

Abstract
Advances in neonatal care have allowed premature infants to survive at earlier gestational ages, but they are often afflicted with neurological delays or deficits. Maternal inflammation has been identified as a major risk factor for premature birth and once born, infants often require supplemental oxygen for survival. Nurr1 (NR4A2) is an orphan nuclear receptor with no known binding site and is essential for the growth of midbrain dopamine neurons. Others have reported that Nurr1 can act as an anti-inflammatory transcription factor in microglia and astrocytes and respond lipopolysaccharide (LPS). We have previously reported decreased numbers of oligodendrocytes and increased numbers of microglia in the mice exposed to both maternal inflammation and neonatal hyperoxia in the perinatal period. These studies tested the hypothesis that the combined exposures to inflammation and hyperoxia would increase Nurr1 expression in microglia in our mouse model and in an immortalized microglia cell line, BV2 cells. Our data indicate that Nurr1 protein expression is increased at postnatal day 0 and postnatal day 28 in whole-brain homogenates from mice exposed to LPS and hyperoxia. Alternatively, Nurr1 message is decreased at postnatal day 60 in isolated microglia, indicating that the increases in whole-brain homogenates may be due to other cell types. In BV2 cells, Nurr1 message in increased by exposure to hyperoxia, but this increase is attenuated in cells exposed to both LPS and hyperoxia. Although Nurr1 regulation is not straightforward, these data indicate that Nurr1 expression is increased in whole-brain homogenates in response to inflammation, but is decreased in isolated primary microglia and BV2 cells in response to similar inflammation. Our data support the hypothesis that Nurr1 expression may play a significant role in regulating inflammation in the brain and understanding the complex regulation of Nurr1 could lead to new therapeutic strategies.
Keywords: BV2 cells, inflammation, lipopolysaccharide, microglia, Nurr1
Introduction
Advances in neonatal intensive care have increased survival rates of premature infants at increasingly earlier gestational ages. However, these vulnerable infants remain at significant risk of the morbidities associated with preterm birth. Neurodevelopmental delays and deficits that can vary widely in type and severity are of principal concern to those who care for these infants and children. These can include cerebral palsy, sensory deficits, cognitive impairments, and behavioral and social dysfunction and their frequency is inversely correlated to the gestational age at birth 1. Such impairments can affect quality of life throughout childhood and potentially throughout an entire life. Maternal inflammation has been identified as a major risk factor for premature birth 2. After birth, premature infants often require supplemental oxygen for survival, and this exposure can lead to additional inflammatory responses. Both of these sources of inflammation, independently and in combination, have the potential to contribute to neurodevelopmental impairments.
Nurr1 (NR4A2) is an orphan nuclear receptor with no known ligand 3, but is described as a transcription factor and is associated with specific physiological roles. The role of Nurr1 has been characterized most thoroughly in midbrain dopaminergic neurons, where it is essential for the growth and maintenance of this neuronal population. Moreover, a Nurr1 knockout is embryonic lethal because of lack of movement in the offspring 4. In human kidney cells exposed to an oxidative stimulus, Nurr1 is exported from the nucleus into the cytoplasm, indicating a potential role for Nurr1 in response to oxidative stress 5. Other studies have found that Nurr1 gene expression is induced by lipopolysaccharide (LPS) in macrophages as well as microglia 6–8. Most relevant to our current studies, Saijo et al. 8 reported that Nurr1 may play an anti-inflammatory role in microglia and astrocytes exposed to LPS by acting as a transrepressor of the NFκB cascade and attenuating the release of inflammatory mediators by these cells. Their data indicated that the release of inflammatory mediators from microglia was significantly increased when Nurr1 was genetically deleted.
Previous studies in our lab have investigated the combined effects of maternal LPS and neonatal hyperoxia exposures in a mouse model, a common clinical scenario for very preterm infants. We have reported pathological changes in the lung, the heart, and the brain of the offspring 9–13. Specifically, we have observed decreased numbers of early cortical oligodendrocytes and increased numbers of microglia in the cortex and hippocampus of mice exposed to both maternal inflammation and neonatal hyperoxia 9. Although inflammation 14,15 and hyperoxia exposure 16–18 have been shown to be particularly harmful to the developing brain, we speculate that the combined exposures will have a more severe effect and potentially alter pathways not affected by either insult individually.
Given the increased numbers of microglia in our model and reports of a potential anti-inflammatory role for Nurr1 in these cells, the aim of our studies was to test the hypothesis that the combined exposures of LPS and hyperoxia would alter Nurr1 expression in microglia in our mouse model and in an immortalized microglia cell line, BV2 cells.
Materials and methods
Animal model
All studies were carried out in strict accordance with animal study protocols that were approved by the Institutional Animal Care and Use Committee at Nationwide Children’s Hospital Research Institute (Columbus, Ohio, USA). All studies were carried out within the guidelines set forth by the ‘Guide for Care and Use of Laboratory Animals’ and within an AAALAC-accredited facility. The animal model utilized in this study has been described extensively in our previous publications 9,10. Briefly, pregnant C3H/HeN mice were injected with 80 µg/kg LPS [EMD Millipore (CalBiochem), Billerica, Massachusetts, USA] or saline intraperitoneally on embryonic day 16. Once the pups are born, they are exposed to room air (RA) or 85% oxygen (O2) for 14 days and then placed back in RA. Mice were anesthetized with an intraperitoneal injection of ketamine and xylazine (150 and 15 mg/kg, respectively). This effectively creates four exposure groups: saline/RA, saline/O2, LPS/RA, and LPS/O2. Brains were collected from pups in each group on postnatal day 0 (PN0), postnatal day 28 (PN28), and postnatal day (PN60).
Microglial enrichment
Microglia were isolated from whole-brain homogenates as described previously by Sawicki et al. 19 and Norden et al. 20. Brains were homogenized and the cell pellets were resuspended in 70% Percoll (17-0891-01; GE-Healthcare, Little Chalfont, UK). Decreasing concentrations of Percoll (50, 35, 0%) were overlaid and the entire sample was centrifuged at 2000g for 20 min. Enriched microglia were collected between the 70 and 50% Percoll layers. Previous studies have shown that this method yields more than 90% CD11b+ cells 21,22.
BV2 cells
BV2 cells (mouse microglial cell line) and related protocols were generously provided by Dr Jonathan Godbout. These cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (S1115 0H; Atlanta Biologicals, Flowery Branch, Georgia, USA) and streptomycin. BV2 cells were plated at a density of 2×106 cells/plate. After 24 h, cells were incubated in 50 ng/ml LPS or saline for 24 h. A portion of the cells was then harvested and the remaining cells were washed 3× with PBS, plated with new media, and incubated in 85% O2 or RA for 48 h and harvested.
Cell counts
Media were collected from BV2 cell plates, and adherent BV2 cells were collected through trypsinization. Both media and cells were centrifuged at 2500 rpm for 5 min at 4°C. The supernatant was removed and the cell pellet was resuspended in 1 ml of media. A small aliquot of the media was diluted with PBS and mixed with trypan blue exclusion dye. Viable and nonviable cells were counted on a hemocytometer.
Real time quantitative PCR
RNA was isolated from brain tissues or cells using TRIzol (Invitrogen, Carlsbad, California, USA) and the RNeasy kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols. RNA (2 μg) was reverse transcribed per instructions using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham Massachusetts, USA). PCR with Maxima SYBR green master mix (Thermo Scientific) was performed on an Eppendorf Realplex Master Cycler using custom DNA primers (Integrated DNA Technologies, Coralville, Iowa, USA).
Western blot analysis
Whole brains were removed from pups on PN0 and PN28 and flash frozen and stored at −80°C until analysis, at which point the tissue was homogenized in lysis buffer. The brain homogenates were centrifuged at 14 000 rpm for 15 min. Cells were suspended with 500 μl lysis buffer (20 mM HEPES, pH 7.4, 50 mM β-glycerophosphate, 2 mM EGTA, 1 mM dithiothreitol, 10 mM NaF, 1 mM NaVO4, 10% glycerol, 1% triton), mixed thoroughly, and centrifuged at 14 000 rpm for 15 min. Supernatants were saved for protein analysis. Protein concentrations were determined using the Bradford assay. Samples were loaded onto SDS-page gels (30 µg for cells, 80 µg for tissue), separated by gel electrophoresis, and transferred onto nitrocellulose membranes. Membranes were probed with rabbit polyclonal anti-Nurr1 (SC-991; Santa Cruz Biotechnology, Santa Cruz, California, USA) at 1–500 concentration. The Nurr1 primary antibody was then detected using a secondary goat antirabbit (170-6515; Bio-Rad Laboratories, Hercules, California, USA) at 1 : 12 000 and developed using enhanced chemiluminescene. The band density was normalized to β-actin protein (ab62676; Abcam, Cambridge, UK).
Immunofluorescence
Coverslips were placed on the bottom of six-well plates and cells were plated as described earlier. After the exposures, the media were removed and the plates were washed 3× with PBS. Cells were fixed with 4% paraformaldehyde in PBS (MP Bio 199983) for 10 min. Cell plates were kept in PBS at 4°C until staining. Cells were permeabilized with ice-cold methanol for 2 min and blocked for 1 h with 10% rabbit serum and subsequently incubated in rabbit anti-Nurr1 (1 : 100) (SC 991; Santa Cruz Biotechnology) at 4°C overnight. After washing, the cells were incubated in AlexaFluor 488 donkey anti-goat for 1 h (A21206; Life Technologies, Carlsbad, California, USA). Coverslips were mounted onto slides with Vectashield containing DAPI (H-1500; Vector Laboratories Inc., Burlingame, California, USA).
Statistics
Statistical analyses were carried out by two-way analysis of variance or Student’s T-test. All data are presented as means±SEM. All analyses were carried out using GraphPad PRISM 5 (Graphpad Software Inc., La Jolla, California, USA).
Results
Nurr1 protein expression in whole-brain homogenates is increased by inflammation
Nurr1 protein expression was measured in whole-brain homogenates from mice exposed to maternal LPS (PN0) or maternal LPS and neonatal hyperoxia (PN28). Western blot analyses indicated that maternal LPS exposure alone increased Nurr1 expression (Fig. 1a). At PN28, after RA or hyperoxia exposure and recovery, the combined LPS and oxygen exposed group also showed a relative increase in Nurr1 protein expression (Fig. 1b). These data suggest that Nurr1 protein expression in the brain is increased in response to inflammatory stress.
Fig. 1.
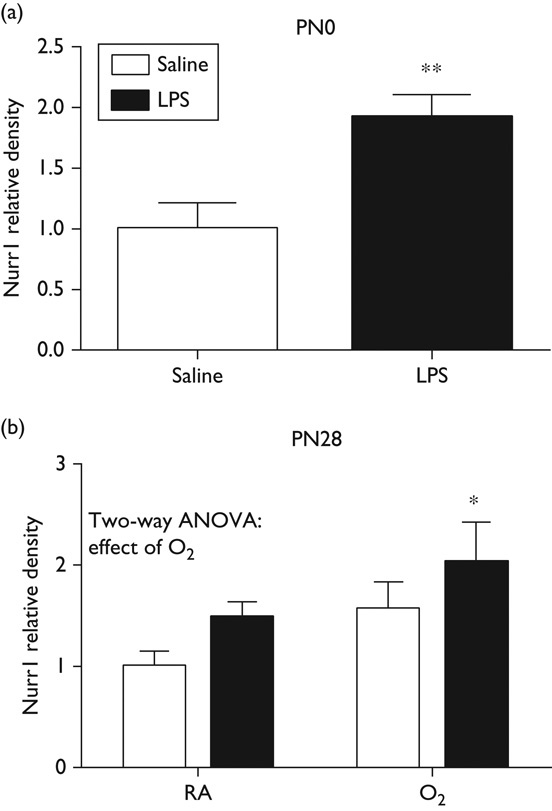
Nurr1 protein expression is increased in whole brain. Whole-brain homogenates were prepared from pups exposed to maternal LPS and/or neonatal hyperoxia at (a) PN day 0 and (b) PN day 28. Protein levels of Nurr1 were measured by Western blot and quantified by densitometry. Data were analyzed by two-way ANOVA, with Tukey’s post-hoc (*P<0.05, **P<0.01, n=5–8), and are presented as mean±SD. ANOVA, analysis of variance; LPS, lipopolysaccharide; PN, postnatal.
Nurr1 expression is suppressed in isolated microglia from mice exposed to maternal lipopolysaccharide and neonatal hyperoxia
Microglia are able to respond to inflammation and often changes in these cells persist long after the initial insult. To determine whether the increases in Nurr1 expression were because of microglia changes and whether these changes persisted, primary microglia were isolated from whole brains taken from mice exposed to maternal LPS and neonatal hyperoxia at PN60. Expressions of IL-1β, iNOS, TGFβ, and Nurr1 were quantified by real time-PCR (RT-PCR) analyses. No differences were observed in IL-1β, iNOS, or TGFβ expression at this late time point. In contrast to the protein measurements at earlier time points (Fig. 1), Nurr1 message in microglia was suppressed in mice exposed to LPS/O2 (Fig. 2).
Fig. 2.

Nurr1 and cytokine levels in microglia isolated from mice exposed to LPS and hyperoxia. Enriched microglia were isolated from mice exposed previously to maternal LPS and neonatal hyperoxia at PN60 as described in the Materials and methods section. RNA was isolated and cytokine expression was measured by RT-qPCR. Data were analyzed using a t-test (*P<0.05, n=4) and are presented as mean±SD. LPS, lipopolysaccharide; RA, room air; RT-qPCR, quantitative reverse transcription PCR.
Microglial BV2 cell model
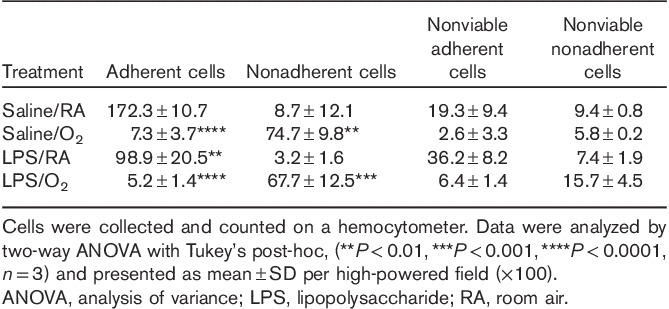
To further investigate the changes in Nurr1 expression, we chose to use a microglial cell line (BV2). Doses of LPS or time of hyperoxia exposure were established in BV2 cells to mimic the mouse model in vivo. BV2 cells were exposed to increasing doses of LPS (5–100 ng/ml) and increasing durations of hyperoxia (0–72 h). Media were collected and cell injury was measured by lactate dehydrogenase (LDH) release. LDH release was increased two-fold at 100 ng/ml, indicating cell death. Similarly, LDH release was increased two-fold at 72 h of hyperoxia exposure. We chose the highest dose that would not result in overt cell death, 50 ng/ml of LPS, followed by 48 h of hyperoxia for the rest of the experiments (data not shown). A substantial number of BV2 cells are nonadherent after oxygen exposure; consequently, adherent and nonadherent cells were collected separately using both trypsinization and centrifugation. The LPS/O2-exposed cells had significantly more nonadherent, dead cells compared with saline/RA-exposed cells; however, hyperoxia exposure alone was associated with considerable loss of adherence (Table 1).
Table 1.
Adherent and nonadherent BV2 cell counts after treatment with lipopolysaccharide and hyperoxia
Lipopolysaccharide and hyperoxia cause increases in cytokine expression in BV2 cells
Cytokine levels were measured by RT-PCR in BV2 cells (nonadherent and adherent cells combined) exposed to LPS and hyperoxia. IL-1β and TNFα expression levels were elevated with exposures to O2 and treatment with LPS (statistical effects of O2, LPS, and an interaction). TGFβ expression levels were also increased, but more specifically in response to O2 (statistical effects of O2) (Fig. 3). These data indicate that BV2 cells responded to inflammatory stimuli by increasing the expression of representative cytokines.
Fig. 3.
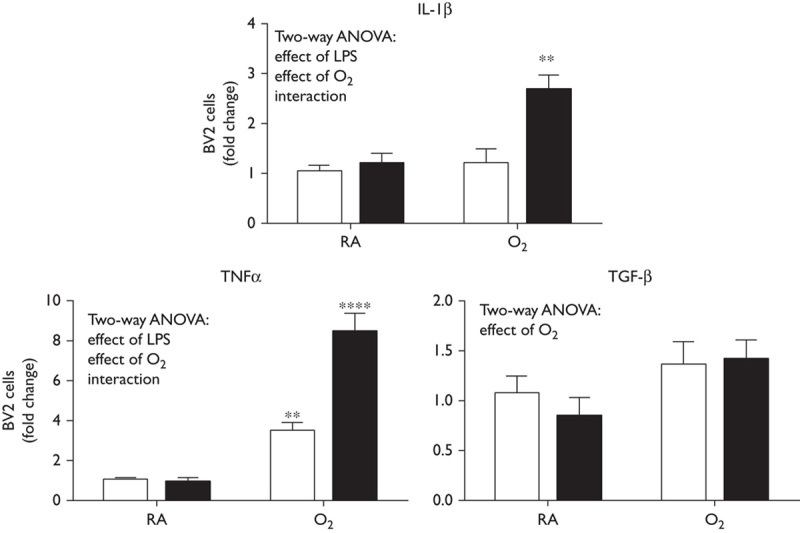
BV2 cell showed increased cytokine expression in response to LPS exposure. BV2 cells were exposed to LPS (50 ng/ml) or saline and exposed to room air or hyperoxia for 48 h (85% O2). Cells were harvested and RNA extracted. Cytokine expression was measured by RT-qPCR. Data were analyzed using a t-test (**P<0.01, ***P<0.001, n=6) and are presented as mean±SD. ANOVA, analysis of variance; LPS, lipopolysaccharide; RA, room air; RT-qPCR, quantitative reverse transcription PCR.
Lipopolysaccharide and hyperoxia cause decreases in Nurr1 levels in BV2 cells
Nurr1 levels were measured by RT-PCR both in nonadherent (Fig. 4a) and in adherent (Fig. 4b) BV2 cells. Oxygen exposure alone induced a marked increase in Nurr1 expression that was attenuated in the LPS/O2-exposed cells (Fig. 4), with both cell groups showing a statistical effect of oxygen.
Fig. 4.
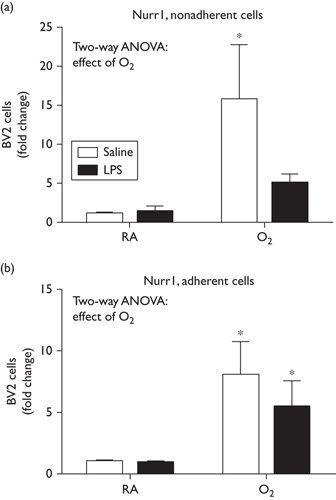
Combined effects of LPS and hyperoxia exposure decrease Nurr1 expression. BV2 cells were exposed to LPS (50 ng/ml) or saline and exposed to room air or hyperoxia for 48 h (85% O2). Nonadherent cells and adherent cells were harvested independently and RNA were extracted. Nurr1 expression was measured by RT-qPCR. Data were analyzed by a two-way ANOVA with Tukey’s post-hoc, (*P<0.05, n=6) and are presented as mean±SD. ANOVA, analysis of variance; LPS, lipopolysaccharide; RT-qPCR, quantitative reverse transcription PCR.
Nurr1 protein expression is decreased in lipopolysaccharide/O2-treated cells
BV2 cell lysates were collected and Nurr1 protein levels were measured by Western blot. Cells exposed to hyperoxia and/or LPS showed significantly reduced Nurr1 protein expression (Fig. 5a). These data are in contrast to the increases in message observed by RT-PCR and may represent RNA or protein instability or turnover in the context of acute inflammation.
Fig. 5.
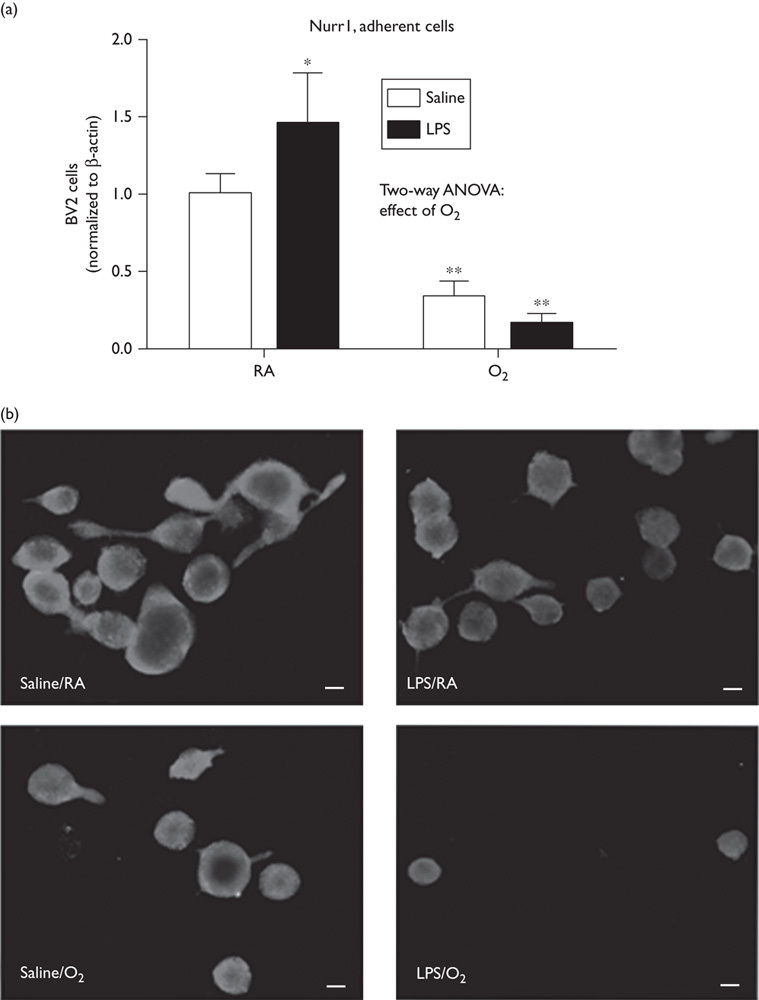
Combined LPS and hyperoxia exposure caused decreases in Nurr1 protein levels in BV2 cells. (a) BV2 cells were exposed to LPS (50 ng/ml) or saline and exposed to room air or hyperoxia for 48 h (85% O2). Cells were harvested and cell lysates were analyzed by Western blot. Data were analyzed by a two-way ANOVA with Tukey’s post-hoc (*P<0.05, **P<0.01, n=6) and are presented as mean±SD. (b) Immunofluorescence was performed on BV2 cells exposed to LPS and hyperoxia. Images were taken at 400×, bar represents 50 µm. ANOVA, analysis of variance; LPS, lipopolysaccharide; RA, room air.
Immunohistochemical analyses of Nurr1 expression in BV2 cells indicate perinuclear expression in the saline/RA-exposed cells and greater nuclear expression in the LPS-exposed and/or O2-exposed cells (Fig. 5b). The cells exposed to LPS/O2 were markedly smaller in size and contained less cytoplasm (a more condensed cell morphology) and fewer of these cells were adherent than in the other exposure groups.
Discussion
In our original report, mice exposed to the combination of maternal LPS and neonatal hyperoxia showed microgliosis in the cerebral cortex and hippocampus 9. Although microglia are essential for the protection of fragile brain tissue from infection or injury, they can produce and excrete substances that are responsible for prolonged, and sometimes misdirected, inflammatory responses. The aim of this study was to determine the role of Nurr1 in microglia in the pathologies observed previously.
Nurr1 is expressed in microglia, but its exact role has not been identified. Studies have suggested that Nurr1 is an anti-inflammatory cytokine that is increased in response to inflammation. Furthermore, Saijo et al. 8, have shown that Nurr1 protein and gene expression increase immediately in response to LPS. Initially, we investigated Nurr1 protein expression in whole-brain homogenates at PN0 in mice born to dams exposed to LPS (Fig. 1a) and at PN28 after maternal LPS and neonatal hyperoxia exposure. Our data were similar to those reported by Saijo and colleagues with considerable increases in Nurr1 in response to LPS, but an additive increase with the additional exposure to hyperoxia (Fig. 1b). The elevated levels of Nurr1 at PN28 were surprising in that these mice were analyzed after 14 days of recovery, indicating that the change in Nurr1 expression persisted after a return to normoxia.
Because of technical limitations, isolation of significant quantities of primary microglia from younger animals is not feasible. Consequently, primary microglia were isolated from mice at PN60. At this late time point, no differences in cytokine expression were observed, suggesting that these cells had recovered from the inflammatory insult by this time (Fig. 2). Interestingly, long after cessation of both exposures, these mice showed decreased Nurr1 mRNA expression in enriched microglia, suggesting that the effect of combined LPS and hyperoxia on the expression of Nurr1 may be long-lasting despite a resolution of the inflammatory response.
To further understand the inconsistencies in our model in vivo, a clinically similar model in vitro was established using the BV2 cell line to study microglial responses to the combined exposures of LPS and hyperoxia. At 24 h, no increases in cytokines were observed with LPS alone and only the expression of TNFα was increased with hyperoxia alone; however, robust increases were observed with combined LPS and hyperoxia (Fig. 3). Interestingly, Nurr1 mRNA levels were increased with hyperoxia alone, but this increase was attenuated in the cells exposed to LPS and hyperoxia (Fig. 4). This same trend was observed when Nurr1 protein levels were assessed by Western blot (Fig. 5a). These findings suggest that there might be transcriptional suppression, increased turnover, or increased degradation of Nurr1 in the LPS/O2 group. Although it is clear that the hyperoxia alone induces an inflammatory state, it is the ‘double hit’ of inflammation and hyperoxia that appears to exacerbate the inflammatory response while also suppressing Nurr1 expression in microglia. Taken together, these data indicate that Nurr1 is increased by LPS/O2 exposure in whole-brain homogenates, but suppressed in both primary microglia and BV2 cells. Our data suggest that the increases in Nurr1 levels observed in whole-brain homogenates are because of Nurr1 expression in a cell type other than microglia.
We do acknowledge the limitations of our studies. First, brains taken at early time points could not be microdissected because of their small size, and testing whole-brain homogenates does not allow for regional analysis. Second, we could not isolate sufficient quantities of primary microglia from younger animals to assess Nurr1 expression; however, studies in BV2 cells did allow us to assess the role of Nurr1 in microglia. In addition, some concerns have been raised about the cross-reactivity of Nurr1 antibodies with Nur77. Because of this, we assayed Nurr1 and Nur77 levels through qPCR. There was markedly less Nur77 expression compared with Nurr1 in our animal model and there was minimal Nurr77 (marginally detectable) expression in our BV2 model.
Collectively, our data reinforce the existing data that Nurr1 expression, regulation, and cell specificity is not straightforward and requires continued study for further elucidation. Others have observed that Nurr1 agonists are effective in treating the symptoms of a mouse model of Parkinson’s disease 23,24 and in curbing inflammation 8,25, indicating that there may be therapeutic potential in modulating Nurr1 expression. Although our data indicate that Nurr1 is increased in one or more cell types in the brain, identification of this (these) cell type(s) is beyond the scope of these studies.
Conclusion
Neurodevelopmental impairments stemming from complications with preterm birth are an understudied problem with wide-ranging impacts. Our study provides novel insight into the possible mechanisms underpinning these neurodevelopmental impairments and Nurr1 as a potential target for further investigation in this field.
Acknowledgements
This work was supported by Nationwide Children’s Hospital (to A.E.G.) and the National Institutes of Health, the National Center for Complementary and Alternative Medicines (NCCAM), and the Office of Dietary Supplements (ODS) (LKR R01AT006880).
The authors would like to thank Dr Jonathan Godbout for providing the BV2 cells.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Allen MC, Cristofalo EA, Kim C. Outcomes of preterm infants: morbidity replaces mortality. Clin Perinatol 2011; 38:441–454. [DOI] [PubMed] [Google Scholar]
- 2.Burd I, Chai J, Gonzalez J, Ofori E, Monnerie H, Le Roux PD, Elovitz MA. Beyond white matter damage: fetal neuronal injury in a mouse model of preterm birth. Am J Obstet Gynecol 2009; 201:271.e1–271.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature 2003; 423:555–560. [DOI] [PubMed] [Google Scholar]
- 4.Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science 1997; 276:248–250. [DOI] [PubMed] [Google Scholar]
- 5.García-Yagüe ÁJ, Rada P, Rojo AI, Lastres-Becker I, Cuadrado A. Nuclear import and export signals control the subcellular localization of Nurr1 protein in response to oxidative stress. J Biol Chem 2013; 288:5506–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barish GD, Downes M, Alaynick WA, Yu RT, Ocampo CB, Bookout AL, et al. A nuclear receptor Atlas: macrophage activation. Mol Endocrinol 2005; 19:2466–2477. [DOI] [PubMed] [Google Scholar]
- 7.Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol 2006; 20:786–794. [DOI] [PubMed] [Google Scholar]
- 8.Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009; 137:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graf AE, Haines KM, Pierson CR, Bolon BN, Houston RH, Velten M, et al. Perinatal inflammation results in decreased oligodendrocyte numbers in adulthood. Life Sci 2014; 94:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velten M, Britt RD, Jr, Heyob KM, Welty SE, Eiberger B, Tipple TE, Rogers LK. Prenatal inflammation exacerbates hyperoxia-induced functional and structural changes in adult mice. Am J Physiol Regul Integr Comp Physiol 2012; 303:R279–R290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velten M, Gorr MW, Youtz DJ, Velten C, Rogers LK, Wold LE. Adverse perinatal environment contributes to altered cardiac development and function. Am J Physiol Heart Circ Physiol 2014; 306:H1334–H1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol (1985) 2010; 108:1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velten M, Hutchinson KR, Gorr MW, Wold LE, Lucchesi PA, Rogers LK. Systemic maternal inflammation and neonatal hyperoxia induces remodeling and left ventricular dysfunction in mice. PLoS One 2011; 6:e24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Normann E, Lacaze-Masmonteil T, Eaton F, Schwendimann L, Gressens P, Thébaud B. A novel mouse model of ureaplasma-induced perinatal inflammation: effects on lung and brain injury. Pediatr Res 2009; 65:430–436. [DOI] [PubMed] [Google Scholar]
- 15.Rousset CI, Kassem J, Olivier P, Chalon S, Gressens P, Saliba E. Antenatal bacterial endotoxin sensitizes the immature rat brain to postnatal excitotoxic injury. J Neuropathol Exp Neurol 2008; 67:994–1000. [DOI] [PubMed] [Google Scholar]
- 16.Ramani M, van Groen T, Kadish I, Bulger A, Ambalavanan N. Neurodevelopmental impairment following neonatal hyperoxia in the mouse. Neurobiol Dis 2013; 50:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yiş U, Kurul SH, Kumral A, Cilaker S, Tuğyan K, Genç S, Yilmaz O. Hyperoxic exposure leads to cell death in the developing brain. Brain Dev 2008; 30:556–562. [DOI] [PubMed] [Google Scholar]
- 18.Zaghloul N, Nasim M, Patel H, Codipilly C, Marambaud P, Dewey S, et al. Overexpression of extracellular superoxide dismutase has a protective role against hyperoxia-induced brain injury in neonatal mice. FEBS J 2012; 279:871–881. [DOI] [PubMed] [Google Scholar]
- 19.Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, et al. Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience 2015; 302:151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norden DM, Fenn AM, Dugan A, Godbout JP. TGFβ produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 2014; 62:881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci 2011; 31:6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 2013; 33:13820–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Miranda BR, Popichak KA, Hammond SL, Jorgensen BA, Phillips AT, Safe S, et al. The Nurr1 activator 1,1-Bis(3′-Indolyl)-1-(p-Chlorophenyl)Methane blocks inflammatory gene expression in BV-2 microglial cells by inhibiting nuclear factor kappaB. Mol Pharmacol 2015; 87:1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CH, Han BS, Moon J, Kim DJ, Shin J, Rajan S, et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson’s disease. Proc Natl Acad Sci USA 2015; 112:8756–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montarolo F, Perga S, Martire S, Bertolotto A. Nurr1 reduction influences the onset of chronic EAE in mice. Inflamm Res 2015; 64:841–844. [DOI] [PubMed] [Google Scholar]