

Epidemiological studies indicate that the origins of high blood pressure initiates in fetal life (1, 2, 3). Recent reviews highlight a role for numerous factors in the pathogenesis of the developmental programming of hypertension including epigenetic processes, glucocorticoids, reduced nephron number, activation of the sympathetic nervous system and the renin angiotensin system (RAS) and endothelial dysfunction (4, 5, 6, 7). Studies published in Hypertension and other journals highlight the complexity of cardiovascular (CV) risk that has its origins in fetal life. The purpose of this mini-review is to present an update on recent studies in Hypertension that investigate the underlying mechanisms that contribute to the developmental programming of hypertension and increased risk for CV disease.
The Sympathetic Nervous System and the Developmental Programming of Hypertension
David Barker first proposed the theory of the fetal origins of coronary heart disease based on the hypothesis that adaptive responses by the fetus to maternal undernutrition would enhance survival to birth at the expense of later CV health (8). The first animal studies to investigate the Barker hypothesis utilized rodent models to demonstrate that maternal undernutrition during gestation programs an increase in blood pressure in the offspring (9). Based on previous studies indicating a role for the renal nerves in experimental models of low birth weight (10, 11), Mizuno and colleagues proposed that increased blood pressure in protein restricted offspring would be associated with an increase in baseline measure of renal sympathetic nerve activity (SNA) (12). Although baseline measurement of renal SNA under anesthesia did not differ in male protein restricted offspring versus male control at 4 to 5 months of age, renal SNA was significantly elevated in response to a secondary hit of physical stress in association with a greater blood pressure response (12) indicating a role for an increase in renal SNA in the developmental programming of increased blood pressure and CV risk. Chronic systemic blockade of the RAS with enalapril, an angiotensin converting enzyme inhibitor, abolishes hypertension in offspring programmed via exposure to a fetal insult such as maternal dietary protein restriction (13) or placental insufficiency (14) suggesting a role for the peripheral RAS in the pathogenesis of increased CV risk programmed by fetal insult. Expression of the angiotensin type 1 receptor is elevated in regions of the brain involved in CV regulation in offspring exposed to maternal dietary protein restriction (15). Intracerebroventricular administration of the angiotensin type 1 receptor antagonist, losartan, at a dose 100 times lower than an effective systemic dose, abolishes hypertension in rat offspring exposed to maternal dietary protein restriction (15) demonstrating a role for the central RAS in the pathogenesis of increased CV risk that has its origins in fetal life. Increases in renal SNA can be modulated by changes in activity of the central RAS (16). Thus, in more a recent study published in Hypertension, Mizuno et al. expanded their investigation to elucidate the mechanism whereby sympathetic responsiveness to physical stress is enhanced in offspring exposed to maternal dietary protein restriction (17). They found that chronic blockade of the RAS with enalapril not only reduced blood pressure in male protein restricted offspring under baseline conditions, it also attenuated the exaggerated increase in renal SNA and diminished the enhanced blood pressure responsiveness to physical stress in male protein restricted offspring with no effect on renal SNA or blood pressure in male control offspring (17). Thus, this study by Mizuno and colleagues suggests that the RAS contributes to the development of enhanced sympathetic and blood pressure responsiveness to physical stress programmed in response to a fetal insult (Figure).
Figure.
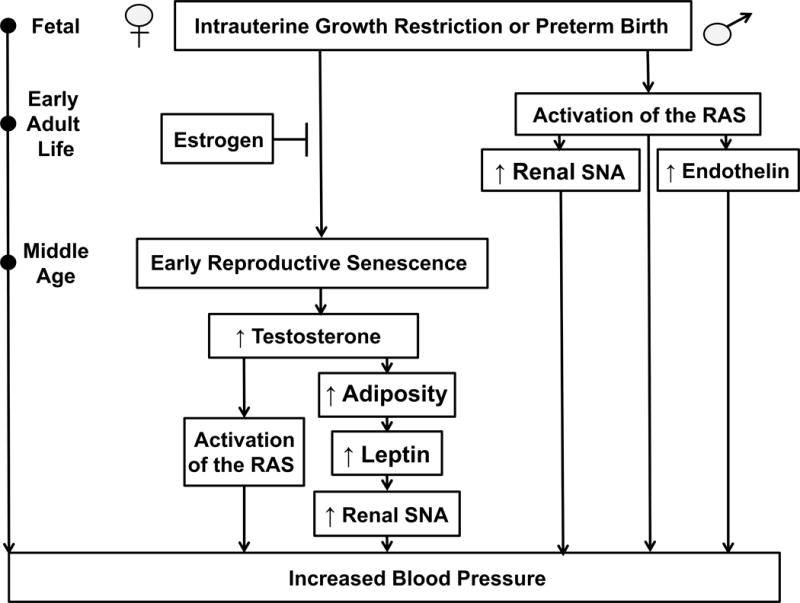
A schematic representation of the proposed pathogenesis of the developmental programming of hypertension. Numerous experimental models of developmental origins including placental insufficiency, moderate maternal protein restriction, fetal exposure to exogenous glucocorticoids, and prenatal exposure to hypoxia or nicotine program a sex difference in adult cardiovascular risk. Several studies indicate that estrogen is protective against the developmental origins of increased cardiovascular risk in female offspring in early adult life. Yet, recent studies report that protection against programmed CV risk in female offspring is lost by one year of age. Despite the method of fetal insult, common mechanistic pathways contribute to programmed CV risk. This review highlights recent advances in the developmental origins of health and disease published in Hypertension. Insight from these studies indicate the importance of the renin angiotensin and endothelin systems, sympathetic nerve activity and sex steroids in the etiology of hypertension that has its origins in early life. Early adult life (3–5 months of age), middle age (12 months of age). Abbreviations: RAS, renin angiotensin system; SNA, sympathetic nerve activity.
Many studies demonstrate an inverse association between birth weight and blood pressure (1, 2, 3) indicating birth weight as a predictor of CV risk. Although malnutrition contributes to fetal growth restriction in many developing countries (18), placental insufficiency is the major cause of low birth weight within the Western world (19). Placental hypoxia that occurs in response to placental insufficiency alters fetal oxygen delivery resulting in intrauterine growth restriction (IUGR) (20). Rook et al. recently utilized a rat model of prenatal hypoxia to examine the role of muscle SNA in the fetal origins of hypertension. Muscle SNA is elevated in healthy young adults of normal stature born small for gestational age who tend to have higher blood pressure relative to appropriate for gestational age counterparts (21) suggesting that an increase in SNA may be a link between birth weight and blood pressure. Although aging in humans is associated with an increase in sympathetic outflow to the peripheral tissues, responsiveness to sympathetic stimulation decreases with aging (22). Therefore, Rook et al. investigated whether temporal changes in muscle SNA were associated with increased blood pressure in male offspring exposed to prenatal hypoxia (23). This study published in Hypertension reported that frequency of single unit muscle SNA and sympathetic innervation of the tibial arteries were increased at 3 months of age in male offspring exposed to prenatal hypoxia (23). Blood pressure was not elevated at this age, but blood pressure was increased by 9 months of age in male offspring exposed to prenatal hypoxia relative to male control (23) suggesting that exposure to hypoxia during fetal life programs an increase in basal muscle SNA that precedes the development of increased blood pressure. Despite the increase in sympathetic nerve density, vasoconstrictor responses to sympathetic nerve stimulation were blunted in male prenatal hypoxia offspring at 3 months of age relative to age-matched control counterparts (23). Blockade of the neurotransmitter neuropeptide Y attenuated these responses in male control offspring at 3 months of age with no effect on vasoconstrictor responses to sympathetic nerve stimulation in prenatal hypoxia counterparts (23). However, responsiveness to sympathetic stimulation was attenuated in male control offspring by 9 months of age (23). Collectively, these data indicate that fetal exposure to prenatal hypoxia programs premature aging of the vasculature; yet, the quantitative importance of muscle SNA in the pathophysiology of increased CV risk following exposure to a fetal insult is not yet clear.
Many experimental models of developmental programming report a sex difference in blood pressure with males exhibiting a significant increase in blood pressure in young adulthood whereas female counterparts remain normotensive (24, 25, 26, 27, 28). Yet, female offspring exposed to an insult during fetal life do not remain protected against an increase in blood pressure in later life (25, 29). Previous studies addressing the importance of the renal nerves and SNA in the developmental programming of hypertension involve studies performed in male offspring (10, 11, 17, 23). Although male IUGR offspring in the model of IUGR induced via placental insufficiency in the rat are smaller in size relative to control counterparts, hypertension in male IUGR offspring is abolished by bilateral renal denervation at 3 months of age (11). In this model blood pressure does not differ between female IUGR relative to female control offspring at 3 months of age (24). However, Intapad and colleagues recently reported in a study published in Hypertension that female IUGR offspring exhibited a significant increase in blood pressure relative to age-matched female counterparts at one year of age (30). Visceral fat mass and circulating leptin levels were increased in conjunction with the significant increase in blood pressure in female IUGR offspring relative to control counterparts at one year of age; increases not present in female IUGR offspring at 6 months of age when they were still normotensive (30). Based on studies by Hall and colleagues suggesting that obesity-related hypertension involves activation of the renal nerves (31), Intapad et al. demonstrated that bilateral renal denervation abolished the increase in blood pressure in female IUGR offspring at one year of age (30). It was previously reported that bilateral renal denervation abolishes hypertension in male IUGR offspring as early as 6 weeks of age, prior to puberty (32). Taken together, data from studies in the model of IUGR induced by placental insufficiency indicate a sex difference in the fetal programming of increased sympathetic outflow to the kidney. The importance of the renal nerves in the pathogenesis of increased blood pressure in male IUGR offspring that is present prior to puberty suggests that activation of the renal nerves is established in utero in male IUGR offspring whereas an additional stimulus must contribute to the activation of the peripheral sympathetic nervous system that occurs in later life in female IUGR littermates. Leptin is associated with hypertension in women as they age (33). Thus, the age-related increase in circulating leptin in female IUGR offspring at one year of age may serve as a secondary stimulus demonstrating sex-specific programming of increased CV risk (Figure).
Maternal pre-pregnancy body mass index is positively associated with blood pressure and total body fat mass in childhood (34) indicating that low birth weight resulting from maternal undernutrition or placental insufficiency is not the only adverse programmable influence on later CV risk. Prior et al. recently reported in Hypertension that fetal exposure to maternal obesity programmed an increase in blood pressure associated with a significant increase in fat mass and leptin in rabbit offspring (35). In this model, renal SNA was elevated in obese offspring in young adulthood relative to control counterparts (35). Obese offspring also exhibited a greater rise in renal SNA in response to air jet stress or intracerebroventricular infusion of leptin relative to control counterparts (36). Samuelson et al. note a similar outcome in mice born to high fat dams (36). Yet, Prior also noted an enhanced increase in renal SNA in response to intracerebroventricular infusion of ghrelin, an activator of neuropeptide Y (35). Thus, sympathetic overactivation following a developmental insult may be influenced by increased adiposity in a manner that is not species-specific (30, 35, 36).
Collectively, studies highlighted in this section suggests that the pathogenesis of elevated sympathetic outflow programmed in response to a developmental insult may be multifactorial. Although the pathogenesis of increased SNA following a developmental insult may involve obesity or increased adiposity, the central RAS may also serve as a potential stimulus for the developmental origins of increased SNA in a manner that is sex- and age-dependent.
Sex Steroids, the Renin Angiotensin System, and the Developmental Programming of Hypertension
Onset of menopause is accelerated in low birth weight women (37) suggesting that the influence of an adverse fetal environment extends beyond the developmental programming of increased blood pressure and CV risk. In the model of IUGR programmed by placental insufficiency, female IUGR offspring exhibit persistent estrous at one year of age unlike control offspring that retain an appropriate age-related pattern of estrous cyclicity (38). Early reproductive senescence in female IUGR at one year of age is also associated with a significant increase in circulating testosterone (38). The transition into menopause is associated with an increase in testosterone in women participants that develop CV disease relative to those women who do not in a longitudinal report from the Chicago site of the Study of Women’s Health Across the Nation (39). Furthermore, testosterone is positively associated with blood pressure after menopause (40). To address the hypothesis that an increase in testosterone contributes to the pathogenesis of elevated blood pressure in female IUGR offspring at one year of age, a recent study published in Hypertension showed that chronic blockade of the androgen receptor with flutamide abolished the significant increase in blood pressure at one year of age in female IUGR rat offspring programmed by fetal exposure to placental insufficiency (41). In this study by Dasinger et al., chronic treatment with enalapril also decreased blood pressure in female IUGR offspring indicating a role for the RAS (41). Testosterone is implicated in the activation of the RAS in the aging female spontaneously hypertensive rat (42). Thus, Dasinger and colleagues also demonstrated that intramedullary expression of the angiotensin type 1 receptor was increased in untreated female IUGR offspring at one year of age, but did not differ in female IUGR treated with flutamide relative to untreated female controls (41). Based on these findings, Dasinger and colleagues proposed that testosterone-mediated activation of the RAS contributes to the development of increased blood pressure in female IUGR offspring at one year of age (Figure). Visceral adiposity increases after menopause and is positively associated with testosterone independent of aging (39). Although this relationship is correlative, these findings suggest that increases in testosterone may contribute to the development of increased visceral adiposity in female IUGR offspring at one year of age (30). Furthermore, increases in visceral fat, and the subsequent increase in circulating leptin, may serve as a stimulus for elevated sympathetic outflow to the renal nerves in the female IUGR (30) (Figure).
Testosterone also contributes to the pathogenesis of hypertension in female offspring programmed in response to fetal exposure to excess maternal testosterone during prenatal life. Blesson and colleagues recently reported that prenatal testosterone excess programs an increase in blood pressure associated with an elevation in testosterone in female offspring mimicking the pathogenesis of polycystic ovary syndrome (PCOS) (43). This study published in Hypertension demonstrated that chronic blockade of the androgen receptor with flutamide abolished the programmed increase in blood pressure in the prenatal testosterone exposed female offspring (43) implicating an important role for hyperandrogenism in the pathogenesis of hypertension in this model (43). It is well established that protein kinase C (PKC) isoforms modulate vascular activity. Thus, Blesson et al. also examined vascular PKC expression and noted that protein expression of the PKCδ isoform was elevated within the mesentery of the prenatal testosterone-exposed rats (43). To elucidate the link between hyperandrogenaemia and PKCδ, they showed that PKCδ expression was increased in a dose-dependent manner in cultured mesenteric artery smooth muscle cells exposed to testosterone. Additional studies identified a testosterone responsive enhancer element located within the PKCδ gene (43). Therefore, this study demonstrated a role for PKCδ in the pathogenesis of increased blood pressure and identified a role for testosterone in the control of PKCδ expression via transcriptional regulation.
The recent study by Dasinger et al. demonstrates that testosterone contributes to the increase in blood pressure that develops in female IUGR offspring at one year of age (41). However, previous studies using the model of IUGR induced via placental insufficiency indicate that estrogen is protective against increased blood pressure and CV risk in female IUGR offspring in young adulthood (44, 45). Specifically, ovariectomy induces a significant increase in blood pressure (44) and enhances the blood pressure response to acute angiotensin II (Ang II) in female IUGR offspring (45). A recent study by Xiao et al. in Hypertension provides further support that estrogen offsets the developmental programming of increased blood pressure and CV risk in female offspring using a model of prenatal nicotine exposure (46). Maternal smoking increases the incidence of low birth weight (47). Although baseline blood pressure is not elevated in rat offspring exposed to nicotine during prenatal life, male prenatal nicotine offspring exhibit an enhanced blood pressure response to acute Ang II at 5 months of age that is not observed in age-matched female littermates (48). A recent publication by Xiao and colleagues in Hypertension demonstrated that the blood pressure response to acute Ang II was enhanced by ovariectomy in female prenatal nicotine offspring at 5 months of age (46). In addition, 17B-estradiol replacement attenuated the heightened blood pressure response to acute Ang II (46) (Figure).
Thus, studies in models of developmental programming indicate a sex difference in CV risk and suggest an age-specific role for sex steroids in the pathogenesis of increased blood pressure in female offspring (41, 44, 48). Experimental studies suggest that estradiol is protective against increased CV risk in female offspring in young adulthood whereas other studies implicate testosterone as a permissive factor in the development of increased blood pressure in female IUGR offspring in later life. Although beyond the scope of this review, testosterone also contributes to increased blood pressure and CV risk in male offspring exposed to a developmental insult (14).
Endothelin, Sex Differences and Programmed Cardiovascular Risk
The National Institutes of Health recently mandated sex as a variable in consideration of how health and disease processes differ. As highlighted above, numerous models of developmental programming report a sex difference in blood pressure (9, 24, 26, 27, 28) with onset of increased CV risk delayed in females (24, 25, 29, 30). Although the importance of endothelin (ET-1) as a potential contributor to the pathogenesis of hypertension and increased CV risk following a fetal insult has undergone limited investigation, recent studies published in Hypertension indicate that the ET-1 system may contribute to sex differences in increased CV risk that has its origins in early life. In a study by Bourque and colleagues, prenatal hypoxia programmed an age-related increase in blood pressure in male, but not female IUGR offspring relative to same-sex counterparts by one year of age (25). Furthermore, blockade of the ET-1 system by the ETA/B receptor antagonist, tezosentan, caused a two-fold decrease in blood pressure in male IUGR rats relative to male control with no effect on blood pressure in female control or IUGR offspring (25) suggesting that female rats were resistant to blockade of the ET-1 system. Another recent study in Hypertension by Intapad and Ojeda et al. also demonstrated a sex-specific response to ET-1 receptor blockade (49). In male IUGR rats programmed by placental insufficiency, blockade of the ETA receptor abolished the enhanced blood pressure response to acute Ang II (Figure) whereas ETA receptor blockade exerted no effect on blood pressure in female control or IUGR offspring (49). Ojeda and colleagues previously reported that the blood pressure response to acute ANG II is exacerbated in ovariectomized female IUGR in young adulthood (45). In the study by Intapad, Ojeda and colleagues, blockade of the ETA receptor had no effect on blood pressure in ovariectomized female IUGR offspring (49) indicating that blood pressure in female offspring, regardless of birth weight, was resistant to ET-1 receptor blockade. Although the stimulus for increased production of ET-1 following a fetal insult remains unknown, Ang II is reported to increase ET-1 production (50). Previous studies indicating that the RAS contributes to increased blood pressure in experimental models of fetal insult (13, 14, 15) suggests that activation of the RAS may serve as a potential mediator of the ET-1 pathway. However, the underlying mechanisms that contribute to the sex difference in the blood pressure response to ET-1 blockade remains to be determined.
Concluding Remarks
Essential hypertension is a complex condition of unknown pathogeneses and recent advances in the field of developmental origins of hypertension add a layer of complexity to our understanding of blood pressure control. Recent studies using experimental models of fetal insult demonstrate that exposure to an adverse environment during fetal life programs an increase in blood pressure via multiple pathways. Yet, despite the method of fetal insult, these experimental studies indicate that similar pathways contribute to the pathogenesis of increased blood pressure that has its origins in early life. Men and women differ in their risk for CV disease in a manner that is altered with age. Recent advances in the field of developmental origins of health and disease also suggest that the pathogenesis of increased blood pressure programmed by exposure to a fetal insult differs in males relative to females and in a manner that is age-specific. Birth weight is a risk factor for hypertension and CV disease; but the clinical significance of birth weight is not yet a consideration in the management of an individual’s CV health. Thus, additional studies are needed to clarify the most effective pharmacological therapeutic approaches for the management of blood pressure in men and women born preterm, IUGR or high birth weight.
Acknowledgments
SOURCES OF FUNDING
Dr. Alexander is supported by the American Heart Grant GRNT19900004 and NIH grants HL074927 and HL51971. Mr. Dasinger is supported by funding from the American Heart Grant 15PRE24700010 and the NIH T32HL105324. Ms. Newsome is supported by funding from the NIH T32HL105324.
Footnotes
DISCLOSURES
None.
References
- 1.de Jong F, Monuteaux MC, van Elburg RM, Gillman MW, Belfort MB. Systematic review and meta-analysis of preterm birth and later systolic blood pressure. Hypertension. 2012;59:226–234. doi: 10.1161/HYPERTENSIONAHA.111.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mu M, Wang SF, Sheng J, Zhao Y, Li HZ, Hu CL, Tao FB. Birth weight and subsequent blood pressure: a meta-analysis. Arch Cardiovasc Dis. 2012;105:99–113. doi: 10.1016/j.acvd.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Li H, Liu SJ, Fu GJ, Zhao Y, Xie YJ, Zhang Y, Wang YX. The associations of high birth weight with blood pressure and hypertension in later life: a systematic review and meta-analysis. Hypertens Res. 2013;36:725–735. doi: 10.1038/hr.2013.33. [DOI] [PubMed] [Google Scholar]
- 4.Morton JS, Cooke CL, Davidge ST. In utero origins of hypertension: Mechanisms and targets for therapy. Physiol Rev. 2016;96:549–603. doi: 10.1152/physrev.00015.2015. [DOI] [PubMed] [Google Scholar]
- 5.Dasinger JH, Alexander BT. Gender differences in the developmental programming of cardiovascular disease. Clin Sci (Lond) 2016;130:337–348. doi: 10.1042/CS20150611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thornburg KL. The programing of cardiovascular disease. J Dev Orig Health Dis. 2015;6:366–375. doi: 10.1017/S2040174415001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Painter RC, Roseboom TJ, De Rooij SR. Long-term effects of prenatal stress and glucocorticoid exposure. Birth Defects Res C Embryo Today. 2012;96:315–324. doi: 10.1002/bdrc.21021. [DOI] [PubMed] [Google Scholar]
- 8.Barker DJ. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langley-Evans SC, Phillips GJ, Jackson AA. In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clin Nutr. 1994;13:319–324. doi: 10.1016/0261-5614(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 10.Dagan A, Kwon HM, Dwarakanath V, Baum M. Effect of renal denervation on prenatal programming of hypertension and renal tubular transporter abundance. Am J Physiol Renal Physiol. 2008;295:F29–F34. doi: 10.1152/ajprenal.00123.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander BT, Hendon AE, Ferril G, Dwyer TM. Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension. 2005;45:754–758. doi: 10.1161/01.HYP.0000153319.20340.2a. [DOI] [PubMed] [Google Scholar]
- 12.Mizuno M, Siddique K, Baum M, Smith SA. Prenatal programming of hypertension induces sympathetic overactivity in response to physical stress. Hypertension. 2013;61:180–186. doi: 10.1161/HYPERTENSIONAHA.112.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manning J, Vehaskari VM. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am J Physiol Regul Integr Comp Physiol. 2005;288:R80–R84. doi: 10.1152/ajpregu.00309.2004. [DOI] [PubMed] [Google Scholar]
- 14.Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, Alexander BT. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2007;292:R758–R763. doi: 10.1152/ajpregu.00311.2006. [DOI] [PubMed] [Google Scholar]
- 15.Pladys P, Lahaie I, Cambonie G, Thibault G, Lê NL, Abran D, Nuyt AM. Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr Res. 2004;55:1042–1049. doi: 10.1203/01.PDR.0000127012.37315.36. [DOI] [PubMed] [Google Scholar]
- 16.DiBona GF. Nervous kidney. Interaction between renal sympathetic nerves and the renin-angiotensin system in the control of renal function. Hypertension. 2000;36:1083–1088. doi: 10.1161/01.hyp.36.6.1083. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno M, Siddique K, Baum M, Smith SA. Enalapril attenuates the exaggerated sympathetic response to physical stress in prenatally programmed hypertensive rats. Hypertension. 2014;63:324–329. doi: 10.1161/HYPERTENSIONAHA.113.02330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.United Nations Children’s Fund. State of the world’s children report, 2003. New York: UNICEF; 2003. Internet: www.unicef.org/publications/pub_sowc03_en.pdf. [Google Scholar]
- 19.Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand. 2002;81:112–114. doi: 10.1034/j.1600-0412.2002.810204.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhu MY, Milligan N, Keating S, Windrim R, Keunen J, Thakur V, Ohman A, Portnoy S, Sled JG, Kelly E, Yoo SJ, Gross-Wortmann L, Jaeggi E, Macgowan CK, Kingdom JC, Seed M. The hemodynamics of late-onset intrauterine growth restriction by MRI. Am J Obstet Gynecol. 2016;214:367.e1–367.e17. doi: 10.1016/j.ajog.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Boguszewski MC, Johannsson G, Fortes LC, Sverrisdóttir YB. Low birth size and final height predict high sympathetic nerve activity in adulthood. J Hypertens. 2004;22:1157–1163. doi: 10.1097/00004872-200406000-00017. [DOI] [PubMed] [Google Scholar]
- 22.Seals DR, Dinenno FA. Collateral damage: cardiovascular consequences of chronic sympathetic activation with human aging. Am J Physiol Heart Circ Physiol. 2004;287:H1895–H1905. doi: 10.1152/ajpheart.00486.2004. [DOI] [PubMed] [Google Scholar]
- 23.Rook W, Johnson CD, Coney AM, Marshall JM. Prenatal hypoxia leads to increased muscle sympathetic nerve activity, sympathetic hyperinnervation, premature blunting of neuropeptide Y signaling, and hypertension in adult life. Hypertension. 2014;64:1321–1327. doi: 10.1161/HYPERTENSIONAHA.114.04374. [DOI] [PubMed] [Google Scholar]
- 24.Alexander BT. Placental insufficiency leads to the development of hypertension in growth-restricted offspring. Hypertension. 2003;41:457–462. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 25.Bourque SL, Gragasin FS, Quon AL, Mansour Y, Morton JS, Davidge ST. Prenatal hypoxia causes long-term alterations in vascular endothelin-1 function in aged male, but not female, offspring. Hypertension. 2013;62:753–758. doi: 10.1161/HYPERTENSIONAHA.113.01516. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz LA, Quan A, Weinberg A, Baum M. Effect of prenatal dexamethasone on rat renal development. Kidney Int. 2001;59:1663–1669. doi: 10.1046/j.1523-1755.2001.0590051663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res. 2001;49:460–467. doi: 10.1203/00006450-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- 29.Pijacka W, Clifford B, Tilburgs C, Joles JA, Langley-Evans S, McMullen S. Protective role of female gender in programmed accelerated renal aging in the rat. Physiol Rep. 2015;3(1–6):e12342. doi: 10.14814/phy2.12342. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Intapad S, Tull FL, Brown AD, Dasinger JH, Ojeda NB, Fahling JM, Alexander BT. Renal denervation abolished the age-dependent increase in blood pressure in female intrauterine growth-restriction rats at 12 months of age. Hypertension. 2013;61:828–834. doi: 10.1161/HYPERTENSIONAHA.111.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ojeda NB, Johnson WR, Dwyer TM, Alexander BT. Early renal denervation prevents development of hypertension in growth-restricted offspring. Clin Exp Pharmacol Physiol. 2007;34:1212–1216. doi: 10.1111/j.1440-1681.2007.04754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma D, Feitosa MF, Wilk JB, Laramie JM, Yu K, Leiendecker-Foster C, Myers RH, Province MA, Borecki IB. Leptin is associated with blood pressure and hypertension in women from the National Heart, Lung and Blood Institute Family Heart Study. Hypertension. 2009;53:473–479. doi: 10.1161/HYPERTENSIONAHA.108.118133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaillard R, Steegers EA, Franco OH, Hofman A, Jaddoe VW. Maternal weight gain in different periods of pregnancy and childhood cardio-metabolic outcomes. The Generation R Study. Int J Obes (Lond) 2015;39:6776–85. doi: 10.1038/ijo.2014.175. [DOI] [PubMed] [Google Scholar]
- 35.Prior LJ, Davern PJ, Burke SL, Lim K, Armitage JA, Head GA. Exposure to a high-fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension. 2014;63:338–345. doi: 10.1161/HYPERTENSIONAHA.113.02498. [DOI] [PubMed] [Google Scholar]
- 36.Samuelsson AM, Morris A, Igosheva N, Kirk SL, Pombo JM, Coen CW, Poston L, Taylor PD. Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension. 2010;55:76–82. doi: 10.1161/HYPERTENSIONAHA.109.139402. [DOI] [PubMed] [Google Scholar]
- 37.Tom SE, Cooper R, Kuh D, Guralnik JM, Hardy R, Power C. Fetal environment and early age at natural menopause in a British birth cohort study. Hum Reprod. 2010;25:791–798. doi: 10.1093/humrep/dep451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Intapad S, Dasinger JH, Brown AD, Fahling JM, Esters J, Alexander BT. Glucose intolerance develops prior to increased adiposity and accelerated cessation of estrous cyclicity in female growth-restricted rats. Pediatr Res. 2016;79:962–970. doi: 10.1038/pr.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen I, Powell LH, Jasielec MS, Kazlauskaite R. Covariation of change in bioavailable testosterone and adiposity in midlife women. Obesity. 2015;23:488–494. doi: 10.1002/oby.20974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rexrode KM, Manson JE, Lee IM, Ridker PM, Sluss PM, Cook NR, Buring JE. Sex hormone levels and risk of cardiovascular events in postmenopausal women. Circulation. 2003;108:1688–1693. doi: 10.1161/01.CIR.0000091114.36254.F3. [DOI] [PubMed] [Google Scholar]
- 41.Dasinger JH, Intapad S, Rudenske BR, Davis GK, Newsome AD, Alexander BT. Chronic blockade of the androgen receptor abolishes age-dependent increases in blood pressure in female growth-restricted rats. Hypertension. 2016;67:1281–1290. doi: 10.1161/HYPERTENSIONAHA.116.07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lima R, Yanes LL, Davis DD, Reckelhoff JF. Roles played by 20-HETE, angiotensin II and endothelin in mediating the hypertension in aging female spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2013;304:R348–R351. doi: 10.1152/ajpregu.00380.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cδ-mediated mechanism. Hypertension. 2015;65:683–690. doi: 10.1161/HYPERTENSIONAHA.114.04521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ojeda NB, Grigore D, Robertson EB, Alexander BT. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension. 2007;50:679–685. doi: 10.1161/HYPERTENSIONAHA.107.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ojeda NB, Intapad S, Royals TP, Black JT, Dasinger JH, Tull FL, Alexander BT. Hypersensitivity to acute angiotensin II in female growth-restricted offspring is exacerbated by ovariectomy. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1199–R1205. doi: 10.1152/ajpregu.00219.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao D, Huang X, Yang S, Zhang L. Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension. 2013;61:1246–1254. doi: 10.1161/HYPERTENSIONAHA.113.01152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ko TJ, Tsai LY, Chu LC, Yeh SJ, Leung C, Chen CY, Chou HC, Tsao PN, Chen PC, Hsieh WS. Parental smoking during pregnancy and its association with low birth weight, small for gestational age, and preterm birth offspring: a birth cohort study. Pediatr Neonatol. 2014;55:20–27. doi: 10.1016/j.pedneo.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension. 2008;51:1239–1247. doi: 10.1161/HYPERTENSIONAHA.107.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Intapad S, Ojeda NB, Varney ET, Royals TP, Alexander BT. Sex-specific effect of endothelin in the blood pressure response to acute angiotensin II in growth-restricted rats. Hypertension. 2015;66:1260–1266. doi: 10.1161/HYPERTENSIONAHA.115.06257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexander BT, Rinewalt AN, Cockrell KL, Bennett WA, Granger JP. Endothelin type A receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension. 2001;37:485–489. doi: 10.1161/01.hyp.37.2.485. [DOI] [PubMed] [Google Scholar]