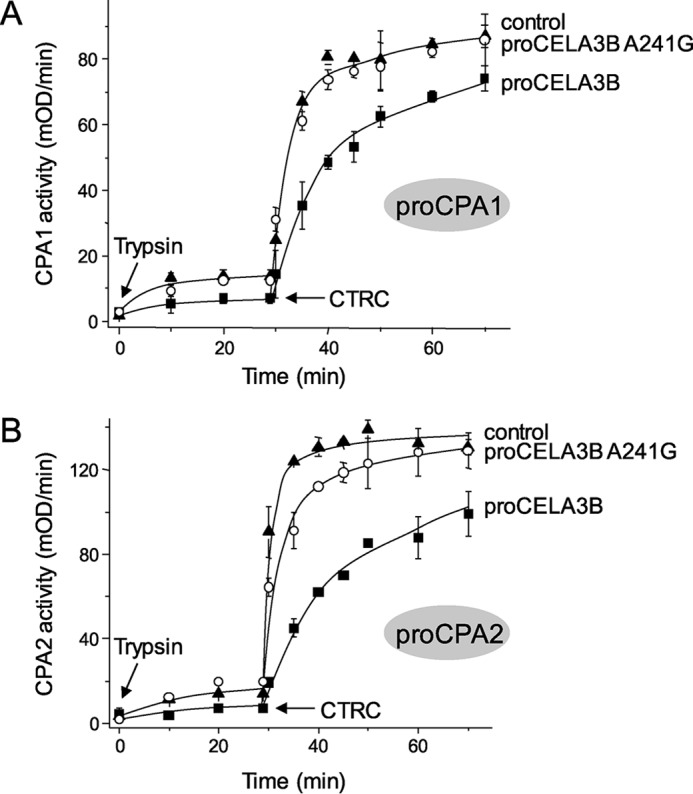
Effect of wild-type proCELA3B and mutant A241G on the activation of proCPA1 (A) and proCPA2 (B) by trypsin and CTRC. Procarboxypeptidases at 200 nm concentration were incubated at 37 °C in the absence (control) or presence of 2 μm wild-type proCELA3B or A241G mutant in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20 (final concentrations) in 200 μl final volume. Where indicated by arrows, 5 nm human cationic trypsin and 5 nm human CTRC were added. At given times, 8- (CPA1) or 15-μl (CPA2) aliquots were withdrawn and carboxypeptidase activity was measured as described under ”Experimental Procedures.“ Data points represent the average of three replicates ± S.E. Error bars may be smaller than symbol sizes. The catalytically inactive S217A mutants of proCELA3B and proCELA3B A241G were used.
Complex Formation Has No Effect on Proelastase Activation or Zymogen Activity
Activation of proCELA3B (150 nm) by trypsin (2.5 nm) was unchanged when performed in the absence or presence of 600 nm proCPA1 or proCPA2 (reviewed but not shown). This finding is consistent with previous observations on the pig binary complex (33). To evaluate whether binding of proCELA3B to procarboxypeptidases alters the intrinsic zymogen activity of the proelastase, we used the non-activable R28H proCELA3B (500 nm) mutant and measured cleavage of a chromogenic peptide substrate in the absence and presence of 500 nm proCPA1. No differences were found (reviewed but not shown).
Complex Formation Stabilizes Binding of the Inhibitory Activation Peptide of Procarboxypeptidases
The activation peptides of proCPA1 and proCPA2 act as tight-binding tethered inhibitors (15–17). After cleavage with trypsin, the activation peptides remain bound to the enzymes with slightly weakened affinity resulting in the appearance of low levels of carboxypeptidase activity (2). The activity versus concentration plots of trypsin-cleaved proCPA1 and proCPA2 follow a square root function; in agreement with the law of mass action (Fig. 8). From these plots we derived KD values of 5.2 ± 0.1 and 6.8 ± 0.4 nm for proCPA1 and proCPA2, respectively. These updated values are somewhat higher than those previously reported (0.8 nm) but still lie in the low nanomolar range (2). When the experiments were repeated in the presence of 500 nm proCELA3B (S217A mutant), we found that complex formation resulted in higher affinity binding of the activation peptides. Thus, KD values of 1.4 ± 0.1 nm were measured for both proCPA1 and proCPA2 complexes, indicating a 4–5-fold increase in binding affinity (Fig. 8). In contrast, 500 nm of the weakly binding A241G proCELA3B (S217A) mutant had only a minimal (1.2-fold) effect on the affinity of the proCPA1 and proCPA2 activation peptides, as judged by the KD values obtained: 4.2 ± 0.1 nm and 5.4 ± 0.2 nm, respectively (Fig. 8). We conclude that the likely physiological role of complex formation is stabilization of procarboxypeptidase zymogens by increasing the affinity of their inhibitory activation peptides.
FIGURE 8.
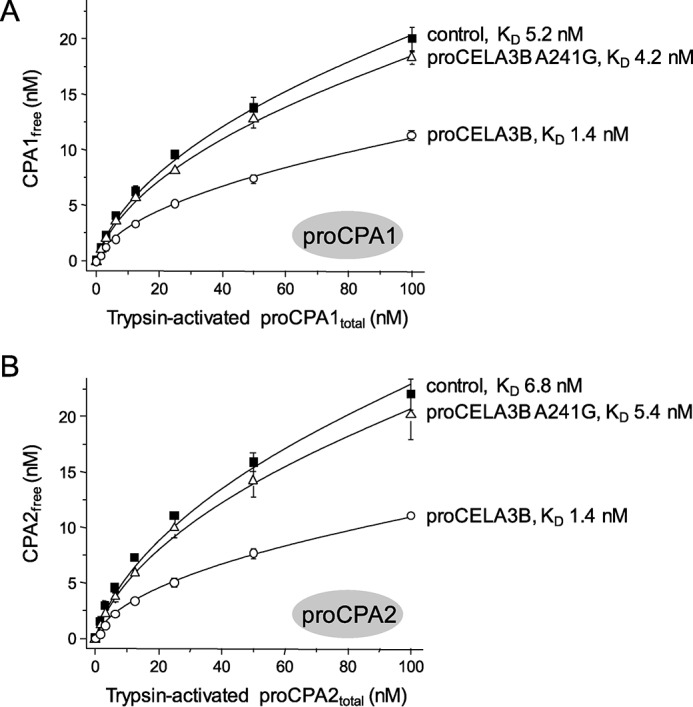
Effect of wild-type proCELA3B and mutant A241G on the binding of the inhibitory activation peptides of proCPA1 (A) and proCPA2 (B). Procarboxypeptidases were incubated at 2 μm concentration with 50 nm human cationic trypsin at 37 °C in 0.1 m Tris-HCl (pH 8.0), 15 mm NaCl, 1 mm CaCl2, and 0.05% Tween 20 (final concentrations) in 100 μl final volume. After 30 min incubation, activated proenzymes were diluted to the indicated concentrations in 100 μl final volume with assay buffer (0.1 m Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm CaCl2, and 0.05% Tween 20) and incubated for 10 min at 22 °C in the absence (control) or presence of 500 nm wild-type proCELA3B or A241G mutant. Carboxypeptidase activity was then measured by adding 10 μl of N-[4-methoxyphenylazoformyl]-l-phenylalanine substrate to 60 μm final concentration, as described under ”Experimental Procedures.“ Carboxypeptidase activities were converted to free CPA1/CPA2 concentrations by dividing the activity values with the slope of the linear concentration-activity plots of CPA1/CPA2 fully activated with trypsin plus CTRC (see supplemental Fig. S1 in Ref. 2). Data points represent the average of three replicates ± S.E. Error bars may be smaller than symbol sizes. Data points were fitted to the equation: y = (−K + sqrt(K∧2 + 4Kx))/2, where K is the equilibrium dissociation constant. The variable x is the total concentration of trypsin-activated proCPA1/proCPA2 present in the reaction and y is the concentration of free CPA1/CPA2 in equilibrium. The catalytically inactive S217A mutants of proCELA3B and proCELA3B A241G were used.
Discussion
Complex formation of mammalian pancreatic procarboxypeptidases with proelastases and chymotrypsinogen C was intensely researched in the pre-genomic era. These investigations identified ternary complexes in ruminants and binary complexes in humans and pigs. The physiological significance of complex formation has never been solved, although coordination of zymogen activation seemed a plausible possibility (14–17). Our interest turned to these older studies when we identified that loss-of-function CPA1 mutations increased risk for chronic pancreatitis in children and we were searching for possible mechanisms to explain pathogenesis (6). We realized that complex formation among pancreatic protease zymogens has never been studied using post-genomic era tools. Today the full complement of digestive proteases, including all isoforms, expressed by the human pancreas has been characterized at the genomic and transcriptomic levels, which sets the stage for recombinant expression of genetically defined proteins and comprehensive, systematic studies at the protein level.
Using qualitative and quantitative binding assays we confirmed that human proCELA3B and, to a lesser degree, proCELA2A bind to proCPA1, and made the new observation that proCELA3B also binds to proCPA2. No other complex formation was observed among human proelastases, chymotrypsinogens, and procarboxypeptidases. KD values for proCELA3B binding were in the ∼20–40 nm range, with stronger binding to proCPA2, whereas proCELA2A showed weaker affinity (KD ∼150 nm). A previous study measured binding affinities for the dissociated components of the bovine ternary complex and found that subunit III (proproteinase E) bound to the binary complex of proCPA and chymotrypsinogen C with a KD value of 160 nm, which, considering the different species and methodology used, seems to be in reasonable agreement with our results (47). The higher affinity of proCELA3B toward proCPA2 versus proCPA1 was an unexpected observation because this complex has never been isolated from human pancreatic juice so far. In fact, the seminal study by Pascual et al. (35), which chromatographically resolved all forms of human procarboxypeptidases, found only monomeric proCPA2. Why this complex escaped detection so far is not readily apparent but it may be related to the protein extraction methods used or some peculiar chromatographic behavior. We also note that proCPA1 is more abundant than proCPA2 in humans (see Fig. 1 in (48)), therefore, proCPA1 complexes may be more easily isolated.
The gene encoding the ortholog of the bovine and pig proproteinase E had duplicated in humans giving rise to the CELA3A and CELA3B genes, which encode proelastases that are 92% identical in their primary structure. Despite the high degree of identity, proCELA3A bound to procarboxypeptidases at least 250–400-fold weaker than proCELA3B. The major determinant of the affinity loss was the Ala241 to Gly evolutionary change in CELA3A relative to CELA3B, which weakened binding more than 50-fold. Inspection of the crystal structure of the bovine ternary complex indicates that replacement of Ala241 with Gly would result in the loss of a hydrophobic contact between the side chain methyl group of Ala and the aromatic phenyl ring of Phe78 in proCPA1. In addition, the Gly substitution may cause local changes in conformation thereby compromising additional interactions. Interestingly, position 241 is polymorphic in a small percentage of the population; both CELA3A G241A and CELA3B A241G variants have been identified (see Table 1). These polymorphic forms are expected to cause individual variations in procarboxypeptidase binding in heterozygous carriers and offer a unique opportunity to investigate the significance of complex formation in different gastrointestinal diseases (e.g. pancreatitis) using genetic association studies. These variations also suggest that complex formation and zymogen activation do not need to be tightly regulated for normal digestive physiology.
The physiological function of complex formation has puzzled researchers since the 1960s (see reviews in Refs. 14–17). The crystal structure of the bovine ternary complex suggests that proCPA activation should be delayed as the binding partners shield the proCPA activation peptide (39–40). Comparing chemically dissociated and re-associated bovine and porcine complexes, earlier studies found relatively small and somewhat conflicting differences in the rate and extent of procarboxypeptidase activation between monomeric and complexed proenzymes (23, 32, 33). In the pig binary complex, which probably resembles the human situation more closely, the initial rate of proCPA activation by trypsin was slower relative to the monomeric enzyme (33). As expected, activation of pig proproteinase E was unaffected by complex formation (33). We recently demonstrated that in the activation of human proCPA1 and proCPA2 trypsin-mediated cleavage serves only as the initiating step and needs to be followed by CTRC-mediated cleavages of the activation peptide (2). In our experiments presented here we studied these two steps in the activation process separately. We found that binding of proCELA3B decreased the initial carboxypeptidase activity generated by trypsin and subsequent addition of CTRC to the trypsin-cleaved procarboxypeptidases resulted in slower development of full carboxypeptidase activity from the complex versus the monomeric proenzyme. More importantly, we demonstrated that binding of proCELA3B to the inhibitory activation peptide of proCPA1 and proCPA2 increased its affinity toward the CPA1 and CPA2 enzymes. This finding strongly suggests that a physiological role of complex formation is the stabilization of the proCPA1 and proCPA2 zymogens and it also explains the less efficient activation of the complexes by trypsin and CTRC. On the other hand, the intrinsic zymogen activity and trypsin-mediated activation of human proCELA3B are unaffected by complex formation.
In summary, using recombinant proenzymes free of contaminating proteases we characterized binding of human proleastases to procarboxypeptidases. Novel findings include the observations that proCELA3B binds not only to proCPA1 but also to proCPA2 and proCELA3A does not form complexes largely due to the evolutionary replacement of Ala241 with Gly. Complex formation increases binding affinity of the inhibitory activation peptide of procarboxypeptidases and thereby stabilizes the zymogen state and hinders activation.
Experimental Procedures
Nomenclature
Amino acid residues were numbered starting with the initiator methionine of the primary translation product; according to the recommendations of the Human Genome Variation Society. The official gene symbols were used to denote human pancreatic proteases with a “pro” prefix to indicate the zymogen form. Note that the ELA (elastase) gene symbol has been recently changed to CELA (chymotrypsin-like elastase). The CELA3A and CELA3B genomic reference sequences on chromosome 1 (primary assembly, NC_000001.11) contain the G241A and W79R variants, respectively (see Table 1). Thus, these reference sequences represent polymorphic minor alleles. The reference sequences in the alternate assembly (NC_018912.2) correspond to the common alleles with respect to these two positions. To be consistent with published literature, we kept the historical proproteinase E designation for the bovine and porcine orthologs of human proCELA3A and proCELA3B.
Plasmid Construction and Mutagenesis
Construction of expression plasmids for human CELA3A, CELA3B, CTRC, CTRB1, CTRB2, CTRL1, CPA1, CPA2, and CPB1 in the pcDNA3.1(−) vector was described previously (2, 6, 8, 9, 49, 50). C-terminal His10 tags were engineered to the proelastase and chymotrypsinogen expression plasmids using gene synthesis (GenScript, Piscataway, NJ). Because the C-terminal amino acid of CELA3A and CELA3B is His, the added tag contained nine His residues. The coding DNA for human CELA2A with a C-terminal His10 tag was custom synthesized (GenScript) according to the sequence of IMAGE clone 6226278 (GenBankTM CA952548) and cloned into pcDNA3.1(−) using XhoI and BamHI restriction sites. Mutations were introduced by overlap extension PCR mutagenesis.
Expression and Purification of Human Pancreatic Protease Precursors
All protease zymogens were expressed in HEK 293T cells via transient transfection and purified from the conditioned medium. HEK 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific, Waltham, MA) with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) and 4 mm l-glutamine at 37 °C, in 5% CO2, in 525 cm2 three-layer tissue culture flasks to 80–90% confluence. Transfections were carried out using 180 μg of plasmid DNA and 450 μl of Lipofectamine 2000 (Thermo Fisher Scientific) reagent in 120 ml of medium. After 16–20 h, cells were washed and covered with 120 ml of Opti-MEM medium (Thermo Fisher Scientific). Conditioned media were harvested after 48 h incubation, replaced with 120 ml of fresh Opti-MEM, and harvested again at 96 h. His-tagged proenzymes were purified from 240 ml of conditioned media using nickel affinity chromatography on a 5-ml Ni-NTA Superflow Cartridge (number 30760, Qiagen, Valencia, CA), with the exception of proCELA2A, which was purified from 720 ml of media on a 1-ml Ni-NTA cartridge (number 30721, Qiagen). The nickel column was equilibrated with buffer NPI-20 (50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazole (pH 8.0)) and the medium was loaded at a flow rate of 4 ml/min using a loading pump. After the column was washed with NPI-20, zymogens were eluted with buffer NPI-250 (50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole (pH 8.0)) at a flow rate of 2 ml/min and 5-ml fractions were collected. When using the 1-ml Ni-NTA cartridge, the elution rate was 1 ml/min and 1-ml fractions were collected. Aliquots (25–50 μl) of the fractions were analyzed by SDS-PAGE and fractions with the highest zymogen concentration were pooled (typically two fractions, 10 ml total volume) and dialyzed in a 8–10-kDa molecular mass cut off (MWCO) dialysis device (Spectra/Por Float-A-Lyzer G2 Dialysis Device, number G235055, 5 ml, Spectrum Labs, Rancho Dominguez, CA) for 36 h against two or three changes of 2–3 liters of 0.1 m Na-HEPES (pH 8.0), 150 mm NaCl (proCELA3A and proCELA3B), or 0.1 m Tris-HCl (pH 8.0), 100 mm NaCl (proCELA2A). Finally, the dialyzed zymogens were concentrated to a volume of 1–3 ml by centrifugation at 1500 × g in a 10-kDa MWCO Vivaspin concentrator.
Non-tagged procarboxypeptidases were purified by anion-exchange chromatography. Conditioned media (240 ml) were first dialyzed against 20 mm Tris-HCl (pH 8.0) and loaded directly onto a MonoQ 5/50 GL column (GE Healthcare Life Sciences) connected to an Äkta Pure chromatography system (GE Healthcare Life Sciences). The column was developed with a NaCl gradient (0–0.5 M) over 30 min at a flow rate of 1 ml/min. Fractions (1 ml) were collected and analyzed by SDS-PAGE and Coomassie Blue staining. The purest fractions (>90%) were used for experiments without further concentration. The fractions contained ∼200 mm NaCl, which became negligible at the dilutions used for the experiments.
Concentration Determination of Zymogen Preparations
Concentrations of purified proenzyme solutions were estimated from their ultraviolet (UV) absorbance at 280 nm, using the following extinction coefficients (in m−1 cm−1 units) calculated with the web-based ProtParam tool: proCELA2A 73,505; proCELA3A 76,025; proCELA3B 74,535; proCELA3B mutant W79R, and triple mutant S77R,S78D,W79L 69,035; proCPA1 72,435; proCPA2 66,600. Typical proelastase preparations were in the 2–10 μm concentration range, whereas proCPA preparations were 20–35 μm. Before equilibrium binding assays, proCELA3B was titrated against proCPA1 and proCPA2 to ensure “self-consistent” concentrations. The titration experiments followed the protocol of the binding assay described below except proCPA concentrations were fixed at 1 μm and proCELA3B was varied between 0 and 1 μm.
Qualitative Binding Assays
To screen for possible binding partners among pancreatic protease zymogens, typically 10–15 ml of conditioned medium containing a given His-tagged proleastase or chymotrypsinogen was mixed with 5 ml of conditioned medium with a non-tagged procarboxypeptidase. After 1 h incubation at 22 °C, the mixture was loaded onto a Bio-Rad polyprep chromatography column containing 0.5 ml of packed Ni-NTA Superflow resin (number 30410, Qiagen), pre-equilibrated with NPI-20 buffer. The column was washed with 10 ml of NPI-20 and eluted with 2.5 ml of NPI-250 buffer. Fractions (0.5 ml) were collected and 150 μl of each was precipitated with 10% trichloroacetic acid (final concentration) and analyzed by 15% SDS-PAGE and Coomassie Blue staining. Co-elution of non-tagged procarboxypeptidases with His-tagged proelastases indicated binding, whereas in the absence of binding procarboxypeptidases were recovered in the flow-through. In control experiments we found that none of the non-tagged procarboxypeptidases bound nonspecifically to the nickel column. Although not directly relevant to the experiments presented, non-tagged forms of proelastases and chymotrypsinogens were also tested for nonspecific adsorption to the nickel column and proCELA3A was found to exhibit some binding, presumably due to His residues located at its N and C termini. This property did not interfere with our binding assays in which His-tagged proCELA3A was used.
Equilibrium Binding Assays
Binding of proelastases to human procarboxypeptidases was characterized by determining the KD value of the reaction in equilibrium. Procarboxypeptidases at a fixed concentration (proCPA1 250 nm, proCPA2 100 nm) were incubated with increasing concentrations of His-tagged proelastases (typically 0–500 or 0–1500 nm, as indicated) in the presence of Ni-NTA Superflow resin. Before use, the Ni-NTA resin beads were washed twice and re-suspended in Tris-HCl (pH 8.0), 150 mm NaCl, and 0.05% Tween 20. Typically, 40 μl of beads were used per 100 μl of the final reaction volume, which was found in preliminary experiments to remove His-tagged proelastases completely in the 0–500 nm concentration range. In the experiments where the proelastase concentration range was 0–1500 nm, beads were increased to 75 μl in 150 μl of reaction volume. Incubations were performed in 0.1 m Tris-HCl (pH 8.0), 150 mm NaCl, and 0.05% Tween 20 (final concentrations) at 22 °C with gentle manual shaking. In preliminary experiments using 15 min, 1- and 16-h incubation times we found no difference in binding, indicating that equilibrium was reached rapidly. Therefore, we routinely used 15-min incubation times after which the nickel beads containing the bound complexes were removed by centrifugation at 1000 rpm for 1 min. The free (unbound) proCPA in the supernatant was activated with 50 nm human cationic trypsin and 25–50 nm human CTRC (final concentrations) for 30 min at 37 °C and carboxypeptidase activity was measured from 10 to 30 μl of supernatant as described below. Residual CPA activities were converted to concentrations using the activity of the proCPA samples incubated in the absence of proleastases as the 100% reference value. The free proCPA concentration was plotted as a function of the total proelastase concentration and the experimental points were fitted with the following equation: y = E − (E + x + K-sqrt((E + x + K)2-4Ex))/2, where the independent variable x represents the total proelastase concentration, the dependent variable y is the free proCPA concentration in equilibrium; K is KD and E designates the total proCPA concentration; sqrt, square root function.
Measurement of Carboxypeptidase Activity
Enzymatic activity of CPA1 and CPA2 were determined using the N-[4-methoxyphenylazoformyl]-l-phenylalanine substrate (number M-2245, Bachem, Torrance, CA) at 60 μm final concentration in 100 or 200 μl final volume (2, 6, 50, 51). Samples were supplemented with assay buffer (0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, 0.05% Tween 20) to 90 or 180 μl volume and the reaction was started by adding 10 or 20 μl of 600 μm substrate. The decrease in absorbance was followed at 350 nm for 5 min in a SPECTRAmax Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA). Rates of substrate cleavage were calculated from fits to the linear portions of the curves and expressed in mOD/min units.
Experimental Uncertainty and Reproducibility
Qualitative binding experiments were carried out from at least two independent transfections; representative gels are shown. Quantitative binding experiments were performed in three technical replicates and average values with S.E. were plotted. The data points were fitted globally and the error of the fit was indicated.
Author Contributions
A. S., C. P., and M. B. designed and performed experiments, analyzed data, prepared figures for publication, and approved the final version of the manuscript. M. S. T. and H. W. designed the project and analyzed data. M. S. T. wrote the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK095753, R01 DK082412, and R01 DK058088. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CPA1
- carboxypeptidase A1
- CELA1
- chymotrypsin-like elastase 1
- CELA2A
- chymotrypsin-like elastase 2A
- CELA2B
- chymotrypsin-like elastase 2B
- CELA3A
- chymotrypsin-like elastase 3A
- CELA3B
- chymotrypsin-like elastase 3B
- CPA2
- carboxypeptidase A2
- CPB1
- carboxypeptidase B1
- CTRB1
- chymotrypsin B1
- CTRB2
- chymotrypsin B2
- CTRC
- chymotrypsin C
- CTRL1
- chymotrypsin-like enzyme 1
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Rinderknecht H. (1986) Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig. Dis. Sci. 31, 314–321 [DOI] [PubMed] [Google Scholar]
- 2. Szmola R., Bence M., Carpentieri A., Szabó A., Costello C. E., Samuelson J., and Sahin-Tóth M. (2011) Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2. J. Biol. Chem. 286, 1819–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K. Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., and Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 [DOI] [PubMed] [Google Scholar]
- 4. Witt H., Luck W., Hennies H. C., Classen M., Kage A., Lass U., Landt O., and Becker M. (2000) Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 25, 213–216 [DOI] [PubMed] [Google Scholar]
- 5. Rosendahl J., Witt H., Szmola R., Bhatia E., Ózsvári B., Landt O., Schulz H. U., Gress T. M., Pfützer R., Löhr M., Kovacs P., Blüher M., Stumvoll M., Choudhuri G., Hegyi P., et al. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Witt H., Beer S., Rosendahl J., Chen J. M., Chandak G. R., Masamune A., Bence M., Szmola R., Oracz G., Macek M. Jr., Bhatia E., Steigenberger S., Lasher D., Bühler F., Delaporte C., et al. (2013) Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 45, 1216–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nemoda Z., and Sahin-Tóth M. (2006) Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 281, 11879–11886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szmola R., and Sahin-Tóth M. (2007) Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szabó A., and Sahin-Tóth M. (2012) Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 287, 20701–20710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szmola R., Kukor Z., and Sahin-Tóth M. (2003) Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J. Biol. Chem. 278, 48580–48589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salameh M. A., Soares A. S., Hockla A., and Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 [DOI] [PubMed] [Google Scholar]
- 12. Kereszturi E., Szmola R., Kukor Z., Simon P., Weiss F. U., Lerch M. M., and Sahin-Tóth M. (2009) Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum. Mutat. 30, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balázs A., Hegyi P., and Sahin-Tóth M. (2016) Pathogenic cellular role of the p.L104P human cationic trypsinogen variant in chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 310, 477–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chapus C., Puigserver A., and Kerfélec B. (1988) The bovine pro-carboxypeptidase A-S6 ternary complex: a rare case of a secreted protein complex. Biochimie 70, 1143–1151 [DOI] [PubMed] [Google Scholar]
- 15. Avilés F. X., Vendrell J., Guasch A., Coll M., and Huber R. (1993) Advances in metallo-procarboxypeptidases. Emerging details on the inhibition mechanism and on the activation process. Eur. J. Biochem. 211, 381–389 [DOI] [PubMed] [Google Scholar]
- 16. Ventura S., Gomis-Rüth F. X., Puigserver A., Avilés F. X., and Vendrell J. (1997) Pancreatic procarboxypeptidases: oligomeric structures and activation processes revisited. Biol. Chem. 378, 161–165 [PubMed] [Google Scholar]
- 17. Vendrell J., Querol E., and Avilés F. X. (2000) Metallocarboxypeptidases and their protein inhibitors. Structure, function and biomedical properties. Biochim. Biophys. Acta 1477, 284–298 [DOI] [PubMed] [Google Scholar]
- 18. Keller P. J., Cohen E., and Neurath H. (1958) Procarboxypeptidase: II. chromatographic isolation, further characterization, and activation. J. Biol. Chem. 230, 905–915 [PubMed] [Google Scholar]
- 19. Brown J. R., Cox D. J., Greenshields R. N., Walsh K. A., Yamasaki M., and Neurath H. (1961) The chemical structure and enzymatic functions of bovine procarboxypeptidase A. Proc. Natl. Acad. Sci. U.S.A. 47, 1554–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown J. R., Greenshields R. N., Yamasaki M., and Neurath H. (1963) The subunit structure of bovine procarboxypeptidase A-S6: chemical properties and enzymatic activities of the products of molecular disaggregation. Biochemistry 2, 867–876 [DOI] [PubMed] [Google Scholar]
- 21. Yamasaki M., Brown J. R., Cox D. J., Greenshields R. N., Wade R. D., and Neurath H. (1963) Procarboxypeptidase A-S6: further studies of its isolation and properties. Biochemistry 2, 859–866 [DOI] [PubMed] [Google Scholar]
- 22. Puigserver A., Vaugoyeau G., and Desnuelle P. (1972) On subunit II of bovine procarboxypeptidase A: properties after alkaline dissociation. Biochim. Biophys. Acta 276, 519–530 [DOI] [PubMed] [Google Scholar]
- 23. Puigserver A., and Desnuelle P. (1977) Reconstitution of bovine procarboxypeptidase A-S6 from the free subunits. Biochemistry 16, 2497–2501 [DOI] [PubMed] [Google Scholar]
- 24. Kerfelec B., Chapus C., and Puigserver A. (1985) Existence of ternary complexes of procarboxypeptidase A in the pancreas of some ruminant species. Eur. J. Biochem. 151, 515–519 [DOI] [PubMed] [Google Scholar]
- 25. Pignol D., Gaboriaud C., Michon T., Kerfelec B., Chapus C., and Fontecilla-Camps J. C. (1994) Crystal structure of bovine procarboxypeptidase A-S6 subunit III, a highly structured truncated zymogen E. EMBO J. 13, 1763–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Avilés F. X., Pascual R., Salva M., Bonicel J., and Puigserver A. (1989) Generation of a subunit III-like protein by autolysis of human and porcine proproteinase E in a binary complex with procarboxypeptidase A. Biochem. Biophys. Res. Commun. 163, 1191–1196 [DOI] [PubMed] [Google Scholar]
- 27. Pascual R., Vendrell J., Avilés F. X., Bonicel J., Wicker C., and Puigserver A. (1990) Autolysis of proproteinase E in bovine procarboxypeptidase A ternary complex gives rise to subunit III. FEBS Lett. 277, 37–41 [DOI] [PubMed] [Google Scholar]
- 28. Gomis-Rüth F. X., Gómez-Ortiz M., Vendrell J., Ventura S., Bode W., Huber R., and Avilés F. X. (1998) Cutting at the right place: the importance of selective limited proteolysis in the activation of proproteinase E. Eur. J. Biochem. 251, 839–844 [DOI] [PubMed] [Google Scholar]
- 29. Brown J. R., Yamasaki M., and Neurath H. (1963) A new form of bovine pancreatic procarboxypeptidase A. Biochemistry 2, 877–886 [DOI] [PubMed] [Google Scholar]
- 30. Kobayashi Y., Kobayashi R., and Hirs C. H. (1981) Identification of zymogen E in a complex with bovine procarboxypeptidase A. J. Biol. Chem. 256, 2466–2470 [PubMed] [Google Scholar]
- 31. Kobayashi R., Kobayashi Y., and Hirs C. H. (1978) Identification of a binary complex of procarboxypeptidase A and a precursor of protease E in porcine pancreatic secretion. J. Biol. Chem. 253, 5526–5530 [PubMed] [Google Scholar]
- 32. Martínez M. C., Avilés F. X., Sansegundo B., and Cuchillo C. M. (1981) Comparison between the monomeric and binary-complex forms of procarboxypeptidase A from whole pig pancreas. Biochem. J. 197, 141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vendrell J., Aviles F. X., San Segundo B., and Cuchillo C. M. (1982) Isolation and re-association of the subunits from the pro-(carboxypeptidase A)-pro-(proteinase E) binary complex from pig pancreas. Biochem. J. 205, 449–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moulard M., Kerfelec B., Mallet B., and Chapus C. (1989) Identification of a procarboxypeptidase A-truncated protease E binary complex in human pancreatic juice. FEBS Lett. 250, 166–170 [DOI] [PubMed] [Google Scholar]
- 35. Pascual R., Burgos F. J., Salva M., Soriano F., Mendez E., and Aviles F. X. (1989) Purification and properties of five different forms of human procarboxypeptidases. Eur. J. Biochem. 179, 609–616 [DOI] [PubMed] [Google Scholar]
- 36. Moulard M., Michon T., Kerfelec B., and Chapus C. (1990) Further studies on the human pancreatic binary complexes involving procarboxypeptidase A. FEBS Lett. 261, 179–183 [DOI] [PubMed] [Google Scholar]
- 37. Oppezzo O., Ventura S., Bergman T., Vendrell J., Jörnvall H., and Avilés F. X. (1994) Procarboxypeptidase in rat pancreas. Overall characterization and comparison of the activation processes. Eur. J. Biochem. 222, 55–63 [DOI] [PubMed] [Google Scholar]
- 38. Yoneda T. (1980) Amino acid composition and the protein form of procarboxypeptidase A purified from the pancreas of the sei whale Balaenoptera bolealis. Comp. Biochem. Physiol. Part B: Comp. Biochem. 67, 81–86 [Google Scholar]
- 39. Gomis-Rüth F. X., Gómez M., Bode W., Huber R., and Avilés F. X. (1995) The three-dimensional structure of the native ternary complex of bovine pancreatic procarboxypeptidase A with proproteinase E and chymotrypsinogen C. EMBO J. 14, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gomis-Rüth F. X., Gómez-Ortiz M., Vendrell J., Ventura S., Bode W., Huber R., and Avilés F. X. (1997) Crystal structure of an oligomer of proteolytic zymogens: detailed conformational analysis of the bovine ternary complex and implications for their activation. J. Mol. Biol. 269, 861–880 [DOI] [PubMed] [Google Scholar]
- 41. Michon T., Granon S., Sauve P., and Chapus C. (1991) The activation peptide of pancreatic procarboxypeptidase A is the keystone of the bovine procarboxypeptidase A-S6 ternary complex. Biochem. Biophys. Res. Commun. 181, 449–455 [DOI] [PubMed] [Google Scholar]
- 42. Tani T., Kawashima I., Furukawa H., Ohmine T., and Takiguchi Y. (1987) Characterization of a silent gene for human pancreatic elastase I: structure of the 5′-flanking region. J. Biochem. 101, 591–599 [DOI] [PubMed] [Google Scholar]
- 43. Rose S. D., and MacDonald R. J. (1997) Evolutionary silencing of the human elastase I gene (ELA1). Hum. Mol. Genet. 6, 897–903 [DOI] [PubMed] [Google Scholar]
- 44. Szepessy E., and Sahin-Tóth M. (2006) Inactivity of recombinant ELA2B provides a new example of evolutionary elastase silencing in humans. Pancreatology 6, 117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tani T., Ohsumi J., Mita K., and Takiguchi Y. (1988) Identification of a novel class of elastase isozyme, human pancreatic elastase III, by cDNA and genomic gene cloning. J. Biol. Chem. 263, 1231–1239 [PubMed] [Google Scholar]
- 46. Shirasu Y., Takemura K., Yoshida H., Sato Y., Iijima H., Shimada Y., Mikayama T., Ozawa T., Ikeda N., and Ishida A. (1988) Molecular cloning of complementary DNA encoding one of the human pancreatic protease E isozymes. J. Biochem. 104, 259–264 [DOI] [PubMed] [Google Scholar]
- 47. Granon S., Kerfelec B., and Chapus C. (1990) Spectrofluorimetric investigation of the interactions between the subunits of bovine pancreatic procarboxypeptidase A-S6. J. Biol. Chem. 265, 10383–10388 [PubMed] [Google Scholar]
- 48. Scheele G., Bartelt D., and Bieger W. (1981) Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology 80, 461–473 [PubMed] [Google Scholar]
- 49. Szabó A., and Sahin-Tóth M. (2012) Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen. FEBS J. 279, 4283–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakano E., Geisz A., Masamune A., Niihori T., Hamada S., Kume K., Kakuta Y., Aoki Y., Matsubara Y., Ebert K., Ludwig M., Braun M., Groneberg D. A., Shimosegawa T., Sahin-Tóth M., and Witt H. (2015) Variants in pancreatic carboxypeptidase genes CPA2 and CPB1 are not associated with chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mock W. L., Liu Y., and Stanford D. J. (1996) Arazoformyl peptide surrogates as spectrophotometric kinetic assay substrates for carboxypeptidase A. Anal. Biochem. 239, 218–222 [DOI] [PubMed] [Google Scholar]