

Abstract
TGF-β inhibits proliferation of prostate epithelial cells. However, prostate cancer cells in advanced stages become resistant to inhibitory effects of TGF-β. The intracellular signaling mechanisms involved in differential effects of TGF-β during different stages are largely unknown. Using cell line models, we have shown that TGF-β inhibits proliferation in normal (RWPE-1) and prostate cancer (DU145) cells but does not have any effect on proliferation of prostate cancer (PC3) cells. We have investigated the role of Jun family proteins (c-Jun, JunB, and JunD) in TGF-β effects on cell proliferation. Jun family members were expressed at different levels and responded differentially to TGF-β treatment. TGF-β effects on JunD protein levels, but not mRNA levels, correlated with its effects on cell proliferation. TGF-β induced significant reduction in JunD protein in RWPE-1 and DU145 cells but not in PC3 cells. Selective knockdown of JunD expression using siRNA in DU145 and PC3 cells resulted in significant reduction in cell proliferation, and forced overexpression of JunD increased the proliferation rate. On the other hand, knockdown of c-Jun or JunB had little, if any, effect on cell proliferation; overexpression of c-Jun and JunB decreased the proliferation rate in DU145 cells. Further studies showed that down-regulation of JunD in response to TGF-β treatment is mediated via the proteasomal degradation pathway. In conclusion, we show that specific Jun family members exert differential effects on proliferation in prostate cancer cells in response to TGF-β, and inhibition of cell proliferation by TGF-β requires degradation of JunD protein.
Keywords: AP-1 transcription factor (AP-1), cell proliferation, prostate cancer, protein degradation, transforming growth factor beta (TGF-B)
Introduction
TGF-β is a secreted cytokine that acts as a major anti-proliferative factor in the initial stages of prostate cancer, whereas in the advanced stages of prostate cancer, it acquires pro-oncogenic and pro-metastatic properties (1–3). The TGF-β cytokine exists in three major isoforms: TGF-β1, TGF-β2, and TGF-β3. TGF-β ligands bind to a heterodimeric receptor complex consisting of two serine-threonine kinase receptors, designated TGF-β type I and type II receptors, and leads to activation of several intracellular pathways (4–7). Concomitant with the switch of TGF-β from growth-inhibitory to growth-promoting signal, expression of TGF-β ligands and receptors is known to be altered in prostate cancer relative to normal prostate cells and is further altered in more aggressive androgen-refractory prostate cancer cells (8, 9). Expression of TGF-β and its family members is also associated with poor prognosis (10–12). It has also been shown that loss of TGF-β type II receptor expression correlates with increasing tumor aggressiveness in prostate cancer (8). However, a significant fraction of prostate cancers become TGF-β-resistant without mutation, deletion, or down-regulation of TGF-β receptors or Smads or other downstream signaling molecules.
Previous studies have shown different effects of TGF-β1 on proliferation of different prostate cancer cell lines; TGF-β inhibits proliferation of DU145 cells but has no effect on proliferation of PC3 cells in the presence of functional TGF-β receptors and Smad signaling (13–16), indicating differences in signaling mechanisms in two cell lines downstream of receptor-dependent Smad activation that are responsible for differential effects of TGF-β on cell proliferation. TGF-β is a pleiotropic cytokine whose signaling outcome is known to depend on the combination of available contributing factors and active pathways in each target tissue. Previous reports have shown that other intracellular proteins influence TGF-β effects (17–19). It has been well established that extensive interactions exist between the TGF-β signaling pathway and other major signaling pathways, including Wnt, Notch, Hedgehog, JNK, MAPK, and AKT/PI3K (20–24). It is also becoming apparent that TGF-β signaling intersects with several transcription factors and regulators, such as GL1, SOX4, Tieg3/Klf11, Id, and AP-1 proteins (25–29). Many studies have implicated AP-1 proteins in TGF-β signaling (30–32). The AP-1 family consists of dimeric protein complexes composed of different Jun proteins (c-Jun, JunB, and JunD) and four Fos proteins (c-Fos, FosB, Fra1, and Fra2). These proteins form Jun-Jun homodimers and Jun-Fos heterodimers and bind to the 12-O-tetradecanoylphorbol-13-acetate response element, TGACTCA palindromic sequence, in the promoters of target genes (33, 34). AP-1 proteins have been shown to be involved in cell proliferation, inflammation, differentiation, apoptosis, wound healing, and carcinogenesis (35–38). Among the AP-1 proteins, there is growing evidence that Jun proteins play a major role in the control of cell proliferation and cell death by regulating the expression of cell cycle regulators (39–42). In prostate cancer, expression of AP-1 proteins has recently been associated with disease recurrence and more aggressive clinical outcome (43, 44).
Within the last few years, several studies have suggested the involvement of Jun proteins in prostate cancer growth, survival, and metastasis. For example, c-Jun has been shown to enhance androgen-dependent cell proliferation and inhibition of apoptosis in LNCaP cells (43, 45), but it mediates the action of a metastasis suppressor gene, KAI1, in PC3 and DU145 cells (46). JunD, along with Fra1 and Fra2, has also been reported to be essential in prostate cancer proliferation and confers protection against radiation-induced cell death (47). In a recent report, JunB was shown to play an important role in maintaining cell senescence that blocks malignant prostate cell transformations (48) and has been shown to be a potent activator of KAI1 (49). Jun proteins by themselves or in combination with members of the Fos proteins have also been implicated in the actions of androgens (50, 51), atmospheric pollutants (52), growth factors (53), phytochemicals (54–56), peroxides (57), isothiocyanates (58), glycoproteins (59), and, most recently, proteasome inhibitors (60). AP-1 proteins form multiple homo- and heterodimers, and the composition of these dimers may dictate expression of specific genes involved in specific biological responses. However, the specific roles of individual AP-1 family members in the development and progression of prostate cancer are still largely unknown. Few reports have shown the effects, if any, of TGF-β on AP-1 in prostate cancer (61–63).
The present study was carried out to determine specific roles of Jun family members in TGF-β effects on proliferation in prostate cancer cells. Our results indicate that JunD is essential for proliferation of prostate epithelial cells, and the inhibitory effects of TGF-β on cell proliferation are dependent on degradation of JunD protein in these cells.
Results
Effects of TGF-β1 on Proliferation of Prostate Cell Lines
We have previously shown that TGF-β1 exerts differential effects on proliferation of different prostate cancer cell lines (15, 64). To confirm these studies, we first determined the effects of TGF-β1 on proliferation of prostate cell lines representing specific stages of prostate cancer progression. Cells were plated overnight (1 × 104 cells), serum-starved for 24 h, and then treated with TGF-β1 (1 and 10 ng/ml) for 18 h. Fig. 1 shows the effects of TGF-β1 on cell proliferation. As measured by [3H]thymidine incorporation, TGF-β1 caused a significant dose-dependent inhibition of cell proliferation in RWPE1 and DU145 cells but not in PC3 and LNCaP cells. Treatment with TGF-β1 resulted in 30% (1 ng/ml) (p < 0.05) and 41% (10 ng/ml) (p < 0.05) inhibition in RWPE1 cells and 24% (1 ng/ml) and 38% (10 ng/ml) (p < 0.05) inhibition of [3H]thymidine incorporation in DU145 cells. LNCaP cells, which do not express TGF-β receptor II, served as negative control (Fig. 1). Next, we treated DU145 and PC3 cells with TGF-β1 (5 ng/ml) to determine the stage of the cell cycle where TGF-β1 exerted its inhibitory effects. TGF-β1 treatment led to an elevated number of cells in the G1 phase with a concomitant decrease in the number of cells in S phase in DU145 cells (Table 1). Similar treatment in PC3 cells did not cause any changes in cell numbers in different stages of the cell cycle.
FIGURE 1.
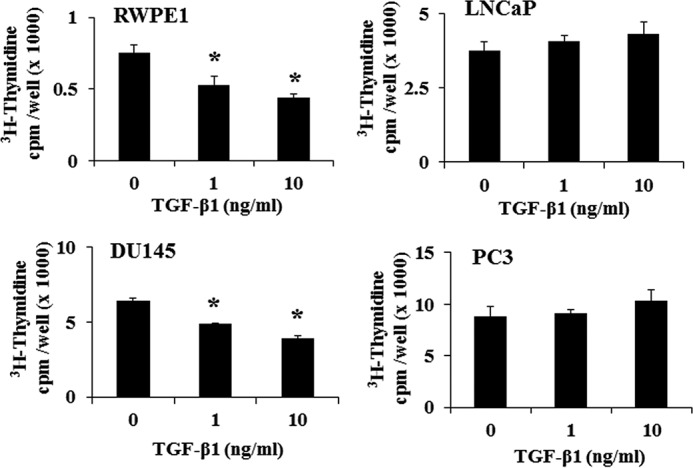
Effects of TGF-β1 on cell proliferation in different prostate cell lines. RWPE1, LNCaP, DU145, and PC-3 cells were treated with different doses of TGF-β1 (5 ng/ml) for 18 h, and [3H]thymidine incorporation into DNA was determined during a 4-h period. Each bar represents mean ± S.D. (error bars) from a representative experiment. *, significantly different from appropriate controls (p < 0.05).
TABLE 1.
Cell cycle phase distributions of DU145 and PC3 cells after treatment with TGF-β1 (5 ng/ml)
| Cell lines | Treatment | G1 | S | G2/M |
|---|---|---|---|---|
| DU145 | Control | 65.5 ± 1.5 | 16.1 ± 1.5 | 19.6 ± 1.8 |
| TGF-β1 | 73.7 ± 0.2a | 11.9 ± 1.9a | 17.0 ± 2.7 | |
| PC3 | Control | 45.4 ± 1.3 | 18.3 ± 2.1 | 36.3 ± 0.8 |
| TGF-β1 | 46.3 ± 2.0 | 20.6 ± 5.4 | 33.1 ± 3.4 |
a p < 0.05, significantly different from appropriate controls.
Expression of Jun Family Members and Their Regulation by TGF-β1 in Prostate Cancer Cells
To establish a prostate cancer model system in which to observe any correlation of Jun expression with prostate cancer progression, we first analyzed expression of Jun family members in four prostate cell lines using semiquantitative RT-PCR. Using gene-specific primers to amplify mRNA encoding each member of this protein family, all members of the Jun family were detectable in all four prostate cell lines (Fig. 2A). To examine the presence of Jun proteins in these prostate cell lines, the total cell lysate proteins were analyzed using Western blotting analysis (Fig. 2B). All Jun proteins were differentially expressed in all prostate cell lines. Lower levels of c-Jun and JunD proteins were detected in RWPE1 and LNCaP cells compared with DU145 and PC3 cells. JunB levels, on the other hand, were higher in RWPE1 and PC3 cells.
FIGURE 2.
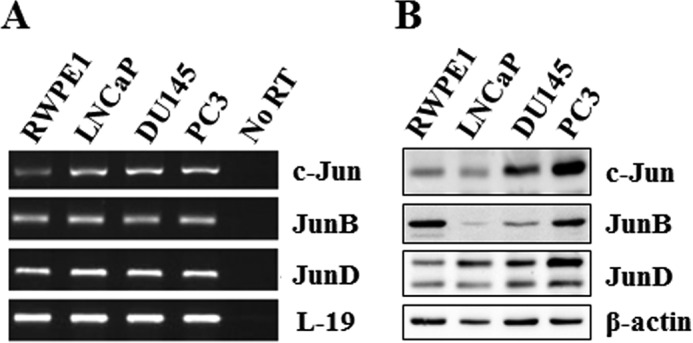
Basal expression of Jun family members in prostate cell lines. Cells were cultured under normal conditions for 24 h and lysed, and total RNA and total proteins were prepared. A, total RNAs were analyzed for expression of mRNA for individual Jun family members by RT-PCR. B, Jun family member protein levels were determined by specific Western blotting analysis. L-19 and β-actin were used as controls in RT-PCR and Western blotting analysis, respectively.
To investigate a possible role of Jun family members in TGF-β effects on cell proliferation, we determined the effects of TGF-β1 on expression of Jun family members in DU145 and PC3 cells. DU145 and PC3 cells (1 × 104) were plated overnight and treated with TGF-β1 (5 ng/ml) for different times. As shown in Fig. 3A, TGF-β1 had only a minor effect, if any, on the mRNA levels of all Jun family members. However, at the protein level, TGF-β1 exerted significant effects on the levels of JunD, c-Jun, and JunB in DU145 and/or PC3 cells in a time-dependent manner (Fig. 3, B and C). JunB was significantly up-regulated in both DU145 and PC3 cells after treatment with TGF-β1 for 2 h (DU145, 4.9 ± 0.56-fold; PC3, 2.8 ± 0.40-fold) (p < 0.05) and 8 h (DU145, 4.0 ± 0.96-fold; PC3, 2.5 ± 0.39-fold) (p < 0.05) (Fig. 3C). Interestingly, TGF-β1 significantly up-regulated c-Jun in PC3 cells starting at 2 h (2.4 ± 0.24-fold; p < 0.05), which stayed elevated for 24 h (1.8 ± 0.46-fold, p < 0.05) but did not have any effect on c-Jun protein levels in DU145 cells. TGF-β1 caused a significant down-regulation of JunD protein in DU145 cells starting at 8 h (0.6 ± 0.18-fold, p < 0.05) but not in PC3 cells. TGF-β induced an increase in the levels of c-Jun and JunB and caused a significant decrease in the levels of JunD in RWPE-1 cells similar to those seen in DU145 cells, but not in PC3 cells (data not shown). These results suggested that down-regulation of JunD protein levels and/or up-regulation of c-Jun and JunB protein levels in DU145 cells may play a role in TGF-β1 effects on inhibition of cell proliferation.
FIGURE 3.

Effects of TGF-β1 on expression of Jun family members in DU145 and PC3 cells. A, levels of mRNA of c-Jun, JunB, and JunD in DU145 and PC3 cell after treatment with TGF-β1 (5 ng/ml) as determined by RT-PCR. B, the protein levels of c-Jun, JunB, and JunD from whole cell lysates of DU145 and PC3 cells after treatment with TGF-β1 (5 ng/ml) at different time points as determined by Western blotting analysis. C, quantitative analysis of relative levels of c-Jun, JunB, and JunD proteins in DU145 and PC3 cells after treatment with TGF-β1. Normalization was performed relative to the signal obtained with β-actin. Each bar represents mean ± S.E. (error bars) (n = 3). *, significantly different from untreated controls (p < 0.05).
Role of Jun Proteins in Proliferation of Prostate Cancer Cells
To determine the possible role of individual Jun proteins in proliferation of prostate cancer cells and whether they play a role in the effects of TGF-β on cell proliferation, we used specific siRNAs to transiently knock down individual Jun proteins in DU145 and PC3 cells (Fig. 4A). Control siRNAs were also transfected to serve as negative control. Expression of Jun proteins was determined by Western blotting analysis, which confirmed a marked down-regulation of the corresponding Jun proteins in comparison with the cells transfected with the control siRNA (Fig. 4A). We analyzed proliferation of DU145 and PC3 cells after knockdown of individual Jun proteins (Fig. 4A). Transfection of JunD siRNA into DU145 or PC3 cells caused a significant reduction in proliferation of both DU145 (82% inhibition, p < 0.05) and PC3 (71% inhibition, p < 0.05) cells. On the other hand, knockdown of either c-Jun or JunB had no significant effect on cell proliferation in both cell lines. These results suggested that JunD is required for proliferation of both DU145 and PC3 cells.
FIGURE 4.

Role of individual Jun family members in proliferation of DU145 and PC3 cells. DU145 and PC3 cells were transfected with either control or specific Jun protein siRNA to knock down expression of individual family members. A, cell proliferation in DU145 and PC3 cells after transfection with control (siControl-A), or JunB, JunD, or c-Jun siRNA. Each bar represents mean ± S.E. (error bars) (n = 3). *, significantly different when compared with appropriate controls (p < 0.05). Levels of Jun proteins after transfection with control siRNA and specific Jun siRNAs were determined by Western blotting analysis (inset). B, cell proliferation in DU145 and PC3 cells after transfection with control or JunD siRNA and treatment with TGF-β1 (5 ng/ml) for 4 days.
To determine whether TGF-β1 can further inhibit cell proliferation after siRNA-mediated repression of JunD, DU145 and PC3 cells were treated with TGF-β1 (5 ng/ml) for 48 h after JunD siRNA transfection (Fig. 4B). As expected, after transfection with control siRNA, we observed a significant decrease (p < 0.05) in proliferation of DU145 cells and no effect in PC3 cells after treatment with TGF-β1. In both cell lines, transfection with JunD siRNA resulted in significant inhibition of proliferation, and TGF-β1 treatment did not cause further decrease in cell proliferation. These results suggest that TGF-β1 effects on proliferation of DU145 cells may be due to its effects on down-regulation of JunD protein in these cells.
Knockdown of JunD and TGF-β Treatment Exert Similar Effects on Cell Cycle Arrest in the G1 Phase
To demonstrate that TGF-β effects on cell cycle arrest are mediated via down-regulation of JunD, we compared the effects of JunD knockdown with TGF-β1 treatment on cell cycle machinery in DU145 cells by FACS analysis. Knockdown of endogenous JunD in DU145 cells resulted in an accumulation of cells in G1 fraction and a corresponding reduction in cells in S and G2/M phases as compared with the siControl (Fig. 5A). These effects are similar to those presented for TGF-β1 treatment shown in Table 1.
FIGURE 5.

Mechanism of inhibition of proliferation in DU145 cells by JunD. A, DU145 cells were transfected with either control (siControl-A) or JunD siRNA. After 72 h, cells were fixed with 70% ethanol and stained with propidium iodide (50 μg/ml). The DNA contents of the cells were measured by flow cytometry, and the percentage distribution of the cells in the G1, S, and G2/M phases was determined. B, DU145 cells were either transfected with JunD siRNA for 72 h with appropriate controls or treated with TGF-β1 (5 ng/ml) for 24 h. A decrease in JunD levels in the JunD knockdown is compared with the decrease in the levels of JunD after treatment with TGF-β1. Protein levels for p21, p27, cyclin D1, Ki-67, c-Myc, and Id-1 were determined in total cell lysates by Western blotting. C, assessment of apoptosis in DU145 cells transfected with JunD siRNA (bottom) and siControl-A (top). DU145 cells were transfected with JunD siRNA and control siRNA for 72 h and then subjected to the TUNEL assay and DAPI staining. The TUNEL apoptosis assay (C1 and C2) was carried out to assess apoptotic cells. The brown stain indicates apoptotic cells, whereas methyl green was used as a nuclear counter stain. DAPI staining (C3 and C4) was carried out to reveal any condensation and formation of dense bodies characteristic of apoptosis.
We also determined the levels of several proteins that play a role in cell cycle regulation in DU145 cells after knockdown of endogenous JunD or after treatment with TGF-β1 for 24 h. We have previously shown that TGF-β1 induces a decrease in c-Myc and Id1 proteins and an increase in p21 in DU145 cells (15) as a part of its inhibitory effects on cell proliferation.
As shown in Fig. 5B, treatment with TGF-β1 for 24 h or knockdown of endogenous JunD exerted identical effects on several cell cycle-associated proteins. There was a significant decrease in the levels of c-Myc, Ki-67, and Id-1 proteins, whereas there was a significant increase in the levels of p21. Both treatments did not affect the levels of p27 in DU145 cells. Interestingly, there was no significant decrease in the levels of cyclin D1, and there was a slight increase in cells treated with TGF-β1.
JunD Knockdown Does Not Affect Cell Viability
To determine whether or not the knockdown of JunD results in decreased cell viability, we used a TUNEL assay to assess apoptosis in JunD siRNA transfected in DU145 cells (Fig. 5, C1 and C2). We observed very few (∼1–2%) apoptotic positive cells in both siControl and JunD siRNA-transfected cells. In addition, we also determined the integrity of total nuclear DNA by DAPI staining in both treatments (Fig. 5, C3 and C4). Again, there was no significant differences in the nuclear DNA between the treatments, indicating that knockdown of JunD has no effect on viability of DU145 cells.
TGF-β1 Does Not Affect JunD Phosphorylation or the SAPK/JNK Pathway
To determine whether TGF-β1 induces phosphorylation of JunD in DU145 cells prior to its degradation, DU145 cells were treated with TGF-β1 (5 ng/ml) at different time points and analyzed for the levels of phospho-JunD and total JunD by Western blotting analysis. As shown in Fig. 6A, TGF-β1 did not induce phosphorylation of JunD. In fact, TGF-β1 degraded basal phospho-JunD, mirroring total JunD degradation. We also examined whether TGF-β1 activates the SAPK/JNK pathway. As shown in Fig. 6B, TGF-β1 did not induce phosphorylation of SAPK/JNKs.
FIGURE 6.

TGF-β1 did not induce JunD or SAPK/JNK phosphorylation in DU145 cells. A, DU145 cells were treated with TGF-β1 (5 ng/ml) for 0 min, 10 min, 30 min, 1 h, 2 h, and 4 h before cell lysis. Protein levels of phosphorylated (p-) and total (t-) JunD and β-actin were determined using Western blotting analysis. B, DU145 cells were treated the same way with TGF-β1 and then analyzed by Western blotting analysis for phosphorylated and total JNK and β-actin. Anisomycin (250 ng/ml) was included as a positive control for JNK activation.
Generation of Stable DU145 Cell Lines Overexpressing Jun Proteins and Effects of TGF-β1 on Their Cell Proliferation
To confirm the role of JunD in proliferation of prostate cancer cells, pcDNA3.1 constructs carrying c-Jun, JunB, or JunD were stably transfected into DU145 cells, and stable transfectants of DU145 overexpressing c-Jun, JunB, or JunD cells were generated. Empty vector (pcDNA3.1) was also transfected into DU145 cells to serve as a vector control. Multiple cell lines were selected for each transfection, and their Jun protein levels were analyzed by Western blotting analyses (Fig. 7A). Three lines overexpressing each Jun protein were selected for cell proliferation assays (Fig. 7B). Cells were plated at an initial density of 1 × 105 and counted manually after 4 days. As shown in Fig. 7B, DU145 cell lines overexpressing either c-Jun or JunB protein exhibited similar or decreased proliferation compared with the controls. In contrast, all cell lines overexpressing JunD exhibited a significant increase in cell proliferation rate (2.7 ± 0.07-fold (D1), 3.4 ± 0.34-fold (D5), 4.1 ± 0.07-fold (D6); p < 0.05). To determine the effects of TGF-β1 on the proliferation of DU145-overexpressing Jun proteins, cells were plated overnight at an initial density of 1 × 105 cells/well and were treated the next day with TGF-β1 (5 ng/ml). After 4 days, cells were counted, and the data were analyzed. As shown in Fig. 7C, TGF-β1 significantly inhibited proliferation of DU145 cells transfected with the empty vector (45% inhibition, p < 0.05). It also caused an inhibition of proliferation of DU145 cells overexpressing JunB and c-Jun proteins, which was not statistically significant. Surprisingly, TGF-β1 also caused a significant inhibition of proliferation in DU145 cells overexpressing JunD protein compared with the untreated cells (40% inhibition, p < 0.05).
FIGURE 7.

Effects of overexpression of individual Jun proteins on cell proliferation in DU145 cells. A, Western blotting analyses showing levels of c-Jun, JunB, and JunD proteins in DU145 subline cells after transfection with control (WT) or individual Jun plasmids. B, -fold change in the cell proliferation rate of selected DU145 lines overexpressing c-Jun (C2, C4, and C5), JunB (B6, B7, and B9), and JunD (D1, D5, and D6). DU145 cells containing the empty vector were used as controls. C, representative stable DU145 cell lines expressing each Jun protein were treated with TGF-β1 (5 ng/ml) for 4 days, and total cell numbers were counted. Each bar represents mean ± S.E. (error bars) (n = 3). *, significantly different when compared with appropriate controls (p < 0.05).
JunD Promotes Colony Formation in DU145 Cells
To determine differential effects of Jun proteins on colony formation, DU145 cells overexpressing Jun proteins and carrying empty vector were cultured in soft agar and allowed to form colonies. Each of the DU145 sublines was able to form colonies; however, DU145 cells that overexpressed JunD showed greater (2.7-fold) ability to grow in soft agar (30.3 ± 3.2 clones, p < 0.001 clones) in comparison with the vector-only DU145 cells (11.0 ± 0.8 clones) or the JunB (15.2 ± 1.6 clones)- and c-Jun (10.2 ± 0.5 clones)-transfected DU145 cells (Fig. 8).
FIGURE 8.

Stable overexpression of JunD in DU145 enhances the anchorage-independent growth of prostate cancer cell colonies on soft agar. DU145 overexpressing c-Jun, JunB, or JunD and DU145 cells containing empty vector, pcDNA3.1, were plated in 0.4% soft agar in complete growth medium for 21 days, and cell colonies were counted. Each bar represents mean ± S.E. (error bars) from three independent wells.
Down-regulation of JunD by TGF-β1 Is Dose-dependent and Is Mediated through TGF-β Receptors and via Ubiquitination and Proteasome Degradation
The effect of TGF-β1 on down-regulation of JunD in DU145 cells was already significant at 1 ng/ml TGF-β1 as shown in (Fig. 9A). No significant additional effect can be observed with higher TGF-β1 dosages. Preincubation with inhibitors of TGF-βRI (SB31542) and RII (LY2157299) blocked TGF-β1 induced reduction in JunD levels in DU145 cells, even potentiating JunD levels (SB31542, 1.3 ± 0.13-fold, p < 0.05; LY2157299, 1.6 ± 0.26-fold, p < 0.05), indicating that JunD degradation was mediated through the classical TGF-β and Smad3 signaling mediated by TGF-β receptors (Fig. 9B).
FIGURE 9.

Down-regulation of JunD by TGF-β1 is dose-dependent and is mediated through TGF-β receptors. A, effects of different doses of TGF-β1 (0, 1, 5, and 10 ng/ml) on JunD levels in DU145 cells treated for 8 h. B, effects of TGF-β1 (5 ng/ml) on JunD levels in DU145 cells in the presence and absence of inhibitors of TGF-βRI (SB431542; 5 μm) and/or TGF-βRII (LY2157299; 10 μm). Quantitative differences in JunD levels after different treatments are presented in the bar graphs. Each bar represents mean ± S.E. (error bars) from three independent experiments. *, significantly different when compared with controls (p < 0.05).
To determine whether down-regulation of JunD protein in DU145 cells in response to TGF-β treatment involves proteasomal degradation, we tested the effect of proteasome inhibitor, MG132, on the levels of JunD protein following TGF-β1 treatment (Fig. 10A). After a 2-h pretreatment with MG132 (25 μm), DU145 cells were treated with TGF-β1 (5 ng/ml) for 8 h. JunD levels in DU145 cells, as expected, declined after 8 h (0.70 ± 0.14-fold, p < 0.05) of TGF-β1 treatment. MG132 itself raised the JunD basal level in DU145 cells (1.74 ± 0.33-fold, p < 0.05). However, TGF-β1 treatment failed to cause a reduction in the levels of JunD in the presence of MG132, suggesting that TGF-β1 down-regulates JunD levels via proteasomal degradation. Experiments described earlier showed that TGF-β1 inhibits proliferation in DU145 cells overexpressing JunD (Fig. 7C), suggesting that TGF-β1 treatment may result in proteasomal degradation of JunD in these cells as well. To confirm this notion, DU145 cells overexpressing JunD (D6) were plated overnight (4 × 105), pretreated with MG132 for 2 h, and then treated with TGF-β1 for 8 h. TGF-β1 was able to down-regulate JunD protein in D6 cells (0.41 ± 0.16-fold, p < 0.05), and MG132 pretreatment inhibited these effects of TGF-β1 (Fig. 10B).
FIGURE 10.

TGF-β1 induces ubiquitination and proteasomal degradation of JunD. Western blotting analyses of JunD and β-actin in DU145 (A) and DU145 cells overexpressing JunD (B). Cells were pretreated with MG132 (25 μm) for 2 h and then treated with TGF-β1 (5 ng/ml) for 8 h. Quantitative analysis of JunD in DU145 and DU145 cells overexpressing JunD after treatment with TGF-β1 in the presence or absence of MG132 is shown in bar graphs. Each bar represents mean ± S.E. (error bars) (n = 3). *, significantly different when compared with controls (p < 0. 05). C, equal amounts of total cell protein from DU145 cells treated with and without TGF-β1 for 8 h in the presence or absence of MG132 (25 μm, 2 h pretreatment) were subjected to SDS-PAGE. All ubiquitinated proteins were detected by Western blotting with anti-ubiquitin antibody. D, total cell lysates from different treatments were immunoprecipitated (Co-IP) using anti-JunD antibody, and the immunoprecipitates were resolved on an SDS-PAGE and immunoblotted with anti-ubiquitin antibody.
To examine the effects of TGF-β on ubiquitination of proteins, total cell lysates from DU145 cells were treated with and without TGF-β1 in the presence and absence of MG132 and analyzed by immunoblotting with anti-ubiquitin antibody (Fig. 10C). The majority of ubiquitinated protein was observed in cells treated with MG132, indicating that all proteins conjugated with ubiquitin were prevented from being degraded by the proteasome. To examine the ubiquitination of JunD, total cell lysates from the same experiment were subjected to immunoprecipitation using JunD antibody, and the resulting precipitates were analyzed by Western blotting analysis with anti-ubiquitin antibody (Fig. 10D). The majority of polyubiquitinated JunD was detected in DU145 samples treated with TGF-β1 in the presence of MG132 compared with those without TGF-β1 treatment. These results confirmed that down-regulation of JunD by TGF-β is achieved through ubiquitination followed by proteasomal degradation.
Discussion
In this study, we demonstrate for the first time that JunD plays an essential role in the proliferation of prostate epithelial cancer cells. We also show that inhibitory effects of TGF-β on the proliferation of prostate epithelial cells depend on proteasomal degradation of JunD, and failure of JunD degradation may induce resistance to inhibitory effects of TGF-β in advanced stages of prostate cancer.
The AP-1 family proteins have been studied for many years, and their involvement in cell proliferation, differentiation, differentiated functions, and apoptosis has been documented extensively (40, 65–67). It has also been suggested by several studies that expression of AP-1 proteins is associated with a more aggressive clinical outcome in prostate cancer (47, 68, 69). Most of these studies have, however, focused on the expression and/or function of activated AP-1 complex containing c-Jun and c-Fos (70). Consequently, the specific functions of individual AP-1 family members and various homo- and heterodimers in the regulation of specific cellular processes remain largely unknown. Therefore, in the current study, we focused on elucidating the comparative roles of Jun family of proteins in prostate cancer cell proliferation. Our results showed that all Jun family members are constitutively expressed in various prostate cell lines at the mRNA level but exhibited differences in the levels of Jun proteins in various cell lines, indicating differential regulation of proteins in different cell lines. The specific knockdown of individual Jun family members by specific siRNA showed that JunD plays an essential role in proliferation of both DU145 and PC3 cells, whereas c-Jun and JunB knockdown had minimal effects on proliferation in these cell lines. The essential role of JunD in proliferation of prostate cancer cells suggests that an AP-1 complex containing a Jun-Jun homodimer or Jun-Fos heterodimer containing JunD is responsible for positive regulation of proliferation of these cells. Because c-Jun and JunB knockdown did not influence cell proliferation, it is logical to assume that they do not form dimers with JunD to induce cell proliferation. Therefore, a dimer containing JunD-JunD or Jun-Fos may be responsible for these effects. Additional studies are needed to identify the JunD dimer partner that is required for induction of cell proliferation in prostate cancer cells. The essential role of JunD was also confirmed by our experiments where we generated DU145 cell lines overexpressing individual Jun family members. Whereas JunD overexpression resulted in a significant increase in the proliferation rate, overexpression of c-Jun or JunB resulted in a reduced proliferation rate. These studies were further supported by increased colony formation in DU145 cells overexpressing JunD compared with DU145 cells overexpressing c-Jun or JunB. These results suggest that individual Jun family members exert distinct and even opposite effects on proliferation of prostate cancer cells. Our results are very similar to recently reported findings indicating that the inhibition of JunD results in induction of several molecules, such as GADD genes and JNKs, ultimately leading to prostate tumor cell death and inhibition of tumor development (71). These results are also supported by recent studies demonstrating that hydrogen peroxide-mediated proliferation is due to binding of Fra-1/JunD and phospho-c-Jun to the HARP promoter sites (57). JunD was also reported to be involved in migration and invasion of prostate cancer cells by inducing the expression of metalloproteinase-1-mediated by the Wnt5a signaling pathway (72). Interestingly, this action of JunD on prostate cancer cell migration was reported to be opposite to the effect of JunB and c-Jun (73). JunD plays important regulatory roles in both androgen-dependent and androgen-independent prostate cancer cells by functioning as a co-activator for the androgen receptor to mediate androgen-induced oxidative stress in LNCaP cells (51, 74) or by interacting with the NFκB pathway to induce IL-6, an important mediator of metastatic, hormone-refractory prostate cancer (75). On the other hand, Church et al. (50) reported that cell growth inhibition in LNCaP cells after treatment with androgens resulted in a concomitant increase in JunD expression. This suggests that AP-1 regulation of cell proliferation depends on the cellular “context” and on the combination of available contributing factors and active pathways in each target tissue.
TGF-β is a multiple-function protein that acts as a tumor suppressor in normal epithelial cells and in early stage cancer cells; cells in later stages of cancer become resistant to its growth-inhibitory effects (13, 76). In advanced stages of the disease, TGF-β acts as a tumor promotor by virtue of its effects on epithelial to mesenchymal transition, cell migration and invasion, angiogenesis, and metastasis (77, 78). Our previous studies and present results show that TGF-β inhibits proliferation in RWPE-1 and DU145 cells, whereas PC3 cells are resistant to these growth-inhibitory effects (15). On the other hand, TGF-β induces cell migration and invasion in PC3 cells but does not affect migration and invasion in DU145 cells (16, 29, 64). Because both DU145 and PC3 cells express functional TGF-β receptors and Smad signaling (15, 29) but exhibit differential responses to TGF-β treatment, we have exploited these cell lines to understand differences in the signaling mechanism that may act either downstream or parallel to Smad signaling and are responsible for their differential responses to TGF-β. Our results show that TGF-β induces a reduction in JunD protein levels in DU145 cells but not in PC3 cells. Because JunD is required for cell proliferation, these results suggest that reduction of JunD levels in DU145 cells in response to TGF-β may lead to reduction in cell proliferation in these cells. On the other hand, the lack of TGF-β effects on PC3 cell proliferation may be due to their resistance to TGF-β-induced reduction of JunD levels. We previously showed that TGF-β caused a down-regulation of Id1, a transcriptional regulator, in DU145 cells but not in PC3 cells (15). Because Id1 knockdown by siRNA in both DU145 and PC3 cells resulted in decreased proliferation in both cell lines, we concluded that inhibitory effects of TGF-β required down-regulation of Id1 in prostate cancer cells (15). Our current study shows that knockdown of JunD affected the expression of several cell cycle regulatory proteins and also caused a reduction in the expression of Id1. These results indicate that JunD may regulate the expression of Id1 which, in turn, may be required for cell proliferation. Our previous studies and our present results identify at least one distinct signaling cascade, downstream of Smad2/3 activation, which may be required for inhibitory effects of TGF-β on cell proliferation and which may be altered in the cells that become resistant to growth-inhibitory effects of this cytokine. Treatment with TGF-β had no effect on JunD mRNA levels in both DU145 and PC3 cells but caused a significant decrease in JunD protein levels in DU145 cells. Furthermore, the effects of TGF-β did not involve phosphorylation of JunD or the activation of the JNK/SAPK pathway, suggesting that reduced intracellular protein levels are primarily responsible for the inhibitory effects of TGF-β on cell proliferation. Interestingly, TGF-β treatment also resulted in down-regulation of JunD protein and inhibition of cell proliferation in DU145 cell lines overexpressing JunD, suggesting post-transcriptional regulation of JunD expression. The reduction in JunD protein levels was inhibited in the presence of proteasomal inhibitor, which also resulted in an increase in the levels of ubiquitinated JunD protein in TGF-β-treated cells, suggesting that TGF-β-induced reduction in JunD levels is due to ubiquitination followed by proteasomal degradation of the protein. The ubiquitin-proteasome pathway for targeted degradation of proteins plays critical roles in a variety of biological processes, such as cell cycle progression, signal transduction, transcriptional regulation, receptor down-regulation, and endocytosis (79, 80). The ubiquitin-proteasome pathway tightly regulates TGF-β family signaling (81–84). In this pathway, E3 ubiquitin ligases play a crucial role in the recognition and degradation of target proteins by the 26S proteasomes (85). Smad degradation regulates TGF-β family signaling (86, 87). Our laboratory has previously shown that TGF-β-induced degradation of Ski protein, a co-repressor of Smad2/3, mediated by the proteasomal pathway is required for TGF-β-induced biological responses in prostate cancer cells (16). The identity of specific E3 ligase(s) and other components involved in the degradation of JunD protein in response to TGF-β is currently not known, and it is plausible to assume that alterations in these components may result in lack of responsiveness to growth-inhibitory effects of TGF-β in advanced stages of prostate cancers.
On the basis of the results of the present study, we conclude that JunD plays a critical role in the proliferation of prostate cancer cells and may provide a significant therapeutic target for the treatment of prostate cancers. Our studies also show that c-Jun and JunB play a minimal role in induction of cell proliferation in prostate cancer. In addition, proteasomal degradation of JunD in response to TGF-β treatment is a prerequisite for growth-inhibitory effects of this cytokine, and alterations in the proteasomal pathway leading to lack of TGF-β effects on degradation of JunD may be partially responsible for resistance to growth-inhibitory effects of TGF-β in advanced stages of cancer. These phenomena may underlie many cases of prostate cancer with an otherwise intact TGF-β signaling mechanism and may represent an event earlier than the loss of TGF-β receptors in prostate cancer progression.
Experimental Procedures
Chemicals and Reagents
Recombinant human TGF-β1 was purchased from PeproTech (Rocky Hill, NJ). Proteasome/calpain inhibitor, MG132, was acquired from Calbiochem. Inhibitors of TGF-βRI (SB431542) and RII (LY2157299) were purchased from Tocris Biosciences (Ellisville, MO) and Xcess Biosciences Inc. (San Diego, CA), respectively. Anisomycin was purchased from Calbiochem.
Human Jun cDNAs (c-Jun, JunB, and JunD) subcloned into the pcDNA3.1(−) Mycstop expression vector along with the empty vector were gifts from Dr. Curt Pfarr (Texas Tech University, El Paso, TX). Antibodies and siRNAs for Jun proteins and antibodies against p21, p27, cyclin D1, c-Myc, and Id1 were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). The antibodies against ubiquitin, phospho-Smad3, phospho-JunD, phospho-SAPK/JNK, and total SAPK/JNK were purchased from Cell Signaling Technology, Inc. (Danvers, MA). The antibody against Ki-67 was purchased from Calbiochem. Anti-β-actin (clone AC-15) antibody was purchased from Sigma-Aldrich. Goat anti-rabbit IgG HRP was purchased from Life Technologies, Inc. Anti-mouse IgG HRP was obtained from GE Healthcare.
Cell Lines and Cell Culture Conditions
Human prostate cell lines (RWPE1, LNCaP, DU145, and PC3) were obtained from American Type Cell Culture Collection (Manassas, VA). All cells were cultured in the recommended growth media at 37 °C with 5% CO2 and 100% humidity as described previously (29, 64).
Cell Proliferation Assays
Cell growth assays were performed using three techniques: thymidine incorporation, flow cytometry, and manual cell counting. Each assay was performed at least three times. For thymidine incorporation, cells were seeded in a 24-well plate at a density of 4 × 104 cells/well and maintained with 5% fetal bovine serum (FBS) overnight. Cells were serum-starved for 24 h and treated with different doses of TGF-β1 (0, 1, and 10 ng/ml) in the presence of 1% FBS for 18 h. Cells were then pulse-labeled for 4 h with 1 μCi/ml [3H]thymidine (GE Healthcare), and the radioactivity incorporated into DNA was determined by liquid scintillation counting as described previously (64). Flow cytometry was used to determine the cell cycle distribution of cells. Cells were plated at an initial density of 1 × 106 cells/well overnight and then serum-starved for 24 h. After treatment with TGF-β1 (5 ng/ml) for 16 h, cells were harvested, washed twice with cold phosphate-buffered saline (PBS), and fixed with cold 70% ethanol for 1 h on ice. Cells were spun down and were incubated with propidium iodide (50 μg/ml) (Molecular Probes, Inc., Eugene, OR) and RNase A (Bio-Rad) at room temperature for 30 min before they were analyzed by flow cytometry. Cell cycle phase distribution was determined from 1 × 104 cells using a BD Accuri Cytometer (Ann Arbor, MI), following the manufacturer's instructions. For manual cell counting, cells were seeded at a density of 1 × 105 cells/well overnight in a 6-well plate and, if needed, treated the next day with TGF-β1 (5 ng/ml) with 1% FBS at specific time points. Cells were then trypsinized and counted using a hemocytometer.
RNA Isolation, cDNA Synthesis, and RT-PCR
Cells were seeded at a density of 5 × 105/well into a 6-well plate overnight in the presence of 5% FBS. For DU145, PC3, and LNCaP cells, their culture media were changed to 1% FBS after 24 h, whereas for RWPE1 cells, culture medium was changed to EpiLife® medium. The cells were then treated with TGF-β1 (5 ng/ml) for 0, 1, and 8 h. Total RNA was isolated from the cells using TRIzol (Life Technologies) as described previously (88). 2 μg of total RNA were reverse-transcribed, and the resulting cDNAs were diluted 4-fold with RNase-free water. 4 μl of the diluted cDNA were added to separate RT-PCR mixtures following established procedures. All gene-specific primers were designed with Beacon-Designer version 5.0 as described previously (29). The following primers were used: c-Jun forward, 5′-TGGAAACGACCTTCTATGACGA-3′; c-Jun reverse, 5′-GTTGCTGGACTGGATTATCAGG-3′; JunB forward, 5′-TACCACGACGACTCATACACA-3′; JunB reverse, 5′-CGCTTTGAGACTCCGGTAGG-3′; JunD forward, 5′-CAAACCCTGCCTTTCCTTTAC-3′; JunD reverse, 5′-GGCGAACCAAGGATTACAAA-3′; L19 forward, 5′-GAAATCGCCAATGCCAACTC-3′; L19 reverse 5′-TCTTAGACCTGCGAGCCTCA-3′. The PCR products were visualized on 1% agarose gels stained with ethidium bromide (Amresco, Solon, OH). The relative intensities of specific PCR bands were determined by ImageJ version 1.48 (National Institutes of Health).
Western Blotting Analysis
Cells from different experiments were washed twice with ice-cold PBS and were lysed in cell lysis buffer (Cell Signaling Technology) containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and 1× protease inhibitor mixture (Calbiochem). Protein concentrations were determined by the Lowry HS assay using the Bio-Rad DC protein assay kit according to the instructions provided by the manufacturer. Cell lysates were mixed with Laemmli's buffer (62.5 mm Tris, pH 6.8, 2% SDS, 5% β-mercaptoethanol, and 10% glycerol), and individual samples (35 μg of protein) were subjected to SDS-PAGE in 10% gels and transferred to PVDF membranes (Millipore, Billerica, MA). The membranes were blocked for 1 h at room temperature in 1× PBS (136 mm NaCl, 2.6 mm KCl, 4.3 mm Na2HPO4, 1.5 mm KH2PO4) and 5% fat-free skim milk. The blots were then incubated overnight at 4 °C with appropriate dilutions of specific primary antibodies (1:2000 dilution for anti-JunD; 1:1000 dilution for anti-c-Jun, anti-JunB, anti-p21, anti-p27, anti-cyclin D1, anti-ubiquitin, anti-phospho-JunD, anti-phospho-SAPK/JNK, anti-total SAPK/JNK, and anti-phospho-Smad3; 1:500 dilution for anti-c-Myc and anti-Ki-67; 1:10,000 dilution for anti-β-actin). After washing, blots were incubated with appropriate immunoglobulin coupled to horseradish peroxidase (dilution 1:20,000) for 1 h. The blots were developed in Millipore Luminata Forte (EMD Millipore, Billerica, MA) for 5 min and exposed to an x-ray film and visualized either by autoradiography or by the Syngene PXI 6 imaging system (Syngene, Frederick, MD). Western blots for β-actin were carried out in parallel as loading controls. The relative intensities of specific protein bands were determined by ImageJ version 1.48 (National Institutes of Health).
Immunoprecipitation
DU145 cells were pretreated with MG132 (25 μm) for 2 h and then treated with TGF-β1 (5 ng/ml) for 8 h. Cells were lysed in cell lysis buffer (Cell Signaling Technology). Total cell lysates containing 500 μg of proteins were used for immunoprecipitation using procedures described previously (16). The resulting supernatants were incubated with 2 μg of anti-JunD antibody overnight at 4 °C. Immunocomplexes were collected by centrifugation after incubation with protein A/G-Sepharose beads (Santa Cruz Biotechnology) and were analyzed by Western blotting analysis with anti-ubiquitin antibody.
Transfection with Specific Jun Protein and Control siRNAs
Cells were seeded at a density of 1.5 × 105 cells in 6-well plates in 2 ml of antibiotic-free normal growth medium supplemented with 5% FBS and incubated overnight at 37 °C. siRNAs (60 nm) for the Jun proteins (c-Jun, JunD, JunB) or control siRNA were transfected in DU145 and PC3 cells using transfection reagent (Santa Cruz Biotechnology) following the manufacturer's recommendations. 48–72 h after transfection, cells were treated with TGF-β1 and/or subjected to different functional analyses.
Generation of DU145 Sublines Overexpressing Jun Proteins
DU145 cells (1.5 × 105) were grown in a serum-free medium in a 6-well plate and then transfected with the empty vector, pcDNA3.1 Mycstop, or with pcDNA3.1 JunB Mycstop, pcDNA3.1 Mycstop JunD, and pcDNA3.1 c-Jun Mycstop using FuGENE® HD transfection reagent (Promega, Madison, WI) following the manufacturer's instructions. For each gene transfection, after 48 h, cells were trypsinized and replated to dilutions of 1:2, 1:5, 1:10, 1:20, 1:40, and 1:80 in a 6-well plate, supplemented with 5% FBS and G418 (800 μg/ml) (Calbiochem). Cells were continuously fed with fresh medium containing G418 every 2–3 days. About 1 week post-transfection, single G418-resistant colonies were picked using sterile cloning discs (Scienceware, Wayne, NJ) and grown to propagate. Total proteins from the transfected lines were extracted, and Western blotting analysis was performed to determine overexpression of the specific Jun proteins. The effects of these transfections on cellular proliferation were determined by cell counting.
TUNEL Assay and DAPI Staining
DU145 cells were seeded onto coverslips at a density of 1.0 × 105 cells in 6-well plates in 2 ml of antibiotic-free normal growth medium supplemented with 5% FBS. Transfection of control and JunD siRNA was performed as described above. Cells were fixed with 3.7% formaldehyde in PBS for 20 min at room temperature. Cells were permeabilized with 0.1% Triton X-100 for 20 min at room temperature and then washed three times with PBS. Fixed cells were evaluated for apoptosis using two independent methods; a TUNEL assay was used to detect DNA fragmentation, which is a hallmark of apoptosis in mammalian cells, and DAPI staining was used to visualize intact total nuclear DNA. For the TUNEL assay, apoptotic cells were detected using the proTUNEL-IHC DNA fragmentation assay kit (GeneTex, Irvine, CA) following the manufacturer's recommendation. For nuclear visualization, fixed cells were stained with DAPI (3 μg/ml; Roche Applied Science) following standard fluorescence staining protocols. Coverslips were mounted on glass slides using Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and then images were captured using ×10 magnification with an Axiovision camera of a Carl Zeiss 200M inverted fluorescence microscope (Carl Zeiss, Thornwood, NY).
Soft Agar Colony Formation
A soft agar colony formation assay was performed on DU145 cells overexpressing Jun proteins in 6-well plates. Each well contained 2 ml of 0.6% agar in complete medium as the bottom layer, 1 ml of 0.4% agar in complete medium and 3000 cells as the feeder layer, and 1 ml of complete medium as the top layer. Cultures were maintained under standard culture conditions. The number of colonies was determined with an inverted phase microscope (Carl Zeiss 200M) at ×100 magnification. A group of ≥2 cells was counted as a colony. The data presented are means ± S.E. of 12 wells (total number of colonies from 4 random fields/well) from two independent experiments at an optimum time of 21 days after cell plating. The experiment was repeated twice, with each experiment using a different Jun-overexpressing DU145 cell line.
Statistical Analysis
All experiments were performed at least three times using different cell preparations. Data from representative experiments are shown in the figures. The significance of the differences among treatments was determined using one-way analysis of variance and Duncan's multiple pairwise comparison tests using the statistical package from SigmaPlot version 11.0 for Windows.
Author Contributions
A. C. M. and B. T. V. performed and analyzed the experiments and contributed the preparation of the manuscript. S. A. K. conceived and coordinated the study and helped in analysis of the data and preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We are grateful to Dr. Curt Pfarr (Texas Tech University, El Paso, TX) for the gift of Jun cDNA plasmids.
Footnotes
This work was supported by NIMHD, National Institutes of Health, Grants G12MD007590 and 5P20MD002285. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This paper is dedicated to the memory of Dr. Natalya Klueva, who was involved in the initiation of these studies but passed away in 2010 after a courageous fight against breast cancer.
References
- 1. Ware J. L. (1993) Growth factors and their receptors as determinants in the proliferation and metastasis of human prostate cancer. Cancer Metastasis Rev. 12, 287–301 [DOI] [PubMed] [Google Scholar]
- 2. Barrack E. R. (1997) TGF β in prostate cancer: a growth inhibitor that can enhance tumorigenicity. Prostate 31, 61–70 [DOI] [PubMed] [Google Scholar]
- 3. Lee C., Sintich S. M., Mathews E. P., Shah A. H., Kundu S. D., Perry K. T., Cho J. S., Ilio K. Y., Cronauer M. V., Janulis L., and Sensibar J. A. (1999) Transforming growth factor-β in benign and malignant prostate. Prostate 39, 285–290 [DOI] [PubMed] [Google Scholar]
- 4. Segarini P. R. (1991) TGF-β receptors. Ciba Found. Symp. 157, 29–40; discussion 41–50 [PubMed] [Google Scholar]
- 5. Yamaguchi Y. (1992) [Function, molecular structure and gene expression regulation of transforming growth factor-β (TGF-β)]. Nihon rinsho 50, 1932–1938 [PubMed] [Google Scholar]
- 6. Axmann A., Seidel D., Reimann T., Hempel U., and Wenzel K. W. (1998) Transforming growth factor-β1-induced activation of the Raf-MEK-MAPK signaling pathway in rat lung fibroblasts via a PKC-dependent mechanism. Biochem. Biophys. Res. Commun. 249, 456–460 [DOI] [PubMed] [Google Scholar]
- 7. Johnson A. N., and Newfeld S. J. (2002) The TGF-β family: signaling pathways, developmental roles, and tumor suppressor activities. ScientificWorldJournal 2, 892–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams R. H., Stapleton A. M., Yang G., Truong L. D., Rogers E., Timme T. L., Wheeler T. M., Scardino P. T., and Thompson T. C. (1996) Reduced levels of transforming growth factor β receptor type II in human prostate cancer: an immunohistochemical study. Clin. Cancer Res. 2, 635–640 [PubMed] [Google Scholar]
- 9. Wikström P., Lindh G., Bergh A., and Damber J. E. (1999) Alterations of transforming growth factor β1 (TGF-β1) and TGFβ receptor expressions with progression in Dunning rat prostatic adenocarcinoma sublines. Urol. Res. 27, 185–193 [DOI] [PubMed] [Google Scholar]
- 10. Djonov V., Andres A., Altermatt H., and Merz V. (1997) TGF-β3 expression correlates with epithelial cell death in normal, hyperplastic and malignant prostate. Int. J. Oncol. 11, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 11. Kim I. Y., Ahn H. J., Lang S., Oefelein M. G., Oyasu R., Kozlowski J. M., and Lee C. (1998) Loss of expression of transforming growth factor-β receptors is associated with poor prognosis in prostate cancer patients. Clin. Cancer Res. 4, 1625–1630 [PubMed] [Google Scholar]
- 12. Wikström P., Stattin P., Franck-Lissbrant I., Damber J. E., and Bergh A. (1998) Transforming growth factor β1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 37, 19–29 [DOI] [PubMed] [Google Scholar]
- 13. Lokeshwar B. L., and Block N. L. (1992) Isolation of a prostate carcinoma cell proliferation-inhibiting factor from human seminal plasma and its similarity to transforming growth factor β. Cancer Res. 52, 5821–5825 [PubMed] [Google Scholar]
- 14. Desruisseau S., Ghazarossian-Ragni E., Chinot O., and Martin P. M. (1996) Divergent effect of TGFβ1 on growth and proteolytic modulation of human prostatic-cancer cell lines. Int. J. Cancer 66, 796–801 [DOI] [PubMed] [Google Scholar]
- 15. Strong N., Millena A. C., Walker L., Chaudhary J., and Khan S. A. (2013) Inhibitor of differentiation 1 (Id1) and Id3 proteins play different roles in TGFβ effects on cell proliferation and migration in prostate cancer cells. Prostate 73, 624–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vo B. T., Cody B., Cao Y., and Khan S. A. (2012) Differential role of Sloan-Kettering Institute (Ski) protein in Nodal and transforming growth factor-β (TGF-β)-induced Smad signaling in prostate cancer cells. Carcinogenesis 33, 2054–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamada D., Kobayashi S., Wada H., Kawamoto K., Marubashi S., Eguchi H., Ishii H., Nagano H., Doki Y., and Mori M. (2013) Role of crosstalk between interleukin-6 and transforming growth factor-β1 in epithelial-mesenchymal transition and chemoresistance in biliary tract cancer. Eur. J. Cancer 49, 1725–1740 [DOI] [PubMed] [Google Scholar]
- 18. Hogan K. A., Ravindran A., Podolsky M. A., and Glick A. B. (2013) The TGFβ1 pathway is required for NFκB dependent gene expression in mouse keratinocytes. Cytokine 64, 652–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seystahl K., Tritschler I., Szabo E., Tabatabai G., and Weller M. (2015) Differential regulation of TGF-β-induced, ALK-5-mediated VEGF release by Smad2/3 versus Smad1/5/8 signaling in glioblastoma. Neuro Oncol. 17, 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Letamendia A., Labbé E., and Attisano L. (2001) Transcriptional regulation by Smads: crosstalk between the TGF-β and Wnt pathways. J. Bone Joint Surg. 83, S31–S39 [PubMed] [Google Scholar]
- 21. Nyhan K. C., Faherty N., Murray G., Cooey L. B., Godson C., Crean J. K., and Brazil D. P. (2010) Jagged/Notch signalling is required for a subset of TGFβ1 responses in human kidney epithelial cells. Biochim. Biophys. Acta 1803, 1386–1395 [DOI] [PubMed] [Google Scholar]
- 22. Javelaud D., Pierrat M. J., and Mauviel A. (2012) Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 586, 2016–2025 [DOI] [PubMed] [Google Scholar]
- 23. Hirota Y., Tsukazaki T., Yonekura A., Miyazaki Y., Osaki M., Shindo H., and Yamashita S. (2000) Activation of specific MEK-ERK cascade is necessary for TGFβ signaling and crosstalk with PKA and PKC pathways in cultured rat articular chondrocytes. Osteoarthritis Cartilage 8, 241–247 [DOI] [PubMed] [Google Scholar]
- 24. Qu X., Shen L., Zheng Y., Cui Y., Feng Z., Liu F., and Liu J. (2014) A signal transduction pathway from TGF-β1 to SKP2 via Akt1 and c-Myc and its correlation with progression in human melanoma. J. Invest. Dermatol. 134, 159–167 [DOI] [PubMed] [Google Scholar]
- 25. Lauth M., and Toftgård R. (2007) Non-canonical activation of GLI transcription factors: implications for targeted anti-cancer therapy. Cell Cycle 6, 2458–2463 [DOI] [PubMed] [Google Scholar]
- 26. Moreno C. S. (2010) The Sex-determining region Y-box 4 and homeobox C6 transcriptional networks in prostate cancer progression: crosstalk with the Wnt, Notch, and PI3K pathways. Am. J. Pathol. 176, 518–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gohla G., Krieglstein K., and Spittau B. (2008) Tieg3/Klf11 induces apoptosis in OLI-neu cells and enhances the TGF-β signaling pathway by transcriptional repression of Smad7. J. Cell. Biochem. 104, 850–861 [DOI] [PubMed] [Google Scholar]
- 28. Chakrabarti L., Wang B. D., Lee N. H., and Sandler A. D. (2013) A mechanism linking Id2-TGFβ crosstalk to reversible adaptive plasticity in neuroblastoma. PLoS One 8, e83521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walker L., Millena A. C., Strong N., and Khan S. A. (2013) Expression of TGFβ3 and its effects on migratory and invasive behavior of prostate cancer cells: involvement of PI3-kinase/AKT signaling pathway. Clin. Exp. Metastasis 30, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birchenall-Roberts M. C., Ruscetti F. W., Kasper J., Lee H. D., Friedman R., Geiser A., Sporn M. B., Roberts A. B., and Kim S. J. (1990) Transcriptional regulation of the transforming growth factor β1 promoter by v-src gene products is mediated through the AP-1 complex. Mol. Cell. Biol. 10, 4978–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han S. H., Yea S. S., Jeon Y. J., Yang K. H., and Kaminski N. E. (1998) Transforming growth factor-β1 (TGF-β1) promotes IL-2 mRNA expression through the up-regulation of NF-κB, AP-1 and NF-AT in EL4 cells. J. Pharmacol. Exp. Ther. 287, 1105–1112 [PubMed] [Google Scholar]
- 32. Verrecchia F., Tacheau C., Schorpp-Kistner M., Angel P., and Mauviel A. (2001) Induction of the AP-1 members c-Jun and JunB by TGF-β/Smad suppresses early Smad-driven gene activation. Oncogene 20, 2205–2211 [DOI] [PubMed] [Google Scholar]
- 33. Hess J., Angel P., and Schorpp-Kistner M. (2004) AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 34. Eferl R., and Wagner E. (2003) AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 [DOI] [PubMed] [Google Scholar]
- 35. FitzGerald U. F., and Barnett S. C. (2000) AP-1 activity during the growth, differentiation, and death of O-2A lineage cells. Mol. Cell Neurosci. 16, 453–469 [DOI] [PubMed] [Google Scholar]
- 36. Jacobs-Helber S. M., Wickrema A., Birrer M. J., and Sawyer S. T. (1998) AP1 regulation of proliferation and initiation of apoptosis in erythropoietin-dependent erythroid cells. Mol. Cell. Biol. 18, 3699–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang D. C., Motwani M., and Fisher P. B. (1998) Role of the transcription factor AP-1 in melanoma differentiation (review). Int. J. Oncol. 13, 1117–1126 [DOI] [PubMed] [Google Scholar]
- 38. Shaulian E. (2010) AP-1: the Jun proteins: oncogenes or tumor suppressors in disguise? Cell. Signal. 22, 894–899 [DOI] [PubMed] [Google Scholar]
- 39. Günthert A. R., Gründker C., Hollmann K., and Emons G. (2002) Luteinizing hormone-releasing hormone induces JunD-DNA binding and extends cell cycle in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 294, 11–15 [DOI] [PubMed] [Google Scholar]
- 40. Shaulian E., and Karin M. (2001) AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400 [DOI] [PubMed] [Google Scholar]
- 41. Shaulian E., and Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 42. Vanhara P., Bryja V., Horváth V., Kozubík A., Hampl A., and Smarda J. (2007) c-Jun induces apoptosis of starved BM2 monoblasts by activating cyclin A-CDK2. Biochem. Biophys. Res. Commun. 353, 92–97 [DOI] [PubMed] [Google Scholar]
- 43. Chen S. Y., Cai C., Fisher C. J., Zheng Z., Omwancha J., Hsieh C. L., and Shemshedini L. (2006) c-Jun enhancement of androgen receptor transactivation is associated with prostate cancer cell proliferation. Oncogene 25, 7212–7223 [DOI] [PubMed] [Google Scholar]
- 44. Uzzo R. G., Crispen P. L., Golovine K., Makhov P., Horwitz E. M., and Kolenko V. M. (2006) Diverse effects of zinc on NF-κB and AP-1 transcription factors: implications for prostate cancer progression. Carcinogenesis 27, 1980–1990 [DOI] [PubMed] [Google Scholar]
- 45. Shimada K., Nakamura M., Ishida E., Kishi M., and Konishi N. (2003) Roles of p38- and c-jun NH2-terminal kinase-mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis. Carcinogenesis 24, 1067–1075 [DOI] [PubMed] [Google Scholar]
- 46. Mashimo T., Bandyopadhyay S., Goodarzi G., Watabe M., Pai S. K., Gross S. C., and Watabe K. (2000) Activation of the tumor metastasis suppressor gene, KAI1, by etoposide is mediated by p53 and c-Jun genes. Biochem. Biophys. Res. Commun. 274, 370–376 [DOI] [PubMed] [Google Scholar]
- 47. Kajanne R., Miettinen P., Tenhunen M., and Leppä S. (2009) Transcription factor AP-1 promotes growth and radioresistance in prostate cancer cells. Int. J. Oncol. 35, 1175–1182 [DOI] [PubMed] [Google Scholar]
- 48. Konishi N., Shimada K., Nakamura M., Ishida E., Ota I., Tanaka N., and Fujimoto K. (2008) Function of JunB in transient amplifying cell senescence and progression of human prostate cancer. Clin. Cancer Res. 14, 4408–4416 [DOI] [PubMed] [Google Scholar]
- 49. Liu W., Iiizumi-Gairani M., Okuda H., Kobayashi A., Watabe M., Pai S. K., Pandey P. R., Xing F., Fukuda K., Modur V., Hirota S., Suzuki K., Chiba T., Endo M., Sugai T., and Watabe K. (2011) KAI1 gene is engaged in NDRG1 gene-mediated metastasis suppression through the ATF3-NFκB complex in human prostate cancer. J. Biol. Chem. 286, 18949–18959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Church D. R., Lee E., Thompson T. A., Basu H. S., Ripple M. O., Ariazi E. A., and Wilding G. (2005) Induction of AP-1 activity by androgen activation of the androgen receptor in LNCaP human prostate carcinoma cells. Prostate 63, 155–168 [DOI] [PubMed] [Google Scholar]
- 51. Mehraein-Ghomi F., Lee E., Church D. R., Thompson T. A., Basu H. S., and Wilding G. (2008) JunD mediates androgen-induced oxidative stress in androgen dependent LNCaP human prostate cancer cells. Prostate 68, 924–934 [DOI] [PubMed] [Google Scholar]
- 52. Kizu R., Okamura K., Toriba A., Kakishima H., Mizokami A., Burnstein K. L., and Hayakawa K. (2003) A role of aryl hydrocarbon receptor in the antiandrogenic effects of polycyclic aromatic hydrocarbons in LNCaP human prostate carcinoma cells. Arch. Toxicol. 77, 335–343 [DOI] [PubMed] [Google Scholar]
- 53. Parra E., Ferreira J., and Saenz L. (2011) Inhibition of Egr-1 by siRNA in prostate carcinoma cell lines is associated with decreased expression of AP-1 and NF-κB. Int. J. Mol. Med. 28, 847–853 [DOI] [PubMed] [Google Scholar]
- 54. Déziel B. A., Patel K., Neto C., Gottschall-Pass K., and Hurta R. A. (2010) Proanthocyanidins from the American cranberry (Vaccinium macrocarpon) inhibit matrix metalloproteinase-2 and matrix metalloproteinase-9 activity in human prostate cancer cells via alterations in multiple cellular signalling pathways. J. Cell. Biochem. 111, 742–754 [DOI] [PubMed] [Google Scholar]
- 55. Hung S. H., Shen K. H., Wu C. H., Liu C. L., and Shih Y. W. (2009) α-Mangostin suppresses PC-3 human prostate carcinoma cell metastasis by inhibiting matrix metalloproteinase-2/9 and urokinase-plasminogen expression through the JNK signaling pathway. J. Agric. Food Chem. 57, 1291–1298 [DOI] [PubMed] [Google Scholar]
- 56. Kwon G. T., Cho H. J., Chung W. Y., Park K. K., Moon A., and Park J. H. (2009) Isoliquiritigenin inhibits migration and invasion of prostate cancer cells: possible mediation by decreased JNK/AP-1 signaling. J. Nutr. Biochem. 20, 663–676 [DOI] [PubMed] [Google Scholar]
- 57. Polytarchou C., Hatziapostolou M., and Papadimitriou E. (2005) Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J. Biol. Chem. 280, 40428–40435 [DOI] [PubMed] [Google Scholar]
- 58. Xu C., Shen G., Yuan X., Kim J. H., Gopalkrishnan A., Keum Y. S., Nair S., and Kong A. N. (2006) ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis 27, 437–445 [DOI] [PubMed] [Google Scholar]
- 59. Zhou W., Janulis L., Park I. I., and Lee C. (2002) A novel anti-proliferative property of clusterin in prostate cancer cells. Life Sci. 72, 11–21 [DOI] [PubMed] [Google Scholar]
- 60. Li W., Zhang X., and Olumi A. F. (2007) MG-132 sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIP(L). Cancer Res. 67, 2247–2255 [DOI] [PubMed] [Google Scholar]
- 61. Chipuk J. E., Cornelius S. C., Pultz N. J., Jorgensen J. S., Bonham M. J., Kim S. J., and Danielpour D. (2002) The androgen receptor represses transforming growth factor-β signaling through interaction with Smad3. J. Biol. Chem. 277, 1240–1248 [DOI] [PubMed] [Google Scholar]
- 62. Park J. I., Lee M. G., Cho K., Park B. J., Chae K. S., Byun D. S., Ryu B. K., Park Y. K., and Chi S. G. (2003) Transforming growth factor-β1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-κB, JNK, and Ras signaling pathways. Oncogene 22, 4314–4332 [DOI] [PubMed] [Google Scholar]
- 63. Song K., Krebs T. L., and Danielpour D. (2006) Novel permissive role of epidermal growth factor in transforming growth factor β (TGF-β) signaling and growth suppression: mediation by stabilization of TGF-β receptor type II. J. Biol. Chem. 281, 7765–7774 [DOI] [PubMed] [Google Scholar]
- 64. Vo B. T., and Khan S. A. (2011) Expression of nodal and nodal receptors in prostate stem cells and prostate cancer cells: autocrine effects on cell proliferation and migration. Prostate 71, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Krosl J., and Sauvageau G. (2000) AP-1 complex is effector of Hox-induced cellular proliferation and transformation. Oncogene 19, 5134–5141 [DOI] [PubMed] [Google Scholar]
- 66. Meggiato T., Calabrese F., De Cesare C. M., Baliello E., Valente M., and Del Favero G. (2003) C-JUN and CPP32 (CASPASE 3) in human pancreatic cancer: relation to cell proliferation and death. Pancreas 26, 65–70 [DOI] [PubMed] [Google Scholar]
- 67. Vleugel M. M., Greijer A. E., Bos R., van der Wall E., and van Diest P. J. (2006) c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 37, 668–674 [DOI] [PubMed] [Google Scholar]
- 68. Fitzgerald K. A., Evans J. C., McCarthy J., Guo J., Prencipe M., Kearney M., Watson W. R., and O'Driscoll C. M. (2014) The role of transcription factors in prostate cancer and potential for future RNA interference therapy. Expert Opin. Ther. Targets 18, 633–649 [DOI] [PubMed] [Google Scholar]
- 69. Ouyang X., Jessen W. J., Al-Ahmadie H., Serio A. M., Lin Y., Shih W. J., Reuter V. E., Scardino P. T., Shen M. M., Aronow B. J., Vickers A. J., Gerald W. L., and Abate-Shen C. (2008) Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 68, 2132–2144 [DOI] [PubMed] [Google Scholar]
- 70. Feng Z., Joos H. J., Vallan C., Mühlbauer R., Altermatt H. J., and Jaggi R. (1998) Apoptosis during castration-induced regression of the prostate is Fos dependent. Oncogene 17, 2593–2600 [DOI] [PubMed] [Google Scholar]
- 71. Zerbini L. F., de Vasconcellos J. F., Czibere A., Wang Y., Paccez J. D., Gu X., Zhou J. R., and Libermann T. A. (2011) JunD-mediated repression of GADD45α and γ regulates escape from cell death in prostate cancer. Cell Cycle 10, 2583–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yamamoto H., Oue N., Sato A., Hasegawa Y., Yamamoto H., Matsubara A., Yasui W., and Kikuchi A. (2010) Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 29, 2036–2046 [DOI] [PubMed] [Google Scholar]
- 73. Selvaraj N., Budka J. A., Ferris M. W., Plotnik J. P., and Hollenhorst P. C. (2015) Extracellular signal-regulated kinase signaling regulates the opposing roles of JUN family transcription factors at ETS/AP-1 sites and in cell migration. Mol. Cell. Biol. 35, 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mehraein-Ghomi F., Basu H. S., Church D. R., Hoffmann F. M., and Wilding G. (2010) Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Res. 70, 4560–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zerbini L. F., Wang Y., Cho J. Y., and Libermann T. A. (2003) Constitutive activation of nuclear factor κB p50/p65 and Fra-1 and JunD is essential for deregulated interleukin 6 expression in prostate cancer. Cancer Res. 63, 2206–2215 [PubMed] [Google Scholar]
- 76. Zhou W., Park I., Pins M., Kozlowski J. M., Jovanovic B., Zhang J., Lee C., and Ilio K. (2003) Dual regulation of proliferation and growth arrest in prostatic stromal cells by transforming growth factor-β1. Endocrinology 144, 4280–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kumano M., Miyake H., Kurahashi T., Yamanaka K., and Fujisawa M. (2008) Enhanced progression of human prostate cancer PC3 cells induced by the microenvironment of the seminal vesicle. Br. J. Cancer 98, 356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li X., Placencio V., Iturregui J. M., Uwamariya C., Sharif-Afshar A. R., Koyama T., Hayward S. W., and Bhowmick N. A. (2008) Prostate tumor progression is mediated by a paracrine TGF-β/Wnt3a signaling axis. Oncogene 27, 7118–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hershko A., and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 80. Hershko A., and Ciechanover A. (1992) The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61, 761–807 [DOI] [PubMed] [Google Scholar]
- 81. Izzi L., and Attisano L. (2004) Regulation of the TGFβ signalling pathway by ubiquitin-mediated degradation. Oncogene 23, 2071–2078 [DOI] [PubMed] [Google Scholar]
- 82. De Boeck M., and ten Dijke P. (2012) Key role for ubiquitin protein modification in TGFβ signal transduction. Ups. J. Med. Sci. 117, 153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang F., and Laiho M. (2003) On and off: proteasome and TGF-β signaling. Exp. Cell Res. 291, 275–281 [DOI] [PubMed] [Google Scholar]
- 84. Wicks S. J., Grocott T., Haros K., Maillard M., ten Dijke P., and Chantry A. (2006) Reversible ubiquitination regulates the Smad/TGF-β signalling pathway. Biochem. Soc. Trans. 34, 761–763 [DOI] [PubMed] [Google Scholar]
- 85. Inoue Y., and Imamura T. (2008) Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 99, 2107–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., and Wrana J. L. (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol. Cell 6, 1365–1375 [DOI] [PubMed] [Google Scholar]
- 87. Tang L. Y., Yamashita M., Coussens N. P., Tang Y., Wang X., Li C., Deng C. X., Cheng S. Y., and Zhang Y. E. (2011) Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 30, 4777–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McDonald C. A., Millena A. C., Reddy S., Finlay S., Vizcarra J., Khan S. A., and Davis J. S. (2006) Follicle-stimulating hormone-induced aromatase in immature rat Sertoli cells requires an active phosphatidylinositol 3-kinase pathway and is inhibited via the mitogen-activated protein kinase signaling pathway. Mol. Endocrinol. 20, 608–618 [DOI] [PubMed] [Google Scholar]