

Abstract
Adverse reactions in normal tissue after radiotherapy (RT) limit the dose that can be given to tumour cells. Since 80% of individual variation in clinical response is estimated to be caused by patient-related factors, identifying these factors might allow prediction of patients with increased risk of developing severe reactions. While inactivation of cell renewal is considered a major cause of toxicity in early-reacting normal tissues, complex interactions involving multiple cell types, cytokines, and hypoxia seem important for late reactions. Here, we review ‘omics’ approaches such as screening of genetic polymorphisms or gene expression analysis, and assess the potential of epigenetic factors, posttranslational modification, signal transduction, and metabolism. Furthermore, functional assays have suggested possible associations with clinical risk of adverse reaction. Pathway analysis incorporating different ‘omics’ approaches may be more efficient in identifying critical pathways than pathway analysis based on single ‘omics’ data sets. Integrating these pathways with functional assays may be powerful in identifying multiple subgroups of RT patients characterized by different mechanisms. Thus ‘omics’ and functional approaches may synergize if they are integrated into radiogenomics ‘systems biology’ to facilitate the goal of individualised radiotherapy.
Keywords: Radiotherapy, Normal-tissue reaction, Predictive tests, Single-nucleotide polymorphisms, Genome-wide association studies, Gene expression microarrays
1. Introduction
Radiation therapy (RT1) is an important component of modern multimodality tumour therapy and is part of the treatment in approximately 60% of cancer patients treated with curative intent (1). Although technological advances in the delivery of RT has reduced the volume of normal tissue receiving critical radiation doses, the tumour dose is still limited by adverse effects in adjacent late-reacting normal tissue. Previous studies on RT-induced telangiectasia of the skin suggested that after considering the effects of absorbed dose and dose per fraction up to 80% of the observed variation in risk was associated with individual patient-related factors (2-4). The identification of patients’ individual susceptibility for the development of adverse effects from RT is an important prerequisite for individualising tumour treatment. Thus the therapeutic window might be widened by increasing the dose to the tumour in patients with relatively radioresistant normal tissue. On the other hand, patients with high risk of developing severe normal-tissue reaction might be candidates for either altered radiotherapy regimens (alternative fractionation schemes, treatment planning, or modalities), changes to surgery (e.g. in breast cancer, mastectomy rather than wide local excision plus RT) or pharmacologic interventions to ameliorate symptoms. Therefore, several approaches to develop a predictive assay for normal-tissue toxicity have been pursued in the past two decades. Here we review the novel high-throughput ‘omics’ technologies and more classical functional assays and discuss how the two approaches may interact synergistically to facilitate the identification of subgroups with different risk of RT-induced toxicity.
2. Clinical endpoints, mechanisms and hypotheses
Before reviewing ‘omics’ approaches and functional assays for radiation-induced normal-tissue reaction, a brief overview of the clinical endpoints and possible mechanisms is given. This is intended to present the context and basic principles of early and late RT-toxicities based on selected representative examples. For a more detailed discussion of mechanistic aspects, the reader is encouraged to consult comprehensive reviews on the special topics.
In tumour therapy, the aim is to prevent proliferation of tumour cells that will inevitably lead to recurrence of the tumour. Thus the clinical endpoint of tumour control is closely related to clonogenic inactivation of tumour cells although the microenvironment and systemic effects may also play a role. Similarly, early reacting normal tissues such as epithelia are characterized by continuous cell renewal with a stem cell niche and a transit-amplifying cell (TAC) compartment forming a differentiation lineage (5, 6). Normally, stem cells divide asymmetrically producing a TAC progenitor cell while retaining a stem cell in its niche. Tissue homeostasis ensures a strict regulation of cell production and loss of terminally differentiated cells by shedding or apoptosis. Thus it is a reasonable assumption that radiation-induced clonogenic cell inactivation is also important for early-reacting tissues (7-9).
An illustrative example is the case of the crypt and villi of the small intestine. Here the stem cell niche consists of 4-6 stem cells protected in the crypts, which produce TACs that form the constantly renewing intestinal lining through amplification and differentiation. If one or more stem cells are lost, the remaining cells temporarily undergo symmetric division until the lost cells are replaced (6, 10). However, if all 4-6 stem cells are lost, the villus will disappear within days because of the high cell turnover. For n stem cells in a crypt, the fraction of inactivated cells after a dose D resulting in a surviving fraction, SF, will be (1-SFD)n. Thus, a dose resulting in SFD=0.01 will inactivate 4-6 stem cells with 94-96% probability. From radiation accidents and experimental animal studies doses D≥10 Gy to the small intestine are known to be lethal (8). In addition to targeted inactivation of stem cells, stromal effects may be involved. Thus high single doses of 15 Gy or more have been reported to induce apoptosis of microvascular endothelial cells owing to release of the second messenger ceramide from membrane sphingo-lipids by acid sphingomyelinase (11). At doses of approximately 20 Gy and above, newly synthesized ceramide may contribute to apoptosis via induction of ceramide synthase (12, 13). Damage to the capillaries of the small intestine will lead to indirect cell death of stem cells in the crypts, although the role of endothelial apoptosis has been disputed (14, 15).
Another experimentally well studied, early reacting system is mouse tongue. Fractionation studies have demonstrated that stem cells are slowly depleted during the early part of a fractionated schedule. However, when depletion reaches a certain level, i.e. the number of stem cells becomes critical for renewal of the epithelium, some of the stem cells switch from asymmetric to symmetric cell division allowing cell renewal to match the rate of cell loss during the remaining part of the fractionated schedule (16-18). The two examples given above show that even for early-reacting tissues with rapid turnover of cells, the role of clonogenic cell inactivation of putative target cells for radiation toxicity is likely to be modified by other biological processes such as a change from asymmetric to symmetric stem-cell division, or between apoptotic pathways. Furthermore, inflammatory reactions stimulated by reactive oxygen species (ROS) and debris from cell death, and mediated by cytokines and immune cells, play an important role in early reaction (19). Immune cells in turn produce ROS that may produce further oxidative DNA damage in the normal tissue, even in cells outside the irradiated region (20, 21).
Based on the role of inflammation and immune cells in the radiation response of tumour and normal tissues, a bioinformatics approach was recently used to identify a molecular network of 24 genes with differential expression common to cancer and healthy tissues (22). Prominent genes coded for members of the NF-κB family and other transcription factors, involved in transcriptional activation of cytokines, and for cytokine receptors, chemokines, cytokines, growth factors, signalling molecules, and cell adhesion molecules involved in the recruitment of immune cells to injured tissues. Genes differentially regulated by irradiation of healthy tissue included radiation response and cell cycle genes TP53, CDKN1A, and CCND1, several genes involved in cytokine expression and signal transduction, but also pro- and anti-apoptotic genes, and genes coding for collagen type 1 and 3 chains. Most of the genes were identified by irradiation of human peripheral blood or brain tissue of experimental animals, and the expression profile is consistent with the role of cytokines and immune cells in normal-tissue reaction, and the apoptotic response of lymphocytes to moderate doses of ionising radiation. However, NF-κB is not limited to blood cells and brain. Thus radiation-induced expression, activation, or translocation, of NF-κB is involved in controlling numerous pro-inflammatory genes, chemokines, chemokine receptors, and cell adhesion molecules, in several different cells and tissues (23).
Late reacting tissues rely much less on cell renewal than early reacting tissue. In many cases, fibrotic changes in connective tissue or organs are involved. Fibroblasts do not readily undergo apoptosis after irradiation but are permanently arrested and continue to be metabolically active for extended periods of time (months and years) (24, 25). Although irradiated fibroblasts are frequently referred to as being senescent, premature terminal differentiation is a more appropriate term as they synthesize increased amounts of extracellular matrix proteins, and show decreased proteolytic activity with downregulated expression of matrix metalloproteases (MMPs) and upregulated expression of tissue inhibitors of MMPs (TIMP) (26-30). Furthermore, some cells differentiate to contractile myofibroblasts expressing α-smooth muscle actin (31, 32). Although true mesenchymal stem cells may only be present in the bone marrow, a mixture of progenitor cells in the TAC compartment, including early stages with high residual capacity for proliferation, can be isolated by outgrowth from skin samples. Because differentiation of progenitor fibroblasts occurs along a long TAC differentiation lineage with exponential expansion, division of rare stem cells or early progenitor cells is sufficient to maintain homeostasis of the tissue (33).
Various studies suggest that different normal-tissue end points do not always correlate closely within individual RT patients. Thus neither erythema nor subdermal fibrosis correlated with telangiectasia of the skin in breast cancer RT patients (3, 34). However, a more recent, larger study showed a significantly increased risk of fibrosis in breast RT patients with telangiectasia although overall, the risk of developing fibrosis was much lower than that of telangiectasia (35). In the RAPPER (“Radiogenomics: Assessment of Polymorphisms for Predicting the Effects of Radiotherapy”) study, comprising 778 breast RT patients enrolled in the Cambridge breast IMRT (intensity-modulated RT) trial, correlation coefficients were generally low though significant correlations were found between several endpoints (e.g. between breast shrinkage and six other endpoints, as well as between breast induration, edema, and telangiectasia) but not between breast induration and telangiectasia (36). The lack of close correlations between different endpoints, in particular between radiation-induced fibrosis and telangiectasia, supports the view that there are differences in the mechanisms underlying the pathogenesis of different endpoints. Thus fibrotic late reactions have been hypothesized to result from complex interactions between different cell types involving inflammatory and profibrogenic reactions with participation of cytokines and immune cells (37-40).
Fibrosis is reminiscent to wound healing with three distinct phases, an inflammatory phase, a proliferative phase, and a remodelling phase (41). Ionising radiation induces expression of inflammatory cytokines and chemokines, including TNF-α, IL-1α, IL-1β, and IL-6 (23, 42, 43). This results in vasodilation, swelling of the injured tissue and recruitment of immune cells, especially macrophages and neutrophils (44). Furthermore, direct irradiation of immune cells induces expression of the inflammasome (45). During the acute inflammatory phase, proteolytic degradation of the damaged extracellular matrix (ECM) components occurs (46). This is followed by the proliferative phase in which T-helper lymphocytes are polarised towards the Th2 cell response and fibrogenic cytokines including TGF-β1 and platelet-derived growth factor (PDGF) are expressed (43, 44). Fibroblasts repair the wound by depositing newly synthesized ECM proteins such as collagen type 1 and 3, and differentiated myofibroblasts contract the wound. During the remodelling phase, the initially deposited ECM is modified by a dynamic process of proteolysis and ECM deposition. Fibrosis develops when these processes are unbalanced in favour of ECM deposition, (47). However, inflammation does not appear to be an absolute prerequisite for the development of induced fibrosis, and in some cases macrophage-derived TGF-β1 is considered to contribute to fibrosis via epithelial-mesenchymal transition (EMT) of epithelial cells (44).
The interactions of ROS with pro- and anti-inflammatory forces in the cytokine network are thought to be crucial for the radiation response (reviewed in (19)), and radiation-induced hypoxia is considered to play a major role in the development of fibrotic reactions (40, 48, 49). Thus disappearance of blood vessels is observed in some forms of fibrosis (50). Various molecular mechanisms have been proposed to explain the persistence and progression of radiation-induced fibrosis in different tissues. TGF-β1 is considered to be a molecular switch responsible for excess synthesis and deposition of collagen and other ECM proteins via the SMAD3 pathway (51-54). However, in radiation enteropathy, the fibrotic response has been found to be maintained by Rho/ROCK-mediated activation of the TGF-β1 target, CCN2 (connective tissue growth factor, CTGF) (42).
In addition to the deterministic endpoints described above, stochastic endpoints involving genetic changes (mutation or malignant transformation) are increasingly relevant as improved cancer treatments have resulted in larger numbers of long-term survivors. Recent meta-analyses estimate relative risks of the order of 1.2 for second cancers after radiotherapy in adult patients though it may be larger in children (55, 56). Genomic instability is considered to be an important factor in the development of radiation-induced cancers (57, 58). A schematic overview of the various processes involved in early and late normal-tissue end points is shown in Figure 1.
Figure 1.
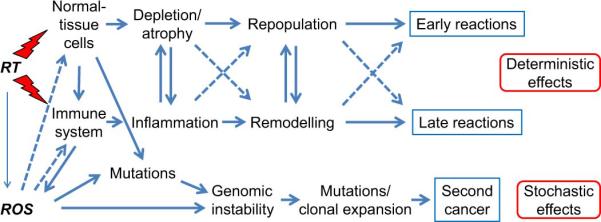
Schematic induction of normal-tissue reaction after radiotherapy (RT). Different cell types (stroma, vascular, parenchymal, and immune cells) in the irradiated tissue interact with each other and with the immune system via cytokines to produce inflammatory and pro-fibrotic reactions. Cell depletion, inflammation, repopulation and remodelling are reminiscent of the wound healing process and lead to deterministic effects. Radiation-induced mutations and genomic instability lead to stochastic effects.
Patients that are extremely hypersensitive to ionizing radiation due to rare mutations such as in the ATM gene (59) are not part of the normal variation of radiotherapy (60). For the vast majority of RT patients, it seems likely that multiple mechanisms and pathways will be involved in determining individual risk of adverse reactions. Therefore, caution has to be taken when characterizing the ‘radiosensitivity’ of patients since the risk of developing a particular normal-tissue reaction is likely dependent on the specific endpoint. In fact, the term ‘radiosensitivity’ implies the measurement of a quantitative dose-response relationship in individual patients which is normally not possible. To the extent that different mechanisms, cells, and pathways are involved in different endpoints, and that external factors such as dose or dose per fraction may confound these mechanisms, approaches based on single genetic or functional tests may not be generally applicable beyond the particular setting that it was developed for. On the one hand it can be argued that an unbiased screening of genetic variants should identify the relevant genes. On the other hand, specific cell systems and functional assays may be required to reflect more closely the mechanism of the endpoint in question.
3. ‘Omics’ approaches
3.1 Candidate gene studies of single nucleotide polymorphisms (SNPs)
Earlier studies attempted correlations of genetic variants with the risk of developing normal-tissue reaction after RT using candidate gene SNPs. The majority of the candidate genes were selected on the basis of their relation with the DNA damage response (DDR), mostly related to DNA repair, or genes involved in inflammatory or pro-fibrotic processes. Various single-centre studies suggested correlations with specific polymorphisms in TGFB1, XRCC3, XRCC1, ATM, GSTP1, GSTA1, or the cumulated number of risk alleles, and subcutaneous fibrosis or changes in breast appearance after breast conserving therapy (61-67). However, a larger study on postmastectomy patients failed to replicate the initial associations for TGFB1, XRCC3, XRCC1, and ATM (68), and a study on gynecological cancer showed no significant correlation with TGFB1 (69). Polymorphisms in XRCC3 and XRCC1 were reported to be associated with telangiectasia in some studies (62, 65) but this was not confirmed in a larger study which found associations with TP53 (70). However, TP53 showed no association with fibrosis after breast cancer radiotherapy (67).
A systematic review of studies on various early and late reactions after radiotherapy of various cancers published up to 2008 reported diverse results with significant correlations in some but not other studies (71). Subsequent larger validation studies and meta-analyses were unable to confirm associations between breast shrinkage and SNPs in TGFB1 and various other genes (36, 72-74). Similarly, TGFB1 polymorphisms were not significantly associated with radiation-induced lung toxicity (pneumonitis) in two of three studies on lung cancer patients (75-77). However, a significant association was found between a SNP in the TNF promoter and combined late reactions (telangiectasia and fibrosis) using the standardized total average toxicity (STAT) score (78) and pooling a discovery and three replication cohorts (79). Furthermore, a significant association was found between TXNRD2 and radiation-induced fibrosis in a study on breast cancer patients (80). Using the STAT score, XRCC1 was associated with skin toxicity (telangiectasia, pigmentation, atrophy) and combined late reaction (81). A test and validation data-set approach in studies on lung cancer patients found SNPs in promoter regions of TGFB1 and HSPB1 to be associated with the risk of radiation esophagitis and pneumonitis (82-84). A summary of candidate SNP studies is shown in Table 1. Further candidate gene studies were reviewed recently (85, 86). Although the candidate gene approach has been considered to be unsuccessful, recent large collaborative radiogenomics studies with a few thousand patients have found replicated associations (79, 81).
Table 1.
Clinical studies associating genetic variation, epigenetic variation, gene expression, and protein expression with adverse normal-tissue reaction to radiotherapy (RT).
| Candidate gene SNP studies | |||||
|---|---|---|---|---|---|
| Gene | RT site | N.T. effect | # pat. | signif. | Reference |
| HSPB1 | lung | esophagitis | n=120+181 | * | [83] Lopez-Guerra et al. 2011 |
| TGFB1 | lung | esophagitis | n=97+101 | * | [82] Guerra et al., 2012 |
| APEX | mastect. | fibrosis | n=41 | n.s. | [62] Andreassen et al., 2003 |
| APEX | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| ATM | mastect. | fibrosis | n=41 | * | [63] Andreassen et al., 2006a |
| ATM | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| GSTA1 | breast | fibrosis | n=257 | * | [67] Terrazino et al., 2012 |
| GSTP1 | breast | fibrosis | n=257 | * | [67] Terrazino et al., 2012 |
| NOS3 | breast | fibrosis | n=257 | n.s. | [67] Terrazino et al., 2012 |
| SOD2 | mastect. | fibrosis | n=41 | * | [62] Andreassen et al., 2003 |
| SOD2 | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| TGFB1 | breast | fibrosis | n=103 | * | [66] Quarmby et al., 2003 |
| TGFB1 | breast | fibrosis | n=257 | * | [67] Terrazino et al., 2012 |
| TGFB1 | breast | fibrosis | n=778 | n.s. | [36] Barnett et al., 2010 |
| TGFB1 | mastect. | fibrosis | n=41 | * | [62] Andreassen et al., 2003 |
| TGFB1 | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| TP53 | breast | fibrosis | n=257 | n.s. | [67] Terrazino et al., 2012 |
| XRCC1 | breast | fibrosis | n=257 | n.s. | [67] Terrazino et al., 2012 |
| XRCC1 | mastect. | fibrosis | n=41 | * | [62] Andreassen et al., 2003 |
| XRCC1 | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| XRCC3 | mastect. | fibrosis | n=41 | * | [62] Andreassen et al., 2003 |
| XRCC3 | mastect. | fibrosis | n=120 | n.s. | [68] Andreassen et al., 2006b |
| XRCC3 | breast | fibrosis | n=257 | n.s. | [67] Terrazino et al., 2012 |
| ATM | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| RAD21 | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| SOD2 | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| TGFB1 | gynaecol | late | n=78 | * | [69] De Ruyck et al., 2006 |
| TGFB1 | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| XCRR1 | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| XRCC3 | various | late | n=34 | n.s. | [64] Azria et al., 2008 |
| HSPB1 | lung | pneumonitis | n=146+125 | * | [84] Pang et al., 2013 |
| TGFB1 | lung | pneumonitis | n=164 | * | [77] Yuan et al., 2009 |
| TGFB11 | lung | pneumonitis | n=209 | n.s. | [76] Wang et al., 2010 |
| TGFB1 | lung | pneumonitis | n=178 | n.s. | [75] Voets et al., 2012 |
| APEX | mastect. | telangiectasia | n=41 | n.s. | [62] Andreassen et al., 2003 |
| APEX1 | breast | telangiectasia | n=409 | n.s. | [70] Chang-Claude et al., 2009 |
| CDKN1A | breast | telangiectasia | n=409 | n.s. | [70] Chang-Claude et al., 2009 |
| SOD2 | mastect. | telangiectasia | n=41 | n.s. | [62] Andreassen et al., 2003 |
| TGFB1 | mastect. | telangiectasia | n=41 | n.s. | [62] Andreassen et al., 2003 |
| TP53 | breast | telangiectasia | n=409 | * | [70] Chang-Claude et al., 2009 |
| XRCC1 | mastect. | telangiectasia | n=41 | n.s. | [62] Andreassen et al., 2003 |
| XRCC1 | breast | telangiectasia | n=409 | n.s. | [70] Chang-Claude et al., 2009 |
| XRCC2 | breast | telangiectasia | n=409 | n.s. | [70] Chang-Claude et al., 2009 |
| XRCC3 | mastect. | telangiectasia | n=41 | n.s. | [62] Andreassen et al., 2003 |
| XRCC3 | breast | telangiectasia | n=409 | n.s. | [70] Chang-Claude et al., 2009 |
| Test/discovery-and-validation gene SNP studies | |||||
|---|---|---|---|---|---|
| Gene(#SNPs) | RT site | N.T. effect | # pat. | signif. | Reference |
| CYP2C8(528) | breast | fibrosis | n=92 | *d.s. | [80] Edvardsen et al., 2013 |
| IL8(528) | breast | fibrosis | n=92 | *d.s. | [80] Edvardsen et al., 2013 |
| MGMT(528) | breast | fibrosis | n=92 | *d.s. | [80] Edvardsen et al., 2013 |
| TDG(305) | breast | fibrosis | n=753 | (*)d.s. | [81] Seibold et al., 2015 |
| TGFB1 | various | fibrosis | n=2782 | n.s. | [73] Barnett et al., 2012 |
| TGFB1 | various | fibrosis | n=2783 | n.s. | [74] Zhu et al., 2013 |
| TGFBR2(528) | breast | fibrosis | n=92 | *d.s. | [80] Edvardsen et al., 2013 |
| TNF(305) | breast | fibrosis | n=753 | (*)d.s. | [81] Seibold et al., 2015 |
| TXN(305) | breast | fibrosis | n=753 | (*)d.s. | [81] Seibold et al., 2015 |
| TXNRD2(528) | breast | fibrosis | n=92 | *d.s. | [80] Edvardsen et al., 2013 |
| TXNRD2(7) | breast | fibrosis | n=433 | *v.s. | [80] Edvardsen et al., 2013 |
| XRCC1(305) | breast | fibrosis | n=753 | *d.s. | [81] Seibold et al., 2015 |
| XRCC1(10) | breast | fibrosis | n=2036 | *v.s. | [81] Seibold et al., 2015 |
| TNF(43) | breast | late(STAT) | n=633 | *d.s. | [79] Talbot et al., 2012 |
| TNF(1) | breast | late(STAT) | n=2036 | *v.s. | [79] Talbot et al., 2012 |
| GWAS | |||||
|---|---|---|---|---|---|
| Gene #SNPs | RT site | N.T. effect | # pat. | signif. | Reference |
| FSHR | prostate | erect. dysfct. | n=79 | * | [93] Kerns et al., 2010 |
| 4 | prostate | erect. dysfct. | n=79 | (*) | [93] Kerns et al., 2010 |
| 12 | prostate | erect. dysfct. | n=235+230 | (*) | [94] Kerns et al., 2013 |
| block of 8 | prostate | urinary | n=346+377 | * | [96] Kerns et al., 2013 |
| 2 | prostate | rectal bleed. | n=368+781 | * | [95] Kerns et al., 2013 |
| TANC1 | prostate | late(STAT) | n=741 | *d.s. | [97] Fachal et al., 2014 |
| TANC1 | prostate | late(STAT) | n=1742 | * | [97] Fachal et al., 2014 |
| 29 | breast | late(STAT) | n=1217 | *d.s. | [98] Barnett et al., 2014 |
| 164 | prostate | late(STAT) | n=633 | *d.s. | [98] Barnett et al., 2014 |
| 11 | br.+prost. | late(STAT) | n=1850+1733 | (*) | [98] Barnett et al., 2014 |
| Epigenetics studies | |||||
|---|---|---|---|---|---|
| Gene | RT site | N.T. effect | # pat. | signif. | Reference |
| XRCC2 | cervix | late | n=54 | * | [129] Paulikova et al. 2013 |
| DGKA | breast | fibrosis | n=24+75 | * | [130] Weigel et al., 2016 |
| miR-99a | prostate | rectal bleed. | n=8+97 | * | [144] Someya et al., 2015 |
| Ku80 mRNA | prostate | rectal bleed. | n=8+97 | * | [144] Someya et al., 2015 |
| Gene expression signatures, fibroblasts | |||||
|---|---|---|---|---|---|
| Gene #genes | RT site | N.T. effect | # pat. | signif. | Reference |
| 18 | mastect. | fibrosis | n=31 | * | [155] Rödningen et al., 2005 |
| [153] Alsner et al., 2007 | |||||
| [154] Rödningen et al., 2008 | |||||
| 9 | H&N | fibrosis | n=160 | * | [156] Andreassen et al., 2013 |
| Gene expression signatures, lymphocytes/blood cells | |||||
|---|---|---|---|---|---|
| Gene #genes | RT site | N.T. effect | # pat. | signif. | Reference |
| 24 | various | early | n=57 | * | [146] Rieger et al., 2004 |
| 19 | prostate | early | n=58 | (*) | [147] Hümmerich et al., 2006 |
| 67 | H&N+Br. | early | n=24 | - | [150] Mayer et al., 2011 |
| 67 | H&N | early | n=24 | - | [149] Greve et al., 2012 |
| 50 | breast | br. appear. | n=59 | * | [151] Finnon et al., 2012 |
| 87 | breast | fibrosis | n=254 | (*) | [157] Landmark-Hoyvik et al., 2011 |
| 72 | prostate | late | n=50 | * | [148] Svensson et al., 2006 |
| RAD51 | prostate | late | n=61 | * | [152] van Oorschot et al., 2014 |
| Cytokines, plasma | |||||
|---|---|---|---|---|---|
| Protein | RT site | N.T. effect | # pat. | signif. | Reference |
| IL-6 | lung | pneumonitis | n=52 | n.s. | [163] Rübe et al., 2009 |
| IL-8 | lung | late | n=55 | * | [166] Hart et al., 2005 |
| IL-8 | lung | late | n=58 | * | [161] Stenmark et al., 2012 |
| TGF-β1 | cervix | late | n=79 | n.s. | [165] Dickson et al., 2000 |
| TGF-β1 | lung | late | n=26 | n.s. | [164] Zhao et al., 2008 |
| TGF-β1 | lung | late | n=58 | (*) | [161] Stenmark et al., 2012 |
| TGF-β1 | lung | pneumonitis | n=73 | (*). | [159] Anscher et al., 1998 |
| TGF-β1 | lung | pneumonitis | n=68 | n.s. | [162] De Jaeger et al., 2004 |
| TGF-β1 | lung | pneumonitis | n=46 | (*) | [160] Novakova-Jiresova et al., 2004 |
| TGF-β1 | lung | pneumonitis | n=52 | n.s. | [163] Rübe et al., 2009 |
| Proteomics, cells | |||||
|---|---|---|---|---|---|
| Protein | RT site | N.T. effect | # pat. | signif. | Reference |
| A2M | lung | late | n=6+20 | * | [168] Oh et al., 2011 |
| C4BPA | lung | late | n=57 | * | [169] Cai et al., 2011 |
| VTN | lung | late | n=57 | * | [169] Cai et al., 2011 |
N.T. (normal tissue); br. (breast); gynaecol. (gynaecological cancer); H&N (head-and-neck); mastect. (post-mastectomy RT); fibr. (fibrosis); br.appear. (breast appearance); # pat. (number of RT patients); signif. (statistical significance); SNP (single-nucleotide polymorphism); GWAS (genome-wide association studies). For candidate SNP studies incorporating a validation cohort, the number of patients in each cohort is given, separated by a ‘+’ sign. For studies using a test/discovery plus validation design, the significant gene is shown with the number of candidate SNPs in the discovery set given in brackets; for validation sets, the number of genes tested is shown. The significance is indicated for the discovery and validation data sets: d.s. (discovery set); v.s. (validation set). For GWAS, a single gene or the number of significant genes identified is given. * (significant); n.s. (not significant). Brackets (*) signify borderline significance or limited inference.
3.2 Genome-wide association studies (GWAS)
Because most candidate gene studies showed only relative low effect sizes associated with single polymorphisms, and because of the complex biological pathways shown to be involved in different adverse endpoints, individual risk of RT toxicity in most patients is probably determined by variation in multiple genes each characterized by low penetrance and small effect size (71, 87). This contrasts with rare mutations in genes conferring extreme radiosensitivity, such as in the ATM gene in the case of ataxia telangiectasia (60). In order to overcome this problem, and to discover new associations not predicted by current hypotheses, unbiased genome-wide association studies (GWAS) were started. Although more than 84 million SNPs have been found following sequencing of the human genome in the 1000 Genomes Project, most are rare leaving approximately 8 million common SNPs with a frequency >5% (88). Most SNPs are shared over a single or all continents but up to 25-30% are unique to a single continent or a population. Many SNPs are in linkage disequilibrium and tag a smaller number of independent loci so it is estimated that screening 0.5-1 million SNPs is sufficient for genotype imputation (89, 90). The ability to detect statistically significant associations depends on the minor allele frequency of the SNP, the prevalence of the phenotype, and the genetic architecture of the phenotype. Therefore, the number of patients required to identify a clinically useful number of SNPs could be very high, going into the thousands. Detailed discussions of the challenges and initial results have been presented in recent reviews (90-92).
Until now, six GWAS on normal-tissue reaction after radiotherapy have been published. Early studies on erectile dysfunction, rectal bleeding and urinary symptoms after radiotherapy for prostate cancer provided evidence for significant associations between SNPs and late reaction but the number of patients in these studies (n=79-1149) only allowed significant inferences about few specific genes (see Table 1) (93-96). A larger study (n=1742) of overall late toxicity combining different endpoints (STAT score) in prostate cancer RT patients implicated a TANC1 locus with a combined P-values of 4.16×10−10 to 4.64×10−11 after inclusion of one and two replication cohorts, respectively (97). The largest GWAS on late reactions published so far (n=3588) used the STAT score to combine breast and prostate cancer cohorts and provided strong evidence that genetic polymorphisms are associated with late toxicity (98). However, stronger associations were observed for specific endpoints than with the generalised STAT score, and associations identified in breast cancer were different from those identified in prostate cancer patients. Thus while GWAS are indeed a promising tool for discovery they may need to be powered to detect significant associations of SNPs with specific endpoints for each tumour type. Furthermore, the identification of a relevant functional gene from a SNP is not trivial because many SNPs are found in non-coding regions and often serve simply as tags owing to the selection strategy mentioned above. While understanding the functional effect of a SNP is not necessary for risk prediction, it is an important step towards better understanding the biologic role of inherited variation in the response to radiotherapy. The Radiogenomics Consortium (RGC) was established in 2010 to bring together the resources and patient numbers for large-scale GWAS on normal-tissue reaction after radiotherapy (99, 100).
In addition to SNPs, the genome shows copy number variations (CNVs) and insertions and deletions (INDELs). The different forms of CNV, and the mechanisms by which they arise, have been reviewed in relation to their risk of various diseases and the genetic risk of radiation (101). High throughput screening of CNVs can be performed by next generation sequencing (NGS) and used in identifying targets for cancer treatment (102). Recently, CNVs in XRCC1 was found to be significantly associated with rectal bleeding in prostate RT patients, and marginally significant for erectile dysfunction, and the integration into a risk model improved prediction of late normal-tissue toxicity (103). Small INDELs from 1-20,000 bp in length are relatively common in the genome and can be detected by NGS (104-106) although the presence of small-scale repeat sequences make the analysis a challenge (107). In all, 3.6 million INDELs have been described (88) which frequently show strong linkage disequilibrium with SNPs providing tags for imputation from GWAS (104, 108). Thus INDELs should be taken into consideration as potentially causative for the phenotype and lend themselves to retrospective analysis of GWAS.
A special case of genetic variation is in mitochondrial DNA (mtDNA). Mitochondria are centres for energy production through oxidative phosphorylation (OXPHOS) linked with glycolysis (Krebs / citric acid cycle). The number of mitochondria per cell varies depending on cell type and physiological state but is typically of the order of 100 or more, each on average containing 2-3 circular mtDNA molecules (109). mtDNA contains 13 genes belonging to the OXPHOS system, as well as genes coding for 22 tRNA and 2 rRNA (110). Other mitochondrial genes, including POLG1, POLG2, and TFAM, involved in replication of mtDNA (111, 112) are found in nuclear DNA. The potential role of changes in mtDNA for cancer therapy has been reviewed recently (113). It is well established that mtDNA influences the sensitivity of cells to chemotherapy (114), and cells depleted of mtDNA, so-called ρ0 (rho-zero) cells, are radioresistant, possibly due to suppression of the G2 checkpoint (115, 116). Interestingly, mutations in TP53 or POLG1 can lead to reduced mtDNA content although the effect on radiosensitivity was not reported (117, 118). NGS or exome sequencing applied to mtDNA may yield information on variation in mtDNA copy number and deletions (119). Changes in mtDNA may also occur in response to chemo- or radiation therapy (reviewed in (113)), and there is evidence for an increased rate of mutations in mtDNA in cells and tissues after irradiation (120). Mitochondria are also sites of ROS production and have developed defence mechanisms which may be important for the response to ionising radiation. Furthermore, the role of mitochondrial membrane potential for the radiation-induced, intrinsic pathway of apoptosis is well known (121). Taken together, mitochondrial function is likely to play an important role in the cellular response to RT by neutralizing ROS, providing energy for cell-cycle regulation and DNA repair, and contributing to the survival/death decision.
3.3 Potential role of epigenetics studies
Gene expression is determined not only by genetic variation in the promoter and other regulatory elements but also by epigenetic modification of transcriptional activity and post-translational modulation. Thus silencing of genes by methylation of cytosine in CpG islands occurs during development and cell differentiation. There is increasing evidence that methylation status may influence cellular radiation response. For example, radiosensitisation of head-and-neck squamous cell carcinoma cells was observed after non-toxic treatment with the demethylating agent 5-aza-2'-deoxycytidine (122). While methylation has received little attention in relation to normal-tissue reaction after radiotherapy, demethylation and hypermethylation may occur in fibrotic disease. A recent review on epigenetics in radiation-induced fibrosis listed nine genes that are hypermethylated and three genes that are hypomethylated in fibrotic tissue, and which are known to be affected by ionising radiation (123). Genome-wide methylation studies on fibroblasts from idiopathic pulmonary fibrosis and nonfibrotic controls showed significant differences in DNA methylation (124-126). Similarly, changes in DNA methylation are associated with systemic sclerosis and with fibrosis in wound healing (127, 128). A methylation study of 18 candidate genes found a marginally significant association (P<0.05) between hypermethylation in the promoter region of the XRCC2 gene and late toxicity in cervical cancer patients treated with chemoradiotherapy (129). In the first genome-wide study of differential methylation, hypomethylation of an enhancer region in the gene coding for the signalling kinase DGKA (diacylglycerol kinase alpha) was found to be associated with the risk for developing late reaction (fibrosis) after adjuvant RT in patients treated with breast conserving therapy (130). It was shown that reduced methylation enabled radiation-induced transcription of DGKA and a fibrogenic response.
The transcriptional activity of genes depends also on access of transcription factors which requires opening of the chromatin structure. This is achieved by acetylation of histones in transcriptionally active euchromatin by histone acetyltransferases (HAT). Deacetylation by histone deacetylases (HDAC) results in a more condensed heterochromatin structure and gene silencing. HDAC inhibitors can activate gene transcription and have been shown to modify radiation sensitivity (131). Furthermore, methylation of histones can repress or activate gene transcription and, together with acetylation, can influence fibrosis (123). Genetic association studies could identify SNPs that affect expression of genes involved in histone modifications [Kerns et al., under review] but high-throughput screening of histone modification has not been used in connection with normal-tissue reaction after radiotherapy. However, it is well established that modifications of histones, including specific phosphorylation of Tyr-139 and dephosphorylation of Tyr-142 in histone H2AX as well as changes in acetylation and methylation of histones H3 and H4, play a central role in the repair of DNA double strand breaks (DSB) by increasing accessibility and providing a scaffold for the DSB repair complexes (132). Thus γH2AX foci representing individual DSB can be detected in cells after irradiation with doses even as low as <0.2 Gy providing a very sensitive method for studying DSB induction and repair (133, 134).
microRNAs (miRNAs) are small (20-22 bases) non-coding RNA with post-transcriptional regulatory functions by mediating mRNA cleavage, destabilisation, or translational repression (135). In many cases they are transcribed as longer primary miRNA (pri-miRNA) sequences which are then processed. Approximately 1,500 miRNAs have been identified which form a complex regulatory network as each miRNA can target many genes (136). Based on sequence analysis, tens or hundreds of potential target genes are identified by various algorithms (137, 138), so each putative target must be validated individually. Ionising radiation changes the expression of at least 23 miRNAs many of which affect radiosensitivity, DNA repair and apoptosis, e.g. let-7, miR-21, miR-34s, miR-181a, and miR-449a (139). Radiation-induced transcription of some miRNAs (e.g. let-7a and let-7b) depends on ATM and p53 but ATM may also phosphorylate BRCA1 and KSRP which affects processing of pri-miRNA (139, 140). miRNAs such as miR-21 and miR-29a have been found to play important roles in renal fibrosis or systemic sclerosis (127, 141). Interestingly, members of the miR-29 family, which are downregulated in systemic sclerosis and target several genes coding for collagen and other ECM proteins, can be downregulated by TGF-β1 (123, 127, 142). In a murine skin model, miR-15a, miR-21, miR-30a, and miR-34a were differentially regulated in radiation-induced fibrosis (143). In a discovery and validation study on lymphocytes, lower levels of DSB repair protein Ku80, and higher levels of miR-99a were significantly associated (P=0.011 and P=0.013, respectively) with late rectal bleeding in prostate RT patients (144). Although the miRNA network is strongly implicated in radiation response and fibrotic disease, global miRNA screening has not, to our knowledge, yet been applied in other clinical studies of normal-tissue reaction after RT. An important caveat to all epigenetic studies mentioned here is the choice of tissue, cells, and culture conditions, since cell differentiation is partly determined by epigenetic factors. Thus while associations which depend only on radiation response may be similar in lymphocytes and fibroblasts, associations which depend on tissue-specific functions are likely to be different and the choice of test system and conditions may be critical.
3.4 Gene expression studies (transcriptome)
The different approaches described above generally focus on different aspects of gene expression. Therefore, there seems to be a case for studying gene expression directly although the conditions under which this should be done is a matter for debate. For example, quiescent cells may react differently from proliferating cells, and transcriptionally activated DDR genes may be expressed only within certain time windows after irradiation. In early breast cancer, predictive gene expression profiles for metastatic risk and treatment outcome have been shown to be clinically useful (145). Expression profiles for normal-tissue reactions have been proposed in a small number of studies using lymphocytes, whole blood, or skin fibroblast cultures established from individual patients. An association between early skin reaction and the transcriptional response of immortalised lymphoblasts 24h after 5 Gy was found in a study on 14 patients with severe early skin reactions and three control groups (146). A predictive 22-gene signature was identified with most genes having roles in DNA repair, stress response, protein degradation, and apoptosis. The expression of 143 DNA repair or repair-related genes in unirradiated lymphocytes identified 19 genes with differential, constitutive expression associated with early reaction to RT in a cohort of 58 prostate patients (147). Late reaction after RT for prostate cancer was correlated with differential gene expression 24h after a 2 Gy irradiation of previously stimulated lymphocytes. Using a 16k microarray mRNA expression from 27 over- and 23 non-responders in a discovery and validation approach, a gene signature correctly discriminated 86% of the patients when combined into a gene set classifier (148). In this study, gene sets were involved in protein turnover, development, stress signalling and apoptosis, the latter being more pronounced in patients without late reaction. Further studies supported associations between lymphocyte gene expression profiles and the risk of early reaction in head-and-neck (n=30) (149) or mixed head-and-neck and breast patients (n=24) (150), and late toxicity in breast (n=59) (151) and prostate cancer RT patients (n=61) (152).
Fibroblast cultures established from unirradiated skin have been used to establish a predictive signature for subcutaneous fibrosis after post-mastectomy radiotherapy. Early-passage fibroblasts were irradiated with 3x3.5 Gy separated by 24h, and gene expression based on 31 patients with none-mild or moderate-severe fibrosis identified an 18-gene predictive signature from a 15k microarray with the majority of genes involved in cell cycle/proliferation, matrix remodelling/cell adhesion, and ROS scavenging (153-155). A reduced signature comprising nine of the 18 genes was validated prospectively in 160 head-and-neck cancer patients of which 46 developed severe (grade 3) fibrosis. None of the 24 patients showing a “resistant” gene expression profile developed severe fibrosis, whereas 46 of the 136 patients with a “sensitive” profile developed this late endpoint (34%) (156). The reason why this gene signature appears to be better at discriminating resistance is unknown. However it seems possible that the RT-techniques used in the two patient groups were associated with relatively high risks of developing late fibrosis so that only resistant patients would stand out whereas patients with average sensitivity would develop fibrosis due to treatment-related factors. Recently, a gene expression profile for late fibrosis was derived in unirradiated blood cells from 254 women treated with breast conserving therapy, 31 of whom developed fibrosis 3-7 years after RT (157). Eighty-seven differentially expressed genes included functions in transcription, intracellular transport and localisation, and development, with down-regulation of most genes but up-regulation of SERPINE1 (coding for plasminogen activator inhibitor-1), in accordance with increased protein levels found in fibrotic conditions (158). Pathway analysis identified the TGF-β1 signalling and interleukin-2 pathways as the two major gene sets (157). Independent replication of these results will be needed and validation in rigorous prospective trials.
3.5 Proteome, kinome, and metabolome
In addition to the ‘omics’ approaches discussed above, translation of mRNA into peptides and proteins, post-translational modification, signal transduction by kinase-mediated protein phosphorylation, and the metabolism and microenvironment of cells, are likely to influence or mediate the development of normal tissue reaction to RT. So far, only few clinical studies have been published in this field, focusing mainly on the role of cytokines, in particular TGF-β1, in the risk of radiation-induced fibrosis. Thus some studies implicated higher circulating plasma levels of TGF-β1 during or towards the end of the treatment (159-161) in patients with radiation-induced late lung complications while other studies found no correlation (162-164). No association was found between TGF-β1 and late toxicity in cervix cancer patients (165), and results in lung cancer patients suggested that the tumour might be the major source of circulating cytokines (163). By contrast, two studies found significantly lower levels of IL-8 in RT patients who developed late lung toxicity (161, 166). Thus, a combination of elevated TGF-β1, lower IL-8, and mean lung dose, was recently reported to be predictive for lung toxicity (161). High throughput proteomic technology has enabled the analysis of large numbers of proteins from a single sample. Most human studies have been performed on serum and plasma samples but the high abundance of albumin and other transporter proteins presents a challenge for the detection of low abundance proteins. Nevertheless, a few studies have examined cytokine profiles in RT patients, which essentially confirmed the results discussed above (for review, see (167)). However, unbiased proteomics approaches have identified A2M (alpha-2-macroglobulin) (168), C4BPA (complement component 4 binding protein, alpha chain), and VTN (vitronectin) (169) as novel biomarkers for RT-induced lung toxicity. Experimental studies on endothelial cells and a fibroblastoid cell line showed changes in proteins involved in metabolism, cell motility, and non-homologous end joining, while studies of the heart proteome identified proteins involved in the acute phase inflammatory response and metabolism (reviewed in (170)).
To our knowledge, no systematic study of the kinome in relation to normal-tissue toxicity has been published. However, experimental studies of the radiation metabolome identified aqueous solutes involved in redox processes and nucleotide metabolism (for reviews, see (171, 172)). The relationship between the different ‘omics’ components, protein levels, post-translational modification, protein localisation and activity leading to effects in cells and tissues, is shown schematically in Figure 2.
Figure 2.

Schematic relationship of ‘omics’ and functional endpoints. Epigenetic factors influence gene expression while protein levels and function are influenced by posttranslational modification. Abbreviations: SNPs (single nucleotide polymorphisms); CNV (copy number variations); INDELs (insertions and deletions); mtDNA (mitochondrial DNA); miRNA (microRNA).
4. Functional assays
A large number of studies have been performed with the purpose of establishing associations between normal-tissue reaction after RT and the functional response of individual patients’ cells to irradiation. Endpoints have been either clonogenic cell survival or processes considered to be related to cellular radiosensitivity, such as DSB repair, chromosome aberrations, or apoptosis.
Normal skin fibroblasts can be readily grown in vitro and are functional cells of connective tissue. Therefore, studies investigated whether the radiosensitivity of patients’ fibroblasts as quantified by surviving fraction (SF) in colony formation assays (CFA) would predict patients’ risk of developing subcutaneous fibrosis after RT. Initial, smaller studies suggested a correlation might exist (173-176). However, the first of these studies was small (n=6) (173) and the correlation was weak (rs= −0.46) in a larger study (n=31) on post-mastectomy RT patients (176). Later studies, including two larger cohorts (n=79 and 104, respectively), were unable to confirm a significant correlation between SF and the risk of radiation-induced fibrosis (177-181) or changes in breast appearance (182). In hindsight, this disappointing result may not be surprising because the turnover of fibroblasts in late-reacting tissues is very slow and, in contrast to epithelial tissues, the function of connective tissue is much less dependent on continuous cell renewal. As mentioned earlier, fibroblasts do not undergo apoptosis after irradiation but differentiate prematurely and become permanently arrested post-mitotic fibrocytes that continue to synthesize extracellular matrix proteins (27, 30, 33) or differentiate into contractile myofibroblasts expressing α-smooth muscle actin (α-sma, ACTA2) (32).
Surrogate endpoints for fibroblast radiosensitivity have been explored. A significant correlation was found between residual DSB 24h after in vitro irradiation and fibrosis in breast RT patients (183) using pulsed field gel electrophoresis (PFGE). However, a validation study did not confirm this (165) although it should be noted that the discovery study included more patients with severe reaction than the validation study and found residual DSB to be a predictor for severity. A smaller study on head-and- neck cancer patients using constant field gel electrophoresis (CFGE) did not find an association either (177). Two studies on micronucleus formation in fibroblasts found no correlation with fibrosis in patients treated for breast or various other cancers (184, 185).
The process of establishing fibroblast cultures in vitro is time consuming and cost intensive whereas peripheral blood lymphocytes (PBLs) are much more readily obtained. Thus various endpoints related to the radiosensitivity of lymphocytes have been studied. Although lymphocytes may not be directly involved in late reaction after RT, the rationale for using PBLs assumes that their response may represent a person's genetically determined individual radiosensitivity. An overview of clinical studies on functional endpoints is given in Table 2. Studies using clonogenic cell survival as endpoint in lymphocytes are rare but an early study suggested that patients with severe reaction to RT were more sensitive to low-dose rate irradiation than untreated controls and RT patient with minor reaction (186). A large prospective study showed that measurements of surviving fraction at 2 Gy in PBLs predicted the risk of late RT toxicity in cervix patients (187). The largest number of lymphocyte studies, however, measured repair of DNA damage. Results from the comet assay (mainly DNA single strand breaks and base damage) suggested that low repair kinetics and higher amounts of residual damage after a dose of 2 Gy increased the risk of early reaction (188) and higher levels of damage were significantly related to severity of the reaction (179). Similar findings were obtained in several recent studies using the more sensitive method of DSB detection by γH2AX repair foci (152, 189-196) though some studies found no significant correlation (149, 151, 197-199). However, in all studies, a large overlap in γH2AX levels was observed between RT sensitive and resistant patients making predictions based on these assays difficult. A possible explanation of the overlap might be if a repair defect is the cause of increased sensitivity to RT only in a subgroup of the sensitive patients. Support for the notion of subgroups of patients with different repair defects comes from the lack of correlation between the chromosomal radiosensitivity of cancer patients measured by the G2 metaphase and the G0 micronucleus assay (200). An association between DSB repair and the risk of adverse reaction is supported by evidence of associations between increased chromosomal aberrations and risk of early (201-203) or late normal tissue reaction (177, 191, 203-205) though evidence for a trend did not always reach significance (206) and was not universally confirmed (151). The micronucleus assay was significant for late but not early reaction in breast RT patients (201) but not for breast appearance (151).
Table 2.
Clinical studies correlating functional in vitro assay data with adverse normal-tissue reaction to radiotherapy (RT).
| Fibroblasts | |||||
|---|---|---|---|---|---|
| Endpoint | RT site | N.T effect | # pat. | signif. | Reference |
| SF | mastect. | early | n=6 | (*) | [173] Burnet et al., 1992 |
| SF | H&N (+br.) | early | n=27 | n.s. | [174] Geara et al., 1993 |
| SF | mastect. | early | n=8 | n.s. | [178] Brock et al. 1995 |
| SF | breast | early | n=23 | n.s. | [179] Oppitz et al., 2002 |
| SF | mastect. | late | n=6 | (*) | [173] Burnet et al., 1992 |
| SF | H&N (+br.) | late | n=27 | * | [174] Geara et al., 1993 |
| SF | mastect. | fibr. | n=12 | * | [175] Johansen et al., 1994 |
| SF | mastect. | late | n=9 | n.s. | [178] Brock et al., 1995 |
| SF | mastect. | fibr. | n=31 | * | [176] Johansen et al., 1996 |
| SF | breast | fibr. | n=79 | n.s. | [181] Russell et al., 1998 |
| SF | H&N | late | n=23 | n.s. | [180] Rudat et al., 1999 |
| SF | breast | br.appear. | n=104 | n.s. | [182] Peacock et al., 2000 |
| SF | H&N | late | n=14 | n.s. | [177] Borgmann et al., 2002 |
| Repair/comet | breast | early | n=24 | n.s. | [179] Oppitz et al., 2002 |
| DSB Repair | breast | fibr., late | n=39 | * | [183] Kiltie et al., 1999 |
| DSB Repair | breast | fibr., late | n=49 | n.s. | [165] Dickson et al., 2002 |
| DSB Repair | H&N | late | n=14 | n.s. | [177] Borgmann et al., 2002 |
| MN | breast | late, | n=36 | n.s. | [184] Johansen et al., 1998 |
| MN | various | early, late | n=4-10 | n.s. | [185] Sprung et al., 2005 |
| Lymphocytes | |||||
|---|---|---|---|---|---|
| Endpoint | RT site | N.T effect | # pat. | signif. | Reference |
| SF | H&N (+br.) | early | n=27 | n.s. | [174] Geara et al., 1993 |
| SF | breast | early | n=15 | (*) | [186] West et al., 1995 |
| SF | H&N (+br.) | late | n=27 | n.s. | [174] Geara et al., 1993 |
| SF | breast | late | n=14 | (*) | [186] West et al., 1995 |
| SF | cervix | late | n=83 | * | [187] West et al., 2001 |
| Apoptosis | various | early | n=30 | n.s. | [149] Greve et al., 2012 |
| Apoptosis | various | early | n=89 | * | [196] Pouliliou et al., 2015 |
| Apoptosis | breast | late | n=31 | n.s. | [207] Barber et al., 2000b |
| Apoptosis | various | late | n=399 | * | [212] Ozsahin et al., 2005 |
| Apoptosis | H&N | late | n=79 | * | [213] Bordon et al., 2010 |
| Apoptosis | breast | late | n=26 | (*) | [194] Henriquez-Hern. et al., 2011 |
| Apoptosis | breast | br.appear. | n=59 | n.s. | [151] Finnon et al., 2012 |
| Apoptosis | breast | br.appear. | n=16 | n.s. | [190] Chua et al., 2014 |
| Repair/comet | breast | early | n=30 | * | [179] Oppitz et al., 2002 |
| Repair/comet | various | early, late | n=30 | * | [188] Müller et al., 2001 |
| DSB repair | various | early | n=18 | n.s. | [199] Vasireddy et al., 2010 |
| DSB repair | H&N | early | n=31 | n.s. | [198] Fleckenstein et al., 2011 |
| DSB repair | H&N | early | n=54 | * | [193] Goutham et al., 2012 |
| DSB repair | various | early | n=30 | n.s. | [149] Greve et al., 2012 |
| DSB repair | various | early | n=57 | * | [192] Djuzenova et al., 2013 |
| DSB repair | breast | early | n=80 | * | [195] Mumbrekar et al., 2014 |
| DSB repair | various | early | n=89 | * | [196] Pouliliou et al., 2015 |
| DSB repair | various | early, late | n=22 | * | [189] Bourton et al., 2011 |
| DSB repair | prostate | early, late | n=50 | n.s. | [197] Brzozowska et al., 2012 |
| DSB repair | various | late | n=22 | n.s. | [199] Vasireddy et al., 2010 |
| DSB repair | breast | late | n=26 | (*) | [194] Henriquez-Hern. et al., 2011 |
| DSB repair | breast | br.appear. | n=59 | n.s. | [151] Finnon et al., 2012 |
| DSB repair | breast | br.appear. | n=16 | * | [190] Chua et al., 2014 |
| DSB repair | prostate | late | n=61 | * | [152] van Oorschot et al., 2014 |
| Chr.aberr. | breast | early | n=123 | * | [201] Barber et al., 2000a |
| Chr.aberr. | various | early | n=52 | n.s. | [202] Borgmann et al., 2008 |
| Chr.aberr. | breast | early | n=87 | * | [202] Borgmann et al., 2008 |
| Chr.aberr. | various | early, late | n=66 | * | [203] Neubauer et al., 1997 |
| Chr.aberr. | prostate | early, late | n=50 | n.s. | [197] Brzozowska et al., 2012 |
| Chr.aberr. | breast | late | n=19 | n.s. | [201] Barber et al., 2000a |
| Chr.aberr. | H&N | late | n=16 | * | [177] Borgmann et al., 2002 |
| Chr.aberr. | breast | fibr. | n=86 | n.s. | [206] Höller et al, 2003 |
| Chr.aberr. | gynaecol. | late | n=29 | * | [205] Werbrouck et al., 2010 |
| Chr.aberr. | breast | late | n=14 | * | [191] Chua et al., 2011 |
| Chr. aberr. | breast | br.appear. | n=59 | n.s. | [151] Finnon et al., 2012 |
| Chr.aberr. | prostate | late | n=30 | * | [204] Beaton et al., 2013 |
| MN | breast | early | n=116 | n.s. | [201] Barber et al., 2000a |
| MN | various | early, late | n=9-22 | n.s. | [185] Sprung et al., 2005 |
| MN | breast | late | n=47 | (*) | [201] Barber et al., 2000a |
| MN | breast | br.appear. | n=59 | n.s. | [151] Finnon et al., 2012 |
Culture conditions, techniques for detecting and quantifying the in vitro endpoints varied between the different studies. Abbreviations: SF (surviving fraction measured by colony formation assay); DSB (DNA double-strand break); MN (micronucleus assay); Chr.aberr. (chromosome aberrations); mastect. (post-mastectomy RT); H&N (head-and-neck); br. (breast); N.T. (normal tissue); fibr. (fibrosis); br.appear. (breast appearance); # pat. (number of RT patients); signif. (statistical significance); * (P<0.05); n.s. (not significant). Brackets (*) signify borderline significance or limited inference.
A small number of studies used apoptosis of lymphocytes as an endpoint for quantifying individual radiosensitivity. An early study found increased apoptosis in breast cancer patients relative to controls but no significant difference between patients with and without late reaction after RT (207). Enhanced apoptosis detected by Annexin V was found to be associated with resistance to late toxicity in 26 breast cancer RT patients (194). A recent study tested early death of lymphocytes by staining with trypan blue 4h after irradiation with 2 Gy and found a significant association with early toxicity grade (196). Over the last decade, an apoptosis assay in T-lymphocyte subsets (CD4+ helper and, primarily, CD8+ cytotoxic) measured by flow cytometry (208-210) has received considerable clinical attention. In this radiation-induced lymphocyte apoptosis (RILA) assay, apoptosis is detected by reduced DNA content (degradation) using a DNA intercalating fluorescent dye (DAPI and later propidium iodide) (211). Thus a significant negative correlation between apoptosis in CD8+ cells measured 48h after a dose of 8 Gy and the risk of late reaction was found in a large cohort (n=399) including a variety of RT patients (212). A significant difference between patients with and without late reaction was not confirmed in a case:control study on breast RT patients but here frozen lymphocytes and a smaller dose were used, and apoptosis was detected by forward/sideward scatter (cell size and granularity) (151). A subsequent smaller study (n=16) using fresh lymphocytes from breast RT patients and a fluorescence-labelled inhibitor of caspases (FLICA) to detect apoptosis also found no difference in mean apoptotic fractions (190). Although not all studies were able to show a significant association of lymphocyte apoptosis with normal-tissue reaction, three studies in which associations with late reactions were significant showed a consistent negative correlation between the frequency of apoptosis and the risk of late reaction (194, 212, 213) while increased early cell death correlated with early toxicity (194, 196, 212). A large multicentre prospective validation study of the RILA assay in breast and prostate patients is currently undertaken as part of the EU-funded REQUITE project (214) initiated by the RGC.
It may appear surprising that the radiation response of lymphocytes should correlate inversely with the risk of developing late reaction to radiotherapy. However, various hypotheses might explain this: 1) a slow apoptotic response of irradiated lymphocytes may enhance the production of cytokines and attract inflammatory immune cells to the irradiated tissue as suggested by Ozsahin, Azria et al. (64, 212). 2) increased inflammation or immune activity in individual patients associated with decreased lymphocyte apoptosis before RT may increase ROS production leading to enhanced genomic instability, terminal differentiation of fibroblasts, and increased risk of fibrogenesis; 3) a genetic defect in the DNA damage response, e.g. in the p53 pathway, may lead to reduced DNA repair, reduced apoptosis, increased genomic instability, and increased premature terminal differentiation of fibroblasts. These hypotheses should be tested in ongoing and new studies.
5. Promise of systems biology for identification of pathways and subgroups
The ‘omics’ approaches used so far focus on identifying genetic factors and changes in transcriptional activity of genes involved in the development of normal-tissue reaction after RT. However, cell and tissue function is also influenced by posttranslational modification, cell signalling networks, and the microenvironment. Thus cell/tissue function depends on the interaction of multiple biological components from the cell to the systemic level. A direct correlation between the level of mRNA transcripts and protein expression does not always exist as the levels of many proteins are regulated at the posttranslational level. An example is p53 (TP53) which is constitutively transcribed and translated in cells but under normal circumstances is present at low levels due to proteosomal degradation by the E3 ubiquitin ligase MDM2. Cellular stresses such as irradiation, lead to phosphorylation and dissociation of p53 and MDM2, which allow p53 protein levels to increase (215). However, total protein levels and function do not always correlate (e.g. increased levels of nonfunctional protein in TP53 mutated cancer cells). A further level of regulation is post-translational modification including phosphorylation, acetylation, methylation at multiple sites specific for different functions of p53 (216). In addition, p53 must translocate into the nucleus and form tetramers to become active as a transcription factor (217).
While SNP haplotyping may be used simply as tags for prediction, a challenge for mechanistic studies is the identification of genes that can be related to the phenotype. Thus SNP tags may not be in immediate proximity to any obvious candidate gene, or the relevant sequences may be at some distance from the coding sequence. In gene expression studies using microarrays, different studies on similar study groups frequently deliver different sets of genes differentially expressed in affected individuals and controls. However, pathway analysis usually provides more stable results even though different studies may identify different genes in a particular pathway. Because of the complexity of gene expression with multiple levels of gene regulation, combining different ‘omics’ approaches should enable more comprehensive identification of important molecular pathways and interactions from unbiased discovery approaches. Bioinformatics software for such analyses is becoming available (51, 218, 219). Integrating pathway analysis with functional assays and experimental studies in a systems biology approach to radiogenomics should help characterize different functional pathways. The relationship between these complementary components is illustrated in Figure 3.
Figure 3.

Integration of ‘omics’ pathway analysis, functional assays, and experimental studies into a Systems Biology approach.
A major challenge in predicting normal-tissue reaction is the potential involvement of multiple pathways leading to increased sensitivity or resistance. For example, and as discussed above, the propensity to produce different types of chromosome aberrations may be independent of each other (200) and might define subgroups for a particular clinical endpoint. Subgroups defined by different mechanisms/pathways would depend on context, i.e. on the cell type and tissue, and might differ for deterministic (early/late toxicity) versus stochastic (genomic instability, second cancer) effects. Furthermore, it is possible that sensitivity and resistance might be mediated by different mechanisms. For example, defects in the DNA damage response or repair systems might confer hypersensitivity to RT but not necessarily radioresistance in non-affected individuals. Other types genetic variants might be more important in conferring resistance, such as increased gene copy numbers and protein expression levels of antioxidants. The identification (and importance) of different subgroups may also depend on treatment-related factors such as dose, dose per fraction, and use of additional therapeutic agents.
In order to unravel the complexity introduced by multiple mechanisms and treatment-related factors, high patient numbers will be required. However, very large multicentre studies or meta-analyses introduce increased heterogeneity so that stratification may be necessary. Multicentre studies composed of large cohorts from individual centres would facilitate stratification, though smaller cohorts from a larger number of centres might be included by grouping them together. Furthermore, genes and pathways that can be identified will depend on the study design. For example, if the risk of developing severe normal-tissue reaction is low in the cohort, genes and pathways associated with hypersensitivity may be identified in affected patients compared with the rest. By contrast, studies on cohorts in which RT produces a relatively high rate of moderate normal-tissue reaction will be more likely to identify genes and pathways associated with resistance. Pooling different types of normal-tissue reaction or cancers may identify genes and pathways that are in common (e.g. DNA damage response) but will miss others that are tissue-specific. Patient selection may also play a role. Thus studies enriched for patients with severe reactions may give different results from studies on unselected cohorts where such patients are rare. Careful consideration of the study design will be important when planning and interpreting large collaborative studies.
6. Conclusions
Candidate SNP and gene expression analyses found some associations with patients’ adverse reactions after radiotherapy though few have been validated for risk prediction. Recent large GWAS and candidate SNP studies including 1000-2000 patients identified significant associations, but heterogeneity with respect to tissues and endpoints limits success. However, the notion of multiple low-effect genes showing additive effects assumes that the effect of each gene is independent. An alternative hypothesis is that risk of toxicity is associated with a smaller number of pathways that can involve multiple genes. Combining different high-throughput, unbiased ‘omics’ technologies may facilitate the identification of such pathways. Some functional assays also suggest correlations with the risk of adverse reaction though large overlaps of in vitro endpoints are frequently observed due in part to assay variability, but possibly an indication of patient subgroups characterized by different underlying mechanisms. Integrating pathway analysis by unbiased ‘omics’ technologies combined with functional assays should be explored and could potentially identify a small number of pathways to yield a useful set of predictors for incorporating into mathematical algorithms for predicting patients’ reaction to RT. Careful consideration of study design is necessary, and large collaborative efforts will be required to assemble homogeneous cohorts with larger numbers of patients and combine expertise in different technologies and bioinformatics to collect and analyse the data. If prediction does not prove possible, a systems biology approach should at least improve our understanding of the cellular and molecular mechanisms and identify potential targets for intervention. Both cases provide a rationale for exploring a multidisciplinary systems biology approach as a means of unravelling the complex pathology of radiation-induced normal-tissue reaction to RT and ultimately improving therapeutic outcome.
Highlights.
Different pathways are involved in normal-tissue reaction after radiotherapy
Combining different ‘omics’ techniques may identify critical pathways
Functional assays may identify patient subgroups with different mechanisms
A systems biology approach promises synergy from ‘omics’ and functional assays
Acknowledgements
This work was supported by funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no 601826 (“REQUITE”). The funding source had no involvement in the writing of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: CFA (colony formation assay); CNV (copy number variation); CFGE (constant-field gel electrophoresis); DSB (double strand break); GWAS (genome-wide association studies); HAT (histone acetyltransferase); HDAC (histone deacetylase); INDELs (insertions and deletions); miRNA (microRNA); NGS (next generation sequencing); PBLs (peripheral blood lymphocytes); PFGE (pulsed-field gel electrophoresis); RILA (radiation-induced lymphocyte apoptosis); RGC (Radiogenomics Consortium); RT (radiotherapy); SF (surviving fraction); SNP (single-nucleotide polymorphism); STAT (standardized total average toxicity).
Conflict of interest statement
All authors declare no conflict of interest
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Safwat A, Bentzen SM, Turesson I, Hendry JH. Deterministic rather than stochastic factors explain most of the variation in the expression of skin telangiectasia after radiotherapy. Int J Radiat Oncol Biol Phys. 2002;52(1):198–204. doi: 10.1016/s0360-3016(01)02690-6. [DOI] [PubMed] [Google Scholar]
- 3.Tucker SL, Turesson I, Thames HD. Evidence for individual differences in the radiosensitivity of human skin. Eur J Cancer. 1992;28A(11):1783–91. doi: 10.1016/0959-8049(92)90004-l. [DOI] [PubMed] [Google Scholar]
- 4.Turesson I. Individual variation and dose dependency in the progression rate of skin telangiectasia. Int J Radiat Oncol Biol Phys. 1990;19(6):1569–74. doi: 10.1016/0360-3016(90)90374-s. [DOI] [PubMed] [Google Scholar]
- 5.Potten CS. Cell cycles in cell hierarchies. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1986;49(2):257–78. doi: 10.1080/09553008514552541. [DOI] [PubMed] [Google Scholar]
- 6.Weinberg RA. The Biology of Cancer. Second Edition ed. Garland Science Publishing; New York, Oxford: 2013. p. 960. [Google Scholar]
- 7.Booth D, Potten CS. Protection against mucosal injury by growth factors and cytokines. Journal of the National Cancer Institute Monographs. 2001;(29):16–20. doi: 10.1093/oxfordjournals.jncimonographs.a003433. [DOI] [PubMed] [Google Scholar]
- 8.Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. 7th ed. Lippincott Williams & Wilkins; Philadelphia: 2012. p. 576. [Google Scholar]
- 9.Hopewell JW. The skin: its structure and response to ionizing radiation. Int J Radiat Biol. 1990;57(4):751–73. doi: 10.1080/09553009014550911. [DOI] [PubMed] [Google Scholar]
- 10.Pajonk F, Vlashi E. Characterization of the stem cell niche and its importance in radiobiological response. Seminars in radiation oncology. 2013;23(4):237–41. doi: 10.1016/j.semradonc.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293(5528):293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 12.Ch'ang HJ, Maj JG, Paris F, Xing HR, Zhang J, Truman JP, et al. ATM regulates target switching to escalating doses of radiation in the intestines. Nat Med. 2005;11(5):484–90. doi: 10.1038/nm1237. [DOI] [PubMed] [Google Scholar]
- 13.Rotolo JA, Mesicek J, Maj J, Truman JP, Haimovitz-Friedman A, Kolesnick R, et al. Regulation of ceramide synthase-mediated crypt epithelium apoptosis by DNA damage repair enzymes. Cancer Research. 2010;70(3):957–67. doi: 10.1158/0008-5472.CAN-09-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendry JH, Dorr W, Hill RP, Potten CS. No apoptotic endothelial cells in irradiated intestine: regarding Schuller et al. (Int J Radiat Oncol Biol Phys 2007;68:205-210). Int J Radiat Oncol Biol Phys. 2008;70(3):801–2. doi: 10.1016/j.ijrobp.2007.07.2395. author reply 3. [DOI] [PubMed] [Google Scholar]
- 15.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science. 2010;327(5965):593–6. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorr W. Three A's of repopulation during fractionated irradiation of squamous epithelia: Asymmetry loss, Acceleration of stem-cell divisions and Abortive divisions. Int J Radiat Biol. 1997;72(6):635–43. doi: 10.1080/095530097142780. [DOI] [PubMed] [Google Scholar]
- 17.Dorr W. Modulation of repopulation processes in oral mucosa: experimental results. Int J Radiat Biol. 2003;79(7):531–7. doi: 10.1080/09553002310001600925. [DOI] [PubMed] [Google Scholar]
- 18.Stewart FA, Dorr W. Milestones in normal tissue radiation biology over the past 50 years: from clonogenic cell survival to cytokine networks and back to stem cell recovery. Int J Radiat Biol. 2009;85(7):574–86. doi: 10.1080/09553000902985136. [DOI] [PubMed] [Google Scholar]
- 19.Schaue D, Kachikwu EL, McBride WH. Cytokines in radiobiological responses: a review. Radiation research. 2012;178(6):505–23. doi: 10.1667/RR3031.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatzi VI, Laskaratou DA, Mavragani IV, Nikitaki Z, Mangelis A, Panayiotidis MI, et al. Non-targeted radiation effects in vivo: a critical glance of the future in radiobiology. Cancer letters. 2015;356(1):34–42. doi: 10.1016/j.canlet.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Sprung CN, Ivashkevich A, Forrester HB, Redon CE, Georgakilas A, Martin OA. Oxidative DNA damage caused by inflammation may link to stress-induced non-targeted effects. Cancer letters. 2015;356(1):72–81. doi: 10.1016/j.canlet.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgakilas AG, Pavlopoulou A, Louka M, Nikitaki Z, Vorgias CE, Bagos PG, et al. Emerging molecular networks common in ionizing radiation, immune and inflammatory responses by employing bioinformatics approaches. Cancer letters. 2015;368(2):164–72. doi: 10.1016/j.canlet.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 23.Hellweg CE. The Nuclear Factor kappaB pathway: A link to the immune system in the radiation response. Cancer Lett. 2015;368(2):275–89. doi: 10.1016/j.canlet.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 24.Bayreuther K, Francz PI, Rodemann HP. Fibroblasts in normal and pathological terminal differentiation, aging, apoptosis and transformation. Archives of Gerontology and Geriatrics. 1992;15(Suppl 1):47–74. doi: 10.1016/s0167-4943(05)80006-8. [DOI] [PubMed] [Google Scholar]
- 25.Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc Natl Acad Sci USA. 1988;85(14):5112–6. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herskind C, Bentzen SM, Overgaard J, Overgaard M, Bamberg M, Rodemann HP. Differentiation state of skin fibroblast cultures versus risk of subcutaneous fibrosis after radiotherapy. Radiother Oncol. 1998;47(3):263–9. doi: 10.1016/s0167-8140(98)00018-8. [DOI] [PubMed] [Google Scholar]
- 27.Herskind C, Johansen J, Bentzen SM, Overgaard M, Overgaard J, Bamberg M, et al. Fibroblast differentiation in subcutaneous fibrosis after postmastectomy radiotherapy. Acta Oncol. 2000;39(3):383–8. doi: 10.1080/028418600750013159. [DOI] [PubMed] [Google Scholar]
- 28.Lara PC, Russell NS, Smolders IJ, Bartelink H, Begg AC, Coco-Martin JM. Radiation-induced differentiation of human skin fibroblasts: relationship with cell survival and collagen production. Int J Radiat Biol. 1996;70(6):683–92. doi: 10.1080/095530096144572. [DOI] [PubMed] [Google Scholar]
- 29.Rodemann HP, Bayreuther K, Francz PI, Dittmann K, Albiez M. Selective enrichment and biochemical characterization of seven human skin fibroblasts cell types in vitro. Exp Cell Res. 1989;180(1):84–93. doi: 10.1016/0014-4827(89)90214-0. [DOI] [PubMed] [Google Scholar]
- 30.Rodemann HP, Peterson HP, Schwenke K, von Wangenheim KH. Terminal differentiation of human fibroblasts is induced by radiation. Scanning Microscopy. 1991;5(4):1135–42. discussion 42-3. [PubMed] [Google Scholar]
- 31.Delanian S, Martin M, Bravard A, Luccioni C, Lefaix JL. Abnormal phenotype of cultured fibroblasts in human skin with chronic radiotherapy damage. Radiother Oncol. 1998;47(3):255–61. doi: 10.1016/s0167-8140(97)00195-3. [DOI] [PubMed] [Google Scholar]
- 32.Dimitrijevic-Bussod M, Balzaretti-Maggi VS, Gadbois DM. Extracellular matrix and radiation G1 cell cycle arrest in human fibroblasts. Cancer Research. 1999;59(19):4843–7. [PubMed] [Google Scholar]
- 33.Herskind C, Rodemann HP. Spontaneous and radiation-induced differentiation of fibroblasts. Exp Gerontol. 2000;35(6-7):747–55. doi: 10.1016/s0531-5565(00)00168-6. [DOI] [PubMed] [Google Scholar]
- 34.Bentzen SM, Overgaard M, Overgaard J. Clinical correlations between late normal tissue endpoints after radiotherapy: implications for predictive assays of radiosensitivity. Eur J Cancer. 1993;29A(10):1373–6. doi: 10.1016/0959-8049(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 35.Lilla C, Ambrosone CB, Kropp S, Helmbold I, Schmezer P, von Fournier D, et al. Predictive factors for late normal tissue complications following radiotherapy for breast cancer. Breast Cancer Res Treat. 2007;106(1):143–50. doi: 10.1007/s10549-006-9480-9. [DOI] [PubMed] [Google Scholar]
- 36.Barnett GC, Coles CE, Burnet NG, Pharoah PD, Wilkinson J, West CM, et al. No association between SNPs regulating TGF-beta1 secretion and late radiotherapy toxicity to the breast: results from the RAPPER study. Radiother Oncol. 2010;97(1):9–14. doi: 10.1016/j.radonc.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6(9):702–13. doi: 10.1038/nrc1950. [DOI] [PubMed] [Google Scholar]
- 38.Rodemann HP, Bamberg M. Cellular basis of radiation-induced fibrosis. Radiother Oncol. 1995;35(2):83–90. doi: 10.1016/0167-8140(95)01540-w. [DOI] [PubMed] [Google Scholar]
- 39.Westbury CB, Yarnold JR. Radiation fibrosis--current clinical and therapeutic perspectives. Clinical oncology. 2012;24(10):657–72. doi: 10.1016/j.clon.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol. 2010;97(1):149–61. doi: 10.1016/j.radonc.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Mancini ML, Sonis ST. Mechanisms of cellular fibrosis associated with cancer regimen-related toxicities. Frontiers in pharmacology. 2014;5:51. doi: 10.3389/fphar.2014.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haydont V, Vozenin-Brotons MC. Maintenance of radiation-induced intestinal fibrosis: cellular and molecular features. World J Gastroenterol. 2007;13(19):2675–83. doi: 10.3748/wjg.v13.i19.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herskind C, Bamberg M, Rodemann HP. The role of cytokines in the development of normal-tissue reactions after radiotherapy. Strahlenther Onkol. 1998;174(Suppl 3):12–5. [PubMed] [Google Scholar]
- 44.Wynn TA. Cellular and molecular mechanisms of fibrosis. The Journal of pathology. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoecklein VM, Osuka A, Ishikawa S, Lederer MR, Wanke-Jellinek L, Lederer JA. Radiation exposure induces inflammasome pathway activation in immune cells. Journal of immunology. 2015;194(3):1178–89. doi: 10.4049/jimmunol.1303051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atkinson MJ. Radiation treatment effects on the proteome of the tumour microenvironment. Advances in experimental medicine and biology. 2013;990:49–60. doi: 10.1007/978-94-007-5896-4_3. [DOI] [PubMed] [Google Scholar]
- 47.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nature reviews Molecular cell biology. 2014;15(12):786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vujaskovic Z, Anscher MS, Feng QF, Rabbani ZN, Amin K, Samulski TS, et al. Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys. 2001;50(4):851–5. doi: 10.1016/s0360-3016(01)01593-0. [DOI] [PubMed] [Google Scholar]
- 49.Westbury CB, Pearson A, Nerurkar A, Reis-Filho JS, Steele D, Peckitt C, et al. Hypoxia can be detected in irradiated normal human tissue: a study using the hypoxic marker pimonidazole hydrochloride. The British journal of radiology. 2007;80(959):934–8. doi: 10.1259/bjr/25046649. [DOI] [PubMed] [Google Scholar]
- 50.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. The Journal of clinical investigation. 2007;117(3):557–67. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alcaraz N, Pauling J, Batra R, Barbosa E, Junge A, Christensen AG, et al. KeyPathwayMiner 4.0: condition-specific pathway analysis by combining multiple omics studies and networks with Cytoscape. BMC systems biology. 2014;8:99. doi: 10.1186/s12918-014-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghosh AK, Yuan W, Mori Y, Varga J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene. 2000;19(31):3546–55. doi: 10.1038/sj.onc.1203693. [DOI] [PubMed] [Google Scholar]
- 53.Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. 2000;47(2):277–90. doi: 10.1016/s0360-3016(00)00435-1. [DOI] [PubMed] [Google Scholar]
- 54.Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochimica et biophysica acta. 2009;1792(8):746–56. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Berrington de Gonzalez A, Curtis RE, Kry SF, Gilbert E, Lamart S, Berg CD, et al. Proportion of second cancers attributable to radiotherapy treatment in adults: a cohort study in the US SEER cancer registries. The Lancet Oncology. 2011;12(4):353–60. doi: 10.1016/S1470-2045(11)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grantzau T, Overgaard J. Risk of second non-breast cancer after radiotherapy for breast cancer: a systematic review and meta-analysis of 762,468 patients. Radiother Oncol. 2015;114(1):56–65. doi: 10.1016/j.radonc.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Aypar U, Morgan WF, Baulch JE. Radiation-induced genomic instability: are epigenetic mechanisms the missing link? Int J Radiat Biol. 2011;87(2):179–91. doi: 10.3109/09553002.2010.522686. [DOI] [PubMed] [Google Scholar]
- 58.Morgan WF. Radiation-induced genomic instability. Health physics. 2011;100(3):280–1. doi: 10.1097/HP.0b013e3182082f12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 60.Burnet NG, Johansen J, Turesson I, Nyman J, Peacock JH. Describing patients’ normal tissue reactions: concerning the possibility of individualising radiotherapy dose prescriptions based on potential predictive assays of normal tissue radiosensitivity. Steering Committee of the BioMed2 European Union Concerted Action Programme on the Development of Predictive Tests of Normal Tissue Response to Radiation Therapy. Int J Cancer. 1998;79(6):606–13. doi: 10.1002/(sici)1097-0215(19981218)79:6<606::aid-ijc9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 61.Andreassen CN, Alsner J, Overgaard J, Herskind C, Haviland J, Owen R, et al. TGFB1 polymorphisms are associated with risk of late normal tissue complications in the breast after radiotherapy for early breast cancer. Radiother Oncol. 2005;75(1):18–21. doi: 10.1016/j.radonc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 62.Andreassen CN, Alsner J, Overgaard M, Overgaard J. Prediction of normal tissue radiosensitivity from polymorphisms in candidate genes. Radiother Oncol. 2003;69(2):127–35. doi: 10.1016/j.radonc.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 63.Andreassen CN, Overgaard J, Alsner J, Overgaard M, Herskind C, Cesaretti JA, et al. ATM sequence variants and risk of radiation-induced subcutaneous fibrosis after postmastectomy radiotherapy. Int J Radiat Oncol Biol Phys. 2006;64(3):776–83. doi: 10.1016/j.ijrobp.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 64.Azria D, Ozsahin M, Kramar A, Peters S, Atencio DP, Crompton NE, et al. Single nucleotide polymorphisms, apoptosis, and the development of severe late adverse effects after radiotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(19):6284–8. doi: 10.1158/1078-0432.CCR-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giotopoulos G, Symonds RP, Foweraker K, Griffin M, Peat I, Osman A, et al. The late radiotherapy normal tissue injury phenotypes of telangiectasia, fibrosis and atrophy in breast cancer patients have distinct genotype-dependent causes. British journal of cancer. 2007;96(6):1001–7. doi: 10.1038/sj.bjc.6603637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quarmby S, Fakhoury H, Levine E, Barber J, Wylie J, Hajeer AH, et al. Association of transforming growth factor beta-1 single nucleotide polymorphisms with radiation-induced damage to normal tissues in breast cancer patients. Int J Radiat Biol. 2003;79(2):137–43. [PubMed] [Google Scholar]
- 67.Terrazzino S, La Mattina P, Gambaro G, Masini L, Franco P, Canonico PL, et al. Common variants of GSTP1, GSTA1, and TGFbeta1 are associated with the risk of radiation-induced fibrosis in breast cancer patients. Int J Radiat Oncol Biol Phys. 2012;83(2):504–11. doi: 10.1016/j.ijrobp.2011.06.2012. [DOI] [PubMed] [Google Scholar]
- 68.Andreassen CN, Alsner J, Overgaard M, Sorensen FB, Overgaard J. Risk of radiation-induced subcutaneous fibrosis in relation to single nucleotide polymorphisms in TGFB1, SOD2, XRCC1, XRCC3, APEX and ATM--a study based on DNA from formalin fixed paraffin embedded tissue samples. Int J Radiat Biol. 2006;82(8):577–86. doi: 10.1080/09553000600876637. [DOI] [PubMed] [Google Scholar]
- 69.De Ruyck K, Van Eijkeren M, Claes K, Bacher K, Vral A, De Neve W, et al. TGFbeta1 polymorphisms and late clinical radiosensitivity in patients treated for gynecologic tumors. Int J Radiat Oncol Biol Phys. 2006;65(4):1240–8. doi: 10.1016/j.ijrobp.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 70.Chang-Claude J, Ambrosone CB, Lilla C, Kropp S, Helmbold I, von Fournier D, et al. Genetic polymorphisms in DNA repair and damage response genes and late normal tissue complications of radiotherapy for breast cancer. Br J Cancer. 2009;100(10):1680–6. doi: 10.1038/sj.bjc.6605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andreassen CN, Alsner J. Genetic variants and normal tissue toxicity after radiotherapy: a systematic review. Radiother Oncol. 2009;92(3):299–309. doi: 10.1016/j.radonc.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 72.Barnett GC, Coles CE, Elliott RM, Baynes C, Luccarini C, Conroy D, et al. Independent validation of genes and polymorphisms reported to be associated with radiation toxicity: a prospective analysis study. The Lancet Oncology. 2012;13(1):65–77. doi: 10.1016/S1470-2045(11)70302-3. [DOI] [PubMed] [Google Scholar]
- 73.Barnett GC, Elliott RM, Alsner J, Andreassen CN, Abdelhay O, Burnet NG, et al. Individual patient data meta-analysis shows no association between the SNP rs1800469 in TGFB and late radiotherapy toxicity. Radiother Oncol. 2012;105(3):289–95. doi: 10.1016/j.radonc.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu ML, Wang M, Shi TY, Li QX, Xi P, Xia KQ, et al. No association between TGFB1 polymorphisms and late radiotherapy toxicity: a meta-analysis. PloS one. 2013;8(10):e76964. doi: 10.1371/journal.pone.0076964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voets AM, Oberije C, Struijk RB, Reymen B, De Ruyck K, Thierens H, et al. No association between TGF-beta1 polymorphisms and radiation-induced lung toxicity in a European cohort of lung cancer patients. Radiother Oncol. 2012;105(3):296–8. doi: 10.1016/j.radonc.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 76.Wang L, Bi N. TGF-beta1 gene polymorphisms for anticipating radiation-induced pneumonitis in non-small-cell lung cancer: different ethnic association. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(30):e621–2. doi: 10.1200/JCO.2010.31.0458. [DOI] [PubMed] [Google Scholar]
- 77.Yuan X, Liao Z, Liu Z, Wang LE, Tucker SL, Mao L, et al. Single nucleotide polymorphism at rs1982073:T869C of the TGFbeta 1 gene is associated with the risk of radiation pneumonitis in patients with non-small-cell lung cancer treated with definitive radiotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(20):3370–8. doi: 10.1200/JCO.2008.20.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barnett GC, West CM, Coles CE, Pharoah PD, Talbot CJ, Elliott RM, et al. Standardized Total Average Toxicity score: a scale- and grade-independent measure of late radiotherapy toxicity to facilitate pooling of data from different studies. Int J Radiat Oncol Biol Phys. 2012;82(3):1065–74. doi: 10.1016/j.ijrobp.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 79.Talbot CJ, Tanteles GA, Barnett GC, Burnet NG, Chang-Claude J, Coles CE, et al. A replicated association between polymorphisms near TNFalpha and risk for adverse reactions to radiotherapy. British journal of cancer. 2012;107(4):748–53. doi: 10.1038/bjc.2012.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edvardsen H, Landmark-Hoyvik H, Reinertsen KV, Zhao X, Grenaker-Alnaes GI, Nebdal D, et al. SNP in TXNRD2 associated with radiation-induced fibrosis: a study of genetic variation in reactive oxygen species metabolism and signaling. Int J Radiat Oncol Biol Phys. 2013;86(4):791–9. doi: 10.1016/j.ijrobp.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 81.Seibold P, Behrens S, Schmezer P, Helmbold I, Barnett G, Coles C, et al. XRCC1 Polymorphism Associated With Late Toxicity After Radiation Therapy in Breast Cancer Patients. Int J Radiat Oncol Biol Phys. 2015;92(5):1084–92. doi: 10.1016/j.ijrobp.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 82.Guerra JL, Gomez D, Wei Q, Liu Z, Wang LE, Yuan X, et al. Association between single nucleotide polymorphisms of the transforming growth factor beta1 gene and the risk of severe radiation esophagitis in patients with lung cancer. Radiother Oncol. 2012;105(3):299–304. doi: 10.1016/j.radonc.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 83.Lopez Guerra JL, Wei Q, Yuan X, Gomez D, Liu Z, Zhuang Y, et al. Functional promoter rs2868371 variant of HSPB1 associates with radiation-induced esophageal toxicity in patients with non-small-cell lung cancer treated with radio(chemo)therapy. Radiother Oncol. 2011;101(2):271–7. doi: 10.1016/j.radonc.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 84.Pang Q, Wei Q, Xu T, Yuan X, Lopez Guerra JL, Levy LB, et al. Functional promoter variant rs2868371 of HSPB1 is associated with risk of radiation pneumonitis after chemoradiation for non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2013;85(5):1332–9. doi: 10.1016/j.ijrobp.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 85.Kerns SL, Kundu S, Oh JH, Singhal SK, Janelsins M, Travis LB, et al. The Prediction of Radiotherapy Toxicity Using Single Nucleotide Polymorphism-Based Models: A Step Toward Prevention. Seminars in radiation oncology. 2015;25(4):281–91. doi: 10.1016/j.semradonc.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.West CM, Barnett GC. Genetics and genomics of radiotherapy toxicity: towards prediction. Genome medicine. 2011;3(8):52. doi: 10.1186/gm268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andreassen CN. Searching for genetic determinants of normal tissue radiosensitivity--are we on the right track? Radiother Oncol. 2010;97(1):1–8. doi: 10.1016/j.radonc.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 88.Genomes Project C. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annual review of genomics and human genetics. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.West CM, Dunning AM, Rosenstein BS. Genome-wide association studies and prediction of normal tissue toxicity. Seminars in radiation oncology. 2012;22(2):91–9. doi: 10.1016/j.semradonc.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 91.Barnett GC, Kerns SL, Noble DJ, Dunning AM, West CM, Burnet NG. Incorporating Genetic Biomarkers into Predictive Models of Normal Tissue Toxicity. Clinical oncology. 2015;27(10):579–87. doi: 10.1016/j.clon.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 92.Kerns SL, Ostrer H, Rosenstein BS. Radiogenomics: using genetics to identify cancer patients at risk for development of adverse effects following radiotherapy. Cancer discovery. 2014;4(2):155–65. doi: 10.1158/2159-8290.CD-13-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kerns SL, Ostrer H, Stock R, Li W, Moore J, Pearlman A, et al. Genome-wide association study to identify single nucleotide polymorphisms (SNPs) associated with the development of erectile dysfunction in African-American men after radiotherapy for prostate cancer. Int J Radiat Oncol Biol Phys. 2010;78(5):1292–300. doi: 10.1016/j.ijrobp.2010.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerns SL, Stock R, Stone N, Buckstein M, Shao Y, Campbell C, et al. A 2-stage genome-wide association study to identify single nucleotide polymorphisms associated with development of erectile dysfunction following radiation therapy for prostate cancer. Int J Radiat Oncol Biol Phys. 2013;85(1):e21–8. doi: 10.1016/j.ijrobp.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kerns SL, Stock RG, Stone NN, Blacksburg SR, Rath L, Vega A, et al. Genome-wide association study identifies a region on chromosome 11q14.3 associated with late rectal bleeding following radiation therapy for prostate cancer. Radiother Oncol. 2013;107(3):372–6. doi: 10.1016/j.radonc.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kerns SL, Stone NN, Stock RG, Rath L, Ostrer H, Rosenstein BS. A 2-stage genome-wide association study to identify single nucleotide polymorphisms associated with development of urinary symptoms after radiotherapy for prostate cancer. The Journal of urology. 2013;190(1):102–8. doi: 10.1016/j.juro.2013.01.096. [DOI] [PubMed] [Google Scholar]
- 97.Fachal L, Gomez-Caamano A, Barnett GC, Peleteiro P, Carballo AM, Calvo-Crespo P, et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nature genetics. 2014;46(8):891–4. doi: 10.1038/ng.3020. [DOI] [PubMed] [Google Scholar]
- 98.Barnett GC, Thompson D, Fachal L, Kerns S, Talbot C, Elliott RM, et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother Oncol. 2014;111(2):178–85. doi: 10.1016/j.radonc.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 99.Rosenstein BS, West CM, Bentzen SM, Alsner J, Andreassen CN, Azria D, et al. Radiogenomics: radiobiology enters the era of big data and team science. Int J Radiat Oncol Biol Phys. 2014;89(4):709–13. doi: 10.1016/j.ijrobp.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.West C, Rosenstein BS, Alsner J, Azria D, Barnett G, Begg A, et al. Establishment of a Radiogenomics Consortium. Int J Radiat Oncol Biol Phys. 2010;76(5):1295–6. doi: 10.1016/j.ijrobp.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 101.Sankaranarayanan K, Nikjoo H. Ionising radiation and genetic risks. XVI. A genome-based framework for risk estimation in the light of recent advances in genome research. Int J Radiat Biol. 2011;87(2):161–78. doi: 10.3109/09553002.2010.518214. [DOI] [PubMed] [Google Scholar]
- 102.Liu B, Morrison CD, Johnson CS, Trump DL, Qin M, Conroy JC, et al. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: principles and challenges. Oncotarget. 2013;4(11):1868–81. doi: 10.18632/oncotarget.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Coates J, Jeyaseelan AK, Ybarra N, David M, Faria S, Souhami L, et al. Contrasting analytical and data-driven frameworks for radiogenomic modeling of normal tissue toxicities in prostate cancer. Radiother Oncol. 2015;115(1):107–13. doi: 10.1016/j.radonc.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 104.McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, et al. Common deletion polymorphisms in the human genome. Nature genetics. 2006;38(1):86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 105.Mullaney JM, Mills RE, Pittard WS, Devine SE. Small insertions and deletions (INDELs) in human genomes. Human molecular genetics. 2010;19(R2):R131–6. doi: 10.1093/hmg/ddq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Natsoulis G, Zhang N, Welch K, Bell J, Ji HP. Identification of Insertion Deletion Mutations from Deep Targeted Resequencing. Journal of data mining in genomics & proteomics. 2013;4(3) doi: 10.4172/2153-0602.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Narzisi G, Schatz MC. The challenge of small-scale repeats for indel discovery. Frontiers in bioengineering and biotechnology. 2015;3:8. doi: 10.3389/fbioe.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu JT, Wang Y, Gibbs RA, Yu F. Characterizing linkage disequilibrium and evaluating imputation power of human genomic insertion-deletion polymorphisms. Genome biology. 2012;13(2):R15. doi: 10.1186/gb-2012-13-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. Journal of cellular physiology. 1988;136(3):507–13. doi: 10.1002/jcp.1041360316. [DOI] [PubMed] [Google Scholar]
- 110.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 111.Davis AF, Ropp PA, Clayton DA, Copeland WC. Mitochondrial DNA polymerase gamma is expressed and translated in the absence of mitochondrial DNA maintenance and replication. Nucleic acids research. 1996;24(14):2753–9. doi: 10.1093/nar/24.14.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rao JU, Engelke UF, Rodenburg RJ, Wevers RA, Pacak K, Eisenhofer G, et al. Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(14):3787–95. doi: 10.1158/1078-0432.CCR-12-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Gisbergen MW, Voets AM, Starmans MH, de Coo IF, Yadak R, Hoffmann RF, et al. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutation research Reviews in mutation research. 2015;764:16–30. doi: 10.1016/j.mrrev.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 114.Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999;18(48):6641–6. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- 115.Cloos CR, Daniels DH, Kalen A, Matthews K, Du J, Goswami PC, et al. Mitochondrial DNA depletion induces radioresistance by suppressing G2 checkpoint activation in human pancreatic cancer cells. Radiation research. 2009;171(5):581–7. doi: 10.1667/RR1395.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang JT, Yamazaki H, Inoue T, Koizumi M, Yoshida K, Ozeki S, et al. Mitochondrial DNA influences radiation sensitivity and induction of apoptosis in human fibroblasts. Anticancer research. 1999;19(6B):4959–64. [PubMed] [Google Scholar]
- 117.Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochimica et biophysica acta. 2009;1787(5):328–34. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Singh KK, Ayyasamy V, Owens KM, Koul MS, Vujcic M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. Journal of human genetics. 2009;54(9):516–24. doi: 10.1038/jhg.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ye F, Samuels DC, Clark T, Guo Y. High-throughput sequencing in mitochondrial DNA research. Mitochondrion. 2014;17:157–63. doi: 10.1016/j.mito.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang SB, Okunieff P. Mitochondrial genetic abnormalities after radiation exposure. Advances in experimental medicine and biology. 2014;812:1–7. doi: 10.1007/978-1-4939-0620-8_1. [DOI] [PubMed] [Google Scholar]
- 121.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological reviews. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 122.De Schutter H, Kimpe M, Isebaert S, Nuyts S. A systematic assessment of radiation dose enhancement by 5-Aza-2'-deoxycytidine and histone deacetylase inhibitors in head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2009;73(3):904–12. doi: 10.1016/j.ijrobp.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 123.Weigel C, Schmezer P, Plass C, Popanda O. Epigenetics in radiation-induced fibrosis. Oncogene. 2015;34(17):2145–55. doi: 10.1038/onc.2014.145. [DOI] [PubMed] [Google Scholar]
- 124.Huang SK, Scruggs AM, McEachin RC, White ES, Peters-Golden M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PloS one. 2014;9(9):e107055. doi: 10.1371/journal.pone.0107055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PloS one. 2012;7(4):e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2012;186(6):525–35. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ciechomska M, van Laar JM, O'Reilly S. Emerging role of epigenetics in systemic sclerosis pathogenesis. Genes and immunity. 2014;15(7):433–9. doi: 10.1038/gene.2014.44. [DOI] [PubMed] [Google Scholar]
- 128.Neary R, Watson CJ, Baugh JA. Epigenetics and the overhealing wound: the role of DNA methylation in fibrosis. Fibrogenesis & tissue repair. 2015;8:18. doi: 10.1186/s13069-015-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paulikova S, Chmelarova M, Petera J, Palicka V, Paulik A. Hypermethylation of RAD51L3 and XRCC2 genes to predict late toxicity in chemoradiotherapy-treated cervical cancer patients. Folia biologica. 2013;59(6):240–5. doi: 10.14712/fb2013059060240. [DOI] [PubMed] [Google Scholar]
- 130.Weigel C, Veldwijk MR, Oakes C, Seibold P, Slynko A, Liesenfeld D, et al. Epigenetic regulation of diacylglycerol kinase alpha (DGKA) promotes radiation-induced fibrosis. Nat Comm. 2016 doi: 10.1038/ncomms10893. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. British journal of cancer. 2013;108(4):748–54. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature cell biology. 2011;13(10):1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 133.Redon CE, Weyemi U, Parekh PR, Huang D, Burrell AS, Bonner WM. gamma-H2AX and other histone post-translational modifications in the clinic. Biochimica et biophysica acta. 2012;1819(7):743–56. doi: 10.1016/j.bbagrm.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100(9):5057–62. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nature reviews Genetics. 2015;16(7):421–33. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 136.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome research. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Oulas A, Karathanasis N, Louloupi A, Pavlopoulos GA, Poirazi P, Kalantidis K, et al. Prediction of miRNA targets. Methods in molecular biology. 2015;1269:207–29. doi: 10.1007/978-1-4939-2291-8_13. [DOI] [PubMed] [Google Scholar]
- 138.Vlachos IS, Hatzigeorgiou AG. Online resources for miRNA analysis. Clinical biochemistry. 2013;46(10-11):879–900. doi: 10.1016/j.clinbiochem.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 139.Mao A, Liu Y, Zhang H, Di C, Sun C. microRNA expression and biogenesis in cellular response to ionizing radiation. DNA and cell biology. 2014;33(10):667–79. doi: 10.1089/dna.2014.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saleh AD, Savage JE, Cao L, Soule BP, Ly D, DeGraff W, et al. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PloS one. 2011;6(10):e24429. doi: 10.1371/journal.pone.0024429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chung AC, Lan HY. MicroRNAs in renal fibrosis. Frontiers in physiology. 2015;6:50. doi: 10.3389/fphys.2015.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Maurer B, Stanczyk J, Jungel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis and rheumatism. 2010;62(6):1733–43. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- 143.Simone BA, Ly D, Savage JE, Hewitt SM, Dan TD, Ylaya K, et al. microRNA alterations driving acute and late stages of radiation-induced fibrosis in a murine skin model. Int J Radiat Oncol Biol Phys. 2014;90(1):44–52. doi: 10.1016/j.ijrobp.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Someya M, Yamamoto H, Nojima M, Hori M, Tateoka K, Nakata K, et al. Relation between Ku80 and microRNA-99a expression and late rectal bleeding after radiotherapy for prostate cancer. Radiother Oncol. 2015;115(2):235–9. doi: 10.1016/j.radonc.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 145.Hornberger J, Alvarado MD, Rebecca C, Gutierrez HR, Yu TM, Gradishar WJ. Clinical validity/utility, change in practice patterns, and economic implications of risk stratifiers to predict outcomes for early-stage breast cancer: a systematic review. Journal of the National Cancer Institute. 2012;104(14):1068–79. doi: 10.1093/jnci/djs261. [DOI] [PubMed] [Google Scholar]
- 146.Rieger KE, Hong WJ, Tusher VG, Tang J, Tibshirani R, Chu G. Toxicity from radiation therapy associated with abnormal transcriptional responses to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(17):6635–40. doi: 10.1073/pnas.0307761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hummerich J, Werle-Schneider G, Popanda O, Celebi O, Chang-Claude J, Kropp S, et al. Constitutive mRNA expression of DNA repair-related genes as a biomarker for clinical radio-resistance: A pilot study in prostate cancer patients receiving radiotherapy. Int J Radiat Biol. 2006;82(8):593–604. doi: 10.1080/09553000600883302. [DOI] [PubMed] [Google Scholar]
- 148.Svensson JP, Stalpers LJ, Esveldt-van Lange RE, Franken NA, Haveman J, Klein B, et al. Analysis of gene expression using gene sets discriminates cancer patients with and without late radiation toxicity. PLoS medicine. 2006;3(10):e422. doi: 10.1371/journal.pmed.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Greve B, Bolling T, Amler S, Rossler U, Gomolka M, Mayer C, et al. Evaluation of different biomarkers to predict individual radiosensitivity in an inter-laboratory comparison--lessons for future studies. PloS one. 2012;7(10):e47185. doi: 10.1371/journal.pone.0047185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mayer C, Popanda O, Greve B, Fritz E, Illig T, Eckardt-Schupp F, et al. A radiation-induced gene expression signature as a tool to predict acute radiotherapy-induced adverse side effects. Cancer letters. 2011;302(1):20–8. doi: 10.1016/j.canlet.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 151.Finnon P, Kabacik S, MacKay A, Raffy C, A'Hern R, Owen R, et al. Correlation of in vitro lymphocyte radiosensitivity and gene expression with late normal tissue reactions following curative radiotherapy for breast cancer. Radiother Oncol. 2012;105(3):329–36. doi: 10.1016/j.radonc.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 152.van Oorschot B, Hovingh SE, Moerland PD, Medema JP, Stalpers LJ, Vrieling H, et al. Reduced activity of double-strand break repair genes in prostate cancer patients with late normal tissue radiation toxicity. Int J Radiat Oncol Biol Phys. 2014;88(3):664–70. doi: 10.1016/j.ijrobp.2013.11.219. [DOI] [PubMed] [Google Scholar]
- 153.Alsner J, Rodningen OK, Overgaard J. Differential gene expression before and after ionizing radiation of subcutaneous fibroblasts identifies breast cancer patients resistant to radiation-induced fibrosis. Radiother Oncol. 2007;83(3):261–6. doi: 10.1016/j.radonc.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 154.Rodningen OK, Borresen-Dale AL, Alsner J, Hastie T, Overgaard J. Radiation-induced gene expression in human subcutaneous fibroblasts is predictive of radiation-induced fibrosis. Radiother Oncol. 2008;86(3):314–20. doi: 10.1016/j.radonc.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 155.Rodningen OK, Overgaard J, Alsner J, Hastie T, Borresen-Dale AL. Microarray analysis of the transcriptional response to single or multiple doses of ionizing radiation in human subcutaneous fibroblasts. Radiother Oncol. 2005;77(3):231–40. doi: 10.1016/j.radonc.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 156.Andreassen CN, Overgaard J, Alsner J. Independent prospective validation of a predictive test for risk of radiation induced fibrosis based on the gene expression pattern in fibroblasts irradiated in vitro. Radiother Oncol. 2013;108(3):469–72. doi: 10.1016/j.radonc.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 157.Landmark-Hoyvik H, Dumeaux V, Reinertsen KV, Edvardsen H, Fossa SD, Borresen-Dale AL. Blood gene expression profiling of breast cancer survivors experiencing fibrosis. Int J Radiat Oncol Biol Phys. 2011;79(3):875–83. doi: 10.1016/j.ijrobp.2010.09.052. [DOI] [PubMed] [Google Scholar]
- 158.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. Journal of cellular physiology. 2012;227(2):493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Anscher MS, Kong FM, Andrews K, Clough R, Marks LB, Bentel G, et al. Plasma transforming growth factor beta1 as a predictor of radiation pneumonitis. Int J Radiat Oncol Biol Phys. 1998;41(5):1029–35. doi: 10.1016/s0360-3016(98)00154-0. [DOI] [PubMed] [Google Scholar]
- 160.Novakova-Jiresova A, Van Gameren MM, Coppes RP, Kampinga HH, Groen HJ. Transforming growth factor-beta plasma dynamics and post-irradiation lung injury in lung cancer patients. Radiother Oncol. 2004;71(2):183–9. doi: 10.1016/j.radonc.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 161.Stenmark MH, Cai XW, Shedden K, Hayman JA, Yuan S, Ritter T, et al. Combining physical and biologic parameters to predict radiation-induced lung toxicity in patients with non-small-cell lung cancer treated with definitive radiation therapy. Int J Radiat Oncol Biol Phys. 2012;84(2):e217–22. doi: 10.1016/j.ijrobp.2012.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.De Jaeger K, Seppenwoolde Y, Kampinga HH, Boersma LJ, Belderbos JS, Lebesque JV. Significance of plasma transforming growth factor-beta levels in radiotherapy for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2004;58(5):1378–87. doi: 10.1016/j.ijrobp.2003.09.078. [DOI] [PubMed] [Google Scholar]
- 163.Rube CE, Palm J, Erren M, Fleckenstein J, Konig J, Remberger K, et al. Cytokine plasma levels: reliable predictors for radiation pneumonitis? PloS one. 2008;3(8):e2898. doi: 10.1371/journal.pone.0002898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao L, Sheldon K, Chen M, Yin MS, Hayman JA, Kalemkerian GP, et al. The predictive role of plasma TGF-beta1 during radiation therapy for radiation-induced lung toxicity deserves further study in patients with non-small cell lung cancer. Lung cancer. 2008;59(2):232–9. doi: 10.1016/j.lungcan.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 165.Dickson J, Magee B, Stewart A, West CM. Relationship between residual radiation-induced DNA double-strand breaks in cultured fibroblasts and late radiation reactions: a comparison of training and validation cohorts of breast cancer patients. Radiother Oncol. 2002;62(3):321–6. doi: 10.1016/s0167-8140(01)00432-7. [DOI] [PubMed] [Google Scholar]
- 166.Hart JP, Broadwater G, Rabbani Z, Moeller BJ, Clough R, Huang D, et al. Cytokine profiling for prediction of symptomatic radiation-induced lung injury. Int J Radiat Oncol Biol Phys. 2005;63(5):1448–54. doi: 10.1016/j.ijrobp.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 167.Guipaud O. Serum and plasma proteomics and its possible use as detector and predictor of radiation diseases. Advances in experimental medicine and biology. 2013;990:61–86. doi: 10.1007/978-94-007-5896-4_4. [DOI] [PubMed] [Google Scholar]
- 168.Oh JH, Craft JM, Townsend R, Deasy JO, Bradley JD, El Naqa I. A bioinformatics approach for biomarker identification in radiation-induced lung inflammation from limited proteomics data. Journal of proteome research. 2011;10(3):1406–15. doi: 10.1021/pr101226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cai XW, Shedden KA, Yuan SH, Davis MA, Xu LY, Xie CY, et al. Baseline plasma proteomic analysis to identify biomarkers that predict radiation-induced lung toxicity in patients receiving radiation for non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6(6):1073–8. doi: 10.1097/JTO.0b013e3182152ba6. [DOI] [PubMed] [Google Scholar]
- 170.Tapio S. Ionizing radiation effects on cells, organelles and tissues on proteome level. Advances in experimental medicine and biology. 2013;990:37–48. doi: 10.1007/978-94-007-5896-4_2. [DOI] [PubMed] [Google Scholar]
- 171.Buettner GR, Wagner BA, Rodgers VG. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell biochemistry and biophysics. 2013;67(2):477–83. doi: 10.1007/s12013-011-9320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Coy SL, Cheema AK, Tyburski JB, Laiakis EC, Collins SP, Fornace A., Jr. Radiation metabolomics and its potential in biodosimetry. Int J Radiat Biol. 2011;87(8):802–23. doi: 10.3109/09553002.2011.556177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Burnet NG, Nyman J, Turesson I, Wurm R, Yarnold JR, Peacock JH. Prediction of normal-tissue tolerance to radiotherapy from in-vitro cellular radiation sensitivity. Lancet. 1992;339(8809):1570–1. doi: 10.1016/0140-6736(92)91833-t. [DOI] [PubMed] [Google Scholar]
- 174.Geara FB, Peters LJ, Ang KK, Wike JL, Brock WA. Prospective comparison of in vitro normal cell radiosensitivity and normal tissue reactions in radiotherapy patients. Int J Radiat Oncol Biol Phys. 1993;27(5):1173–9. doi: 10.1016/0360-3016(93)90540-c. [DOI] [PubMed] [Google Scholar]
- 175.Johansen J, Bentzen SM, Overgaard J, Overgaard M. Evidence for a positive correlation between in vitro radiosensitivity of normal human skin fibroblasts and the occurrence of subcutaneous fibrosis after radiotherapy. Int J Radiat Biol. 1994;66(4):407–12. doi: 10.1080/09553009414551361. [DOI] [PubMed] [Google Scholar]
- 176.Johansen J, Bentzen SM, Overgaard J, Overgaard M. Relationship between the in vitro radiosensitivity of skin fibroblasts and the expression of subcutaneous fibrosis, telangiectasia, and skin erythema after radiotherapy. Radiother Oncol. 1996;40(2):101–9. doi: 10.1016/0167-8140(96)01777-x. [DOI] [PubMed] [Google Scholar]
- 177.Borgmann K, Roper B, El-Awady R, Brackrock S, Bigalke M, Dork T, et al. Indicators of late normal tissue response after radiotherapy for head and neck cancer: fibroblasts, lymphocytes, genetics, DNA repair, and chromosome aberrations. Radiother Oncol. 2002;64(2):141–52. doi: 10.1016/s0167-8140(02)00167-6. [DOI] [PubMed] [Google Scholar]
- 178.Brock WA, Tucker SL, Geara FB, Turesson I, Wike J, Nyman J, et al. Fibroblast radiosensitivity versus acute and late normal skin responses in patients treated for breast cancer. Int J Radiat Oncol Biol Phys. 1995;32(5):1371–9. doi: 10.1016/0360-3016(95)00068-A. [DOI] [PubMed] [Google Scholar]
- 179.Oppitz U, Schulte S, Stopper H, Baier K, Muller M, Wulf J, et al. In vitro radiosensitivity measured in lymphocytes and fibroblasts by colony formation and comet assay: comparison with clinical acute reactions to radiotherapy in breast cancer patients. Int J Radiat Biol. 2002;78(7):611–6. doi: 10.1080/09553000210126466. [DOI] [PubMed] [Google Scholar]
- 180.Rudat V, Dietz A, Nollert J, Conradt C, Weber KJ, Flentje M, et al. Acute and late toxicity, tumour control and intrinsic radiosensitivity of primary fibroblasts in vitro of patients with advanced head and neck cancer after concomitant boost radiochemotherapy. Radiother Oncol. 1999;53(3):233–45. doi: 10.1016/s0167-8140(99)00149-8. [DOI] [PubMed] [Google Scholar]
- 181.Russell NS, Grummels A, Hart AA, Smolders IJ, Borger J, Bartelink H, et al. Low predictive value of intrinsic fibroblast radiosensitivity for fibrosis development following radiotherapy for breast cancer. Int J Radiat Biol. 1998;73(6):661–70. doi: 10.1080/095530098141915. [DOI] [PubMed] [Google Scholar]
- 182.Peacock J, Ashton A, Bliss J, Bush C, Eady J, Jackson C, et al. Cellular radiosensitivity and complication risk after curative radiotherapy. Radiother Oncol. 2000;55(2):173–8. doi: 10.1016/s0167-8140(00)00173-0. [DOI] [PubMed] [Google Scholar]
- 183.Kiltie AE, Ryan AJ, Swindell R, Barber JB, West CM, Magee B, et al. A correlation between residual radiation-induced DNA double-strand breaks in cultured fibroblasts and late radiotherapy reactions in breast cancer patients. Radiother Oncol. 1999;51(1):55–65. doi: 10.1016/s0167-8140(99)00030-4. [DOI] [PubMed] [Google Scholar]
- 184.Johansen J, Streffer C, Fuhrmann C, Bentzen SM, Stausbol-Gron B, Overgaard M, et al. Radiosensitivity of normal fibroblasts from breast cancer patients assessed by the micronucleus and colony assays. Int J Radiat Biol. 1998;73(6):671–8. doi: 10.1080/095530098141924. [DOI] [PubMed] [Google Scholar]
- 185.Sprung CN, Chao M, Leong T, McKay MJ. Chromosomal radiosensitivity in two cell lineages derived from clinically radiosensitive cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(17):6352–8. doi: 10.1158/1078-0432.CCR-04-1931. [DOI] [PubMed] [Google Scholar]
- 186.West CML, Elyan SAG, Berry P, Cowan R, Scott D. A comparison of the radiosensitivity of lymphocytes from normal donors, cancer patients, individuals with ataxiatelangiectasia (A-T-) and A-T heterozygotes. Int J Radiat Biol. 1995;68(2):197–203. doi: 10.1080/09553009514551101. [DOI] [PubMed] [Google Scholar]
- 187.West CM, Davidson SE, Elyan SA, Valentine H, Roberts SA, Swindell R, et al. Lymphocyte radiosensitivity is a significant prognostic factor for morbidity in carcinoma of the cervix. Int J Radiat Oncol Biol Phys. 2001;51(1):10–5. doi: 10.1016/s0360-3016(01)01575-9. [DOI] [PubMed] [Google Scholar]
- 188.Muller WU, Bauch T, Stuben G, Sack H, Streffer C. Radiation sensitivity of lymphocytes from healthy individuals and cancer patients as measured by the comet assay. Radiation and environmental biophysics. 2001;40(1):83–9. doi: 10.1007/s004110000087. [DOI] [PubMed] [Google Scholar]
- 189.Bourton EC, Plowman PN, Smith D, Arlett CF, Parris CN. Prolonged expression of the gamma-H2AX DNA repair biomarker correlates with excess acute and chronic toxicity from radiotherapy treatment. Int J Cancer. 2011;129(12):2928–34. doi: 10.1002/ijc.25953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chua ML, Horn S, Somaiah N, Davies S, Gothard L, A'Hern R, et al. DNA double-strand break repair and induction of apoptosis in ex vivo irradiated blood lymphocytes in relation to late normal tissue reactions following breast radiotherapy. Radiation and environmental biophysics. 2014;53(2):355–64. doi: 10.1007/s00411-014-0531-z. [DOI] [PubMed] [Google Scholar]
- 191.Chua ML, Somaiah N, A'Hern R, Davies S, Gothard L, Yarnold J, et al. Residual DNA and chromosomal damage in ex vivo irradiated blood lymphocytes correlated with late normal tissue response to breast radiotherapy. Radiother Oncol. 2011;99(3):362–6. doi: 10.1016/j.radonc.2011.05.071. [DOI] [PubMed] [Google Scholar]
- 192.Djuzenova CS, Elsner I, Katzer A, Worschech E, Distel LV, Flentje M, et al. Radiosensitivity in breast cancer assessed by the histone gamma-H2AX and 53BP1 foci. Radiation oncology. 2013;8:98. doi: 10.1186/1748-717X-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Goutham HV, Mumbrekar KD, Vadhiraja BM, Fernandes DJ, Sharan K, Kanive Parashiva G, et al. DNA double-strand break analysis by gamma-H2AX foci: a useful method for determining the overreactors to radiation-induced acute reactions among head-and-neck cancer patients. Int J Radiat Oncol Biol Phys. 2012;84(5):e607–12. doi: 10.1016/j.ijrobp.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 194.Henriquez-Hernandez LA, Carmona-Vigo R, Pinar B, Bordon E, Lloret M, Nunez MI, et al. Combined low initial DNA damage and high radiation-induced apoptosis confers clinical resistance to long-term toxicity in breast cancer patients treated with high-dose radiotherapy. Radiation oncology. 2011;6:60. doi: 10.1186/1748-717X-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mumbrekar KD, Fernandes DJ, Goutham HV, Sharan K, Vadhiraja BM, Satyamoorthy K, et al. Influence of double-strand break repair on radiation therapy-induced acute skin reactions in breast cancer patients. Int J Radiat Oncol Biol Phys. 2014;88(3):671–6. doi: 10.1016/j.ijrobp.2013.11.218. [DOI] [PubMed] [Google Scholar]
- 196.Pouliliou SE, Lialiaris TS, Dimitriou T, Giatromanolaki A, Papazoglou D, Pappa A, et al. Survival Fraction at 2 Gy and gammaH2AX Expression Kinetics in Peripheral Blood Lymphocytes From Cancer Patients: Relationship With Acute Radiation-Induced Toxicities. Int J Radiat Oncol Biol Phys. 2015;92(3):667–74. doi: 10.1016/j.ijrobp.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 197.Brzozowska K, Pinkawa M, Eble MJ, Muller WU, Wojcik A, Kriehuber R, et al. In vivo versus in vitro individual radiosensitivity analysed in healthy donors and in prostate cancer patients with and without severe side effects after radiotherapy. Int J Radiat Biol. 2012;88(5):405–13. doi: 10.3109/09553002.2012.666002. [DOI] [PubMed] [Google Scholar]
- 198.Fleckenstein J, Kuhne M, Seegmuller K, Derschang S, Melchior P, Graber S, et al. The impact of individual in vivo repair of DNA double-strand breaks on oral mucositis in adjuvant radiotherapy of head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2011;81(5):1465–72. doi: 10.1016/j.ijrobp.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 199.Vasireddy RS, Sprung CN, Cempaka NL, Chao M, McKay MJ. H2AX phosphorylation screen of cells from radiosensitive cancer patients reveals a novel DNA double-strand break repair cellular phenotype. British journal of cancer. 2010;102(10):1511–8. doi: 10.1038/sj.bjc.6605666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Scott D, Barber JB, Spreadborough AR, Burrill W, Roberts SA. Increased chromosomal radiosensitivity in breast cancer patients: a comparison of two assays. Int J Radiat Biol. 1999;75(1):1–10. doi: 10.1080/095530099140744. [DOI] [PubMed] [Google Scholar]
- 201.Barber JB, Burrill W, Spreadborough AR, Levine E, Warren C, Kiltie AE, et al. Relationship between in vitro chromosomal radiosensitivity of peripheral blood lymphocytes and the expression of normal tissue damage following radiotherapy for breast cancer. Radiother Oncol. 2000;55(2):179–86. doi: 10.1016/s0167-8140(99)00158-9. [DOI] [PubMed] [Google Scholar]
- 202.Borgmann K, Hoeller U, Nowack S, Bernhard M, Roper B, Brackrock S, et al. Individual radiosensitivity measured with lymphocytes may predict the risk of acute reaction after radiotherapy. Int J Radiat Oncol Biol Phys. 2008;71(1):256–64. doi: 10.1016/j.ijrobp.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 203.Neubauer S, Dunst J, Gebhart E. The impact of complex chromosomal rearrangements on the detection of radiosensitivity in cancer patients. Radiother Oncol. 1997;43(2):189–95. doi: 10.1016/s0167-8140(97)01932-4. [DOI] [PubMed] [Google Scholar]
- 204.Beaton LA, Marro L, Malone S, Samiee S, Grimes S, Malone K, et al. Investigating gamma H2AX as a Biomarker of Radiosensitivity Using Flow Cytometry Methods. ISRN radiology. 2013;2013:704659. doi: 10.5402/2013/704659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Werbrouck J, De Ruyck K, Beels L, Vral A, Van Eijkeren M, De Neve W, et al. Prediction of late normal tissue complications in RT treated gynaecological cancer patients: potential of the gamma-H2AX foci assay and association with chromosomal radiosensitivity. Oncology reports. 2010;23(2):571–8. [PubMed] [Google Scholar]
- 206.Hoeller U, Borgmann K, Bonacker M, Kuhlmey A, Bajrovic A, Jung H, et al. Individual radiosensitivity measured with lymphocytes may be used to predict the risk of fibrosis after radiotherapy for breast cancer. Radiother Oncol. 2003;69(2):137–44. doi: 10.1016/j.radonc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 207.Barber JB, West CM, Kiltie AE, Roberts SA, Scott D. Detection of individual differences in radiation-induced apoptosis of peripheral blood lymphocytes in normal individuals, ataxia telangiectasia homozygotes and heterozygotes, and breast cancer patients after radiotherapy. Radiation research. 2000;153(5 Pt 1):570–8. doi: 10.1667/0033-7587(2000)153[0570:doidir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 208.Crompton NE, Miralbell R, Rutz HP, Ersoy F, Sanal O, Wellmann D, et al. Altered apoptotic profiles in irradiated patients with increased toxicity. Int J Radiat Oncol Biol Phys. 1999;45(3):707–14. doi: 10.1016/s0360-3016(99)00256-4. [DOI] [PubMed] [Google Scholar]
- 209.Crompton NE, Shi YQ, Emery GC, Wisser L, Blattmann H, Maier A, et al. Sources of variation in patient response to radiation treatment. Int J Radiat Oncol Biol Phys. 2001;49(2):547–54. doi: 10.1016/s0360-3016(00)01477-2. [DOI] [PubMed] [Google Scholar]
- 210.Ozsahin M, Ozsahin H, Shi Y, Larsson B, Wurgler FE, Crompton NE. Rapid assay of intrinsic radiosensitivity based on apoptosis in human CD4 and CD8 T-lymphocytes. Int J Radiat Oncol Biol Phys. 1997;38(2):429–40. doi: 10.1016/s0360-3016(97)00038-2. [DOI] [PubMed] [Google Scholar]
- 211.Menz R, Andres R, Larsson B, Ozsahin M, Trott K, Crompton NE. Biological dosimetry: the potential use of radiation-induced apoptosis in human T-lymphocytes. Radiation and environmental biophysics. 1997;36(3):175–81. doi: 10.1007/s004110050069. [DOI] [PubMed] [Google Scholar]
- 212.Ozsahin M, Crompton NE, Gourgou S, Kramar A, Li L, Shi Y, et al. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(20):7426–33. doi: 10.1158/1078-0432.CCR-04-2634. [DOI] [PubMed] [Google Scholar]
- 213.Bordon E, Henriquez-Hernandez LA, Lara PC, Ruiz A, Pinar B, Rodriguez-Gallego C, et al. Prediction of clinical toxicity in locally advanced head and neck cancer patients by radio-induced apoptosis in peripheral blood lymphocytes (PBLs). Radiation oncology. 2010;5:4. doi: 10.1186/1748-717X-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.West C, Azria D, Chang-Claude J, Davidson S, Lambin P, Rosenstein B, et al. The REQUITE project: validating predictive models and biomarkers of radiotherapy toxicity to reduce side-effects and improve quality of life in cancer survivors. Clinical oncology. 2014;26(12):739–42. doi: 10.1016/j.clon.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 215.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13(6):941–50. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 216.Gu B, Zhu WG. Surf the post-translational modification network of p53 regulation. International journal of biological sciences. 2012;8(5):672–84. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harbor perspectives in biology. 2010;2(8):a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Eichner J, Rosenbaum L, Wrzodek C, Haring HU, Zell A, Lehmann R. Integrated enrichment analysis and pathway-centered visualization of metabolomics, proteomics, transcriptomics, and genomics data by using the InCroMAP software. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2014;966:77–82. doi: 10.1016/j.jchromb.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 219.Hanson NW, Konwar KM, Hawley AK, Altman T, Karp PD, Hallam SJ. Metabolic pathways for the whole community. BMC genomics. 2014;15:619. doi: 10.1186/1471-2164-15-619. [DOI] [PMC free article] [PubMed] [Google Scholar]