

Abstract
The relative contribution of self-perpetuating- vs. hemodynamic-induced fibrosis to the progression of chronic kidney disease (CKD) following acute kidney injury (AKI) is unclear. In the present study, male Sprague-Dawley rats underwent right uninephrectomy and were instrumented with a blood pressure radiotelemeter. Two weeks later separate groups of rats were subjected to 40 minutes renal ischemia-reperfusion or sham surgery and followed for 4 or 16 weeks to determine the extent to which glomerulosclerosis and tubulointerstitial fibrosis as a result of the AKI – CKD transition (i.e., at 4 weeks post AKI) change over time during the progression of CKD (i.e., at 16 weeks post AKI). On average, tubulointerstitial fibrosis was ~3-fold lower (P<0.05) whereas glomerulosclerosis was ~6-fold higher (P<0.05) at 16 vs. 4 weeks post AKI. At 16 weeks post AKI, marked tubulointerstitial fibrosis was only observed in rats exhibiting marked glomerulosclerosis, proteinuria, and kidney weight consistent with a hemodynamic pathogenesis of renal injury. Moreover, quantitative analysis between blood pressure and renal injury revealed a clear and modest blood pressure threshold (average 16 week systolic blood pressure of ~127 mmHg) for the development of glomerulosclerosis. In summary, very modest levels of blood pressure may be playing a substantial role in the progression of renal disease following AKI in settings of preexisting CKD associated with 50% loss of renal mass. In contrast, these data do not support a major role of self-perpetuating tubulointerstitial fibrosis in the progression CKD following AKI in such settings.
Keywords: hypertension, acute kidney injury, chronic kidney disease, ischemia-reperfusion, autoregulation, glomerular filtration rate, tubulointerstitial fibrosis
INTRODUCTION
The incidence of acute kidney injury (AKI) is rising1,2 and continues to be associated with elevated acute mortality rates3. Over the last decade, it has been clearly established that AKI is also associated with an increased risk of developing chronic kidney disease (CKD), end-stage renal disease (ESRD), and the associated increases in long-term morbidity and mortality4–7. Moreover, the risk of developing ESRD following recovery from AKI is substantially greater in patients with preexisting CKD7. Yet, considerable uncertainty exists regarding the relative contribution of the various mechanisms that have been proposed to accelerate progression of renal disease following AKI, especially in preexisting CKD states.
AKI can impact many different cell types along the entire length of the nephron and lead to a heterogeneous injury pattern; however, the main pathology associated with AKI is tubulointerstitial fibrosis (TIF), especially within the inner cortical and outer medullary regions, with only minimal to modest levels of glomerular injury8. In the acute stages following AKI, severely injured tubular epithelial cells and microvascular endothelial cells can undergo both necrosis and apoptosis and lead to the entire loss of their associated nephrons with ensuing TIF8. Although most sublethally injured tubules do recover normal structure and function following AKI, a subset of tubules remain in a dedifferentiated state and continue to release profibrotic and proinflammatory molecules9–11. The impaired recovery of such tubules is thought to be related to pathological processes initiated during AKI, such as, hypoxia, cellular senescence, maladaptive repair, and inflammation that can also contribute to the loss of nephrons and development of TIF following AKI6,12–18. Of note, the loss of nephrons and ensuing TIF that develops acutely following AKI (i.e., 1–4 weeks following injury), due either to lethally injured cells or impaired recovery of sublethally injured cells, denotes the AKI – CKD transition11.
In contrast to the AKI – CKD transition, the progression of CKD following AKI requires injury to and loss of nephrons that were either uninjured during AKI or had essentially recovered from the AKI episode. In all forms of CKD progression, the loss of renal function correlates best with the level of TIF19. Yet, there is considerable uncertainty regarding the pathogenesis of de novo TIF production over extended periods following AKI6. Such mechanisms of TIF production can be broadly separated into those pathways initiated during AKI vs. remote pathways that are independent of the AKI event, per se. The persistence of pathological processes initiated during AKI (e.g., hypoxia, cellular senescence, maladaptive repair, inflammation, etc.) have been proposed to contribute to CKD progression primarily via a self-perpetuating TIF pathway6,12–18. According to this theory, the TIF commonly observed as part of the AKI – CKD transition (i.e., 2–4 weeks post AKI in rodents) should expand over time15–17. Conversely, hemodynamic mechanisms similar to those responsible for the progression of CKD in rats with 5/6 renal ablation, which are, per se, independent of AKI, are also thought to contribute to CKD progression following AKI6,9–11,20,21. Such pathways are activated following a sufficient loss of renal mass, in this case due to AKI, and include systemic and glomerular hypertension, impaired renal blood flow (RBF) autoregulation, hyperfiltration and hypertrophy10,11,22. In this setting, CKD progression primarily occurs as a result of barotrauma-mediated glomerulosclerosis (GS)-induced TIF23,24. As described by Kriz and colleagues23,24, this injury pathway results from adhesions of injured glomerular capillaries to areas of the Bowman’s capsule as a result of glomerular hypertension coupled with reduced podocyte density. The resulting glomerular tuft – Bowman’s capsule synechia leads to misdirected filtration to the tubulointerstitium resulting in an inflammatory and TIF response that encapsulates the glomerular tuft and early proximal tubule. This process can ultimately lead to atubular glomeruli and the complete loss of the nephron. According to this theory, elevated levels of TIF at extended time points following AKI should primarily be observed in kidneys exhibiting renal morphologic and histologic manifestations of hypertension-induced renal injury (e.g., glomerular hypertrophy, proteinuria, focal segmental GS, etc.). Currently, the relative importance of these two pathways of TIF production to the progression of CKD following AKI is vague and we are not aware of any previous study that has rigorously evaluated the blood pressure (BP) – renal injury relationships at extended time points following AKI in rats.
The major goal of this study was to investigate the potential contribution of self-perpetuating- vs. hemodynamic-induced TIF production to the progression of CKD following ischemia-reperfusion (IR)-induced AKI in uninephrectomized rats. We focused on uninephrectomized rats because previous studies have shown that significant progression of CKD does occur over several months in this model of preexisting renal mass reduction as opposed to the very modest progression of CKD following IR injury in rats with two kidneys over the same time period13,25–27. The pattern and magnitude of renal injury, consisting of TIF, GS, and vascular injury, was assessed at 4 and 16 weeks post IR in different groups of rats to determine the extent to which such pathologies observed as a result of the AKI – CKD transition (i.e., 4 weeks post IR) change during the progression of CKD (i.e., 16 weeks post IR). To investigate the potential role of hemodynamic mechanisms in the progression of CKD following AKI, we examined the quantitative relationships between radiotelemetrically-measured BP and renal injury at 16 weeks post IR. In addition, we also assessed the effects of IR on renal hemodynamics and injury at an early time point (i.e., 4 weeks) following AKI in chronically instrumented uninephrectomized rats. We hypothesized that if self-perpetuating TIF was playing a major role in the progression of CKD following AKI, then levels of TIF at 16 weeks post AKI would be significantly greater than that observed at 4 weeks post AKI.
METHODS
A detailed description of the surgical procedures and methods are provided in the online-only Data Supplement.
Experimental Animals
A total of 71 male Sprague-Dawley rats (Charles River) were obtained at 8 weeks of age and provided a standard rat chow (1.0% NaCl, Purina) and drinking water ad libitum. The animals were cared for in accordance with National Institutes of Health and institutional guidelines and studies were approved by the Institutional Animal Care and Use Committee at Edward Hines Veterans Administration Hospital and Loyola University.
Experimental Design
Assessment of BP and renal injury 16 weeks post AKI
Rats recovered for 2 weeks following uninephrectomy (UNX) + BP radiotelemeter implantation to allow for completion of compensatory increases in renal size and function28. Two weeks post UNX, a 24-hr urine collection and blood sample were obtained to assess baseline proteinuria and serum creatinine (SCr). Rats were then subjected to 40 minutes IR (n=36) or sham IR (n=8) as described previously9. A blood sample was obtained at 48 hours post IR for the assessment of SCr, which was used as an index of AKI severity. Proteinuria and SCr were assessed every 4 weeks over the 16 week protocol. At 16 weeks post IR, renal injury was assessed in a blinded fashion.
Assessment of renal injury, hemodynamics and RBF autoregulation 4 weeks post AKI
These rats underwent similar surgical procedures as those described above with respect to UNX, radiotelemeter implantation, and IR (n=17) or sham IR (n=10). At 3 weeks post IR, all rats were chronically instrumented with a RBF transducer and an osmotic minipump containing FITC-inulin for the determination of glomerular filtration rate (GFR). One week later, BP and RBF were obtained (200 Hz) for 2–3 hours/day over 3 days in conscious rats. A blood sample and a 24-hour urine collection were then obtained for the measurement of Hct and GFR. Following measurements in the conscious state, rats were prepared for steady-state step RBF autoregulation experiments. At the completion of these studies, renal injury was assessed in a blinded fashion.
Statistical Analysis
Results are means ± SE. Statistical comparisons between groups over time were performed using two-way repeated measures ANOVA. Post hoc comparisons were made using a Student-Newman-Keuls test. A nonparametric Mann-Whitney test was used to evaluate differences in renal injury parameters between IR and sham IR groups at each time point and within IR and sham IR groups at different time points. Linear regression analysis was used to calculate the slope of the relationship between glomerular injury and BP and between autoregulatory index and GFR. Unpaired t-tests were used to evaluate differences among all other variables between IR and sham IR groups. P values of <0.05 were considered statistically significant.
RESULTS
BP and renal injury over 16 weeks post AKI
Of the 36 rats subjected to IR, 12 were euthanized within 3 days and 2 were euthanized within 4–8 weeks following IR for humane reasons and were not included in the analysis. There were no significant differences at baseline between IR and sham IR groups with respect to body weight (271 ± 7 vs. 247 ± 14 grams), SCr (Figure S1), systolic BP (Figure 1), and proteinuria (Figure 2). SCr at 48 hours post IR was significantly greater in rats subjected to IR vs. sham IR (3.6 ± 0.5 vs. 0.5 ± 0.1 mg/dl, respectively); however, by 4 weeks post IR and for the remainder of the protocol, no significant differences were noted between groups (Figure S1). Systolic BP was significantly higher in IR vs. sham IR groups at 1 and 2 weeks post IR, but subsequently returned toward baseline levels such that no differences were noted between groups at 3 weeks post IR (Figure 1). Yet, a modest but steady increase in BP occurred in the IR group beginning at 5 weeks post IR which became significantly higher vs. sham IR rats by 9 weeks post IR and for the remainder of the protocol. A similar pattern was observed in proteinuria with significantly greater levels observed in rats subjected to IR vs. sham IR at 12 and 16 weeks post IR.
Figure 1.
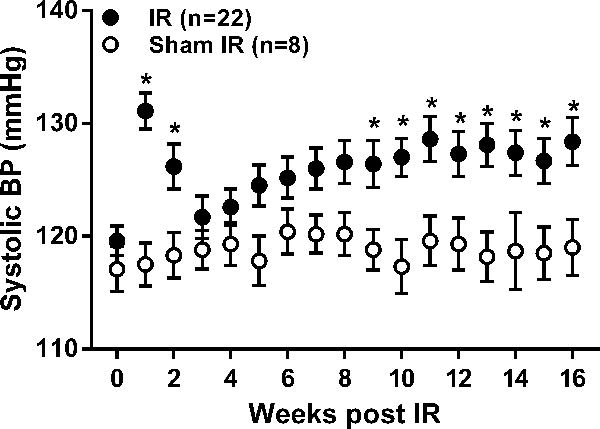
Weekly averages of radiotelemetrically measured systolic BP (mean ± SEM) at baseline (time 0) and over the subsequent 16 weeks in uninephrectomized rats subjected to 40 min. renal ischemia-reperfusion (IR) or sham IR. * p<0.05 maximum vs. rats subjected to sham IR at respective time point.
Figure 2.
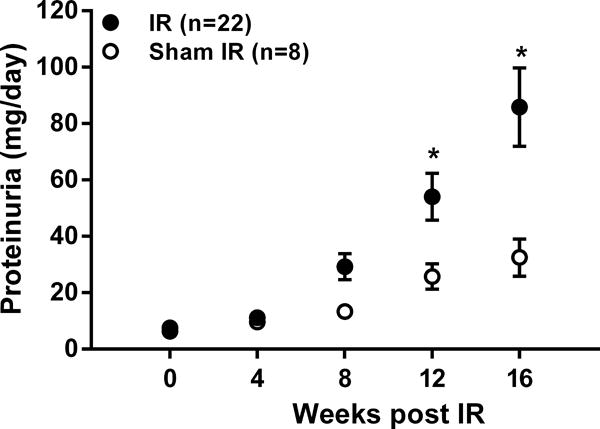
Proteinuria, as determined from 24-hour urine collections, at baseline (time 0) and every 4 weeks for 16 weeks in uninephrectomized rats subjected to 40 min. renal ischemia-reperfusion (IR) or sham IR. * p<0.05 maximum vs. rats subjected to sham IR at respective time point.
At 16 weeks post IR, no significant differences in body weight (585 ± 14 vs. 626 ± 21 grams) or Hct (41 ± 1 vs. 42 ± 1 %) were observed between IR and sham IR groups, respectively. Kidney weight was greater in the IR vs. sham IR group when expressed either as absolute weight (5.3 ± 0.1 vs. 4.0 ± 0.2 grams, P<0.001) or relative to body weight (9.2 ± 0.4 vs. 6.6 ± 0.2 grams/kilogram, P<0.001). Renal injury was significantly greater in the IR vs. sham IR group as evidenced by a ~5-fold greater level of GS and the observance of TIF only in the IR group (Figure 3b). The GS phenotype in all rats consisted of segmental/global GS as opposed to ischemic GS. Vascular injury was not observed in either group. Figure 4a illustrates the quantitative relationships between BP and GS among all rats. In general, while a significant positive correlation between BP and GS was observed, there was a clear BP threshold (~127 mmHg) for the development of substantial GS within the IR group. For example, the level of GS in rats (n=11) whose average 16-week systolic BP was greater than 127 mmHg was 15.8 ± 3.5% as compared to the very modest 3.6 ± 0.8% observed in rats (n=11) whose BP was less than 127 mmHg after IR (P < 0.05). Similar quantitative relationships were noted between BP and TIF (Figure 4b). The TIF score for rats whose 16-week average systolic BP was greater than 127 mmHg was 1.3 ± 0.4 vs. 0.2 ± 0.1 in rats whose 16-week average systolic BP was less than 127 mmHg after IR (P < 0.05). Of note, there was a very strong correlation between GS and TIF within the IR group (Figure 4c) indicating that the progression of CKD following AKI, manifest as high levels of TIF, was only observed in those rats that developed substantial levels of GS (i.e., greater than 10% GS) because GS was very modest at 4 weeks post AKI (see next paragraph). Similar robust and significant relationships were also observed between GS and TIF vs. proteinuria and renal mass at 16 weeks post IR (Figure S2 and S3), consistent with a hemodynamic pathogenesis of renal injury. In contrast, weaker, albeit significant, relationships between SCr 48 hours post IR and GS (r2 = 0.21, P < 0.05) and SCr 48 hours post IR and TIF (r2 = 0.33, P < 0.05) were observed, indicating that the severity of AKI, per se, was not likely a major determinant in the progression of renal disease after IR (Figure S4). Moreover, no clear 48 hour SCr threshold for elevated levels of GS or TIF at 16 weeks post IR was apparent, unlike that observed with the relationships among BP, GS, and TIF.
Figure 3.
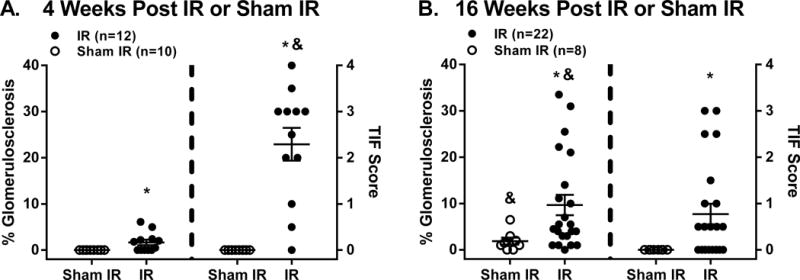
Semi-quantitation of glomerulosclerosis (% of 100 examined glomeruli exhibiting any degree of sclerosis) and tubulointerstitial fibrosis (TIF, 0–4 scale with 0=no TIF, 1=25%, 2=50%, 3=75%, and 4=100% of section exhibiting TIF) at A: 4 weeks and B: 16 weeks post renal ischemia-reperfusion (IR) or sham IR in different groups of rats. Data are mean ± SEM. * p<0.05 maximum vs. sham IR group. & p<0.05 maximum vs. respective score at 4 or 16 weeks post IR.
Figure 4.
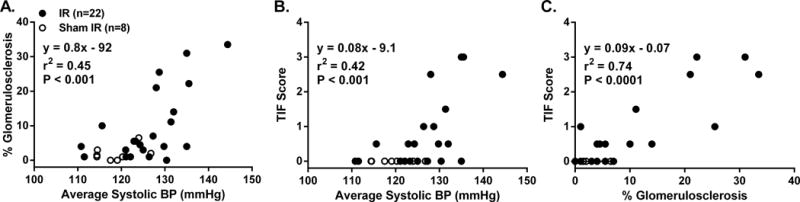
Quantitative relationships between A: % glomerulosclerosis and 16-week average systolic BP, B: tubulointerstitial fibrosis (TIF) and 16-week average systolic BP, and C: TIF and % glomerulosclerosis at 16 weeks post renal ischemia-reperfusion (IR) or sham IR.
Renal injury, hemodynamics and RBF autoregulation 4 weeks post AKI
Of the 17 rats subjected to IR, 5 were euthanized within 3 days following IR for humane reasons. Hemodynamic measurements and injury were successfully obtained in the remaining 12 rats in the IR group. At 48 hours post IR, SCr was significantly elevated in the IR vs. sham IR group (4.1 ± 0.6 vs. 0.6 ± 0.1 mg/dl, respectively). Of note, the 48 hour SCr values in the rats subjected to IR and studied at 4 weeks post IR were similar in the group of rats subjected to IR and studied at 16 weeks post IR, suggesting a comparable severity of AKI (Figure S1). At 4 weeks post IR, no significant differences in body weight were noted between the IR and sham IR groups (460 ± 11 vs. 452 ± 13 grams). Kidney weight was significantly greater in the IR vs. sham IR groups when expressed as absolute weight (3.4 ± 0.2 vs. 2.4 ± 0.1 grams, P < 0.001) or relative to body weight (7.5±0.6 vs. 5.3±0.1 grams/kilogram, P < 0.002). In the sham IR group, no GS or TIF was observed. In contrast, rats subjected to IR exhibited substantial TIF but very modest GS (Figure 3a). Moreover, there was a very strong relationship (r2 = 0.82, P < 0.0001) between SCr 48 hours post IR and the level of TIF at 4 weeks post IR (Figure S5), indicating that the severity of AKI was likely a major determinant of the level of TIF observed during the AKI – CKD transition.
Of note, the level of GS was significantly greater at 16 vs. 4 weeks post IR whereas the level of TIF was significantly greater at 4 vs. 16 weeks post IR (Figure 3b). Thus, GS increased whereas TIF decreased over time. Indeed, the evidence that the majority of kidneys (15 of 22) examined at 16 weeks post IR had TIF scores of 0–0.5 on a scale of 0–4, indicating minimal to very modest levels of TIF, whereas the majority of kidneys (9 of 12) examined at 4 weeks post IR had TIF scores greater than 2, indicating the presence of TIF in 50% or more of the kidney, despite a similar severity of AKI is consistent with the notion that TIF, per se, is a contractile process that tends to shrink over time11,23,24,29.
Table 1 presents the results of the hemodynamic assessment at 4 weeks post IR (n=12) or sham IR (n=10) in conscious rats. No significant differences in MAP were observed. However, a significantly elevated RVR (P < 0.05) and consequently reduced RBF (P < 0.005) was observed in rats subjected to IR vs. sham IR. Rats subjected to IR also had a lower RPF (P < 0.05), despite their substantially lower (P < 0.005) Hct. levels as compared to the sham IR group. Although GFR was lower in the IR vs. sham IR group, the difference was not significant (P = 0.24). Similarly, FF was not significantly different between groups (P = 0.30). A summary of the conventional step RBF autoregulatory assessment is presented in Table 1 and Figure 5. Autoregulatory assessments were made in 7 of the 10 chronically instrumented rats in the sham IR group. In the IR group, autoregulatory capacity was not assessed in 1 chronically instrumented rat due to technical issues. In both groups, RBF did not significantly change across MAP steps of ~ 100, 120 and 140 mmHg, indicating that autoregulatory capacity, on average, was not substantially impaired in either group. Indeed, the autoregulatory indices were similar between groups (Table 1); however, a wide range was seen in both the IR (0.07 – 0.89) and sham IR (0.03 – 0.80) groups. Given that the severity of and recovery from IR within individual groups is highly variable, we examined the relationship between autoregulatory index and GFR in rats subjected to IR. As shown in Figure 5, those rats with the lowest GFR values, and likely greatest loses of functional renal mass following AKI, had the greatest impairment in RBF autoregulation.
Table 1.
Assessment of renal hemodynamics in conscious rats at 4 weeks post IR or sham IR.
| Group | MAP (mmHg) |
RVR (mmHg/ml/min) |
RBF (ml/min) |
Hct. (%) |
RPF (ml/min) |
GFR (ml/min) |
FF (%) |
AR index |
|---|---|---|---|---|---|---|---|---|
|
IR (n=12) |
116±3 | 9.4±1* | 13.4±1* | 0.38±0.01* | 8.4±1* | 2.2±0.2 | 27±2 | 0.45±0.1 |
|
Sham IR (n=10) |
116±3 | 6.5±1 | 18.7±1 | 0.45±0.01 | 10.4±1 | 2.5±0.2 | 25±1 | 0.35±0.1 |
IR = ischemia-reperfusion, MAP = mean arterial pressure, RBF = renal blood flow, Hct. = hematocrit, RPF = renal plasma flow, GFR = glomerular filtration rate, FF = filtration fraction, AR = autoregulatory.
P<0.05 vs. Sham IR.
Figure 5.
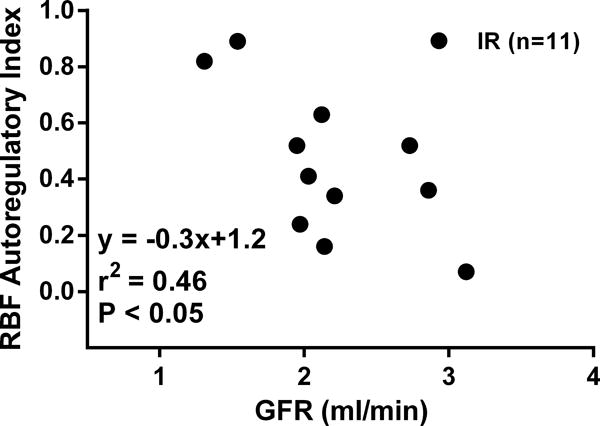
Relationship between renal blood flow (RBF) autoregulatory index (0–1 with 0 indicating perfect and 1 indicating completely impaired autoregulation) and glomerular filtration rate (GFR) at 4 weeks post renal ischemia-reperfusion (IR) or sham IR in uninephrectomized rats. GFR was determined in conscious chronically instrumented rats the day before RBF autoregulatory indices were assessed under anesthesia.
DISCUSSION
A substantial amount of evidence has accumulated over the last decade suggesting that AKI accelerates the progression of CKD to ESRD6. Yet, much uncertainty exists regarding the relative contribution of the two predominant mechanisms of renal injury, self-perpetuating TIF vs. hemodynamically-mediated GS-induced TIF, widely believed to primarily contribute to the progression of CKD following AKI. Such uncertainty can be partly explained by the fact that these pathways of renal injury have not been rigorously assessed and compared within the same study, until now. The novel findings of the present study are that 1) on average, the level of TIF significantly decreases over time following AKI, 2) significant progression of CKD after AKI, manifest by extensive TIF 16 weeks post IR, was only observed in those rats with prominent levels of GS, proteinuria, and kidney weight, 3) elevated levels of TIF and GS were only observed in those rats whose average 16-week systolic BP was greater than 127 mmHg, and 4) at 4 weeks following AKI, those rats who exhibited the most impaired functional recovery also had the greatest impairment in RBF autoregulation. Collectively, these data strongly support the concept that even very modest levels of BP following AKI in the setting of preexisting renal mass reduction may nevertheless substantially contribute to the subsequent progression of CKD. In contrast, these data do not support a major role of self-perpetuating TIF in the progression of CKD following AKI in such settings.
TIF is Significantly Lower at 16 vs. 4 weeks Post AKI
At 4 weeks post AKI, renal injury primarily consisted of TIF with only minimal levels of glomerular injury. Numerous physiologic and cellular changes such as reduced RBF, capillary rarefaction, hypoxia, cell senescence, maladaptive repair, inflammation, etc., are not only thought to contribute to the TIF observed at such early stages following AKI (i.e., the AKI – CKD transition), but also have been proposed to persist and contribute to the subsequent progression of CKD via a self-perpetuating TIF pathway6,12–17. That is AKI-induced TIF should beget more TIF15,16. However, a novel finding in the present study was that the level of TIF was substantially lower in rats studied at 16 vs. 4 weeks post IR. Although this may seem at odds with current views, our data is nevertheless consistent with previous studies that evaluated TIF at similar time points following IR in rats with a single kidney and did not observe a worsening of TIF26,27. In contrast, Forbes et al.30 did observe an increase in the fractional area of collagen III staining at 2 and 6 months as compared to 4 weeks post IR. Surprisingly, no histological or morphological changes in glomeruli were observed in their study, contrary to the majority of previous studies that have reported significant levels of GS over similar extended periods following IR injury in rats with a single kidney13,20,25. Reasons for the differences between the Forbes et al. study and ours is unclear, but may be due to the use of female rats, smaller group sizes and different quantitative methods of histological assessments in their study.
Although TIF was assessed in different groups of rats at 4 and 16 week time points in the present study, there is no reason to suggest that the severity of AKI was greater in the group studied at 4 weeks. Both groups exhibited similar increases in 48 hour SCr. Moreover, the levels of SCr 48 hours post IR and TIF at 4 weeks post IR is similar to what we have recently observed9, which suggests that this model of renal IR injury results in a reproducible severity of AKI in our laboratory. The demonstration that TIF as a result of the AKI – CKD transition at 4 weeks post IR was very strongly correlated with the severity of AKI, but the TIF observed at 16 weeks post IR during CKD progression was only modestly correlated with the severity of AKI suggests that different pathophysiological mechanisms are contributing to TIF production at these two time points.
Additional evidence supporting a lack of a major role of self-perpetuating TIF in the progression of CKD following AKI in the present study was the pattern of glomerular injury. If self-perpetuating TIF were mediating such progression of CKD, we would have expected that glomerular injury at 16 weeks post AKI would primarily consist of shrunken tufts with significant periglomerular fibrosis as the TIF expanded into the renal cortex. In contrast, the only glomerular lesion observed was segmental or global GS and such lesions were mainly seen in larger glomeruli and hypertrophied kidneys. This phenotype of glomerular injury is consistent with an intravascular origin and is commonly seen in hyperperfused and hypertrophied glomeruli exposed to modestly elevated levels of glomerular capillary pressures over extended time periods31.
The significantly lower level of TIF at 16 vs. 4 weeks post AKI is nevertheless consistent with the notion that a fundamental feature of TIF is that it shrinks. Kriz and colleagues have postulated, that TIF, per se, is a protective mechanism that serves to anatomically confine lethally injured nephrons to maintain renal parenchymal architecture and preserve normal renal structure and function in uninjured nephrons23,24. Venkatachalam and colleagues have also suggested that AKI-induced TIF should shrink over time as long as other mechanisms which promote the development of TIF are not initiated after AKI11. Finally, our data are in agreement with those recently reported in kidney transplant recipients in which fibrosis and transcript expression of disease processes were longitudinally evaluated for over a year29. They reported that kidneys exhibiting extensive fibrosis over extended time periods after receiving a kidney transplant were not dependent on an autonomous fibrosis response, but rather on new disease processes that developed after AKI and were independent of the acute transplant-associated fibrotic response.
Potential Role of Modest Elevations in BP in the Progression of CKD Following AKI
A novel finding in the present study was the modest, yet significant, increase in BP following AKI in uninephrectomized rats fed a normal 1% NaCl diet over a 16 week period. Previous studies have demonstrated the significance of nephron loss32 as well as TIF21,33, both consequences of AKI in the setting of preexisting CKD, in increasing the susceptibility to salt-sensitive hypertension, in part via an impairment in the pressure natriuresis mechanism34. Indeed, rats previously subjected to bilateral IR exhibit significant increases in BP when switched from a 1% to 4% NaCl diet21,34. However, in the few studies that have assessed BP following IR in uninephrectomized rats fed a normal NaCl diet, significant increase in BP were not detected21,26,27. However, the small sample sizes, limited follow-up time and/or the use of inadequate tail-cuff methods35 to accurately detect modest changes in BP in these previous studies provide likely explanations for the different results. Of note, our study using gold-standard techniques to chronically assess BP following AKI are in agreement with a recent clinical study demonstrating that AKI predisposes to the development of hypertension36.
Importantly, our study suggests that modest levels of BP may nevertheless be playing a major role in the progression of CKD following AKI in rats with preexisting 50% renal mass reduction. The quantitative relationships between BP and renal injury revealed a considerable amount of variability in the extent of GS and TIF; nevertheless, such analysis led to the important observation that substantial CKD progression, manifest by robust levels of TIF, was only observed in those rats whose who developed significant levels of GS. These data suggest that development of GS following AKI may be essential to the subsequent progression of CKD. Indeed, the phenotype of glomerular injury (i.e., focal segmental or global GS), the strong correlations between BP, GS, TIF, proteinuria and kidney weight, and the very clear systolic BP threshold for the development of GS and TIF (~ 127 mmHg) is consistent with a hemodynamically-mediated pathogenesis of CKD progression22. In contrast to the very modest susceptibility to hypertension-induced renal injury in the majority of patients and experimental models with intact renal mass22,31, the risk is substantially greater in CKD states, such as that observed following AKI in the presence of preexisting renal mass reduction. We have demonstrated that such differences in susceptibility can be explained by differences in RBF autoregulation22,31. Impairment in RBF autoregulation in CKD states allows a greater degree of systemic BP to be transmitted to the renal microvasculature, including the glomerular capillaries. The strong correlation between GS and proteinuria, a robust index of glomerular capillary hypertension37,38, observed in the present study is supportive of these concepts. Such differences in the renal transmission of systemic BP to the glomerular capillaries may also explain, in part, the greater rate of CKD progression following IR in rats with a single kidney as compared to those with intact kidneys13,25. The significant negative correlation between autoregulatory index and GFR at 4 weeks post IR, before the development of significant GS, is also supportive of the concept that further reductions of renal mass following AKI in rats with preexisting CKD increase the susceptibility to hypertensive renal injury. The general importance of impaired RBF autoregulation associated with reductions in renal mass in increasing the susceptibility to CKD progression has also recently been described in clinical populations39. Thus, our study highlights the potential importance of even very modest levels of BP, what would be considered normotensive by current guidelines40, to nevertheless substantially contribute to the progression renal disease following AKI in the presence of preexisting CKD.
Potential Role of Hemodynamic Factors in the Contribution to the AKI – CKD Transition
Despite no significant differences in GFR between rats subjected to IR vs. sham IR at 4 weeks post AKI, RBF was significantly lower in the IR group. These data may have important implications with respect to the role of hypoxia in contributing to the extent of acute nephron loss as a direct result of the AKI event or recovery from it (i.e., the AKI – CKD transition). As GFR is a major determinant of renal oxygen consumption, chronic reductions in RBF in the IR group suggest that oxygen demand may be greater than oxygen supply. The mechanisms that mediate such potential mismatches in oxygen supply and demand are likely related to microvascular rarefaction and impairments in renal metabolism following IR11,13,41,42. It has been suggested that improving oxygen supply or the impairments in renal metabolism could mitigate the loss of nephrons following IR by improving recovery of sublethally injured cells, but the cellular pathways have not been identified11,13,42.
PERSPECTIVES
We conclude that self-perpetuating TIF is not playing a major role in CKD progression following AKI in uninephrectomized rats. In contrast, our data suggest that even very modest elevations in BP following AKI in such settings may nevertheless be substantially contributing to CKD progression via hemodynamically-mediated GS-induced TIF. Of particular relevance to our study, early nephrology follow-up following severe AKI has been shown to be associated with a significant reduction in mortality over a two year period43. It is possible that such early follow-up is associated with a more aggressive lowering of BP and greater attention to preventing increases in proteinuria, which is indicative of glomerular hypertension, following AKI. Similar striking benefits of aggressive BP lowering on reducing cardiovascular morbidity and mortality have recently been reported in the Systolic Blood Pressure Intervention Trial (SPRINT)44. Future studies using the experimental paradigm described in this study will examine whether aggressive (i.e., systolic BP < 120) vs. conventional (i.e., systolic BP < 140 mmHg) antihypertensive therapy is required to prevent CKD progression following AKI in preexisting CKD states. Moreover, such studies will also examine whether specific antihypertensive classes (e.g., renin-angiotensin system inhibitors) confer greater protection against CKD progression following AKI via BP–independent pathways. It is unclear at this time whether the progression of CKD following AKI in various preexisting CKD states is largely dependent on BP lowering, as observed in the majority of hypertensive CKD models22, or if it is associated with a greater BP–independent component.
Supplementary Material
Novelty and Significance.
What is New?
The quantitative relationships between radiotelemetrically-measured BP and renal injury over extended time periods following AKI in uninephrectomized rats and suggest that even very modest elevations in BP may nevertheless be substantially contributing to progression of renal disease via GS-induced TIF production.
Self-perpetuating TIF is not likely playing a major role in the progression of renal disease following AKI in uninephrectomized rats.
What is Relevant?
These data indicate that intensive antihypertensive therapy following AKI in patients with preexisting CKD may offer substantial benefits in reducing the subsequent progression of renal disease.
Summary.
This study examined the potential contribution of self-perpetuating TIF vs. hemodynamically-mediated GS-induced TIF in the progression of CKD following AKI in uninephrectomized rats. While TIF was significantly lower at 16 vs. 4 weeks post AKI, GS was significant greater and a very modest BP threshold (average systolic BP ~ 127 mmHg) for the development of such GS was observed. These data indicate that modest BP elevations, as opposed to self-perpetuating TIF, may be largely responsible for the development of de novo TIF during the progression of CKD following AKI in rats with preexisting 50% renal mass reduction.
Acknowledgments
The authors acknowledge Theresa Herbst for technical support and Martha Prado for secretarial support. Dr. Polichnowski also wishes to acknowledge Dr.’s Anil Bidani and Karen Griffin for their careful review of the manuscript and helpful comments.
SOURCE OF FUNDING
This work was supported by a Career Development Award 1K2BX001285 from the Office of Research and Development of the Department of Veterans Affairs and a Young Investigator Award from the National Kidney Foundation of IL to A.J. Polichnowski.
Footnotes
DISCLOSURES
None
References
- 1.Hsu CY, McCulloch CE, Fan D, Ordonez JD, Chertow GM, Go AS. Community-based incidence of acute renal failure. Kidney Int. 2007;72:208–212. doi: 10.1038/sj.ki.5002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nature Reviews Nephrology. 2014;10:193–207. doi: 10.1038/nrneph.2013.282. [DOI] [PubMed] [Google Scholar]
- 3.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 4.Bagshaw SM. Short- and long-term survival after acute kidney injury. Nephrology, Dialysis, Transplantation. 2008;23:2126–2128. doi: 10.1093/ndt/gfn300. [DOI] [PubMed] [Google Scholar]
- 5.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011;79:1361–1369. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of esrd among elderly. J Am Soc Nephrol. 2009;20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharfuddin AA, Weisbord SD, Palevsky PM, Molitoris BA. Acute kidney injury. In: Shorecki K, Chertow GM, Marsden PA, Taal MW, Yu ASL, editors. Brenner & Rector’s The Kidney. Philadelphia, PA: Elseiver, Inc; 2016. pp. 958–1011. [Google Scholar]
- 9.Polichnowski AJ, Lan R, Geng H, Griffin KA, Venkatachalam MA, Bidani AK. Severe renal mass reduction impairs recovery and promotes fibrosis after aki. J Am Soc Nephrol. 2014;25:1496–1507. doi: 10.1681/ASN.2013040359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: A springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298:F1078–1094. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, aki-ckd transition, and kidney disease progression. J Am Soc Nephrol. 2015;26:1765–1776. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:F887–899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- 13.Basile DP, Donohoe DL, Roethe K, Mattson DL. Chronic renal hypoxia after acute ischemic injury: Effects of l-arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol. 2003;284:F338–348. doi: 10.1152/ajprenal.00169.2002. [DOI] [PubMed] [Google Scholar]
- 14.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after aki leading to accelerated kidney ageing and ckd. Nature Reviews Nephrology. 2015;11:264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int Suppl. 2000;75:S22–26. [PubMed] [Google Scholar]
- 16.Nangaku M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 17.Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 18.Zager RA. ‘Biologic memory’ in response to acute kidney injury: Cytoresistance, toll-like receptor hyper-responsiveness and the onset of progressive renal disease. Nephrology, Dialysis, Transplantation. 2013;28:1985–1993. doi: 10.1093/ndt/gft101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20:1–17. doi: 10.1016/s0272-6386(12)80312-x. [DOI] [PubMed] [Google Scholar]
- 20.Azuma H, Nadeau K, Takada M, Mackenzie HS, Tilney NL. Cellular and molecular predictors of chronic renal dysfunction after initial ischemia/reperfusion injury of a single kidney. Transplantation. 1997;64:190–197. doi: 10.1097/00007890-199707270-00002. [DOI] [PubMed] [Google Scholar]
- 21.Spurgeon-Pechman KR, Donohoe DL, Mattson DL, Lund H, James L, Basile DP. Recovery from acute renal failure predisposes hypertension and secondary renal disease in response to elevated sodium. Am J Physiol Renal Physiol. 2007;293:F269–278. doi: 10.1152/ajprenal.00279.2006. [DOI] [PubMed] [Google Scholar]
- 22.Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and ckd progression. Curr Opin Nephrol Hypertens. 2013;22:1–9. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaissling B, Lehir M, Kriz W. Renal epithelial injury and fibrosis. Biochimica Et Biophysica Acta. 2013;1832:931–939. doi: 10.1016/j.bbadis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 25.Cruzado JM, Torras J, Riera M, Herrero I, Hueso M, Espinosa L, Condom E, Lloberas N, Bover J, Alsina J, Grinyo JM. Influence of nephron mass in development of chronic renal failure after prolonged warm renal ischemia. Am J Physiol Renal Physiol. 2000;279:F259–269. doi: 10.1152/ajprenal.2000.279.2.F259. [DOI] [PubMed] [Google Scholar]
- 26.Pagtalunan ME, Olson JL, Meyer TW. Contribution of angiotensin ii to late renal injury after acute ischemia. J Am Soc Nephrol. 2000;11:1278–1286. doi: 10.1681/ASN.V1171278. [DOI] [PubMed] [Google Scholar]
- 27.Pagtalunan ME, Olson JL, Tilney NL, Meyer TW. Late consequences of acute ischemic injury to a solitary kidney. J Am Soc Nephrol. 1999;10:366–373. doi: 10.1681/ASN.V102366. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain RM, Shirley DG. Time course of the renal functional response to partial nephrectomy: Measurements in conscious rats. Exp Physiol. 2007;92:251–262. doi: 10.1113/expphysiol.2006.034751. [DOI] [PubMed] [Google Scholar]
- 29.Venner JM, Famulski KS, Reeve J, Chang J, Halloran PF. Relationships among injury, fibrosis, and time in human kidney transplants. JCI Insight. 2016;1:e85323. doi: 10.1172/jci.insight.85323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forbes JM, Hewitson TD, Becker GJ, Jones CL. Ischemic acute renal failure: Long-term histology of cell and matrix changes in the rat. Kidney Int. 2000;57:2375–2385. doi: 10.1046/j.1523-1755.2000.00097.x. [DOI] [PubMed] [Google Scholar]
- 31.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: Implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 32.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 33.Franco M, Sanchez-Lozada LG, Bautista R, Johnson RJ, Rodriguez-Iturbe B. Pathophysiology of salt-sensitive hypertension: A new scope of an old problem. Blood Purification. 2008;26:45–48. doi: 10.1159/000110563. [DOI] [PubMed] [Google Scholar]
- 34.Pechman KR, De Miguel C, Lund H, Leonard EC, Basile DP, Mattson DL. Recovery from renal ischemia-reperfusion injury is associated with altered renal hemodynamics, blunted pressure natriuresis, and sodium-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1358–1363. doi: 10.1152/ajpregu.91022.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurement in humans and experimental animals. Part 2: Blood pressure measurement in experimental animals. Hypertension. 2005;45:299–310. doi: 10.1161/01.HYP.0000150857.39919.cb. [DOI] [PubMed] [Google Scholar]
- 36.Hsu CY, Hsu RK, Yang J, Ordonez JD, Zheng S, Go AS. Elevated bp after aki. J Am Soc Nephrol. 2016;27:914–923. doi: 10.1681/ASN.2014111114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffin KA, Polichnowski A, Litbarg N, Picken M, Venkatachalam MA, Bidani AK. Critical blood pressure threshold dependence of hypertensive injury and repair in a malignant nephrosclerosis model. Hypertension. 2014;64:801–807. doi: 10.1161/HYPERTENSIONAHA.114.03609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshioka T, Shiraga H, Yoshida Y, Fogo A, Glick AD, Deen WM, Hoyer JR, Ichikawa I. “Intact nephrons” as the primary origin of proteinuria in chronic renal disease. Study in the rat model of subtotal nephrectomy. J Clin Invest. 1988;82:1614–1623. doi: 10.1172/JCI113773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fotheringham J, Odudu A, McKane W, Ellam T. Modification of the relationship between blood pressure and renal albumin permeability by impaired excretory function and diabetes. Hypertension. 2015;65:510–516. doi: 10.1161/HYPERTENSIONAHA.114.04656. [DOI] [PubMed] [Google Scholar]
- 40.Taler SJ, Agarwal R, Bakris GL, Flynn JT, Nilsson PM, Rahman M, Sanders PW, Textor SC, Weir MR, Townsend RR. Kdoqi us commentary on the 2012 kdigo clinical practice guideline for management of blood pressure in ckd. Am J Kidney Dis. 2013;62:201–213. doi: 10.1053/j.ajkd.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basile DP. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007;72:151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka S, Tanaka T, Nangaku M. Hypoxia as a key player in the aki-to-ckd transition. Am J Physiol Renal Physiol. 2014;307:F1187–1195. doi: 10.1152/ajprenal.00425.2014. [DOI] [PubMed] [Google Scholar]
- 43.Harel Z, Wald R, Bargman JM, Mamdani M, Etchells E, Garg AX, Ray JG, Luo J, Li P, Quinn RR, Forster A, Perl J, Bell CM. Nephrologist follow-up improves all-cause mortality of severe acute kidney injury survivors. Kidney Int. 2013;83:901–908. doi: 10.1038/ki.2012.451. [DOI] [PubMed] [Google Scholar]
- 44.Wright JT, Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–2116. doi: 10.1056/NEJMoa1511939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.