

Abstract
Olfactory receptors, which belong to the family of G-protein-coupled receptors, are found to be ectopically expressed in non-sensory tissues mediating a variety of cellular functions. In this study we detected the olfactory receptor OR51E2 at the transcript and the protein level in human epidermal melanocytes. Stimulation of primary melanocytes with the OR51E2 ligand β-ionone significantly inhibited melanocyte proliferation. Our results further showed that β-ionone stimulates melanogenesis and dendritogenesis. Using RNA silencing and receptor antagonists, we demonstrated that OR51E2 activation elevated cytosolic Ca2+ and cAMP, which could mediate the observed increase in melanin synthesis. Co-immunocytochemical stainings using a specific OR51E2 antibody revealed subcellular localization of the receptor in early endosomes associated with EEA-1 (early endosome antigen 1). Plasma membrane preparations showed that OR51E2 protein is present at the melanocyte cell surface. Our findings thus suggest that activation of olfactory receptor signaling by external compounds can influence melanocyte homeostasis.
Keywords: calcium imaging, cell differentiation, cell proliferation, cell signaling, G protein-coupled receptor (GPCR)
Introduction
Olfactory receptor (OR)5 genes constitute the basis for the sense of smell and were first described as being expressed exclusively in the olfactory epithelium (1). Later on, a subset of human OR genes have been found to be expressed in various non-olfactory tissues (2–4). This ectopic expression of OR genes raised the possibility that a subset of ORs may have physiological functions in non-olfactory tissues in addition to their canonical role in odor detection in the sensory neurons. Functionality of olfactory receptors in non-olfactory cell types is becoming better understood. Initially, a lack of correlation in expression levels in different tissues between human-mouse orthologous pairs and between intact and pseudogenized ORs was taken as an indication that functional interpretation of ectopic OR expression cannot be based on transcriptional information (5). Analysis of non-olfactory tissues in human and chimpanzee later revealed that OR orthologous genes are expressed in the same tissues more often than expected by chance alone. Orthologous OR genes with conserved ectopic expression have evolved under stronger evolutionary constraints than OR genes expressed exclusively in the olfactory epithelium (6), indicating that ectopically expressed OR genes have conserved functions in non-olfactory tissues. In the studies performed so far, functional analysis of ectopically expressed ORs revealed a possible role in sperm chemotaxis (7–9). Human ORs were also shown to mediate serotonin release in enterochromaffin cells (10) and to influence proliferation of prostate cancer cells (11, 12). The OR2A4 that localizes to the cytokinetic structures in cells was shown to participate in cytokinesis (13). The mouse OR MOR23 exerts a role in skeletal muscle regeneration by regulating cell adhesion and migration (14). In the murine kidney, ORs and olfactory signaling proteins control the glomerular filtration rate, renin secretion, and blood pressure (15, 16). A synthetic sandalwood compound was shown to accelerate wound healing processes by activation of OR2AT4 expressed in human keratinocytes (17).
In contrast to olfactory neurons, where activation of the receptors elicits a receptor current, activation of ectopically expressed receptors can have diverse effects. ORs are G-protein-coupled receptors (GPCRs), which can couple to different intracellular signaling cascades depending on the activation of different types of heterotrimeric G-proteins (18), the interaction with other cellular partners such as arrestins (19) and scaffolding proteins (20), (hetero-)dimerization with other receptors (21), lipid-protein interactions (22), and intracellular localization (23). GPCRs, which are activated by chemical ligands such as small amines, peptide hormones, chemokines, and odorants, can elicit a huge variety of cellular responses, among them cell shape changes and altered adhesion (24) as well as changes in cell proliferation (25).
In the present study we functionally characterized the signaling pathway and the implication of activation of OR51E2 in human melanocytes. We found that OR activation in these cells elicits Ca2+ signals and ultimately regulates cellular proliferation and differentiation, similar to that observed previously in prostate epithelial cells (11).
Experimental Procedures
Cell Culture and Transfection
Reagents for cell culture use were purchased from Life Technologies unless stated otherwise. Hana3a cells were maintained under standard conditions in DMEM supplemented with 10% FBS, 100 units/ml penicillin and streptomycin and 2 mm l-glutamine. Hana3a cells were transiently transfected (3 μg of OR cDNA per dish) in 35-mm dishes (Falcon, BD Bioscience) using a standard calcium phosphate precipitation technique. Primary neonatal normal human epidermal melanocytes were a generous gift from Dr. G. Neufang (Beiersdorf, Hamburg, Germany). Melanocytes were maintained under standard conditions in melanocyte growth medium (Cell Systems, St. Katharinen, Germany). For siRNA experiments, melanocytes were transiently transfected with either targeted or scrambled siRNAs using Lipofectamine 2000 (Life Technologies) according to manufacturers' instructions. The transfection rates were <1% with both OR51E2 siRNA and control siRNA as detected by co-expression of GFP. Cells were analyzed in Ca2+ imaging experiments 2 days after transfection.
Antibodies
The following primary antibodies were used: (a) rabbit polyclonal antibody against OR51E2 (Eurogentec, Seraing, Belgium); (b) mouse polyclonal antibody against human EEA-1 (Abcam); (c) rabbit polyclonal antibodies against human p44/42 MAPK and against human p38 MAPK (Cell Signaling, Danvers, MA); (d) rabbit polyclonal antibodies against human-phosphorylated p44/42 MAPK and against human phosphorylated p38 MAPK (Cell Signaling); (e) mouse monoclonal antibody against glycerinaldehyde-3-phosphate-dehydrogenase (GAPDH) (Abcam); (f) mouse monoclonal antibody against human tyrosinase (Santa Cruz Biotechnology, Dallas, TX); (g) mouse monoclonal antibody against rhodopsin, 4D2 (Abcam); (h) rabbit monoclonal against proliferating cell nuclear antigen (PCNA; Abcam); (i) mouse monoclonal alkaline phosphatase-coupled antibody against digoxin (Sigma); (j) secondary goat-anti-rabbit and goat-anti-mouse antibodies conjugated to HRP (Bio-Rad); (k) secondary goat-anti-rabbit and goat-anti-mouse antibodies conjugated to Alexa Fluor 546 or Alexa Fluor 488 (Life Technologies).
Western Blotting
80% confluent cells were harvested by scraping, and proteins were prepared by standard methods. Sample aliquots of fractionated melanocytes were mixed with Laemmli buffer (30% glycerol, 3% SDS, 125 mm Tris/Cl, pH 6.8), resolved by 10% SDS-PAGE, and transferred to nitrocellulose membrane (Protan, Schleicher and Schuell, Dassel, Germany). The nitrocellulose membranes were stained with Ponceau S (Sigma), blocked with TBST (150 mm NaCl, 50 mm Tris-Cl, Tween 20, pH 7.4) containing 5% nonfat dried milk (Bio-Rad), and incubated with the primary antibody diluted in 3% dry milk in TBST. After washing and incubation with horseradish peroxidase-coupled secondary antibodies, detection was performed with ECL plus (Amersham Biosciences) and the Fusion-SL image acquisition system (Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany). Signal intensities were quantified using ImageJ (v1.4.3.67, Broken Symmetry Software).
Cell Surface Protein Isolation
Biotinylation and isolation of cell surface proteins for Western blotting analysis were performed using the Thermo ScientificTM PierceTM Cell Surface Protein Isolation kit according to the manufacturer's instructions (Thermo Fisher Scientific, Pierce, Rockford, IL).
Single Cell Ca2+ Imaging
Melanocytes were incubated for 45 min, and Hana3a cells were incubated for 30 min in loading buffer, pH 7.4, containing Ringers' solution and 7.5 μm Fura-2-AM (Life Technologies). After removal of extracellular Fura-2 by washing, ratiofluorometric Ca2+ imaging was performed using an inverted microscope equipped for ratiometric live cell imaging (IX71, Olympus, Hamburg, Germany) with a 150-watt xenon arc lamp, a motorized fast change filter wheel illumination system for multiwavelength excitation (MT20, Olympus), and a 12-bit 1376 × 1032-pixel charge-coupled device (CCD) camera (F-View II, Olympus). WinNT-based cellRTM imaging software (Olympus) served to collect and quantify spatiotemporal Ca2+-dependent fluorescence signals (excited at 340 and 380 nm, measured at 510 nm, and calculated as a f340/f380 intensity ratio) of the cells. Compounds were diluted in Ringer's solution or for Ca2+free experiments in Ringer's solution containing 50 μm EGTA and applied using a specialized pressure-driven microcapillary perfusion system designed for instantaneous solution change and focal application. Images were acquired in randomly selected fields of view. Ultrapure α-ionone and β-ionone were a generous gift of Dr. J. Panten (Symrise, Holzminden, Germany). Steroids were purchased from Steraloids (Steraloids Inc., Newport, RI), aminoethoxydiphenyl borate (2-APB), SKF 96365, GdCl3, and BTP2 were from Tocris (Tocris Bioscience, Bristol, UK), and endothelin-1, thapsigargin, and ATP were from Sigma. Odorants were prediluted in DMSO (Sigma) so that the DMSO concentration did not exceed 1‰ (v/v), which was well tolerated by melanocytes. All odorants assayed for potential inhibition of OR51E2 were tested in at least three different series of transfection experiments.
Immunocytochemistry
Cells were seeded on coverslips and maintained as described above. The cells were fixed by incubation with 4% paraformaldehyde at 4 °C for 30 min. After blocking with 1% gelatin for 1 h, cells were incubated overnight with the primary antibody. For visualization, secondary fluorescent IgGs (Life Technologies) (1:1000) were used. Micrographs were taken by using a LSM510 Meta confocal microscope (Zeiss, Jena, Germany).
Apoptosis Detection
50–70% confluent melanocytes were stimulated for 72 h with different concentrations of β-ionone or solvent only. Apoptotic activity was detected by Caspase-Glo® 3/7 Assay (Promega, Madison, WI). Apoptotic cells were quantified using the terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) based In Situ Cell Death Detection kit (Hoffmann-La Roche) according to the manufacturer's instructions.
Cell Proliferation
Growing melanocytes were plated in 96-well plates at a density of 5 × 103 cells/well. After 24 h at 37 °C with 5% CO2, cells were treated with different concentrations of β-ionone. Cell proliferation was assessed after 6 days using CyQUANT cell proliferation assay kit (Life Technologies). For the visualization of proliferating cells via PCNA staining, cells were stimulated for 6 days with β-ionone (50 μm) or solvent only. Afterward, cells were stained with anti-PCNA antibody (1:500) as described under “Immunocytochemistry” and with Alexa Fluor® 546 phalloidin (Life Technologies; 1:200).
DNA and siRNA Constructs
Prostate-specific G-protein-coupled receptor targeted and scrambled hairpin siRNA designs were carried out with siRNA Target Designer-Version 1.51 (Promega) as described previously (11). The best working siRNA sequence of OR51E2 was gctgcctcctgtcatcaat; the oligonucleotide sequences to generate 5′-target-loop-reverse-complement-3′ hairpins were: 5′tctcgtgcctctgtcatcaataagttctctattcatcacaggaggcagcct-3′, 5′-ctgcaggctgcctcctgtcatcaatagagaacttattcatgacaggaggcagc-3′. The following scrambled versions of the siRNA sequence were used as control: 5′-tctcgtacactgaccccctttgtaagttctctacaaagggggtcagtgtacct-3′, 5′-ctgcaggtacactgacccccttctagagaacttacaaagggggtcagtgtac-3′.
Reverse-transcriptase PCR (RT-PCR)
RNA of melanocytes was isolated with the RNeasy Mini kit including on-column DNase digestion (Qiagen, Hilden, Germany) according to manufacturers' instruction. RNA concentration and quality (A260/A280 ratio) were analyzed using Spectrophotometer NanoDrop ND-1000 (Thermo Scientific, Waltham, MA). cDNA synthesis and semiquantitative RT-PCR cDNA synthesis was performed by using the iScript cDNA Synthesis kit (Bio-Rad). Control reactions with no reverse transcriptase (−RT) served to exclude contamination with genomic DNA. RT-PCR was performed using GoTaq qPCR Master Mix (Promega) with 2 ng of template cDNA and 10 pmol of specific primers designed for OR amplification. The amplifications were done for 40 cycles (45°s, 94 °C; 45 s, 60 °C; 45 s, 72 °C). Primers used for RT-PCR were: actin 5′-tcttccagccttccttcctg-3′ and 5′-cacggagtacttgcgctcag-3′; Tyr-P1 5′-ggcccgggctcaattcccaa-3′ and 5′-gcaagggccagacctcccga-3′; OR1J2 5′gaacaccatcccccatgtct-3′ and 5′-tggatccctttggttgaagg-3′; OR2D2 5′-agtgcgcccttcttgcagtga-3′ and 5′-actgcctcggtagggtagcct-3′; OR4N5 5′-ctgcacttgcctttctgtgg-3′ and 5′-atatgggtggtgcatgtgga-3′; OR5C1 5′-ctatgtcgcagcgtctatgc-3′ and 5′-gcagtggagggatatcgcag-3′; OR6K3 5′-gaggggatcttgctgaccac-3′ and 5′-aggcttagcacagggaccaa-3′; OR6T1 5′-ttccccagtagccacctcat-3′ and 5′-caagcatcttgggaaccaca-3′; OR9G1 5′-ctggctgcctgtgtcagttc-3′ and 5′-accagcaatgcacacagctt-3′; OR10A4 5′-agcttctcagccctgtccac-3′ and 5′-gggcacaatgaccaagttga-3′; OR10H5 5′-gctcactgtcatgggctacg-3′ and 5′-ccaccaccagcacatcatct-3′; OR11H4 5′-gacccatgaacaggtcagca-3′ and 5′-aggcaaaatttcccagcaga-3′; OR51E2 5′-actg-ccttccaagtcagagc-3′ and 5′-cttgcctcccacagcctg-3′; OR2A4/7 5′-ggtgccccggatgctggtg-3′ and 5′-ggggtggcagatggccacg-3′; OR51B5 5′-caatggcaccctccttcttc-3′ and 5′-caagcagaatgccagactcg-3′; OR5P3 5′-tggtagagtttactcttttggggt-3′ and 5′-tgagcatgacaggtgtgact-3′; OR6B3 5′-cagctccctggtgtgcaccg-3′ and 5′-caccagctggaggcacagcc-3′; OR10AD1 5′-actgcctggttctttgggctga-3′ and 5′-gccaatcactatgggggcctca-3′; OR51E2 (intron-overspanning) 5′-cctcagccttctgagtcagc-3′ and 5′-gagactgtgacaa-gccctgg-3′.
Semiquantitative real-time PCR was performed using GoTaq® qPCR Master Mix (Promega) with the Mastercycler Realplex2 (Eppendorf, Hamburg, Germany) (20 μl total volume; 40 cycles of 95 °C, 60 °C and 72 °C at 45 s each). All experiments were conducted in triplicate. PCR was performed with cDNA (equivalent of 100 ng total RNA) and specific primer pairs for the amplification of tyrosinase and GAPDH. The housekeeping gene GAPDH was used for relative quantification by the ΔCt (cycle threshold) method. Results are reported as -fold changes in gene expression compared with control as calculated by the 2−ΔΔCt method. The following primer pairs were used: tyrosinase (1) 5′-ggctgttttgtactgcctgc-3′ and 5′-aggaacctctgcctgaaagc-3′; tyrosinase(2) 5′-cttcacaggggtggatgacc-3′ and 5′-tctctgtgcagtttggtccc-3′; GAPDH 5′-accacagtccatgccatcac-3′ and 5′-tcccaccaccctgttgctgta-3′.
cAMP Assay
cAMP concentration was analyzed using a cAMP kit from R&D systems (Minneapolis, MI). In brief, 7 × 104 melanocytes were stimulated with growth medium containing odorant or forskolin and 100 μm 3-isobutyl-1-methylxanthine (IBMX) as cAMP-phosphodiesterase inhibitor. Afterward, the cells were lysed in 0.1 m HCl in ice-cold ethanol. After 30 min of incubation on ice, cells lysates were desiccated for 5 h at 37 °C. Samples were reconstituted in 400 μl of 0.1 m HCl. Sample volumes of 100 μl per well were transferred to a microtiter plate, and cAMP concentrations per well were measured according to the manufacturers' instructions with the following minor changes. To exclude differences in anti-cAMP antibody binding efficiency, all samples and standards were adjusted to identical ionic conditions; i.e. standards provided by the manufacturer in 10 mm HCl/ethanol were desiccated and reconstituted in 0.1 m HCl. For each tested condition and standard, three replicate experiments were performed, and results were averaged.
Melanin Content Assay
Melanocytes were cultured for 72 h in basal medium containing β-ionone (50 μm), forskolin (20 μm), α-ionone (200 μm), H89 (10 μm), or the solvent only (0.1% DMSO). Melanin contents of stimulated melanocytes were measured according to the method of Oka et al. (26) with a slight modification. After stimulation, cells were harvested by scraping, and cell numbers were counted using a counting chamber (Blaubrand Neubauer improved, Sigma). To take the anti-proliferative effect of β-ionone into account, cell numbers were adjusted to 1 × 105 before determination of the melanin content. Cell pellets were solubilized in boiling 1 m NaOH for 10 min. Spectrophotometric analysis of melanin content was performed at 400 nm absorbance.
Differentiation Assay
Melanocytes were cultured for 6 days in basal medium containing β-ionone (50 μm), forskolin (20 μm), or the solvent only (0.1% DMSO). Cell morphology was checked by bright field microscopy using a Zeiss Axioskop2 microscope with 20× magnification. Undifferentiated (bipolar morphology, small cell bodies, less pigmentation) and differentiated melanocytes (multiple dendrite, large cell body, high pigmentation) were quantified.
Synthesis of Fluoresceine-5-isothiocyanate (FITC)-labeled Steroids
FITC-labeled steroids were obtained by chemical synthesis from dihydrotestosterone and dehydrotestosterone. In brief, the 17-β-OH group of the respective steroid was activated by acylation with carbonyldiimidazole (27–29) in tetrahydrofuran. The resulting monoimidazolide was treated with excess 4,7,10-trioxa-1,13-tridecanediamine in acetonitrile followed by evaporation and washing with aqueous NaHCO3. Treatment of the resulting primary amine intermediate with fluorescein-5-isothiocyanate in DMF in the presence of Hünig's base (iPr2NEt) provided the fluorescently labeled steroid ligands. The final products were purified to homogeneity by using preparative HPLC and obtained as orange powders after lyophilization in 12–50% overall yield. Details of synthesis procedures and characterization data for all new compounds α (m.p., high resolution mass spectrometry, 1H and 13C NMR, IR) was described in the following section.
All solvents, when not purchased in suitable purity or dryness, were distilled using standard methods. Alternatively, solvents (HPLC grade) were passed through activated alumina columns under dry argon atmosphere (solvent purification system, M. Braun Inertgas-Systeme GmbH, Garching, Germany). Deionized water was used for all experiments. All reagents were purchased from commercial suppliers (Acros, Novabiochem, Sigma) and used without purification. Analytical thin layer chromatography (TLC) was carried out on Merck precoated silica gel plates (60F-254) using ultraviolet light irradiation at 254 nm or phosphomolybdic acid solution as staining reagent (1 wt% in EtOH). Flash column chromatography was performed using silica gel (J. T. Baker, particle size 40 μm; pore size 60 Å) under a pressure of 0.3–0.5 bar. Analytical HPLC was performed on an Agilent 1100 system using a C18 gravity 3-μm reverse phase column (Macherey & Nagel, Düren, Germany). The separations were started at 10% MeCN (with 0.1% HCOOH) in H2O (with 0.1% trifluoroacetic acid) with a flow of 1 ml/min, and the MeCN proportion was linearly increased after 1 min to 100% over a period of 10 min and then kept at constant ratio for a period of 5 min. Preparative HPLC was performed on a Varian system using a C18 gravity 5-μm reversed phase column (Macherey & Nagel) using a MeCN in H2O gradient. 1H and 13C NMR spectra were recorded on a Varian Mercury VX 400 (400.1 MHz (1H) and 100.6 MHz (13C)) spectrometer. Chemical shifts are expressed in parts per million (ppm), and the spectra were calibrated to residual solvent signals of CDCl3 (7.26 ppm (1H) and 77.0 ppm (13C)), CD3OD (3.31 ppm (1H), and 49.0 ppm (13C)), respectively. Coupling constants are given in Hertz (Hz), and the following notations indicate the multiplicity of the signals: s (singlet), d (doublet), t (triplet), q (quartet), qui (quintet), sext (sextet), sept (septet), m (multiplet), app (apparent), br (broad signal). Unless otherwise stated, NMR spectra were recorded at 27 °C. Low resolution mass spectra were recorded on a Thermo Finnigan LCQ ESI spectrometer (source voltage 70 keV). High resolution FAB (fast atom bombardment) spectra were recorded on a Jeol SX 102 A (matrix: meta-nitrobenzyl alcohol). High resolution electrospray ionization spectra were recorded on a Thermo Electron LTQ Orbitrap (source voltage 3.8 kV, resolution: 60000) spectrometer.
Dihydrotestosterone-derived imidazolide (7):
SCHEME 1.
Structure 7.
Dihydrotestosterone (3, 300 mg, 1.00 mmol) and carbonyl diimidazole (330 mg, 2.00 mmol) were dissolved in THF (15 ml) and stirred overnight at room temperature. After the disappearance of dihydrotestosterone on TLC (Rf = 0.4, EtOAc/hexanes 1:1), the solvent was evaporated. The residue was partitioned with ethyl acetate (50 ml) and water (20 ml). The layers were separated, and the organic extract was washed with saturated NaHCO3 solution and brine (20 ml each), dried with MgSO4, and evaporated to obtain imidazolide 7 a colorless solid (350 mg). The crude product was purified by Flash column chromatography (EtOAc/hexanes 1:9 → 1:2) to obtain the title compound as a colorless solid (280 mg, yield 73%). TLC: Rf = 0.25 (EtOAc/hexanes 1:1). HPLC: tR = 10.6 min. ESI-MS: calc. for C23H33N2O3 385.3, found 385.1 [M+H+]. 1H NMR (400 MHz, CDCl3): δ = 8.13 (1H, t, J = 0.78 Hz, imidazole), 7.41 (1H, t, J = 1.39 Hz, imidazole), 7.08 (1H, dd, J = 1.4 Hz and 0.79 Hz, imidazole), 4.83 (1H, dd, J = 7.3 Hz and 9.3Hz, H-17 of dihydrotestosterone), 2.37-2.17 (4H, m), 2.3 (1H, ddd, J = 15 Hz, 3.98 Hz and 2.18 Hz), 1.95 (1H, ddd, J = 13.2 Hz, 6.4 Hz and 2.8 Hz), 1.85 (1H, td, J = 11.9 Hz and 2.9 Hz), 1.70–1.54 (4H, m), 1.53-1.43 (2H, m), 1.41–1.16 (7H, m), 1.12-1.01 (1H, m), 0.97 (3H, s, Me), 0.93-0.81 (1H, m), 0.85 (3H, s, Me). 13C NMR (100 MHz, CDCl3): δ = 211.8 (CO), 148.8 (carbamate), 137.2, 130.8, 117.3 (3C, imidazole), 87.1 (C-17 of dihydrotestosterone), 53.9, 50.6, 46.8, 44.8, 43.3, 38.7, 38.3, 37.0, 35.9, 35.4, 31.4, 28.9, 27.6, 23.7, 21.1, 12.6, 11.7.
Dihydrotestosterone-derived PEG-carbamate (9):
SCHEME 2.

Structure 9.
Imidazolide 7 (50 mg, 0.13 mmol) was added to a solution of 4,7,10-trioxatridecan-1,13-diamine (0.085 ml, 0.39 mmol) in acetonitrile (3 ml) followed by one drop of DMF to get a homogeneous solution. The reaction mixture was stirred overnight at room temperature under argon atmosphere. After complete disappearance of the starting material on TLC the solvent was evaporated. The residue was partitioned with ethyl acetate (20 ml) and water (10 ml). The organic layer was washed with saturated NaHCO3 solution and brine (10 ml each), dried with MgSO4, and evaporated to obtain carbamate 9 a colorless oil (50 mg). The product was used for the next reaction without further purification. HPLC: tR = 6.4 min. ESI-MS: calc. for C30H53N2O6 537.4, found 537.5 [M+H+].
Dihydrotestosterone-derived FITC conjugate (11):
SCHEME 3.

Structure 11.
Compound 9 (50 mg crude, 0.13 mmol) was dissolved in DMF (5 ml), and FITC (162 mg, 0.39 mmol) and EtNiPr2 (116 μl, 0.65 mmol) were added. The mixture was stirred, and turnover was monitored by HPLC. After 4 h, the solvent was evaporated, and the residue was purified using preparative HPLC. Pure fractions were combined and lyophilized to give the title compound 11 as an orange solid (14 mg, 12% yield after 2 steps), m.p.: 155–157 °C. HPLC: tR = 7.9 min. High resolution mass spectrometry (ESI): calc. for C51H64O11N3S 926.4262, found 926.4263. IR (undiluted, cm−1): ν = 3068 (br), 2930, 2871, 1676, 1633. 1H NMR (400 MHz, DMSO-d6): δ 10.14 (1H, br), 9.96 (1H, br), 9.05 (1H, s), 8.22 (1H, s), 8.11 (1H, br), 7.73 (1H, br), 7.68 (1H, br), 7.24 (1H, s), 7.16 (1H, d, J = 8.3 Hz), 7.11 (1H, s), 6.98 (1H, s), 6.91 (1H, t, J = 5.8 Hz), 6.67 (2H, d, J = 2 Hz), 6.60-6.53 (3H, m), 4.37 (1H, t, J = 8.5 Hz), 3.55-3.45 (12H, m), 2.99 (2H, m), 2.42-2.24 (1H, m), 2.08-1.84 (4H, m), 1.84-1.77 (2H, m), 1.61-0.96 (17H, m), 0.95 (3H, s, Me), 0.92-0.79 (1H, m), 0.72 (3H, s).
Dehydrotestosterone-derived imidazolide (6):
SCHEME 4.
Structure 6.
Dehydrotestosterone (2, 30 mg, 0.10 mmol) was treated similarly to dihydrotestosterone 7. Imidazolide 6 was obtained as a colorless solid (33 mg, 0.087 mmol, 87% yield) and used directly for the next step. TLC: Rf = 0.27 (EtOAc/hexanes 1:1). HPLC: tR = 9.5 min. ESI-MS: calc. for C23H29N2O3 381.2, found 381.1 [M+H+].
Dehydrotestosterone-derived PEG-carbamate (8):
SCHEME 5.

Structure 8.
Imidazolide 6 (33 mg, 0.087 mmol) was converted to the PEG-carbamate similar to 7. The carbamate 8 such obtained was directly used for the next transformation (yield 48 mg, quantitative). HPLC: tR = 6.5 min. ESI-MS: calc. for C30H49N2O6 533.4, found 533.5 [M+H+].
Dehydrotestosterone-derived FITC conjugate (10):
SCHEME 6.

Structure 10.
Compound 8 (0.087 mmol) was dissolved in DMF (1 ml) and EtNiPr2 (31 μl, 0.17 mmol) was added. In another flask, 72 mg of FITC (0.17 mmol) was dissolved in DMF (1 ml), and EtNiPr2 (31 μl, 0.17 mmol) was added. The solutions were combined, and more DMF (2 ml) was added. The orange mixture was stirred at 20 °C for 12 h. The solvent was evaporated, and the residue was subjected to purification by preparative HPLC. Appropriate fractions were lyophilized to obtain the title compound 10 as an orange solid (20 mg, 25% over three steps). m.p.: 157–158 °C. HPLC: tR = 8.7 min. High resolution mass spectrometry (ESI): calc. for C51H60O11N3S 922.3943, found 922.3949 [M+H+]. IR (undiluted, cm−1): 3065 (br), 2943, 2871, 1675, 1636. 1H NMR (400 MHz, DMSO-d6): δ 10.09 (2H, br), 9.89 (1H, br), 8.19 (1H, s), 8.05 (1H, br), 7.70 (1H, br), 7.15 (1H, d, J = 8.4 Hz), 6.96 (1H, t, J = 7Hz), 6.6557 (1H, s), 6.6512 (1H, s), 6.60-6.52 (4H, m), 6.12 (2H, m), 5.59 (1H, m), 4.42 (1H, t, J = 7.9 Hz), 3.53-3.43 (13H, m), 3.36 (2H, t, J = 6.2 Hz), 3.02 (2H, qui, J = 6Hz), 2.25-2.14 (2H, m), 2.09-1.98 (1H, m), 1.94-1.86 (1H, m), 1.85-1.72 (3H, m), 1.70-1.56 (4H, m), 1.51-1.26 (4H, m), 1.26-1.07 (3H, m), 1.04 (3H, s), 0.80 (3H, s). 13C NMR (100 MHz, CDCl3): δ 198.7, 169.2, 163.8, 160.2, 156.9, 152.6, 141.0, 129.7, 128.3, 123.7, 113.3, 110.4, 102.9, 81.8, 70.4, 70.3, 70.2, 68.9, 68.7, 50.8, 48.0, 43.6, 38.3, 38.2, 37.6, 36.8, 36.2, 34.3, 34.0, 30.3, 29.3, 28.0, 23.2, 20.5, 16.7, 12.5.
Results
Olfactory Receptors Are Expressed in Human Melanocytes
Frog melanophores have previously been shown to disperse their melanosomes in response to odorants, although the concurrent increase in intracellular cAMP levels on pigment dispersion was discussed controversially (30, 31). Moreover, screening for naturally occurring compounds with antiproliferative activity on melanoma cells has led to the identification of the odorant 4-allyl-2-methoxyphenol (eugenol) (32, 33).
We, therefore, analyzed whether ORs are expressed in melanocytes. We started by analyzing receptor expression in cultured human melanocytes by RT-PCR (Fig. 1A) and found different ORs to be expressed, among them OR51E2, an olfactory receptor that was previously also detected in prostate cells and in the olfactory epithelium (11). Because the OR51E2 ligand, the isoprenoid β-ionone (with an odor of violet), was included in the earlier studies on the effect of odorants on pigment dispersion (30, 31), we specifically studied the expression of this receptor in detail. RT-PCR with intron-spanning primers to exclude amplification from genomic DNA contamination revealed that OR51E2 transcripts are expressed in human epidermal melanocytes (NHEM) from donors of ethnic origins with differing pigmentation levels (Fig. 1B). We also detected OR51E2 protein in human epidermal melanocytes by Western blotting and immunocytochemical analysis using an OR51E2-specific antibody (Fig. 1, C and D). Specificity of the antibody was demonstrated by co-immunocytochemical staining of Hana3a cells heterologously expressing rho-tagged OR51E2 (Fig. 1E).
FIGURE 1.

OR51E2 is expressed in human epidermal melanocytes. A, RT-PCR detection of 13 different ORs in normal human epidermal melanocytes (NHEM). Gel electrophoresis of OR amplicons from melanocyte cDNA (+) and no reverse transcriptase cDNA controls (−) to exclude genomic DNA contamination is shown. Amplification of actin and tyrosinase-related protein 1 (Tyr-P1) served as positive controls. OR51B5, OR5P3, and OR6B3 transcripts were not detected. B, detection of OR51E2 transcripts in melanocytes (normal human epidermal melanocytes) by RT-PCR together with a schematic representation of the intron over-spanning primers for this receptor. OR51E2 transcripts could be detected in melanocytes of different pigmentation levels (Caucasian and Negroid origin). LNCaP cells served as the positive control and negative control: genomic DNA (gDNA). The size of the OR51E2 amplicon was 1050 bp (cDNA) or >15 000 bp (gDNA). C, detection of OR51E2 protein in primary melanocytes by Western blotting; the size of the OR51E2 monomeric (51E2-M) protein was 35 kDa and dimeric protein was 70 kDa (51E2-D). D, immunocytochemical staining using an OR51E2-specific antibody; shown are confocal micrographs of an OR51E2 staining in melanocytes. The control consisted of omission of the primary antibody. Bars indicate 15 μm. E, co-immunocytochemical staining of Hana3a cells transiently expressing OR51E2 with an N-terminal rho tag, which consists of the first 20 amino acids of rhodopsin. Detection of the recombinant rho-OR51E2- protein was performed using an antibody against OR51E2 and an antibody against the N-terminal rho tag. Shown are confocal micrographs of the OR51E2 staining in OR51E2 expressing Hana3a cells. Mock-transfected Hana3a cells served as negative control. Bars indicate 20 μm. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Co-labeling (yellow) of OR51E2-expressing Hana3a cells indicates specificity of the used OR51E2 antibody.
Effect of β-Ionone on Proliferation and Apoptosis
Because OR51E2 has been shown to be involved in the regulation of prostate cancer cell growth (11), we investigated the effect of the OR51E2 ligand β-ionone on melanocyte proliferation. Melanocytes were treated for 6 days in basal medium containing different concentrations of β-ionone, and the DNA content was determined to reveal the number of cells. β-Ionone stimulation decreased the cell number in a significant manner, even at submicromolar concentrations (Fig. 2A). The maximal decrease in proliferation (∼30%) was observed after 6 days of treatment with 50 μm β-ionone. This result was verified via immunocytochemical staining with an antibody against the PCNA as a marker for cell proliferation (Fig. 2B).
FIGURE 2.

Activation of OR51E2 promotes differentiation. A, β-ionone stimulation affects melanocytes proliferation. Proliferation of melanocytes after treatment with increasing concentrations of β-ionone for 6 days compared with control conditions. The relative cell number was determined by measurement of the DNA content. The data are shown as the mean of three independent experiments with five technical replicates and were normalized to the cell number in control experiments, and significance was calculated by Student's t test referring to cell number of untreated control cells (**, p < 0.01; ***, p < 0.001). B, immunocytochemical staining using an antibody against PCNA reveals significantly reduced numbers of proliferating cells after stimulation with β-ionone compared with control cells. Left panel, immunofluorescence confocal micrographs of melanocytes labeled with a PCNA-specific antibody (green) and Alexa Fluor® 546 phalloidin (red). Melanocytes were treated with 50 μm β-ionone or solvent only (control) for 6 days. The bar indicates 50 μm. Right panel, the data are shown as the mean of four independent experiments each with 500 quantified cells that were normalized to the cell number in control experiments. Significance was calculated by Student's t test referring to cell number of untreated control cells (*, p < 0.05). C, β-ionone stimulation had no significant effect on melanocyte apoptosis. Left panel, induction of apoptotic pathways was assessed by the Caspase-Glo 3/7 assay. Significance was calculated by Student's t test referring to cells treated with the solvent only. Right panel, representative photomicrographs of melanocytes stained by TUNEL assay after treatment with 50 μm β-ionone for 72 h. DNAse I treatment (20 min) served as the positive control for the detection of apoptosis. n.s., not significant. D, β-ionone stimulation induces melanogenesis. Melanin content was measured 72 h after β-ionone treatment (50 μm). The β-ionone-induced effect on pigmentation was suppressed by co-stimulation with α-ionone (200 μm) or prestimulation with H89 (10 μm) for 2 h. The data are the average of the results from three biological replicates each performed in triplicate and were normalized to the melanin content in solvent-only-treated cells (0.1% DMSO, control). Forskolin (20 μm) served as a positive control. Error bars indicate the S.E. Significance was calculated by Student's t test referring to the melanin content in control cells. E, RT-qPCR quantification of tyrosinase gene expression in melanocytes stimulated for 6 h with β-ionone (50 μm), forskolin (20 μm), and solvent only (0.1% DMSO, control). Right panel, gel electrophoresis of tyrosinase amplicons using two different primer pairs (1 and 2) from melanocyte cDNA (+) and no reverse transcriptase controls (−). Amplification of GAPDH served as internal control. Left panel, relative tyrosinase gene expression of β-ionone-stimulated melanocytes using two different primer pairs (1 and 2). Tyrosinase mRNA levels were normalized by GAPDH in each experiment, and changes in mRNA expression were calculated compared with control cells. Bars represent the mean of three experiments, each performed in triplicate. Error bars represent the S.E. F, right panel, Western blotting analysis reveals an β-ionone-induced increase in tyrosinase protein. Cells were stimulated with β-ionone (50 μm) and forskolin (20 μm, positive control) for 72 h. Detection of GAPDH served as the internal standard. Left panel: quantification of tyrosinase signal intensities. Bars represent the mean of four independent experiments; tyrosinase signal intensities were normalized by GAPDH in the same experiment. G, left panel, phase-contrast image of an undifferentiated and differentiated melanocyte in culture. Undifferentiated melanocytes are bipolar, less pigmented, and have small cell bodies, whereas differentiated melanocytes exhibited an increased size of the cell body, increased pigmentation, and multiple dendrites. The bar indicates 10 μm. Right panel, exposure to β-ionone induced melanocyte dendritogenesis. Cells were cultured for 6 days in basal medium containing β-ionone (50 μm) and 0.1% DMSO as negative control. Forskolin (20 μm) served as a positive control. Undifferentiated (gray part of the bar) and differentiated cells (black part of the bar) were counted and are shown as the percentage of total cells. The data are the mean of three independent experiments each with 500 cells quantified. Statistics were performed by Student's t test, referring to the cell number of control cells (*, p < 0.05). H, activation of OR51E2 resulted in phosphorylation of p38 MAPK and p44/42 MAPK (ERK1/2) as shown by Western blotting analyses. Melanocytes were stimulated with 50 μm β-ionone for 30 min. Detection of p38 MAPK and ERK total protein is shown to control loaded protein amounts.
As we observed decreased cell numbers upon exposure to β-ionone, we investigated the influence of β-ionone on cell apoptosis rates in primary human melanocytes. Primary melanocytes were stimulated for 72 h before detection of apoptosis by assessment of Caspase 3/7 activity, as indicative of an activated apoptotic signaling pathway, and TUNEL staining to quantify apoptotic cells (Fig. 2C). The Caspase 3/7 assay showed a nominal increase of the caspase activity after stimulation with 50 μm β-ionone, which was not significant. However, TUNEL staining implicated that long term β-ionone stimulation does not induce apoptosis in melanocytes. We, therefore, conclude that the observed decrease in cell number after stimulation with 50 μm β-ionone is a result of a declining proliferation rate.
β-Ionone Induces Melanogenesis and Differentiation
We next investigated the effect of β-ionone on melanogenesis and dendritogenesis as indicators of pigment cell differentiation (34). To observe whether β-ionone stimulation induces melanogenesis in normal melanocytes, the melanin content of normal human melanocytes was determined. Forskolin, a potent stimulator of adenylate cyclases, which cause melanogenesis and dendritogenesis of mammalian pigment cells (35, 36), served as the positive control. The β-ionone-induced increase of the melanin content was similar to that of forskolin treatment after 72 h cultivation in basal medium containing the respective stimuli (Fig. 2D). Co-stimulation with the OR51E2 competitive antagonist α-ionone (11) demonstrates that the observed increase in melanogenesis depends on activation of OR51E2. We furthermore identified PKA as a key mediator of OR51E2-triggered melanogenesis by using the specific PKA inhibitor H89 (37), which inhibited the β-ionone-induced melanogenesis when the cells were pre-stimulated. Concordantly, qPCR analysis revealed that β-ionone stimulation for 72 h induced tyrosinase expression (Fig. 2E), a promoter of melanocyte melanogenesis. This was confirmed with Western blotting analysis that showed an increase in tyrosinase protein levels (Fig. 2F).
Moreover, we examined the effect of β-ionone on melanocyte morphology as an indicator for cell differentiation. Melanocyte differentiation involves branching and extension of the dendrites. Primary melanocytes cultured in basal medium were treated for 6 days with 50 μm β-ionone, and the number of dendrites was quantified. The majority of control cells exhibited a typical bipolar morphology, whereas most of the β-ionone-treated cells had multiple dendrites. Quantification of this effect showed that treatment with β-ionone induced a morphological change in dendrites and significantly increased the percentage of cells with more than two dendrites (Fig. 2G).
As melanocyte cellular responses to external growth and differentiation signals can be mediated by MAP kinases (38–40), we studied activation (phosphorylation) of prominent members of the MAP kinase family in β-ionone-treated melanocytes. Western blotting analysis using antibodies specific for phosphorylated and non-phosphorylated extracellular stress-regulated kinase (p44/42 MAPK) and p38 MAPK revealed that both kinases are activated in β-ionone-treated melanocytes (Fig. 2H). β-Ionone stimulation did not result in a marked increase in phosphorylation of SAPK/JNK (data not shown). In summary, we could show that activation of OR51E2 affects pigment cell melanogenesis and differentiation by activation of a cAMP-PKA-mediated pathway.
OR51E2 Activation in Melanocytes
To investigate if β-ionone exerts its effects on melanocytes by activation of OR51E2, we first examined the effects of short term β-ionone stimulation on the level of intracellular Ca2+ in melanocytes via the Ca2+ imaging method. Activation of an OR expressed in olfactory sensory neurons as well as in heterologous cell systems, induces intracellular Ca2+ signals as a result of OR-initiated signaling. Stimulation of Fura-2-loaded melanocytes with β-ionone also caused a rise in cytosolic Ca2+ (Fig. 3A). However, this β-ionone-induced Ca2+ signal was different in response kinetic compared with olfactory sensory neurons and Hana3a cells expressing OR51E2. Olfactory sensory neurons and OR-expressing Hana3a cells show a fast transient Ca2+ signal upon odorant-ligand stimulation, whereas application of β-ionone in melanocytes caused a slow but robust increase in cytosolic Ca2+ that started rising after 1 min from the begin of the application (Fig. 3B). When the ligand was supplied continuously, the maximal amplitude was reached after 5 min application of β-ionone. The signal decreased afterward in the presence of the ligand, indicating a desensitization of the signaling cascade (Fig. 3C). The β-ionone-induced rise in intracellular Ca2+ was found to be dose-dependent, and the threshold concentration to trigger a cellular response to β-ionone was ∼10 μm (Fig. 3D).
FIGURE 3.

OR51E2 activation induces a Ca2+ signal. A, representative picture of Fura-2-loaded melanocytes. Intracellular Ca2+ levels are indicated in pseudocolors, and images of changes in cytosolic Ca2+ were captured during a representative experiment. B, representative Ca2+ imaging trace of a melanocyte. In a randomly selected field of view, β-ionone (50 μm) application induced a transient, slow rise in intracellular Ca2+ (black) in individual cells. The gray Ca2+ imaging trace is representative for the vehicle controls (0.1% DMSO). Application of the solvent did not result in any changes in cytosolic Ca2+. Cytosolic Ca2+ levels were monitored as integrated f340/f380 fluorescence ratio expressed as a function of time. C, in prolonged stimulation the maximal signal amplitude was reached after a 5-min application of β-ionone. After reaching the maximum peak, the cytosolic Ca2+ concentration decreases until a plateau phase was sustained. β-Ionone was applied for 9 min. D, β-ionone induced Ca2+ increase is dose-dependent. β-ionone was applied for 9 min at different concentrations to ensure maximal signal amplitude generation. Signal amplitude was quantified, normalized to the maximal peak height, and is displayed as a function of the applied β-ionone concentration (n = 49–129 cells). E, Ca2+ imaging on primary melanocytes transfected with a plasmid encoding for siRNA directed against OR51E2. As the plasmid encodes for GFP under the same promoter, siRNA-expressing cells can be identified via GFP fluorescence (left panel); pseudocolor images were captured of Fura-2 loaded cells during Ca2+ imaging experiment (right panel). OR51E2-siRNA/GFP-expressing melanocytes (turquoise labeling) showed no significant changes in the Fura-2 ratio upon application of β-ionone, whereas non-transfected cells (blue labeling) did. F, β-ionone-induced Ca2+ signals in siRNA transfected (turquoise labeling) and control melanocytes (blue labeling) displayed as a function of time. 50 μm β-ionone was applied for 5 min. G, quantification of the relative signal amplitudes of the β-ionone evoked Ca2+ signals, normalized to the β-ionone response of control cells (n = 109 cells). n = 4 independent transfections were analyzed. Shown are OR51E2-siRNA-expressing melanocytes (n = 42 siRNA-expressing cells) and melanocytes transfected with a plasmid encoding for scrambled OR51E2-siRNA (control siRNA, n = 27). OR51E2-siRNA-transfected (n = 23) and control cells (n = 64) were stimulated with 40 nm endothelin-1 (ET-1) to control that siRNA expression did not affect cell viability. Error bars represent the S.E. Significance was calculated by Student's t test for each sample group referring to the signal amplitude in control melanocytes (***, p < 0.001). H, representative Ca2+ imaging traces of Fura-2-loaded melanocytes. α-Ionone (250 μm) was applied for 5 min. Cytosolic Ca2+ levels were monitored as integrated f340/f380 fluorescence ratio expressed as a function of time. I, co-application of the OR51E2-specific inhibitor α-ionone reduced the β-ionone-induced Ca2+ signals (n = 48). β-Ionone (50 μm) and the α-ionone and β-ionone 2:1 mix (100 μm and 50 μm, respectively) were applied for 4 min.
We next aimed to confirm that the β-ionone induced increase in cytosolic Ca2+ is dependent on activation of OR51E2. We, therefore, performed Ca2+ imaging experiments with reduced OR51E2 expression rate due to RNA silencing (Fig. 3E). β-Ionone-induced Ca2+ signal amplitudes were quantified and compared with those in non-transfected control cells within the same experiment. The average of the β-ionone-mediated Ca2+ signal amplitude was found to be significantly reduced (∼85%) in siRNA-expressing melanocytes compared with control cells (Fig. 3, F and G). Expression of scrambled control siRNA did not show an effect on the β-ionone-induced Ca2+ responses in transfected cells; neither did the transfection procedure alter Ca2+ responses of cells to endothelin-1 (Fig. 3G), which was reported to induce a Ca2+ rise in melanocytes through action on the endothelin-1B receptor (41). We further showed that the β-ionone-evoked Ca2+ rise could be prevented by co-application of the OR51E2 competitive antagonist α-ionone (11) (Fig. 3I), whereas α-ionone alone did not produce Ca2+ signals (Fig. 3H). Together, these data show that β-ionone produces an increase in the cytosolic Ca2+ concentration in melanocytes and that the β-ionone induced Ca2+ rise is mediated by the activation of OR51E2.
OR51E2 Signaling in Melanocytes
We next aimed to elucidate the signaling mechanism of OR51E2 in primary human melanocytes. To determine the origin of the agonist-evoked Ca2+ rise, we used Ringer's solution with varying Ca2+ concentrations. The β-ionone-induced Ca2+ increase still occurred after removal of the extracellular Ca2+, suggesting that release from intracellular stores contributes to the β-ionone-induced Ca2+ signal (Fig. 4, A and G). The signal amplitude was significantly reduced (by ∼30%), the signal onset was delayed, and the rise in intracellular Ca2+ was slower under Ca2+-free conditions. Extracellular Ca2+ is, therefore, considered to significantly account for the β-ionone-induced Ca2+ response in melanocytes. The observation that thapsigargin prestimulation suppresses the β-ionone-induced Ca2+ response (Fig. 4, B and G) might reflect a reciprocal regulation between the OR51E2-initiated pathway and store-operated calcium entry (SOCE). To identify potential Ca2+ channels mediating the Ca2+ influx, we tested pharmacological inhibitors of SOCE and transient receptor potential (TRP) channels. Whereas ORAI and TRP channel blocker 2-APB significantly diminished the β-ionone-induced Ca2+ signal (Fig. 4, C and G), TRPC and SOCE channel blocker SKF 96365 (Fig. 4, D and G) and SOCE inhibitor BTP2 (Fig. 4, E and G) as well as stretch-activated calcium channel, TRPA1, and TRPC blocker GdCl3 (Fig. 4, E and G) did not influence the β-ionone induced Ca2+ rise in melanocytes when co-applied with β-ionone.
FIGURE 4.
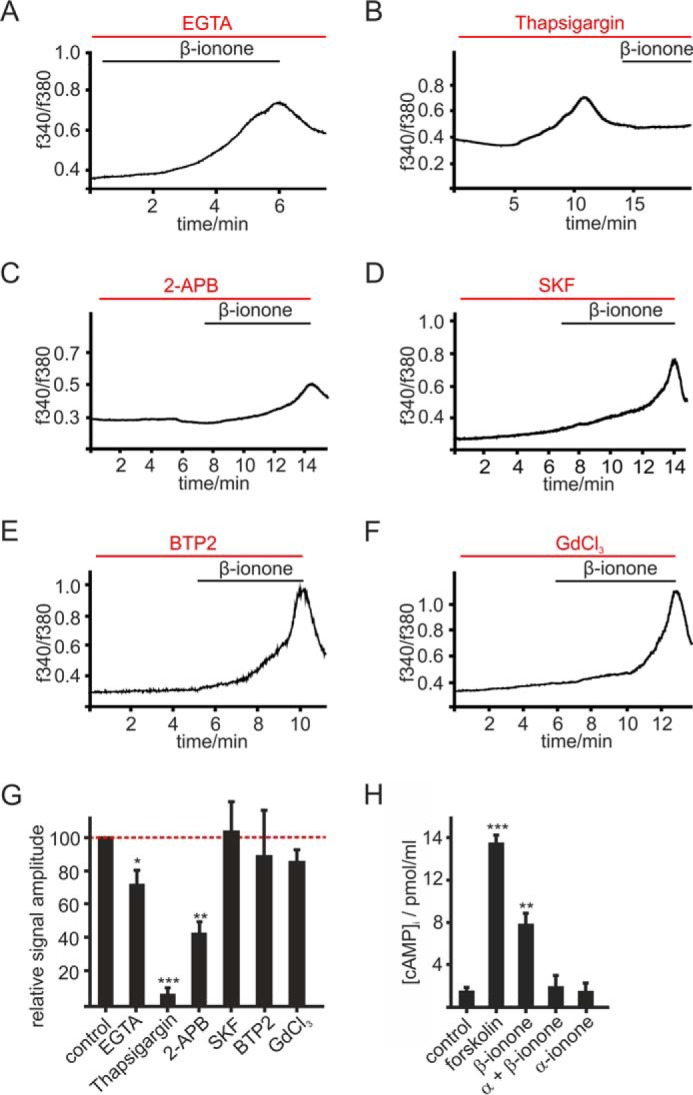
OR51E2 signaling in melanocytes. A–F, representative recordings of Fura-2-loaded melanocytes. Cytosolic Ca2+ concentration is monitored as integrated fluorescence ratio as a function of time. A, Ringer's solution containing 50 μm EGTA decreases the β-ionone (50 μm) induced response. Pretreatment with thapsigargin (10 μm) (B) or 2-APB (25 μm) (C) reduced the β-ionone (50 μm)-induced Ca2+ signals in melanocytes. SKF 96365 (SKF; 10 μm) (D), BTP2 (20 μm) (E), and GdCl3 (100 μm) (F) did not influence the β-ionone (50 μm)-induced Ca2+ response in melanocytes. G, quantification of β-ionone-induced Ca2+ signals in blocker experiments relative to control measurements (β-ionone only) or quantification of Ca2+ signal amplitudes in Ca2+ free conditions (+50 μm EGTA), normalized to the melanocyte responses measured in Ca2+ containing buffer (control). Data significance was calculated using Student's t test referring to the β-ionone induced Ca2+ signal in controls. The data are shown as the means ± S.E. (n ≥ 65 cells) (*, p < 0.05; **, p < 0.01; ***, p < 0.001). H, OR51E2 signaling involves activation of adenylate cyclases. β-Ionone (50 μm, 30 min) stimulation resulted in a 4-fold increase in intracellular cAMP, which was suppressed by co-stimulation with α-ionone (200 μm). Stimulation of melanocytes by α-ionone alone did not increase intracellular cAMP. Forskolin (20 μm) served as a positive control. Experiments were performed as duplicates in four independent experiments. Significance was calculated by Student's t test referring to the intracellular cAMP concentration in control cells. Error bars represent the S.E. (**, p < 0.01; ***, p < 0.001).
In olfactory sensory neurons, ORs couple to Gαolf, a protein homologous to Gαs, resulting in activation of an adenylate cyclase and generation of cAMP. We here investigated whether OR51E2 activation in melanocytes also modulates cellular cAMP levels. We determined cAMP levels in primary melanocytes and found that β-ionone stimulation increased the intracellular cAMP concentration 4-fold. This effect was suppressed by co-stimulation with the OR51E2 inhibitor α-ionone, indicating that the elevation of cAMP levels upon β-ionone treatment is caused by activation of OR51E2 (Fig. 4H). Usage of pharmacological tools that specifically inhibit key enzymes typically activated by GPCR stimulation (adenylate cyclase, phospholipase C) did not allow for straightforward interpretation of the Ca2+ data, probably due to the observed cytotoxic side effects of the tested inhibitors MDL-12,330A, edelfosine and U73122 in melanocytes. However, the pharmacological characterization using inhibitors of Ca2+ signaling (thapsigargin, 2-APB, SKF, BTP2, GdCl3) indicate that the OR51E2-triggered Ca2+ signal is constituted by Ca2+ release from intracellular stores and an additive Ca2+ influx from the extracellular space. Ca2+ influx may be mediated by members of the TRP ion channel family that are expressed in melanocytes such as TRPM family members (42), TRPA1 (43), or TRPV1 (44) or calcium release-activated channels, such as ORAI1/2 (45). However, an involvement of ORAI channels, TRPC channels, and TRPA1 could be excluded because SKF, BTP2, and GdCl3 did not block the β-ionone-induced Ca2+ signal at tested concentrations. Therefore, we suggest that members of the TRPM family mediate the observed Ca2+ influx. This assumption is supported by the finding that 2-APB, which blocks inter alia TRPM3, TRPM7, and TRPM8, significantly reduced the β-ionone-induced Ca2+ signal.
Subcellular Localization of OR51E2
A well characterized GPCR that is involved in biogenesis of the pigment producing organelles, the melanosomes, is the ocular albinism type 1 (46–51). Ocular albinism type 1, an atypical GPCR, primarily localizes to intracellular endolysosomes and the melanosomes of retinal pigment epithelial cells rather than the cell surface (52, 53).
We, therefore, investigated where OR51E2 resides in human epidermal melanocytes. To determine the subcellular localization of the receptor, we performed immunofluorescence studies to visualize OR51E2 protein. Melanocytes were labeled with an antibody against OR51E2. In epidermal melanocytes, immunofluorescence staining revealed that OR51E2 is localized in a vesicular pattern throughout the cytosol (Fig. 5A). OR51E2-containing vesicles accumulated at the extremity of the dendrites. Localization at the plasma membrane was not apparent in immunocytochemical stainings. Co-staining with antibodies targeting several cellular organelles revealed that OR51E2 did not localize to the endoplasmic reticulum, the Golgi apparatus, melanosomes, and lysosomes (data not shown) but showed co-localization with EEA-1 (Fig. 5B), an early endosomal marker. Analysis of numerous cells revealed that there was clear OR51E2 labeling at the plasma membrane.
FIGURE 5.

Intracellular localization of OR51E2. A, immunofluorescence confocal micrographs of a melanocyte labeled with an OR51E2-specific antibody (green) and DAPI to visualize the nucleus (blue). Intracellular, vesicular staining of OR51E2 can be observed. The bar indicates 5 μm. B, immunofluorescence confocal micrographs of a melanocyte co-labeled with an OR51E2-specific antibody (green) and an antibody to EAA-1 (red). Co-staining of cellular organelles appears in yellow in the merged image (arrows). The bar indicates 5 μm. C, surface localization of OR51E2 in melanocytes verified by surface biotinylation and detection by Western blotting. A representative Western blotting of biotinylated membrane is shown (n = 3 independent experiments). GAPDH served to control of purification of cell surface proteins only. D, Ca2+ responses of melanocytes to steroid ligands of OR51E2. Shown are representative Ca2+ imaging traces of melanocytes stimulated with the OR51E2 steroid ligands 6-dehydrotestosterone-(s1,2),4,6-androstadien-17α-ol-3-one (s2, 4) and 1,4,6-androstadien-3,17-dione (s3, 5). Ca2+ signals are displayed as integrated fluorescence ratio as a function of time. Steroids were applied for 5 min. E, heterologously expressed OR51E2 responds to FITC-tagged 6-dehydrotestosterone. Shown is a representative Ca2+ imaging trace of a Hana3a cell transiently expressing OR51E2. ATP (20 μm, 2 s) served to control the ability of the cells to produce Gq-mediated Ca2+ signals. The integrated fluorescence ratio (f340/f380) of the Fura-2 loaded cell is shown as a function of time. FITC-6-dehydrotestosterone (100 μm) was applied for 10 s. F, representative Ca2+ imaging recording of melanocytes treated with FITC-steroids (100 μm) for 5 min. Only application of FITC-6-dehydrotestosterone (FITC-s1) induced a Ca2+ signal in the cells; application of FITC-DHT showed no effect on cytosolic Ca2+ levels. G, quantification of cytosolic Ca2+ signal amplitudes upon stimulation of melanocytes with β-ionone (50 μm), 6-dehydrotestosterone (s1, 100 μm), dihydrotestosterone (DHT, 100 μm), FITC-6-dehydrotestosterone (FITC-s1, 100 μm), and FITC-DHT (100 μm). Signal amplitudes were quantified and normalized by the β-ionone-induced maximal signal amplitudes. Three independent experiments (each with >70 cells) were performed in quadruplicates for each steroid, and data are shown as the mean. Significance was calculated by Student's t test for each sample group referring to the β-ionone-induced signal amplitude. Error bars represent the S.E. (**, p < 0.01; ***, p < 0.001). H, repetitive stimulation with β-ionone evoked increasing Ca2+ signals. Representative Ca2+ imaging trace of a Fura-2-loaded melanocyte. β-Ionone (50 μm) was applied for 5 min in each application. I, left panel, different durations of pretreatment with 50 μm β-ionone resulted in increased responses to consecutive stimulation with β-ionone. Shown are representative Ca2+ imaging traces of Fura-2-loaded primary melanocytes; β-ionone (50 μm) was consecutively applied for 5 min. Right panel, quantification of signal amplitudes in melanocytes pretreated with β-ionone (50 μm) for different durations; bars represent the mean normalized to control amplitudes (from cells not pretreated). Three independent experiments were performed for each prestimulation, each with >50 cells. Significance was calculated by Student's t test for each sample group referring to the β-ionone induced Ca2+ signal in control cells. Error bars represent the S.E. (**, p < 0.01).
Because immunocytochemical methods failed to give clear information about membrane localization of OR51E2, we next aimed to resolve whether receptor activation occurs also at the plasma membrane or only at intracellular membranes. To provide evidence for plasma membrane localization of OR51E2, we detected the protein by Western blotting in surface preparations of primary melanocytes (Fig. 5C).
To distinguish whether the β-ionone induced Ca2+ signal results from activation of surface or cytosolic OR51E2, we generated ligands for receptor activation, which are less membrane-permeable than the unmodified ligands. β-Ionone exhibits hydrophobic structural properties and may, therefore, be membrane-permeable, making it possible that the receptor is activated by the ligand inside an intracellular compartment. Other OR51E2 activating ligands described previously are the steroids 4,6-androstadien-17α-ol-3-one, 1,4,6-androstadien-3,17-dione, and 6-dehydrotestosterone (11). For chemical structures, see Scheme 7. Activation of OR51E2 by these steroid ligands (100 μm) could be shown in melanocytes (Fig. 5D), whereas stimulation with dihydrotestosterone (DHT), which does not activate recombinant OR51E2, failed to induce Ca2+ signals in melanocytes. Notably, the Ca2+ signal induced by OR51E2-activating steroid ligands was significantly higher compared with the β-ionone induced Ca2+ responses (Fig. 5D).
SCHEME 7.

Chemical structures of ligands and synthesis of FITC-labeled steroids. A, molecular structures of ligands used in this study. Steroids are labeled according to IUPAC. The dienone substructure common to active ligands is highlighted. B, synthesis of FITC labeled steroids. i, alcohol 2 or 3, THF (70 mm), carbonyldiimidazole (2.0 eq.), 12 h, 20 °C. ii, monoimidazolide 6 or 7, acetonitrile/DMF (99:1), 4,7,10-trioxatridecan-1,13-diamine (3.0 eq.), 12 h, 20 °C. iii, amine 8 or 9, DMF (0.015 m), FITC (3.0 eq.); diisopropylethylamine (6.0 equiv), 4 h, then preparative HPLC, 12–25% yield overall.
These data suggested that β-ionone might be a weaker agonist and that the A/B-ring region of the steroids was important for ligand activity, whereas the D ring should be amenable to modification. To study the cellular site of activation of OR51E2, we synthesized an OR51E2-activating dehydrotestosterone appended with the membrane-impermeable anionic dye molecule FITC by means of a water-soluble PEG linker. As control, we used DHT that was shown to be inactive on OR51E2 (11). The FITC label was attached to the OH-group of dihydrotestosterone and 6-dehydrotestosterone on position 17 (see Scheme 7 and “Experimental Procedures”). Due to the change in physicochemical properties these dye-labeled, hydrophilic steroid ligands are expected to exhibit a reduced cell membrane permeability compared with the parental steroids.
Interestingly, the FITC-tagged dehydrotestosterone derivative was found to activate recombinant OR51E2 as shown by Ca2+ imaging experiments on Hana3a cells transiently expressing OR51E2 (Fig. 5E). Similarly, application of the same FITC-tagged steroid ligand to primary human melanocytes triggered Ca2+ responses (Fig. 5, F and G). Although the maximal signal amplitude was smaller compared with untagged 6-dehydrotestosterone, it resembled the Ca2+ response amplitude evoked by β-ionone. Neither treatment of melanocytes with the inactive steroid DHT nor with FITC-DHT induced a comparable Ca2+ response in the cells (Fig. 5, F and G), indicating that OR51E2 protein, which is present at the plasma membrane of melanocytes, received the external signal.
Repetitive β-ionone stimulation of melanocytes resulted in an increase in the Ca2+ response amplitudes (Fig. 5H). When we exposed melanocytes to β-ionone before re-stimulation in Ca2+ imaging experiments, we observed significant increases in the β-ionone induced Ca2+ levels that were dependent on the duration of preexposure (Fig. 5, H and I). We assume that these effects could be caused by increased insertion of OR51E2 into the plasma membrane upon activation of the receptor, thereby increasing the low amounts of functional OR51E2 at the cell surface of melanocytes.
Discussion
Functionality of an Ectopically Expressed Odorant Receptor in Melanocytes
The first evidence for an influence of odorants on pigment cells was obtained in the 1980s, when the superfamily of odorant receptors was unknown. Frog melanophores were shown to disperse their melanosomes in response to odorants, and odorant-induced pigment dispersion was accompanied by rises in intracellular cAMP levels (30). Corresponding studies discovered cinnamaldehyde and β-ionone to have pigment dispersing activity in fish melanophores (31), but neither substances resulted in a measurable rise in cAMP levels. We describe here that β-ionone led to an increase in the Ca2+ concentration and elevated cAMP levels. In addition, melanin content increased in primary epidermal melanocytes, possibly due to up-regulated expression of tyrosinase. Moreover, we describe that OR51E2 mediated β-ionone-induced effects in melanocytes. By gene-silencing and by using a specific antagonist, we could clearly show that the β-ionone-induced Ca2+ signal in melanocytes depends on the expression of OR51E2.
OR51E2 belongs to the family of odorant receptors and is expressed in prostate cancer cells (11). Analysis of the role of OR51E2 in melanocytes revealed that the receptor affects melanocyte proliferation, differentiation, and melanogenesis and indicates that ectopically expressed ORs have important cellular functions. As OR51E2 expression is up-regulated in human prostate cancer cells compared with healthy prostate epithelial cells, the receptor may be used as a potential tumor biomarker (54–57). Therefore, it would be interesting to investigate OR51E2 expression and function in melanoma cells derived from epidermal melanocytes. OR51E2 acts as a cell surface steroid receptor that mediates rapid, non-genomic, steroidal signaling in prostate cancer cells (11). The previously identified steroid ligands of OR51E2 activate Ca2+ responses in melanocytes in a similar fashion. The exquisite selectivity of this receptor with respect to the molecular structure of the agonist suggests that endogenous ligands should exist to regulate the proliferation and differentiation of melanocytes. It is tempting to speculate that this yet elusive endogenous ligand for OR51E2 may be a keratinocyte-derived factor that is chemically similar to the identified agonists, possibly a steroidal compound.
OR51E2 Localization
Intracellular localization of GPCRs was previously shown in retinal pigment epithelial cells. The atypical GPCR ocular albinism type 1 is localized to membranes of melanosomes and late endosomes/lysosomes, although low amounts of the receptor were detected also at the plasma membrane (58). Ocular albinism type 1 is considered to regulate melanosome biogenesis by transducing signals through activation of heterotrimeric G-proteins on the cytoplasmatic side of the organelle membrane (46, 48, 59).
OR51E2 was mainly localized in early endosomes associated with EEA-1 in human epidermal melanocytes. Endosomal organelles are direct precursors to premelanosomes (stage I melanosomes) (60). As in other cells, two distinct early endosomal domains can be distinguished, tubulovesicular structures containing the bulk of EEA-1 and globular structures, which hold enriched concentrations of the melanoma-associated protein melan-A. However, we could exclude localization of OR51E2 to the globular endosomal domains by co-labeling OR51E2 and melan-A. Although immunocytochemical signals were not apparent in immunocytochemical analysis, we clearly demonstrated plasma membrane localization of OR51E2 by cell surface preparations and Western blotting analysis. Activation of OR51E2 in melanocytes still occurred when using a steroid agonist for OR51E2 with reduced membrane permeability, indicating that the observed Ca2+ signals result from activation of OR51E2 protein at the cell surface rather than cytosolic OR51E2.
Signaling Cascade of OR51E2 in Normal Human Epidermal Melanocytes
The present study shows that signaling of OR51E2 in melanocytes involves cAMP, potentially TRPM family members, Ca2+, and activation of PKA and MAPKs. The OR51E2-induced Ca2+ signal was found to be partially dependent on extracellular Ca2+, as the amplitude of the response was significantly reduced in the absence of Ca2+. However, pharmacological characterization revealed that the influx of extracellular Ca2+ is neither mediated via store-operated Ca2+ channels of the ORAI family, nor by TRPC, TRPV, and TRPA1 channels but indicate an involvement of other TRP channels. Based on previous reports and our results (with given limitations of the employed Ca2+ imaging technique), we conclude that the molecular identity of the TRP channels is in the TRPM family as members of this TRP family are expressed in melanocytes and can be blocked by 2-APB while remaining unaffected by the other tested inhibitors.
The proposed OR51E2 initiated signal transduction pathway in melanocytes may involve PLC-mediated signaling as well. Unfortunately, common inhibitors of PLC appeared unsuitable for application on melanocytes. In classical olfactory tissues, olfactory receptors can couple to both adenylyl cyclase- and PLC-mediated pathways (61).
In prostate cancer cells, OR51E2 activates TRPV6 via Src-kinase in a G-protein-independent manner (62). However, comparison of the OR51E2 signaling in prostate cancer cells and melanocytes reveals differences, such as an involvement of adenylate cyclase (11). Thus, the signal transduction cascade of ectopically expressed ORs seems to be majorly determined by the cellular background and not by the properties of the receptor protein.
Role of OR51E2 on Melanogenesis and Pigment Cell Differentiation
Pigment production in vertebrates is modulated by a variety of hormones and neurotransmitters acting on transmembrane receptors located on the cell surface (63, 64). Activation of the ET-1 receptor increases cytosolic Ca2+ concentrations via ORAI1 channels, thereby stimulating melanogenesis (45, 65). The intracellular Ca2+ concentration itself was found not to be essential for melanogenesis but to play an important role in modulating the responses of melanocytes to melanogenic stimuli (67, 68). Activation of OR51E2 resulted in a prolonged increase in the intracellular Ca2+ concentration and led to an induction of melanogenesis via the cAMP pathway. Regulation of melanogenesis was shown to involve stimulation of adenylate cyclase followed by an increase in the intracellular cAMP level and activation of cAMP-dependent protein kinases (PKA) (69). Activated PKA is involved in the phosphorylation of cAMP-responsive element-binding protein (CREB) and CREB-binding protein. Phosphorylated CREB leads to an activation of microphthalmia-associated transcription factor (MITF), whereas MITF regulates the expression (but not activity) of melanogenic enzymes (tyrosinase, TYRP1, and TYRP2) (for review, see Ref. 70). Thus, the observed up-regulation of tyrosinase likely results from OR51E2 triggered activation of PKA.
Downstream signaling of OR51E2 in epidermal melanocytes also involves activation of p38 MAPK and p44/42 MAPK (ERK1/2) as possible modulators of melanogenesis. These MAPKs are described to control pigment cellular responses to melanogenetic stimuli by induction of tyrosinase proteasomal degradation, thus antagonizing activated cAMP signaling (71).
Effects of OR51E2 activation on melanocyte morphology and melanogenesis implicate that OR51E2 is also involved in regulation of melanocyte differentiation. Many coat color genes, known for their ability to regulate melanosome formation, control in addition melanoblast migration, proliferation, and differentiation and melanosome distribution. Thus, melanocyte proliferation and differentiation is not only regulated by typical growth factor receptors but also by genes classically known for their role in pigment formation.
Interestingly, repetitive cell stimulation in Ca2+ imaging experiments showed an increased responsiveness of melanocytes pretreated with β-ionone. We hypothesize that OR51E2 that is localized in the early endosome may translocate to the plasma membrane upon activation of cell surface receptors. A new paradigm is emerging in some cellular contexts, in which stocks of functional GPCRs retained within intracellular compartments can be rapidly mobilized to the plasma membrane to maintain sustained physiological responsiveness (66). However, further data must be acquired to confirm such a role for OR51E2 in human melanocytes.
The present study constitutes a new example for the functionality of ectopic ORs. It also contributes to the understanding the molecular processes involved in regulation of skin pigmentation by showing that the ectopically expressed olfactory receptor OR51E2 is functionally expressed in melanocytes. Pigment cells respond to the OR51E2 ligand with decreased proliferation rates and increased melanogenesis. As OR51E2-induced signaling mechanisms could influence melanocyte homeostasis, receptor-activating steroids or terpenoids might provide novel compounds for the treatment of pigmentation disorders and proliferative pigment cell disorders such as melanoma.
Author Contributions
L. G., N. J., and S. V. performed the experimental research. H.-D. A. and B. M. performed synthesis of the FITC-steroids. L. G., E. M. N., and H. H. conceived and designed the research. L. G., N. J., and E. M. N. wrote the manuscript.
Acknowledgments
We thank F. Moessler and H. Bartel for technical assistance, Dr. J. Panten (Symrise, Holzminden, Germany) for providing odorants, Dr. G. Neufang and W. Gerwat (Beiersdorf) for providing primary melanocytes, Dr. W. Zhang (Bayer HealthCare) for contributions to the preliminary experiments in this study, and Dr. S. Sinclair (Ruhr-University Bochum, Germany) for proofreading and helpful suggestions on the manuscript.
This study was supported by grants from the Deutsche Forschungsgemeinschaft (SFB642; to E. M. N. and H. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- OR
- olfactory receptor
- 2-APB
- aminoethoxydiphenyl borate
- CREB
- cAMP-responsive element-binding protein
- DHT
- dehydrotestosterone
- EEA-1
- early endosome antigen 1
- GPCR
- G-protein-coupled receptor
- ORAI
- calcium release-activated calcium channel
- PCNA
- proliferating cell nuclear antigen
- TRP
- transient receptor potential ion channel
- tR
- retention time
- SOCE
- store-operated calcium entry
- DMF
- N,N-dimethylformamide
- ESI
- electrospray ionization
- calc.
- calculated
- qPCR
- quantitative PCR.
References
- 1. Buck L., and Axel R. (1991) A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65, 175–187 [DOI] [PubMed] [Google Scholar]
- 2. Flegel C., Manteniotis S., Osthold S., Hatt H., and Gisselmann G. (2013) Expression profile of ectopic olfactory receptors determined by deep sequencing. PloS ONE 8, e55368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang N., and Koo J. (2012) Olfactory receptors in non-chemosensory tissues. BMB Rep. 45, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang X., De la Cruz O., Pinto J. M., Nicolae D., Firestein S., and Gilad Y. (2007) Characterizing the expression of the human olfactory receptor gene family using a novel DNA microarray. Genome Biol. 8, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feldmesser E., Olender T., Khen M., Yanai I., Ophir R., and Lancet D. (2006) Widespread ectopic expression of olfactory receptor genes. BMC Genomics 7, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De la Cruz O., Blekhman R., Zhang X., Nicolae D., Firestein S., and Gilad Y. (2009) A signature of evolutionary constraint on a subset of ectopically expressed olfactory receptor genes. Mol. Biol. Evol. 26, 491–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fukuda N., Yomogida K., Okabe M., and Touhara K. (2004) Functional characterization of a mouse testicular olfactory receptor and its role in chemosensing and in regulation of sperm motility. J. Cell Sci. 117, 5835–5845 [DOI] [PubMed] [Google Scholar]
- 8. Spehr M., Gisselmann G., Poplawski A., Riffell J. A., Wetzel C. H., Zimmer R. K., and Hatt H. (2003) Identification of a testicular odorant receptor mediating human sperm chemotaxis. Science 299, 2054–2058 [DOI] [PubMed] [Google Scholar]
- 9. Veitinger T., Riffell J. R., Veitinger S., Nascimento J. M., Triller A., Chandsawangbhuwana C., Schwane K., Geerts A., Wunder F., Berns M. W., Neuhaus E. M., Zimmer R. K., Spehr M., and Hatt H. (2011) Chemosensory Ca2+ dynamics correlate with diverse behavioral phenotypes in human sperm. J. Biol. Chem. 286, 17311–17325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braun T., Voland P., Kunz L., Prinz C., and Gratzl M. (2007) Enterochromaffin cells of the human gut: sensors for spices and odorants. Gastroenterology 132, 1890–1901 [DOI] [PubMed] [Google Scholar]
- 11. Neuhaus E. M., Zhang W., Gelis L., Deng Y., Noldus J., and Hatt H. (2009) Activation of an olfactory receptor inhibits proliferation of prostate cancer cells. J. Biol. Chem. 284, 16218–16225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez M., Luo W., Weng J., Zeng L., Yi Z., Siwko S., and Liu M. (2014) PSGR promotes prostatic intraepithelial neoplasia and prostate cancer xenograft growth through NF-kappaB. Oncogenesis 3, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X., Bedigian A. V., Wang W., and Eggert U. S. (2012) G protein-coupled receptors participate in cytokinesis. Cytoskeleton 69, 810–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffin C. A., Kafadar K. A., and Pavlath G. K. (2009) MOR23 promotes muscle regeneration and regulates cell adhesion and migration. Dev. Cell 17, 649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pluznick J. L., Protzko R. J., Gevorgyan H., Peterlin Z., Sipos A., Han J., Brunet I., Wan L. X., Rey F., Wang T., Firestein S. J., Yanagisawa M., Gordon J. I., Eichmann A., Peti-Peterdi J., and Caplan M. J. (2013) Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pluznick J. L., Zou D. J., Zhang X., Yan Q., Rodriguez-Gil D. J., Eisner C., Wells E., Greer C. A., Wang T., Firestein S., Schnermann J., and Caplan M. J. (2009) Functional expression of the olfactory signaling system in the kidney. Proc. Natl. Acad. Sci. U.S.A. 106, 2059–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Busse D., Kudella P., Grüning N. M., Gisselmann G., Ständer S., Luger T., Jacobsen F., Steinsträßer L., Paus R., Gkogkolou P., Böhm M., Hatt H., and Benecke H. (2014) A synthetic sandalwood odorant induces wound-healing processes in human keratinocytes via the olfactory receptor OR2AT4. J. Invest. Dermatol. 134, 2823–2832 [DOI] [PubMed] [Google Scholar]
- 18. Luttrell L. M. (2006) Transmembrane signaling by G protein-coupled receptors. Methods Mol. Biol. 332, 3–49 [DOI] [PubMed] [Google Scholar]
- 19. Defea K. (2008) Beta-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br. J. Pharmacol. 153, S298–S309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andreeva A. V., Kutuzov M. A., and Voyno-Yasenetskaya T. A. (2007) Scaffolding proteins in G-protein signaling. J. Mol. Signal. 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vilardaga J. P., Agnati L. F., Fuxe K., and Ciruela F. (2010) G-protein-coupled receptor heteromer dynamics. J. Cell Sci. 123, 4215–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Escribá P. V., Wedegaertner P. B., Goñi F. M., and Vögler O. (2007) Lipid-protein interactions in GPCR-associated signaling. Biochim. Biophys. Acta 1768, 836–852 [DOI] [PubMed] [Google Scholar]
- 23. Hanyaloglu A. C., and von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 24. Cotton M., and Claing A. (2009) G protein-coupled receptors stimulation and the control of cell migration. Cell. Signal. 21, 1045–1053 [DOI] [PubMed] [Google Scholar]
- 25. New D. C., and Wong Y. H. (2007) Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oka M., Ichihashi M., and Chakraborty A. K. (1996) Enhanced expression of protein kinase C subspecies in melanogenic compartments in B16 melanoma cells by UVB or MSH. J. Invest. Dermatol. 106, 377–378 [DOI] [PubMed] [Google Scholar]
- 27. Staab H. (1956) Transacylierungen I: N-Acyl-Verbindungen stickstoffhaltiger heterozyklen. Chem. Ber. 89, 1927–1940 [Google Scholar]
- 28. Staab H. (1962) Syntheses using heterocyclic amides (Azolides). Angew. Chem. Int. Ed. Engl. 1, 351–367 [Google Scholar]
- 29. Silva M. R., Beja A. M., Moreira V. M., Santos R. C., and Salvador J. A. R. (2007) 3-Oxoandrost-4-en-17b-yl-1H-imidazole-1-carboxylate. Acta Cryst. Sect. E 63, o4824 [Google Scholar]
- 30. Lerner M. R., Reagan J., Gyorgyi T., and Roby A. (1988) Olfaction by melanophores: what does it mean? Proc. Natl. Acad. Sci. U.S.A. 85, 261–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karlsson J. O., Svensson S. P., Mårtensson L. G., Odman S., Elwing H., and Lundström K. I. (1994) Effects of odorants on pigment aggregation and cAMP in fish melanophores. Pigment Cell Res. 7, 61–64 [DOI] [PubMed] [Google Scholar]
- 32. Ghosh R., Nadiminty N., Fitzpatrick J. E., Alworth W. L., Slaga T. J., and Kumar A. P. (2005) Eugenol causes melanoma growth suppression through inhibition of E2F1 transcriptional activity. J. Biol. Chem. 280, 5812–5819 [DOI] [PubMed] [Google Scholar]
- 33. Pisano M., Pagnan G., Loi M., Mura M. E., Tilocca M. G., Palmieri G., Fabbri D., Dettori M. A., Delogu G., Ponzoni M., and Rozzo C. (2007) Antiproliferative and pro-apoptotic activity of eugenol-related biphenyls on malignant melanoma cells. Mol. Cancer 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirobe T. (1992) Control of melanocyte proliferation and differentiation in the mouse epidermis. Pigment Cell Res. 5, 1–11 [DOI] [PubMed] [Google Scholar]
- 35. Englaro W., Rezzonico R., Durand-Clément M., Lallemand D., Ortonne J. P., and Ballotti R. (1995) Mitogen-activated protein kinase pathway and AP-1 are activated during cAMP-induced melanogenesis in B-16 melanoma cells. J. Biol. Chem. 270, 24315–24320 [DOI] [PubMed] [Google Scholar]
- 36. Hearing V. J., and Tsukamoto K. (1991) Enzymatic control of pigmentation in mammals. FASEB J. 5, 2902–2909 [PubMed] [Google Scholar]
- 37. Lee J., Kim Y. S., and Park D. (2007) Rosmarinic acid induces melanogenesis through protein kinase A activation signaling. Biochem. Pharmacol. 74, 960–968 [DOI] [PubMed] [Google Scholar]
- 38. Imokawa G., Yada Y., and Kimura M. (1996) Signalling mechanisms of endothelin-induced mitogenesis and melanogenesis in human melanocytes. Biochem. J. 314, 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imokawa G., Kobayasi T., and Miyagishi M. (2000) Intracellular signaling mechanisms leading to synergistic effects of endothelin-1 and stem cell factor on proliferation of cultured human melanocytes: cross-talk via trans-activation of the tyrosine kinase c-kit receptor. J. Biol. Chem. 275, 33321–33328 [DOI] [PubMed] [Google Scholar]
- 40. Smalley K., and Eisen T. (2000) The involvement of p38 mitogen-activated protein kinase in the α-melanocyte stimulating hormone (α-MSH)-induced melanogenic and anti-proliferative effects in B16 murine melanoma cells. FEBS Lett. 476, 198–202 [DOI] [PubMed] [Google Scholar]
- 41. Kang H. Y., Kang W. H., and Lee C. (1998) Endothelin-B receptor-mediated Ca2+ signaling in human melanocytes. Pflugers Arch. 435, 350–356 [DOI] [PubMed] [Google Scholar]
- 42. Guo H., Carlson J. A., and Slominski A. (2012) Role of TRPM in melanocytes and melanoma. Exp. Dermatol. 21, 650–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bellono N. W., Kammel L. G., Zimmerman A. L., and Oancea E. (2013) UV light phototransduction activates transient receptor potential A1 ion channels in human melanocytes. Proc. Natl. Acad. Sci. U.S.A. 110, 2383–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi T. Y., Park S. Y., Jo J. Y., Kang G., Park J. B., Kim J. G., Hong S. G., Kim C. D., Lee J. H., and Yoon T. J. (2009) Endogenous expression of TRPV1 channel in cultured human melanocytes. J. Dermatol. Sci. 56, 128–130 [DOI] [PubMed] [Google Scholar]
- 45. Stanisz H., Stark A., Kilch T., Schwarz E. C., Müller C. S., Peinelt C., Hoth M., Niemeyer B. A., Vogt T., and Bogeski I. (2012) ORAI1 Ca2+ channels control endothelin-1-induced mitogenesis and melanogenesis in primary human melanocytes. J. Invest. Dermatol. 132, 1443–1451 [DOI] [PubMed] [Google Scholar]
- 46. Schiaffino M. V., d'Addio M., Alloni A., Baschirotto C., Valetti C., Cortese K., Puri C., Bassi M. T., Colla C., De Luca M., Tacchetti C., and Ballabio A. (1999) Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat. Genet. 23, 108–112 [DOI] [PubMed] [Google Scholar]
- 47. Innamorati G., Piccirillo R., Bagnato P., Palmisano I., and Schiaffino M. V. (2006) The melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Res. 19, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schiaffino M. V., and Tacchetti C. (2005) The ocular albinism type 1 (OA1) protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Pigment Cell Res. 18, 227–233 [DOI] [PubMed] [Google Scholar]
- 49. Staleva L., and Orlow S. J. (2006) Ocular albinism 1 protein: trafficking and function when expressed in Saccharomyces cerevisiae. Exp. Eye Res. 82, 311–318 [DOI] [PubMed] [Google Scholar]
- 50. Palmisano I., Bagnato P., Palmigiano A., Innamorati G., Rotondo G., Altimare D., Venturi C., Sviderskaya E. V., Piccirillo R., Coppola M., Marigo V., Incerti B., Ballabio A., Surace E. M., Tacchetti C., Bennett D. C., and Schiaffino M. V. (2008) The ocular albinism type 1 protein, an intracellular G protein-coupled receptor, regulates melanosome transport in pigment cells. Hum. Mol. Genet. 17, 3487–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Falletta P., Bagnato P., Bono M., Monticone M., Schiaffino M. V., Bennett D. C., Goding C. R., Tacchetti C., and Valetti C. (2014) Melanosome-autonomous regulation of size and number: the OA1 receptor sustains PMEL expression. Pigment Cell Melanoma Res. 27, 565–579 [DOI] [PubMed] [Google Scholar]
- 52. Piccirillo R., Palmisano I., Innamorati G., Bagnato P., Altimare D., and Schiaffino M. V. (2006) An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J. Cell Sci. 119, 2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schiaffino M. V., Baschirotto C., Pellegrini G., Montalti S., Tacchetti C., De Luca M., and Ballabio A. (1996) The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc. Natl. Acad. Sci. U.S.A. 93, 9055–9060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu L. L., Stackhouse B. G., Florence K., Zhang W., Shanmugam N., Sesterhenn I. A., Zou Z., Srikantan V., Augustus M., Roschke V., Carter K., McLeod D. G., Moul J. W., Soppett D., and Srivastava S. (2000) PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res. 60, 6568–6572 [PubMed] [Google Scholar]
- 55. Xia C., Ma W., Wang F., Hua S. b., and Liu M. (2001) Identification of a prostate-specific G-protein coupled receptor in prostate cancer. Oncogene 20, 5903–5907 [DOI] [PubMed] [Google Scholar]
- 56. Xu L. L., Sun C., Petrovics G., Makarem M., Furusato B., Zhang W., Sesterhenn I. A., McLeod D. G., Sun L., Moul J. W., and Srivastava S. (2006) Quantitative expression profile of PSGR in prostate cancer. Prostate Cancer Prostatic. Dis. 9, 56–61 [DOI] [PubMed] [Google Scholar]
- 57. Rigau M., Morote J., Mir M. C., Ballesteros C., Ortega I., Sanchez A., Colás E., Garcia M., Ruiz A., Abal M., Planas J., Reventós J., and Doll A. (2010) PSGR and PCA3 as biomarkers for the detection of prostate cancer in urine. Prostate 70, 1760–1767 [DOI] [PubMed] [Google Scholar]
- 58. Lopez V. M., Decatur C. L., Stamer W. D., Lynch R. M., and McKay B. S. (2008) l-DOPA is an endogenous ligand for OA1. PLoS Biol. 6, e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Giordano F., Simoes S., and Raposo G. (2011) The ocular albinism type 1 (OA1) GPCR is ubiquitinated and its traffic requires endosomal sorting complex responsible for transport (ESCRT) function. Proc. Natl. Acad. Sci. U.S.A. 108, 11906–11911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raposo G., and Marks M. S. (2002) The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic 3, 237–248 [DOI] [PubMed] [Google Scholar]
- 61. Klasen K., Corey E. A., Kuck F., Wetzel C. H., Hatt H., and Ache B. W. (2010) Odorant-stimulated phosphoinositide signaling in mammalian olfactory receptor neurons. Cell. Signal. 22, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Spehr J., Gelis L., Osterloh M., Oberland S., Hatt H., Spehr M., and Neuhaus E. M. (2011) G protein-coupled receptor signaling via Src kinase induces endogenous human transient receptor potential vanilloid type 6 (TRPV6) channel activation. J. Biol. Chem. 286, 13184–13192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yamaguchi Y., and Hearing V. J. (2009) Physiological factors that regulate skin pigmentation. Biofactors 35, 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Park H. Y., Kosmadaki M., Yaar M., and Gilchrest B. A. (2009) Cellular mechanisms regulating human melanogenesis. Cell. Mol. Life Sci. 66, 1493–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Imokawa G., Yada Y., and Miyagishi M. (1992) Endothelins secreted from human keratinocytes are intrinsic mitogens for human melanocytes. J. Biol. Chem. 267, 24675–24680 [PubMed] [Google Scholar]
- 66. Achour L., Labbé-Jullié C., Scott M. G., and Marullo S. (2008) An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol. Sci. 29, 528–535 [DOI] [PubMed] [Google Scholar]
- 67. Carsberg C. J., Jones K. T., Sharpe G. R., and Friedmann P. S. (1995) Intracellular calcium modulates the responses of human melanocytes to melanogenic stimuli. J. Dermatol. Sci. 9, 157–164 [DOI] [PubMed] [Google Scholar]
- 68. Bellono N. W., and Oancea E. V. (2014) Ion transport in pigmentation. Arch. Biochem. Biophys. 563, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Buscà R., and Ballotti R. (2000) Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 13, 60–69 [DOI] [PubMed] [Google Scholar]
- 70. Slominski A., Tobin D. J., Shibahara S., and Wortsman J. (2004) Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 84, 1155–1228 [DOI] [PubMed] [Google Scholar]
- 71. Bellei B., Maresca V., Flori E., Pitisci A., Larue L., and Picardo M. (2010) p38 regulates pigmentation via proteasomal degradation of tyrosinase. J. Biol. Chem. 285, 7288–7299 [DOI] [PMC free article] [PubMed] [Google Scholar]