

Abstract
Kunitz-type (KT) protease inhibitors are low molecular weight proteins classically defined as serine protease inhibitors. We identified a novel secreted KT inhibitor associated with the gut and parenchymal tissues of the infective juvenile stage of Fasciola hepatica, a helminth parasite of medical and veterinary importance. Unexpectedly, recombinant KT inhibitor (rFhKT1) exhibited no inhibitory activity toward serine proteases but was a potent inhibitor of the major secreted cathepsin L cysteine proteases of F. hepatica, FhCL1 and FhCL2, and of human cathepsins L and K (Ki = 0.4-27 nm). FhKT1 prevented the auto-catalytic activation of FhCL1 and FhCL2 and formed stable complexes with the mature enzymes. Pulldown experiments from adult parasite culture medium showed that rFhKT1 interacts specifically with native secreted FhCL1, FhCL2, and FhCL5. Substitution of the unusual P1 Leu15 within the exposed reactive loop of FhKT1 for the more commonly found Arg (FhKT1Leu15/Arg15) had modest adverse effects on the cysteine protease inhibition but conferred potent activity against the serine protease trypsin (Ki = 1.5 nm). Computational docking and sequence analysis provided hypotheses for the exclusive binding of FhKT1 to cysteine proteases, the importance of the Leu15 in anchoring the inhibitor into the S2 active site pocket, and the inhibitor's selectivity toward FhCL1, FhCL2, and human cathepsins L and K. FhKT1 represents a novel evolutionary adaptation of KT protease inhibitors by F. hepatica, with its prime purpose likely in the regulation of the major parasite-secreted proteases and/or cathepsin L-like proteases of its host.
Keywords: cysteine protease, enzyme inhibitor, enzyme kinetics, host-pathogen interaction, molecular modeling, parasite, protease inhibitor, protein-protein interaction, serine protease
Introduction
Fasciola hepatica is a zoonotic parasitic helminth common in temperate and sub-tropical regions of the world. The parasite is responsible for causing the disease fasciolosis in hundreds of millions of livestock, principally sheep and cattle. This results in reduced feed conversion, decreased dairy production, inferior meat quality and parasite-related mortality, thus costing the agricultural industry an estimated United States $3 billion annually (1, 2). F. hepatica is also estimated to infect up to 17 million people throughout the world, primarily in developing countries, with ∼180 million at risk of infection (3, 4). The mammalian host becomes infected following ingestion of grass or other vegetation contaminated with F. hepatica cysts (metacercariae). The parasites then exocyst in the host duodenum and penetrate through the intestinal wall before migrating to the liver and bile ducts. To facilitate this journey, the parasite excretes and secretes an array of molecules that come into contact with host cells and tissues, the most abundant being proteases and protease inhibitors (5, 6). These molecules are important for the parasite's survival within its host and perform roles in immunomodulation, immune evasion, feeding, parasite development, and protein regulation (5–9).
Transcriptomic data analysis of the infective newly excysted juvenile (NEJ) 5 stage of F. hepatica identified a cDNA sequence that encodes a protein with homology to a Kunitz-type (KT) serine protease inhibitor (10). In their monomeric form, KT protease inhibitors are typically low molecular mass proteins of 6–8 kDa. They contain six cysteine residues that form three conserved disulfide bonds in a 1–6, 2–4, and 3–5 arrangement that maintains structural integrity of the inhibitor and allows presentation of a protease-binding loop at its surface (see Fig. 2) (11–13). A highly exposed P1 active site residue at position 15, which inserts into the S1 site of the cognate protease, is located at the peak of the binding loop and is of prime importance in determining the specificity of serine protease inhibition (14). The P1 site residue is usually arginine (Arg) or lysine (Lys), both of which have a positively charged side chain (11) and are the preferential site of interaction for the digestive protease trypsin; thus, KT protease inhibitors are classically associated with trypsin inhibition (e.g. bovine pancreatic trypsin inhibitor, BPTI) (15–17). Other serine proteases often inhibited by KT inhibitors include the digestive enzyme chymotrypsin, neutrophil elastase, and several serine proteases involved in the blood coagulation cascade, such as thrombin, kallikrein, and various other tissue factors (12, 17–21). The P1 residue in the F. hepatica KT is a leucine (Leu), which has been found in certain KT inhibitors that have a greater specificity for chymotrypsin over trypsin (11).
FIGURE 2.

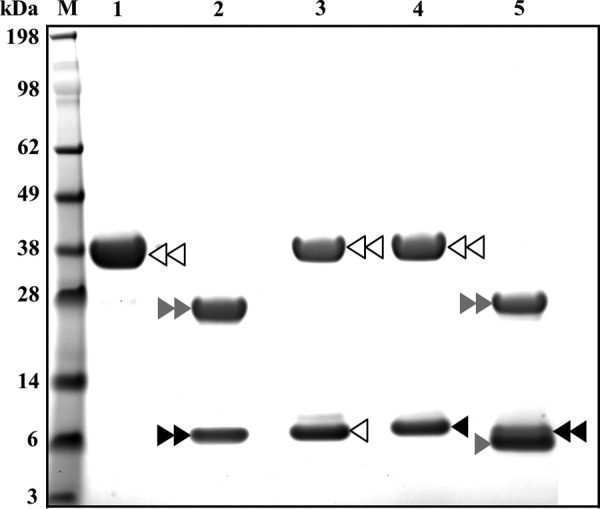
Structural representation of FhKT1 and FhKT1Leu15/Arg15 and their recombinant expression. A, sequence alignment of BPTI, FhKT1, and FhKT1Leu15/Arg15. The asterisk denotes the P1 site at position 15. Lines indicate the conserved disulfide bonds that occur between Cys1 and Cys6, Cys2 and Cys4, and Cys3 and Cys5, with cysteine residues highlighted in black bars. B, recombinant forms of FhKT1 and FhKT1Leu15/Arg15 were expressed as secretory proteins in the methylotrophic yeast P. pastoris with a yield of ∼5–10 mg of soluble protein from each 1 liter of culture. rFhKT1 (lane 1) and rFhKT1Leu15/Arg15 (lane 2) were isolated by NTA-affinity chromatography and analyzed by NuPAGE Novex 4–12% BisTris protein gel, stained with Biosafe Coomassie (Bio-Rad). C, homology model of FhKT1 built based on BPTI (PDB code 3OTJ) displaying the three disulfide bonds (yellow), the α-helix (red), and anti-parallel β-sheets (blue) characteristic of KT protease inhibitors. The P1 Leu15 located at the peak of the reactive loop is also shown in gray.
Although genes encoding KT protease inhibitors have been identified in a number of parasitic organisms (11), only a few have been characterized functionally. However, because of their putative protease-inhibition properties, they are proposed to be important in parasite defense (11). For example, in intestinal nematodes such as hookworms (22–24) and cestodes such as Echinococcus granulosus (25, 26), KT protease inhibitors may protect the parasite by inhibiting potentially harmful host digestive enzymes. However, in the guts of blood-feeding schistosome parasites (27, 28) and the secretions of biting insects (29) and ticks (30–32), KT inhibitors are suggested to obstruct blood coagulation enzymes to prevent blood clotting. A KT inhibitor with trypsin inhibitory activity has previously been identified in soluble extracts of adult F. hepatica taken from the bile ducts of ruminants (33), but its function remains obscure as it exhibited no activity against the major coagulation enzymes and only low activity (Ki = 143 nm) against trypsin.
Here, we report the discovery of a KT protease inhibitor (FhKT1) from the infective and tissue invasive stage of F. hepatica that exhibits no inhibitory activity against serine proteases but has potent activity against cathepsin L-like cysteine proteases. Substitution of the atypical P1 Leu15 that projects from the exposed reactive loop of the inhibitor with an Arg residue modestly reduces its cysteine protease inhibition profile but endows the protein with trypsin-inhibitory activity. Molecular modeling of the interaction of FhKT1 with the major secreted parasite cathepsin L cysteine proteases and human cathepsin L-like cysteine proteases indicated the importance of the P1 Leu15 in docking the inhibitor into the S2 active site of these enzymes and suggested key inhibitor-enzyme interactions that impart FhKT1 its unique inhibitory properties.
Results
Expression of fhkt1 in Newly Excysted Juveniles in F. hepatica
Transcriptome analysis of the newly excysted juvenile stage showed that fhkt1 is expressed by the early F. hepatica stages that invade the host. Differential gene expression analysis during the first 24 h post-excystment shows that fhkt1 gene is up-regulated in NEJs 24 h post-excystment compared with NEJs 3 h post-excystment (Fig. 1A). Western blot analysis using anti-FhKT1 antibodies revealed an ∼7-kDa band in medium in which NEJs were cultured for 24 h (Fig. 1B, lane 2), confirming that FhKT1 is secreted by this parasite the 1st day after excystment. Mass spectrometry analysis of NEJ-secreted products revealed that FhKT1 is released into medium by both 3- and 24-h post-excystment NEJs (Fig. 1C and supplemental file 1).
FIGURE 1.

FhKT1 is a temporally regulated and secreted protein in F. hepatica NEJ. A, graphical representation of fhkt1 expression within NEJs 3 and 24 h post-excystment in transcripts per kilobase million (TPM). Error bars indicate standard deviation of two separate experiments. B, Western blot analysis of RPMI 1640 culture medium from F. hepatica NEJs cultured for 24 h. Following the transfer of protein from LDS-polyacrylamide gel onto a nitrocellulose membrane by Western blotting, blots were spliced and probed with pre-immune mouse antiserum (lane 1) and anti-FhKT1 monoclonal antibodies raised in mice (lane 2). The immunoreactive band in lane 2 corresponding to FhKT1 is indicated by a gray arrow. M, molecular mass marker. C, graphical representation of mass spectrometry analysis of FhKT1 present in RPMI 1640 culture medium from F. hepatica NEJs cultured for 3 and 24 h, shown as unique peptide number. Error bars indicate standard deviation of three separate experiments. D, immunolocalization of FhKT1 in NEJs by confocal scanning laser microscopy. NEJs were fixed and stained as described under “Experimental Procedures.” FhKT1 expression in NEJs from two time points were compared, 3 h post-excystment (panels i–iii) and 24 h post-excystment (panels iv–vi). Fixed NEJs were probed with either anti-FhKT1 polyclonal antibodies raised in rabbit (panels ii, iii, v, and vi) or rabbit pre-immune antiserum (negative controls, panels i and iv), followed by the secondary antibody, fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG. No FITC staining was observed in the negative controls (panels i and v). Very little FITC staining (green fluorescence) was evident in the 3-h NEJs (panels ii and iii). FITC staining was observed in 24-h NEJs (panels v and vi), with FhKT1 being evident in the parenchyma (particularly concentrated in parenchymal cell bodies, white arrowheads) and gut (black arrowheads). Panels iii and vi and high power images of panels ii and v, respectively. All specimens were counter-stained with phalloidin-tetramethylrhodamine isothiocyanate (TRITC) to stain muscle tissue (red fluorescence) and provide structure. OS, oral sucker; VS, ventral sucker. Scale bars, 20 μm. M, molecular mass markers.
To localize FhKT1 expression within the NEJ, confocal microscopy was carried out on 3- and 24-h post-excystment NEJs. These were probed with polyclonal anti-FhKT1 antibodies and counter-stained with TRITC-labeled phalloidin to provide NEJ structural visualization of muscle tissue under fluorescence microscopy. Little fluorescence was observed in NEJs 3 h post-excystment using anti-FhKT1 (Fig. 1D, panels ii and iii). By contrast, in NEJs 24 h post-excystment, bright fluorescence was observed in the parenchymal tissue and particularly concentrated in parenchymal cell bodies (white arrowheads) (Fig. 1D, panels v and vi). Diffuse staining was observed within the NEJ gut (black arrowheads) (Fig. 1D, panels v and vi). This increase in fluorescence in 24-h NEJs compared with 3-h NEJs is consistent with the up-regulated transcript and protein expression observed in 24-h NEJs (Fig. 1, A and C). No FITC fluorescence was observed in negative control NEJs (3 and 24 h post-excystment) probed with pre-immune sera (Fig. 1D, panels i and iv).
F. hepatica Inhibitor FhKT1 Exhibits a Unique Specificity for Cysteine Proteases
Surrogate expression of the fhkt1 gene in the yeast Pichia pastoris allowed us to produce and purify a recombinant form of the inhibitor (rFhKT1, Fig. 2). Surprisingly, in our initial screens we found that the purified rFhKT1 did not exhibit any inhibition against a variety of standard serine proteases, including trypsin, chymotrypsin, elastase, thrombin, kallikrein, and human cathepsin G (Fig. 3A). This was not due to the recombinant protein being functionally inactive as we subsequently discovered that rFhKT1 was a potent inhibitor of two F. hepatica cathepsin L-like cysteine proteases, FhCL1 and FhCL2. In further screens, rFhKT1 was shown to inhibit human cathepsin L and cathepsin K (Fig. 3A). In these initial inhibition screens, rFhKT1 was tested at 300 nm, and the activity of FhCL1, FhCL2, and human cathepsins K and L was reduced by 98.86% (±0.205), 90.29% (±0.025), 99.54% (±0.08), and 96.79% (±0.09), respectively (Fig. 3A). Of further relevance was our observations that at a 300 nm final concentration rFhKT1 did not show any inhibition of F. hepatica cathepsin B1 (FhCB1) and B2 (FhCB2) or of human cathepsin B and cathepsin S (Table 1). In parallel assays, we showed that the activity of trypsin, chymotrypsin, neutrophil elastase, and cathepsin G was completely inhibited by 300 nm BPTI and that thrombin and kallikrein were completely inhibited by 0.002 units of hirudin (data not shown). Given that the concentration of FhKT1 inhibitor required for the inhibition of the cathepsin L and K cysteine proteases was found to be lower than the concentration of enzyme required to produce a reliable activity curve in fluorogenic assays, these were considered to be tight-binding inhibitors, and thus enzymatic data were fitted to the Morrison equation (Fig. 4). Resulting Kiapp values were fitted to Equation 2 mentioned above to determine Ki values. Kinetic inhibition constants determined at pH 5.5 for rFhKT1 against the two parasite proteases differed substantially; the Ki for FhCL1 was 0.4 (±0.1) nm and for FhCL2 was 10 (±0.3) nm. The inhibition constant was 1.6 (±0.1) nm for human cathepsin L and 5 (±0.3) nm for human cathepsin K (Table 1).
FIGURE 3.

Inhibition profile of FhKT1 and FhKT1Leu15/Arg15 against cysteine and serine proteases. A, inhibitory activity of rFhKT1 (black bars) and rFhKT1Leu15/Arg15 (gray bars) at 300 nm was screened using a range of cysteine proteases, including F. hepatica cathepsin L1 (FhCL1), F. hepatica cathepsin L2 (FhCL2), F. hepatica cathepsin B1 (FhCB1), F. hepatica cathepsin B2 (FhCB2), human cathepsin K (HsCK), human cathepsin L (HsCL), human cathepsin B (HsCB), human cathepsin S (HsCS), and serine proteases, including trypsin, chymotrypsin, neutrophil elastase (NE), cathepsin G, thrombin, and kallikrein. Inhibition is presented relative to the activity of each enzyme in the absence of inhibitors. B, activity of FhCL1 in the presence of 300 nm rFhKT1 (black bars) and rFhKT1Leu15/Arg15 (gray bars) at different pH values (see “Experimental Procedures” for details of buffer systems) relative to FhCL1 activity in the absence of inhibitors. C, activity of FhCL2 in the presence of 300 nm rFhKT1 (black bars) and rFhKT1Leu15/Arg15 (gray bars) at different pH values, relative to FhCL2 activity in the absence of inhibitors. Error bars indicate standard deviation of three separate experiments.
TABLE 1.
rFhKT1 and rFhKT1Leu15/Arg15 inhibition specificity against a panel of biologically relevant serine and cysteine proteases and relative inhibition constants
| Enzyme (concentration) | rFhKT1 |
rFhKT1Leu15/Arg15 |
||
|---|---|---|---|---|
| Inhibition | Ki | Inhibition | Ki | |
| Cysteine proteases | ||||
| F. hepatica cathepsin L1 (2.7 nm) | + | 0.4 nm (± 0.1 nm) | + | 0.7 nm (± 0.04 nm) |
| F. hepatica cathepsin L2 (5 nm) | + | 10 nm (± 0.3 nm) | + | 27 nm (± 1.5 nm) |
| F. hepatica cathepsin B1 (180 nm) | − | Not inhibited | − | Not inhibited |
| F. hepatica cathepsin B2 (180 nm) | − | Not inhibited | − | Not inhibited |
| Human cathepsin L (0.2 nm) | + | 1.6 nm (± 0.1 nm) | + | 3 nm (± 0.1 nm) |
| Human cathepsin K (2 nm) | + | 5 nm (± 0.3 nm) | + | 10 nm (± 0.2 nm) |
| Human cathepsin S (2 nm) | − | Not inhibited | − | Not inhibited |
| Human cathepsin B (3.6 nm) | − | Not inhibited | − | Not inhibited |
| Serine proteases | ||||
| Trypsin (42 nm) | − | Not inhibited | + | 1.5 nm (± 0.7 nm) |
| Chymotrypsin (40 nm) | − | Not inhibited | − | Not inhibited |
| Neutrophil elastase (50 nm) | − | Not inhibited | − | Not inhibited |
| Cathepsin G (100 nm) | − | Not inhibited | − | Not inhibited |
| Thrombin (43 pm) | − | Not inhibited | − | Not inhibited |
| Kallikrein (150 nm) | − | Not inhibited | − | Not inhibited |
FIGURE 4.
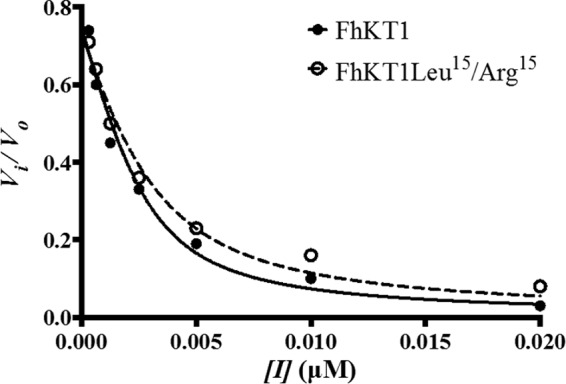
Plot of F. hepatica cathepsin L1 proteolytic activity with the substrate Z-Leu-Arg-NHMec, as a function of inhibitor concentration for rFhKT1 (solid circles) and rFhKT1Leu15/Arg15 (clear circles). The curves represent the best fit to the Morrison equation (solid line = rFhKT1; dashed line = rFhKT1Leu15/Arg15). Error bars indicate standard deviation of three separate experiments.
Impact of Substituting the FhKT1 P1 Leu Residue to Arg on Protease Inhibition
Given the importance of the P1 residue at position 15 within the exposed loop in KT protease inhibitors, we examined the impact of substituting this residue in FhKT1 from a leucine to the more commonly found arginine. This variant form, termed rFhKT1Leu15/Arg15, retained its ability to inhibit F. hepatica FhCL1 and FhCL2 and human cathepsin L and K, but as with the wild-type rFhKT1, it did not inhibit FhCB1 and FhCB2 and human cathepsin B and S (Table 1). However, several key observations were made, including the following: (a) rFhKT1Leu15/Arg15 was less inhibitory than rFhKT1 with Ki 2–3-fold higher against the parasite and mammalian cysteine proteases (Table 1); (b) although the inhibition of FhCL1 and FhCL2 by both rFhKT1 and rFhKT1Leu15/Arg15 was effective over a broad pH range of 4.5–8.0 and optimal at neutral pH, the variant inhibitor was less effective in the lower pH range (Fig. 3, B and C); (c) in contrast to rFhKT1, the variant rFhKT1Leu15/Arg15 possessed potent inhibitory activity against the serine protease trypsin, with trypsin activity reduced by 88.92% (±0.89) in assays performed at 300 nm inhibitor (Fig. 3A). rFhKT1Leu15/Arg15 exhibited a Ki of 1.5 nm (±0.7 nm) against trypsin (Table 1), which demonstrates that the substitution of Leu to Arg is sufficient to endow this inhibitor with the ability to bind within the active site of this serine protease. However, it is noteworthy that this inhibitory constant is still 10 times higher than that recorded for BPTI (0.15 nm), and even at 300 nm, rFhKT1Leu15/Arg15 did not show any inhibition toward the other serine proteases tested, including chymotrypsin, elastase, thrombin, kallikrein, and human cathepsin G (Fig. 3A).
rFhKT1 Prevents the Auto-catalytic Activation of Parasite Cysteine Proteases
The parasite cysteine proteases FhCL1 and FhCL2 are produced as inactive 37-kDa zymogens (Fig. 5, lane 1) that can be auto-catalytically activated in vitro at pH 4.5 (34). To determine whether rFhKT1 and rFhKT1Leu15/Arg15 could regulate the activation of the parasite proteases, these inhibitors were mixed with zymogens of FhCL1 and FhCL2 at pH 4.5, and the activation was monitored by LDS-PAGE. After 45 min of incubation at 37 °C in citrate phosphate buffer, pH 4.5, FhCL1 auto-activates to give rise to an ∼25-kDa mature active form and a released 6-kDa propeptide (Fig. 5, lane 2). Incubation of FhCL1 with rFhKT1 or rFhKT1Leu15/Arg15 prevented the appearance of both of these bands indicating that auto-catalytic cleavage was prevented (Fig. 5, lanes 3 and 4). Similar results were observed for FhCL2 (data not shown). For comparison, we showed that co-incubation of FhCL1 with the KT inhibitor BPTI did not prevent auto-catalytic activation of the zymogen (Fig. 5, lane 5).
FIGURE 5.
Inhibition of the auto-catalytic activation of FhCL1 by rFhKT1 and rFhKT1Leu15/Arg15. M, molecular mass marker; lane 1, FhCL1 zymogen; lane 2, mature FhCL1 following auto-catalytic activation at pH 4.5 for 1 h; lane 3, FhCL1 and rFhKT1 following incubation at pH 4.5 for 1 h; lane 4, FhCL1 and rFhKT1Leu15/Arg15 following incubation at pH 4.5 for 1 h; lane 5, FhCL1 and bovine pancreatic trypsin inhibitor following incubation at pH 4.5 for 1 h. Double white arrowhead indicates FhCL1 zymogen; double gray arrowhead indicates mature FhCL1, and double black arrowhead indicates FhCL1 pro-peptide removed following auto-catalytic activation. Single white arrowhead indicates position of FhKT1; single black arrowhead indicates FhKT1Leu15/Arg15, and single gray arrowhead shows position of BPTI. Proteins were separated on a NuPAGE Novex 4–12% BisTris protein gel and stained with Biosafe Coomassie (Bio-Rad). M, molecular mass markers.
To examine the stability of inactivation of FhCL1 by rFhKT1 over time, FhCL1 was first auto-catalytically activated at pH 4.5 and then incubated with rFhKT1 for 6 h at 37 °C in citrate phosphate buffer, pH 5.5, in the presence of substrate; the reaction was monitored by the release of the fluorophore (7-amino-4-methylcoumarin), and samples of the reactions were analyzed by LDS-PAGE (Fig. 6). rFhKT1 completely inactivated the FhCL1, and this inactivation was stable for the entire 6 h (Fig. 6A). Furthermore, the rFhKT1 remained intact over this time and was observed as a band at ∼6 kDa. However, a minor band at ∼5 kDa was observed at the 2- and 6-h time points (Fig. 6B). This band was not immunoreactive with anti-His tag monoclonal antibodies (Fig. 6D), but it was reactive with anti-rFhKT1 polyclonal antibodies (Fig. 6C), showing that this protein results from proteolytic clipping at the C terminus of rFhKT1 by FhCL1. A similar cleavage pattern was observed with rFhKT1Leu15/Arg15 and also when FhCL2 was used in the reactions (data not shown).
FIGURE 6.
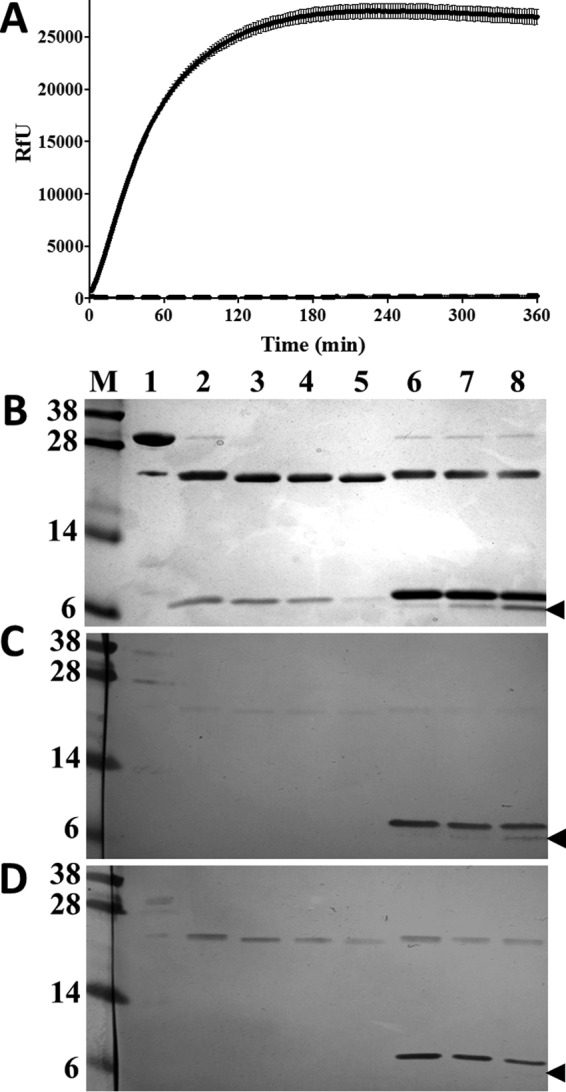
rFhKT1 inhibits mature FhCL1 and forms a stable complex. A, progress curve of fully activated FhCL1 inhibited by rFhKT1 over 360 min. Activity is represented by relative fluorescent units. Activity of uninhibited FhCL1 is represented by the solid line, and rFhKT1-inhibited FhCL1 is represented by the dashed line. Error bars indicate standard deviation of three separate experiments. B, FhCL1 zymogen (lane 1) was auto-catalytically activated at pH 4.5 for 1 h to the mature active enzyme (lane 2). The mature enzyme was then incubated in the absence (lanes 3–5) or presence (lanes 6–8) of rFhKT1 for 1 h (lanes 3 and 6), 2 h (lanes 4 and 7), and 6 h (lanes 5 and 8), and then the reaction mixes were analyzed by LDS-PAGE. The protein gel was stained with Biosafe Coomassie (Bio-Rad). Replicate gels were used for Western blot analysis by probing with polyclonal anti-rFhKT1 (C) and polyclonal antibody to His6-tagged proteins polyclonal antibodies (D). The arrowhead indicates the position of a cleaved product of ∼5-kDa band that reacts with polyclonal anti-rFhKT1 but not anti-His6-tagged antibodies indicating that it represents a cleavage product of rFhKT1. M, molecular mass markers.
Inhibition assays that were extended for up to 7 days showed that FhCL1 activity remained inhibited by rFhKT1 over this prolonged period. However, the activity of mature FhCL1 (in the absence of inhibitor) depleted by >99% over the course of the experiment, with activity reduced from 21,271.27 (± 961.52) RfU/min at 0 days to 65.73 (± 14.49) RfU/min at day 7 (data not shown).
Inhibitory Activity of rFhKT1 Is Dependent on Internal Disulfide Bridges
Reduction and alkylation of both rFhKT1 and rFhKT1Leu15/Arg15 were performed to break internal disulfide bridges and to disrupt the tertiary structure of the protein. This procedure depleted the inhibitory activity of both inhibitors against FhCL1 (Fig. 7A). Similarly, this treatment ablated the inhibitory activity of rFhKT1Leu15/Arg15 against trypsin (Fig. 7B). Interestingly, we discovered that both rFhKT1 and rFhKT1Leu15/Arg15 could partially re-nature to their functional form during a 10-day incubation period at 4 °C; over this time, the inhibitors regained between 60 and 80% of their inhibitory activity (Fig. 7A). Likewise, rFhKT1Leu15/Arg15 regained inhibitory activity against trypsin (Fig. 7B).
FIGURE 7.
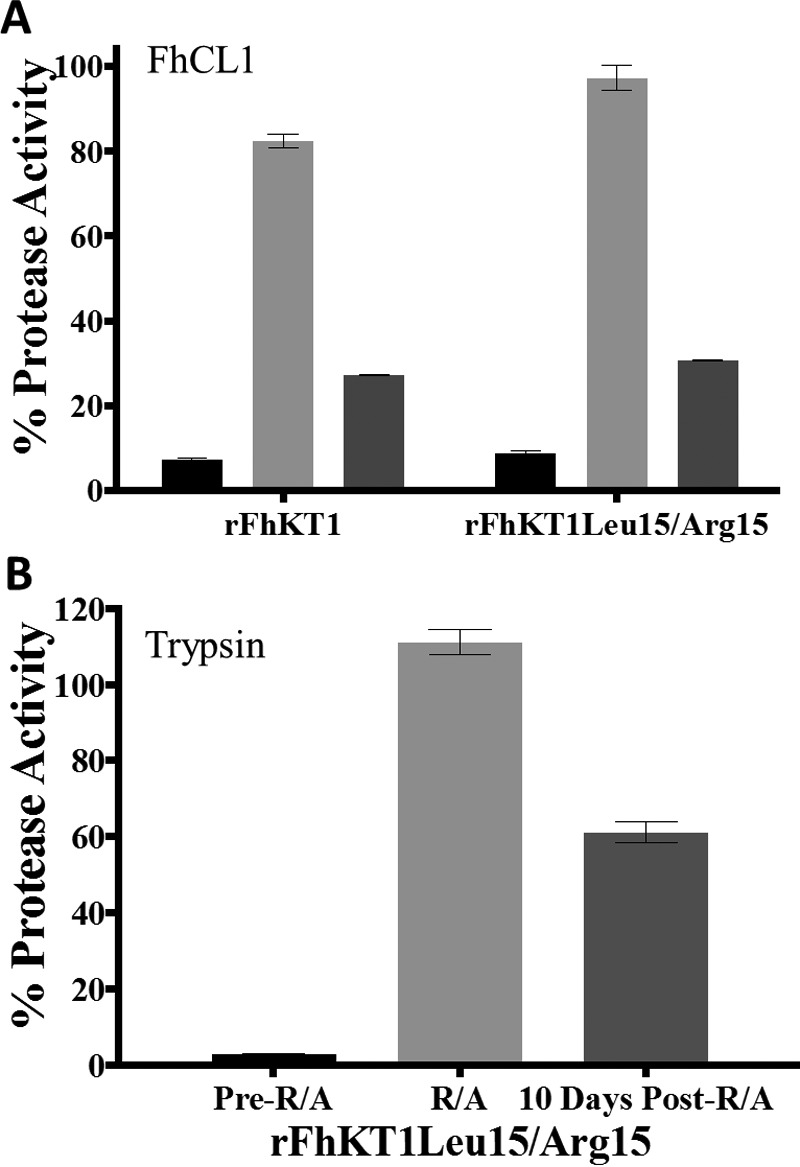
Inhibitory activity of rFhKT1 and rFhKT1Leu15/Arg15 is dependent on disulfide bridges. A, inhibition of FhCL1 activity by rFhKT1 and rFhKT1Leu15/Arg15 before (black bars) and after reduction (R) and alkylation (A) (light gray bars) and with reduced and alkylated inhibitor following incubation for 10 days at 4 °C (dark gray bars). B, inhibition of trypsin activity by rFhKT1Leu15/Arg15 before (pre-reduction and alkylation (R/A), black bars) and after reduction and alkylation (R/A, light gray bars), and with reduced and alkylated inhibitor following incubation for 10 days at 4 °C (post-R/A, dark gray bars). Error bars indicate standard deviation.
rFhKT1 Interacts with Several Native F. hepatica Cysteine Proteases
Adult F. hepatica parasites secrete several cysteine proteases, both in vivo and in vitro. To demonstrate that FhKT1 can bind and inhibit native parasite proteases, the inhibitors were mixed with culture medium in which adult F. hepatica parasites had been maintained (F. hepatica excretory-secretory proteins), and activity was monitored using Z-Phe-Arg-NHMec as substrate; both inhibitors blocked >80% of total secreted cathepsin L-like activity (Fig. 8A).
FIGURE 8.

rFhKT1 binds and inhibits native F. hepatica-secreted cysteine proteases. A, cysteine protease activity (presented as RfU) in adult F. hepatica RPMI 1640 culture media measured with the fluorogenic peptide substrate, Z-Phe-Arg-NHMec, in the absence (circles) or presence of rFhKT1 (squares) or rFhKT1Leu15/Arg15 (triangles). B, cysteine protease activity in the adult F. hepatica RPMI 1640 culture media following pulldown using Ni-NTA beads alone (circles), rFhKT1 and Ni-NTA beads (squares), or rFhKT1Leu15/Arg15 and Ni-NTA beads (triangles). C, LDS-PAGE analysis of pulldown from adult F. hepatica RPMI 1640 culture media with Ni-NTA beads alone (lane 1) and with rFhKT1 and Ni-NTA beads (lane 2), stained with Biosafe Coomassie (Bio-Rad). LC-MS/MS analysis of the 26-kDa band observed in lane 2 revealed that it consisted of FhCL1, FhCL2, and FhCL5 in their fully active mature forms. M, molecular mass marker.
To identify the native cysteine proteases to which rFhKT1 bound, this inhibitor was added to media in which adult parasites were cultured for 5 h. Following the parasite culture, the media were removed, and rFhKT1 was pulled down with Ni-NTA beads; fluorogenic assays demonstrated that removal of the inhibitors from each medium, respectively, also resulted in the depletion of >95% of the cysteine protease activity secreted by the adult parasites (Fig. 8B). LDS-PAGE analysis of the proteins bound to the Ni-NTA beads revealed a 6-kDa band representing the rFhKT1 and another band of ∼25 kDa, which is in keeping with the size of mature cathepsin L cysteine proteases (Fig. 8C). The 6- and ∼25-kDa proteins were not detected when Ni-NTA beads were added to the culture media containing no recombinant inhibitor. LC-MS/MS analysis verified that the ∼25-kDa band pulled down by rFhKT1 consisted of a mix of F. hepatica cysteine proteases in their mature forms, including FhCL1, FhCL2, and another cathepsin L protease known as FhCL5 (supplemental file 2) (6, 35).
FhKT1 Binds to the Cysteine Protease Active Site in a Unique Manner
To understand the molecular basis of FhKT1 binding to cathepsins, a homology model of FhKT1 was built using the crystal structure of BPTI as a template. Although protein sequence identity between BPTI and FhKT1 is only 23%, both possess the six conserved cysteine residues that form three disulfide bonds (11), suggesting similar folding (root mean square deviation = 1.3 Å). The homology model of FhKT1 was docked to the crystal structure of FhCL1 previously reported by us (36). The rigid body protein-protein docking protocol was employed, and the selected docking solution was optimized using molecular dynamic simulations. A docking solution in which the P1 Leu15-containing side of FhKT1 faces the active site of FhCL1, similar to the BPTI binding orientation in trypsin in the crystal structure, was used for optimization. The resultant protein-protein complex indicates that FhKT1 binds sufficiently tight within the active site groove of FhCL1 to block access of small substrates (e.g. Z-Phe-Arg-NHMec) to the reactive cysteine (Cys132) in the S1 active site pocket (Fig. 9, A and B).
FIGURE 9.

Molecular model of FhKT1-F. hepatica cathepsin L1 interactions. The homology model of FhKT1 (A and B) and FhKT1Leu15/Arg15 (C and D) built based on BPTI (PDB code 3OTJ) and the crystal structure of F. hepatica cathepsin L (PDB code 2O6X) are shown in pink and gray schematics, respectively. The interacting residues of FhKT1 and FhCL1, the residues of the FhCL1 S1 pocket (Cys132, His269, and Asn289), and the residues of the S2′ pocket (Gln126, Gly127, and Asn128) and Glu178 are visualized in stick-like representation. The residues of the S2 pocket, the residues of S2′ pocket, and other interacting residues of FhCL1 are shown in purple, green, and gray, respectively. Hydrogen bonds and hydrophobic interactions are in black and green, respectively. Residue labels are in blue for FhKT1 and black for FhCL1. A and B, Leu15 of FhKT1 is predicted to sit in the hydrophobic S2 pocket of FhCL1, forming contacts with Leu176, Met177, and Val267. In addition, it is indicated that FhKT1 forms H-bonds with Asn128 of the S2′ pocket. It is also predicted that Gly16 and Gly17 block substrate access to the S1 active site pocket by being positioned across it, without actually penetrating the pocket. C and D, Arg15 of FhKT1Leu15/Arg15 is predicted to sit in the hydrophobic S2 pocket of FhCL1, forming contacts with Leu176, Met177, and Val267. The guanidine group of Arg15 is predicted to interact with Glu178 via H-bonds. In addition, as for FhKT1 it is also indicated that Gly16 and Gly17 sit across the S1 active site pocket without penetrating the pocket itself, thus blocking substrate access. FhKT1Leu15/Arg15 is also predicted to form H-bonds with Asn128 of the S2′ pocket. Panels at right are enlarged versions of the left, with focus toward the FhCL1 active site.
In this model of interactions, the residue Leu15 of the exposed loop of FhKT1 forms strong hydrophobic interactions with residues Leu176, Met177, and Val267 of the S2 pocket of FhCL1 (Fig. 9, A and B). This places Gly16 and Gly17 across the S1 pocket, but neither penetrates this subsite. The Ile18 is not predicted to make any interactions with the FhCL1, but Arg19 forms an H-bond with the Asn128 of the S2′ pocket. Relevantly, the sequence alignment of the cysteine proteases (Tables 2 and 3 and Fig. 10) used in this study shows that residues of the S2 and S1′-S2′ pockets are highly similar in physicochemical properties in FhCL1, FhCL2, human cathepsins L and K, which are all inhibited by FhKT1. By contrast, these residues are diverse in F. hepatica cathepsins FhCB1 and FhCB2, and in human cathepsins B and S, which are not inhibited by FhKT1 (Tables 2 and 3 and Fig. 10). This observation suggests that the S2 and S1′-S2′ pockets play a crucial role in selective binding of FhKT1 to FhCL1, FhCL2, and human cathepsins L and K.
TABLE 2.
Comparison of residues forming the S2 active site of cysteine proteases from F. hepatica and H. sapiens
| S2 residuesa |
||||||
|---|---|---|---|---|---|---|
| 176 | 177 | 242 | 267 | 270 | 316 | |
| F. hepatica CL1 | Leu | Met | Ala | Val | Ala | Leu |
| F. hepatica CL2 | Tyr | Met | Ala | Leu | Ala | Leu |
| Human cathepsin K | Tyr | Met | Ala | Leu | Ala | Leu |
| Human cathepsin L | Leu | Met | Ala | Met | Gly | Ala |
| F. hepatica CB1 | Gln | Cys | Gly | Gly | Ala | N/A |
| F. hepatica CB2 | Lys | Cys | Thr | Gly | Ala | N/A |
| Human cathepsin S | Phe | Met | Gly | Val | Gly | Phe |
| Human cathepsin B | Pro | Cys | Ala | Gly | Ala | Val |
a Residue numbering is based on FhCL1 (shown in pink in Fig. 10).
TABLE 3.
Comparison of residues surrounding the S1 active site region of cysteine proteases from F. hepatica and H. sapiens (based on sequence alignment)
| Residue no.a |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 125 | 126 | 127 | 128 | 129 | 130 | 131 | 132b | 133 | 134 | 135 | 136 | |
| F. hepatica CL1 | Asp | Gln | Gly | Asn | Cys | Gly | Ser | Cys | Trp | Ala | Phe | Ser |
| F. hepatica CL2 | Asp | Gln | Gly | Gln | Cys | Gly | Ser | Cys | Trp | Ala | Phe | Ser |
| Human cathepsin K | Asn | Gln | Gly | Gln | Cys | Gly | Ser | Cys | Trp | Ala | Phe | Ser |
| Human cathepsin L | Asn | Gln | Gly | Gln | Cys | Gly | Ser | Cys | Trp | Ala | Phe | Ser |
| F. hepatica CB1 | Ser | Ala | Ile | Asp | Leu | Val | Ser | Cys | Cys | Ser | Tyr | Cys |
| F. hepatica CB2 | Ala | Ala | Ala | Asp | Pro | Leu | Ser | Cys | Cys | Thr | Tyr | Cys |
| Human cathepsin S | Tyr | Gln | Gly | Ser | Cys | Gly | Ala | Cys | Trp | Ala | Phe | Ser |
| Human cathepsin B | Ser | Ala | Glu | Asp | Leu | Leu | Thr | Cys | Cys | Gly | Ser | Met |
a Residue numbering is based on FhCL1.
b Active site cysteine residues are shown in boldface.
FIGURE 10.

Sequence alignment of F. hepatica and Homo sapiens cysteine proteases. F. hepatica cysteine protease sequences include FhCL1 (cathepsin L1, AAB41670), FhCL2 (cathepsin L2, AAC47721), FhCB1 (cathepsin B1, ABF85678), and FhCB2 (cathepsin B2, ABF85679). H. sapiens cysteine protease sequences include HsCK (cathepsin K, NP_000387), HsCL (cathepsin L, NP_001903), HsCS (cathepsin S, NP_004070), and HsCB (cathepsin B, NP_001899). Residues surrounding the S1 active site region are shown in green, and residues forming the S2 active site are shown in pink. Residues that form interactions with FhKT1 and FhKT1Leu15/Arg15 are shown in bold.
The binding orientation we obtained for the P1 Leu15 helps explain the interactions of Arg at this position in the variant rFhKT1Leu15/Arg15. The hydrophobic tail of Arg15 can form hydrophobic interactions with the S2 pocket, whereas the guanidine group could form H-bonds with Glu178 of FhCL1 (Fig. 9, C and D). Intriguingly, the other cathepsins that we have shown to bind FhKT1Leu15/Arg15, i.e. FhCL2 and human cathepsins L and K, also have a residue capable of forming H-bonds at this position (Table 2 and Fig. 10).
Unlike BPTI, FhKT1 does not bind to trypsin and other serine proteases tested in this study. In the available crystal structures of the BPTI-trypsin complex, the P1 Lys15 of BPTI forms H-bonds with Asp194 and Ser195 of the trypsin S1 (37). However, the hydrophobic P1 Leu15 of FhKT1 is unlikely to bind to the hydrophilic S1 pocket of serine proteases preventing the formation of the protein-protein complex. Binding of rFhKT1Leu15/Arg15 to trypsin, however, suggests that the P1 Arg15 can form these crucial H-bonds in the S1 pocket.
Kunitz inhibitors with P1 Leu often exhibit an increased specificity for chymotrypsin over trypsin (11). In this study, however, we found that FhKT with a P1 Leu15 does not inhibit chymotrypsin. Our modeling of these two molecules suggests that this could be due to different structuring of surface polypeptide loops of FhKT1 and chymotrypsin that are unable to establish a complementary interface to form a complex and allow the P1 Leu15 to enter the active site.
Discussion
We have shown that the infective stage of the helminth parasite F. hepatica expresses a temporally regulated and secreted Kunitz-type (KT) protease inhibitor (FhKT1) with novel inhibition properties. Unlike any previously described KT protease inhibitors, FhKT1 exhibits exclusive and stable inhibition of cysteine proteases, specifically cathepsin L- and K-like cysteine proteases. Recombinant FhKT1 inhibits these cysteine proteases across a wide pH range, from pH 4.5 to 8.5, with optimal inhibition at neutral pH. Our studies suggest that the six conserved cysteine residues that maintain the structural integrity of the central domain of KT-type inhibitors perform a similar function in FhKT1 as reduction and alkylation of the disulfide cross-links abrogate inhibitory activity.
Previously, we resolved the three-dimensional structure of FhCL1 and showed that the active site is typical of the cathepsin L cysteine proteases, consisting of a deep open groove created between the right and left domains of the enzyme (36). Here, we built a homology model of FhKT1 on the basis of the known BPTI structure and docked it into the FhCL1 active site to provide insights into the interactions responsible for the binding of FhKT1 to cathepsin L-like proteases. The P1 Leu15 in FhKT1, which projects from the peak of the inhibitor reactive loop, is predicted to form crucial hydrophobic interactions with residues in the FhCL1 S2 active site pocket (Leu176, Met177, and Val267), whereas residue Arg19 of FhKT1 is predicted to form H-bond interactions with Asn128 within the S2′ pocket, respectively. Because FhKT1 spans these S and S′ subsites, it places the Gly16 and Gly17 residues in a position whereby they block access of even small molecules, such as Z-Phe-Arg-NHMec, to the reactive Cys132 residue in the S1 pocket that is essential for forming a thiol intermediate with the substrate before the scissile bond is cleaved.
It is intriguing that FhKT1 also exhibits specificity within the cathepsin-like cysteine proteases themselves by not inhibiting parasite or human cathepsin B and human cathepsin S. This specificity of inhibition was explained by our computational modeling and sequence comparison that revealed a distinct difference in the composition of the S2 and S1′-S2′ active site pockets within this group of proteases; these changes reduce their hydrophobicity and alter charge arrangements such that they cannot interact with the P1 Leu15 (see Tables 2 and 3 and Fig. 10). It is noteworthy that the occluding loop that is common in cathepsin B proteases and is responsible for conferring these enzymes with carboxypeptidase activity (38) does not prevent access of FhKT1 to the active site.
Although the P1 Leu15 of FhKT1 was predicted to be important for interactions with the hydrophobic S2 pocket of the cathepsin L-like proteases, this residue could be replaced by Arg (FhKT1Leu15/Arg15). The P1 Arg15 did exert some adverse effects, especially at low pH, on the inhibitor's activity, but it did not alter its specificity against cysteine proteases. Molecular docking with FhCL1 indicated that the hydrophobic tail of Arg15 could still form hydrophobic interactions with the S2 pocket, similar to those predicted for Leu15. In addition, the guanidine group of Arg15 is predicted to form H-bonds with Glu178. These interactions could not take place in the cathepsin B and S proteases, and therefore, the variant showed no activity against these enzymes. Other amino acid replacements at position 15, however, could likely have a more dramatic effect on the FhKT1's inhibitory properties.
There are no previous reports of a KT-type protease inhibitor that exclusively inhibits cysteine proteases like FhKT1, although there are several that describe members with low cross-activity against other proteases. For example, a KT inhibitor from the hard tick Boophilus microplus (BmTIsint) blocks serine proteases (trypsin and plasma kallikrein, Ki = 3.3 and 16.5 nm, respectively), as well as the cysteine protease human cathepsin L (Ki = 108 nm) (39), and the KT inhibitor (ShPI-1A) from the sea anemone Stichodactyla helianthus exhibits broad inhibitory specificity toward serine (trypsin Ki = 0.1 nm, chymotrypsin Ki = 2.3 nm, human plasmin Ki = 2.7 nm, and porcine kallikrein Ki = 85 nm), cysteine (Ki for papain not determined), and aspartic proteases (porcine pepsin Ki = 33 nm) (40, 41). Like rFhKT1Leu15/Arg15, these KT inhibitors possess at P1 Arg15 that accounts for their serine protease activity; however, apart from BmTIsint having a Glu39 residue at a similar position to FhKT1, it is not possible to fully explain their cysteine protease inhibitory activity from primary sequence comparisons alone (39). Interestingly, a KT inhibitor reported from the blood feeding horn fly Hematobia irritans irritans has a P1 Arg15 and residues Glu10 and Arg44 similar to FhKT1 as well as an uncommon residue at position 39 (His39) (42), which implies that it could inhibit both serine and cysteine proteases. Clarification of the residues involved in restricted and broad protease inhibition will emerge as this and other KT inhibitors are more fully characterized and will inform the development of methods for functional predictions. For now, however, we suggest that KT inhibitors should be classified into subgroups based on their inhibition profile, including KT serine-specific protease inhibitors, KT cysteine-specific protease inhibitors, KT serine/cysteine protease inhibitors, and even KT serine/cysteine/aspartic protease inhibitors. Our study also points toward the opportunity of designing specific and broad inhibitors against proteases of various mechanistic classes by making defined alterations to the basic 68-amino acid scaffold of the KT inhibitor.
To facilitate penetration of the host intestine and migration through the liver, the infective juvenile parasite secretes an array of temporally regulated cathepsin L proteases (36). We have shown that the expression of FhKT1 is also highly up-regulated in the first 24 h after excystment. Protein expression was shown by immunolocalization studies to be associated most particularly with cells bodies within the parenchymal tissue. The parenchymal tissues fill the spaces between the internal organs of the parasite and are suggested to function as a flexible cytoskeleton and an important transport system (43, 44). The cell bodies, which are separated from each other within the parenchymal tissue, are highly metabolically active and contain numerous mitochondria and Golgi complexes (44, 45). They act as a store for carbohydrate reserves, such a glycogen, and are important in parasite metabolism and growth as they become depleted as the parasite develops in the first 24 h (46). The cell bodies have contacts with the gastrodermal cells lining the parasite gut where we detected more diffuse labeling of the FhKT1. The up-regulation of the FhKT1 in the cell bodies and gut correlates with the parasite's penetration of the duodenum wall after excystment, which is facilitated by the secretion of gut cysteine proteases suggesting that the inhibitor plays a role in controlling proteolytic activity at this crucial phase of infection.
We also showed by pulldown experiments that FhKT1 binds and inactivates native FhCL1, FhCL2, and FhCL5 secreted by the adult parasite and thus has broad specificity for the parasite's cathepsin Ls. Importantly, FhKT1 prevents the auto-catalytic activation of the inactive cathepsin zymogen to the mature active enzyme. In a similar manner to the function of BPTI, which regulates trypsin activation and activity to prevent excessive tissue damage in the pancreas (47), FhKT1 may control the activation of the parasite-secreted cathepsin Ls, preventing aberrant activation within the parasite itself (9, 48). Limiting cysteine protease activity would be important to the parasite's survival in the host, because although the enzymes are necessary for degrading host blood and tissues, they also make significant contributions to the clinical manifestations of the disease, including tissue damage, hemorrhaging, and induction of inflammatory immune responses. By complexing and inactivating excess secreted proteases, FhKT1 could also restrict this pathology and prevent transport of damaging proteases via the circulation that could cause tissue inflammation at sites distant from the parasite. In a previous study, Bozas et al. (33) isolated a fraction from adult F. hepatica containing a KT inhibitor with activity against trypsin. Furthermore, an F. hepatica KT inhibitor was detected among a preparation that was isolated by affinity purification of adult F. hepatica excretory-secretory products using anti-cysteine protease monoclonal antibodies (MM3) (49). These studies show that the mature parasite also expresses an FhKT and suggest that the parasite produces more than one KT inhibitor that is possibly temporally regulated alongside the secreted cathepsin Ls. It is noteworthy that the parasite does not secrete any serine proteases during its development in the mammalian host and, therefore, would not require a specific serine-protease inhibitor.
Cysteine protease inhibitors (e.g. cystatins) have also been implicated in the modulation of host immune responses (50, 51). We have shown that FhKT1 inhibits human cathepsins L and K, both of which are important proteases of the host immune system. Human cathepsin L functions in the proteolytic processing of antigens in the lysosome for presentation on the surface of antigen-presenting cells (52–54). If taken up by phagocytic cells, FhKT1 inhibition of human cathepsin L could impair this processing and thus dampen adaptive cellular and antibody responses to the parasite. Indeed, previous work in our laboratory has shown that a similarly sized molecule (6 kDa) termed helminth defense molecule secreted by F. hepatica is readily taken up by macrophages and impairs antigen processing and presentation (55). Human cathepsin K also plays a critical immune regulatory function as it is involved in Toll-like receptor 9 signaling in dendritic cells (56), and therefore its inhibition would significantly downplay dendritic cell activation. Interestingly, Falcón et al. (57) recently reported that a fraction isolated from adult F. hepatica that prevented dendritic cell activation in mice contained a KT protease inhibitor, leading the authors to propose that the inhibitor may have an important role in the immune evasion mechanisms of the parasite. Given the potential role of FhKT1 in parasite infection, through the regulation of the major parasite cysteine proteases and cysteine proteases implicated in the host immune system, FhKT1 represents a novel vaccine and drug target.
In summary, the juvenile infective stage of the helminth F. hepatica secretes a KT protease inhibitor that atypically lacks the ability to inhibit serine proteases while simultaneously possessing a unique capacity to inhibit cathepsin L-like cysteine proteases. The particular arrangement of functional amino acids predicted to confer this molecule with its unique properties has provided valuable new information and understanding of protease inhibition by KT protease inhibitors. Furthermore, it has opened up the possibility of designing new inhibitors that could target proteases of various mechanistic classes that are important in infectious and non-infectious diseases.
Experimental Procedures
fhkt1 Gene and Protein Expression in F. hepatica Newly Excysted Juveniles
RNA sequencing and gene expression analysis were carried out in duplicate for 3 and 24 h post-excystment of NEJs as described by Cwiklinski et al. (6). The fhkt1 gene was identified by putative annotation of the F. hepatica gene models using in silico tools (Uniprot, Gene Ontology (GO), and InterProScan) (6) and BLAST analysis against the F. hepatica genome using a previously identified adult F. hepatica Kunitz sequence (Fh_Contig2704, Young et al. (10)). Differential fhkt1 gene expression at the 3- and 24-h NEJ life stages was investigated using transcriptome data (European Nucleotide Archive accession number PRJEB6687; Cwiklinski et al. (6)) mapped to the gene models identified within the genome (represented as transcripts per kilobase million).
NEJs (North American isolate; Baldwin Aquatics Inc.) were cultured in RPMI 1640 medium for 24 h to obtain their secretory products as described previously (5). These were concentrated by ultrafiltration to 1 mg/ml and stored at −80 °C. Approximately 5 μg of protein was subjected to LDS-PAGE and Western blotting (5) and probed with anti-FhKT1 (1:1000 dilution) raised in mice against the peptide sequence Cys-Glu-Gly-Asn-Asp-Asn-Arg-Phe-Asp-Ser-Lys-Ser-Ser-Cys. As a negative control, another sample was probed with pre-immune mouse antiserum at the same dilution.
NEJ-secreted products were characterized using the proteomics platform of the Quebec Genomics Center, Quebec City, Quebec, Canada. Samples of RPMI 1640 medium in which NEJs (North American isolate; Baldwin Aquatics Inc.) were cultured for 3 and 24 h were analyzed by LC/MS-MS, performed on a TripleTOF 5600 mass spectrometer fitted with a nanospray III ion source (ABSciex Concord, Ontario, Canada), and coupled to an Agilent 1200 HPLC. FhKT1 was identified in MS/MS samples (three separate samples for both time points) analyzed using Mascot version 2.4.0 (Matrix Science) set to search a database composed of gene models identified within the F. hepatica genome (version 1.0, 101,780 entries; Cwiklinski et al. (6)) and validated using Scaffold (version 4.3.2) as per Cwiklinski et al. (7).
Immunolocalization of FhKT1 in Newly Excysted Juveniles by Confocal Microscopy
F. hepatica metacercariae (Italian isolate; Ridgeway Research, UK) were excysted as described (5), and NEJs were cultured in RPMI 1640 medium (ThermoFisher Scientific) for 3 and 24 h, respectively. The parasites were fixed with 4% paraformaldehyde in 0.1 m PBS (Sigma) overnight at 4 °C and then washed three times with antibody diluent (AbD: 0.1 m PBS containing 0.1% (v/v) Triton X-100, 0.1% (w/v) bovine serum albumin, and 0.1% (w/v) sodium azide). NEJs were then incubated in 0.1 m PBS containing anti-FhKT1 antiserum (prepared in rabbit against the recombinant FhKT1) at a 1:500 dilution, overnight at 4 °C, followed by three washes in AbD. As a negative control, separate samples of 3- and 24-h NEJs were incubated in 0.1 m PBS containing rabbit pre-immune antiserum at a 1:500 dilution. Following washing, all NEJ samples were incubated in a 1:200 dilution of the secondary antibody, fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (Sigma), overnight at 4 °C, followed by three washes in AbD. To counter-stain muscle tissues, NEJs were incubated in AbD containing 200 μg/ml phalloidin-TRITC overnight at 4 °C. Following three final washes in AbD, NEJs were whole-mounted in a 9:1 glycerol solution containing 0.1 m propyl gallate and viewed using confocal scanning laser microscopy (Leica TCS SP5) under the HCX PL APO CS ×100 oil objective lens. Leica type F immersion oil was used in viewing, and all images were taken at room temperature.
Expression of Recombinant FhKT1 and FhKT1Leu15/Arg15 in the Methylotrophic Yeast P. pastoris
The wild-type Fhepatica KT protease inhibitor gene identified by transcriptomic analysis (Fh_Contig2704, Young et al. (10); fhkt1, Cwiklinski et al. (6)) was codon-optimized for expression in P. pastoris and synthesized in the puc57 vector (GenScript). A variant of this gene (fhkt1leu15/arg15), whereby the P1 leucine residue at position 15 was substituted to an arginine residue (see Fig. 2) was similarly synthesized and codon-optimized for expression in P. pastoris. Both synthesized genes did not include the hypothetical N-terminal signal peptide (based on SignalP data). A C-terminal His tag was incorporated into each gene, as well as an N-terminal KpnI restriction site and a C-terminal MlyI restriction site for cloning into the yeast expression vector, Both fhkt1 and fhkt1leu15/arg15 were transformed into One Shot® TOP10 chemically Competent Escherichia coli cells (ThermoFisher Scientific) for propagation and ligated into the pPinkα-HC vector before transformation into PichiaPinkTM Strain 1 cells (ThermoFisher Scientific) by electroporation. (1500 V for 5 ms), following the manufacturer's protocol. Transformed cells were streaked onto Pichia adenine dropout plates and selected for protein expression, as previously described (58).
fhkt1 and fhkt1leu15/arg15-transformed yeast cells were grown in 1 liter of buffered glycerol-complex medium at 28 °C to an A600 of 20 and then centrifuged at 3000 × g for 5 min at room temperature. Pellets were resuspended in 200 ml of buffered methanol-complex medium, and protein expression was induced at 16 °C for a further 3 days by daily addition of 2% methanol. Cells were then pelleted by centrifuging at 3000 × g, and the supernatant was collected. The supernatant was diluted 1:4 with column buffer (sodium phosphate buffer, pH 8, 10 mm imidazole) and passed over a column containing a 500-μl bed volume of Ni-NTA beads (Qiagen) at a rate of 0.5 ml/min. The column was washed with 10 ml of wash buffer (50 mm sodium phosphate buffer, pH 8, 20 mm imidazole), and bound recombinant protein was eluted from the column using 5 ml of elution buffer (50 mm sodium phosphate buffer, pH 7, 250 mm imidazole). The eluted protein was buffer-exchanged by dialysis (with a 3-kDa molecular mass cutoff) into PBS over 24 h at 4 °C, aliquoted, and stored at −80 °C until needed.
A Bradford assay was carried out to determine protein yield, and protein purity was visualized by gel electrophoresis on precast NuPAGE Novex 4–12% BisTris protein gels (ThermoFisher Scientific). For Western blot analysis, proteins were transferred to a nitrocellulose membrane and blocked with blocking solution (5% semi-skimmed milk powder in TBST buffer). Blots were probed with the primary antibody, anti-histidine tag (His6) monoclonal antibody (ThermoFisher Scientific), which had been diluted 1:10,000 in 1% blocking solution. Blots were then washed three times with TBST (15 min for each wash) before the secondary antibody, alkaline phosphatase-conjugated goat anti-mouse IgG (Sigma) diluted 1:15,000 in TBST buffer, was applied. Antibodies were left to incubate with the membrane for 1 h before being washed three times with TBST. Bound antibody was visualized with the substrate SIGMA FASTTM 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium) (Sigma) and imaged using a G:BOX Chemi XRQ imager (Syngene).
Inhibition Assays to Determine rFhKT1 and rFhKT1Leu15/Arg15 Specificity
The inhibitory specificity of rFhKT1 and rFKT1Leu15/Arg15 was screened using a panel of mammalian serine and cysteine proteases and F. hepatica cysteine proteases. Enzymes screened for inhibition included the following: bovine trypsin (Sigma); bovine chymotrypsin (Sigma); human neutrophil elastase (Elastin Products Co.); bovine thrombin (Sigma); porcine pancreatic kallikrein (Sigma); human cathepsin G (Elastin Products Co.); human cathepsin B (Sigma); human cathepsin K (Enzo Life Sciences); human cathepsin L (Sigma); human cathepsin S (Sigma). Purified F. hepatica cathepsin L1 (FhCL1), F. hepatica cathepsin L2 (FhCL2), F. hepatica cathepsin B1 (FhCB1), and F. hepatica cathepsin B2 (FhCB2) were expressed as functionally active recombinant forms in P. pastoris in our laboratory (36).
Reaction conditions and substrates employed for measuring the activity of each protease are presented in Table 4. rFhKT1 and rFhKT1Leu15/Arg15 (300 nm) were first incubated with each protease in a 100-μl volume reaction buffer for 15 min at 37 °C. Reaction volumes were then brought to 200 μl with the addition of fluorogenic substrate dissolved in reaction buffer, and the proteolytic activity was measured as RfU in a PolarStar Omega spectrophotometer (BMG LabTech, UK). Enzyme inhibition studies were performed with FhCL1 and FhCL2 at different pH values ranging from 4.5 to 8.5 using citrate phosphate buffer. All assays were carried out in triplicate.
TABLE 4.
Fluorogenic assay conditions for each protease that inhibition was screened against
| Enzyme | Assay Buffer | Substrate |
|---|---|---|
| F. hepatica cathepsin L1 (2.7 nm) | 50 mm citrate phosphate buffer, pH 5.5, 2 mm dithiothreitol, 0.01% Brij L23 | Z-Leu-Arg-NHMec (20 μm) |
| F. hepatica cathepsin L2 (5 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z-Leu-Arg-NHMec (20 μm) |
| F. hepatica cathepsin B1 (34 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z-Phe-Arg-NHMec (20 μm) |
| F. hepatica cathepsin B2 (180 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z−Gly−Pro−Arg−NHMec (20 μm) |
| Human cathepsin L (0.2 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z-Phe-Arg-NHMec (20 μm) |
| Human cathepsin K (2 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z-Phe-Arg-NHMec (20 μm) |
| Human cathepsin S (2 nm) | 100 mm sodium acetate buffer, pH 5.6, 1 mm EDTA, 1 mm DTT, 0.01% Brij L23 | Z-Val-Val-Arg-NHMec (20 μm) |
| Human cathepsin B (3.6 nm) | 50 mm citrate phosphate buffer pH 5.5, 2 mm DTT, 0.01% Brij L23 | Z-Phe-Arg-NHMec (20 μm) |
| Trypsin (42 nm) | 100 mm Sodium Phosphate Buffer pH 7.6, 1 mm HCl, 20 mm Ca, 0.01% Brij L23 | Z-Leu-Arg-NHMec (20 μm) |
| Chymotrypsin (40 nm) | 100 mm sodium phosphate buffer, pH 7.6, 1 mm HCl, 20 mm calcium, 0.01% Brij L23 | Suc-Ala-Ala-Pro-Phe-NHMec (20 μm) |
| Neutrophil elastase (50 nm) | 100 mm Hepes, 500 mm NaCl, pH 7.5, 0.01% Brij L23 | MeOSuc-Ala-Ala-Pro-Val-NHMec (20 μm) |
| cathepsin G (100 nm) | 100 mm Tris pH 7.5, 0.01% Brij L23 | Suc-Ala-Ala-Pro-Pro-NHMec (100 μm) |
| Thrombin (43 pm) | 100 mm Tris, 150 mm NaCl, pH 7.4, 0.01% Brij L23 | Z-Gly-Pro-Arg-NHMec (2 μm) |
| Kallikrein (150 nm) | 20 mm Tris-HCl, pH 7.8, 100 mm NaCl, 0.01% Brij L23 | Z-Phe-Arg-NHMec (20 μm) |
To determine inhibition constants, decreasing concentrations (nm) of rFhKT1 and rFhKT1Leu15/Arg15 were exposed to each protease by serial dilution. Substrate concentrations were equal to the known Km values of the cysteine proteases. Ki values were determined using the Morrison equation for tight-binding inhibition (built into GraphPad Prism 5.0). Briefly, initial velocities were fitted to Morrison's equation (Equation 1), and the resulting apparent Ki (Kiapp) was fitted to Equation 2 to determine the Ki for FhKT1 and FhKT1Leu15/Arg15 with each cysteine protease inhibited (59).
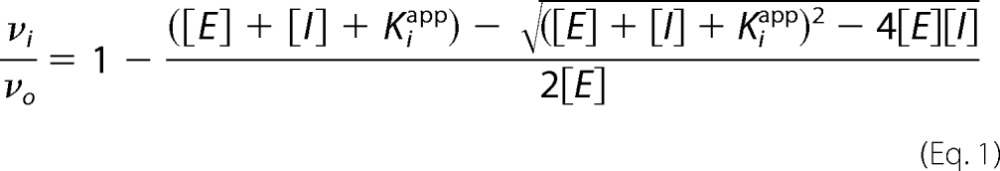 |
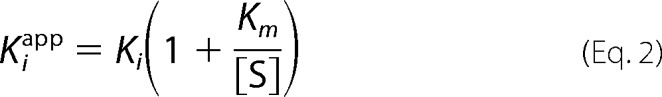 |
υi is the initial velocity of a reaction containing the inhibitor, and υ0 is the initial velocity of a reaction without the inhibitor; [E] is the concentration of enzyme catalytic sites; [I] is the inhibitor concentration; [S] is the substrate concentration, and Km is the known Michaelis-Menten constant for a given substrate with a given enzyme. Inhibitor concentrations ranged from 20 to 0.1 nm, providing a range of concentrations above and below the enzyme concentrations used for the cysteine proteases inhibited (see Table 1).
To determine the importance of the tertiary structure for the inhibitory activity of rFhKT1 and rFhKT1Leu15/Arg15, samples of both inhibitors were reduced and alkylated using tributylphosphine (Sigma) and acrylamide (Sigma), and their inhibition of FhCL1 and trypsin was measured. Briefly, the inhibitors were diluted in 40 mm Tris, 5 mm TBP, and 10 mm acrylamide and incubated for 2 h at room temperature before quenching the alkylation process by addition of 10 mm DTT (Sigma). For inhibition assays, untreated and reduced/alkylated rFhKT1 and rFhKT1Leu15/Arg15 were added to the fluorogenic assays (performed as described above) at a final concentration of 25 nm (sufficient for >95% protease inhibition by the non-treated inhibitors). Samples of rFhKT1 and rFhKT1Leu15/Arg15 taken after quenching of the alkylating reagent samples were kept at 4 °C for 10 days and then assayed for inhibitory activity as above.
Inhibition of rFhCL1 and rFhCL2 Auto-catalytic Activation by rFhKT1 and rFhKT1Leu15/Arg15
Unprocessed FhCL1 zymogen (∼37 kDa, 1.35 μm, ∼1 μg) and rFhKT1 or rFhKT1Leu15/Arg15 (6.5 μm, ∼1 μg) were premixed in citrate phosphate buffer, pH 4.5, and incubated at 37 °C for 45 min. For comparison, FhCL1 zymogen was also incubated in citrate phosphate buffer, pH 4.5, in the presence of 1 μg of BPTI. As a control for auto-catalytic activation of FhCL1, activation was also carried out in the absence of inhibitor, which has previously been shown to be successful at pH 4.5 (34). Following incubation at 37 °C for 45 min, the cysteine protease inhibitor E-64 (100 μm) was immediately added to the samples to prevent any further potential proteolysis by the cysteine protease. A sample of the FhCL1 zymogen was also used for comparison, whereby E-64 was immediately added to prevent any auto-catalytic activation. Samples were analyzed by LDS-electrophoresis using precast NuPAGE Novex 4–12% BisTris protein gels (ThermoFisher Scientific), stained with Biosafe Coomassie (Bio-Rad), and imaged using a G:BOX Chemi XRQ imager (Syngene).
Stability of the rFhKT1-Cysteine Protease Complex
F. hepatica cathepsin L1 that was fully activated at pH 4.5 for 1 h (25 kDa, 2 μm, ∼1 μg) was incubated in the presence or absence of rFhKT1 (6.5 μm, ∼1 μg) in citrate phosphate buffer, pH 5.5, and incubated at 37 °C. The reaction was monitored for 6 h using the peptide substrate Z-Phe-Arg-NHMec. Samples of the reaction were removed at 1, 2, and 6 h and added immediately to the cysteine protease inhibitor E-64 (100 μm) to prevent any further potential proteolysis by the cysteine protease. These samples were then analyzed by LDS-electrophoresis and by Western blot analysis with rabbit anti-rFhKT1 polyclonal antibodies (prepared by Eurogentec, Liège, Belgium) and anti-His tag monoclonal antibodies (Sigma). To determine the long term stability of FhCL1 inhibition by rFhKT1, these were mixed as described above, and enzyme activity in samples with and without inhibitor was monitored over the course of 7 days.
Pulldown of Cysteine Protease from Adult F. hepatica Excretory-Secretory Products
Adult F. hepatica parasites (flukes) were obtained from lamb livers sourced from an abattoir in Northern Ireland, United Kingdom. Upon collection, adult F. hepatica were washed with PBS (containing 0.1% glucose) and maintained in RPMI 1640 culture medium (ThermoFisher Scientific) containing 2 mm l-glutamine (ThermoFisher Scientific), 30 mm Hepes (ThermoFisher Scientific), and 0.1% glucose at two flukes/ml. Ten μg of rFhKT1 was added to one culture of media, although, as a control, no inhibitor was added to a similar parasite culture. After 5 h of incubation at 37 °C, the parasite culture media were collected and centrifuged at 300 × g for 10 min and then at 700 × g for 30 min (both at 4 °C) to remove large debris (e.g. parasite eggs). Then, a 500-μl bed volume of pre-washed Ni-NTA beads was added to each media and mixed by rotating for 1 h at 4 °C. The Ni-NTA beads were then centrifuged in a bench top microcentrifuge at 100 × g, and the supernatant was decanted and stored. The pelleted Ni-NTA beads were washed twice with wash buffer before elution with 100 μl of elution buffer (see above). Column flow-through and eluted samples were analyzed on precast NuPAGE Novex 4–12% BisTris protein gels (ThermoFisher Scientific), stained with Biosafe Coomassie, and imaged using a G:BOX Chemi XRQ imager (Syngene).
The cysteine protease activity of the media before and after the pulldown with Ni-NTA beads was determined by fluorogenic assays using the cathepsin L substrate Z-Phe-Arg-NHMec. An ∼26-kDa protein band that had been pulled down from the medium to which rFhKT1 was added was characterized by LC-MS/MS analysis at the Fingerprints Proteomics Facility, University of Dundee, Scotland, United Kingdom.
Molecular Modeling of rFhKT1 and rFhKT1Leu15/Arg15 Protease Inhibition
The crystal structure of BPTI with the PDB code of 3OTJ (60) was used to build the homology model of FhKT1 using Prime 4.0 (61, 62) with the energy-based setting. The obtained homology model was first docked to the crystal structure of FhCL1 previously reported by us (PDB code 2O6X) (36) using the HEX 6.1 protein-protein docking program (63) and subjected to molecular dynamics simulations using MacroModel 10.8. In docking, the shape-based and electrostatic-based assessment of binding was conducted with over 100 generated and analyzed docking solutions. The docking solution in which the P1 Leu15 of FhKT1 is pointed to the active site of FhCL1 was selected for molecular dynamics simulations. 10 ns of molecular dynamics simulations and 5000 steps of followed minimization were conducted in implicit solvent at a temperature of 300 K to obtain the final complex. OPLS_2005 force field was used in MacroModel calculations. A similar docking and simulation procedure was used to study FhKT1 binding with human cathepsins K, L, S, and B (PDB codes 1YK7, 4AXM, 3N3G, and 3K9M, respectively). The image with the molecular models was prepared with Maestro 10.2.
Author Contributions
D. S. conceived the ideas, conducted most of the experiments, analyzed the results, and was involved in experimental design and wrote the paper. I. G. T. performed the molecular modeling and docking simulations and was involved in writing and editing the manuscript. H. L. J. performed the immunolocalization studies. O. D. performed enzymatic assays and characterizations. M. W. R. and J. D. were involved in experimental design, provided materials, and were involved in the writing and editing of the manuscript. K. C. was involved in sequence analysis and interpretation of results and was involved in writing and editing the manuscript. J. P. D. conceived the ideas, contributed to data analysis, was involved in writing the manuscript, was the principle investigator, was involved in designing the study, and provided the majority of the resources utilized in the study.
Supplementary Material
Acknowledgment
We thank T. Barbour for providing the recombinant F. hepatica cathepsins B1 and B2 used in the inhibition screening assays.
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Files 1 and 2.
- NEJ
- newly excysted juvenile
- KT
- Kunitz-type
- E-64
- trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane
- NHMec
- 7-amino-4-methylcoumarin
- Ni-NTA
- nickel-nitrilotriacetic acid
- BPTI
- bovine pancreatic trypsin inhibitor
- RfU
- relative fluorescent unit
- PDB
- Protein Data Bank
- RPMI 1640 medium
- Roswell Park Memorial Institute 1640 medium
- LDS-PAGE
- lithium dodecyl sulfate-PAGE
- AbD
- antibody diluent
- TRITC
- tetramethylrhodamine isothiocyanate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Z
- benzyloxycarbonyl.
References
- 1. Howell A., Baylis M., Smith R., Pinchbeck G., and Williams D. (2015) Epidemiology and impact of Fasciola hepatica exposure in high-yielding dairy herds. Prev. Vet. Med. 121, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torgerson P., and Claxton J. (1999) in Fasciolosis (Dalton J. P., ed) pp. 113–139, CABI Publishing, New York [Google Scholar]
- 3. Molina-Hernández V., Mulcahy G., Pérez J., Martínez-Moreno Á., Donnelly S., O'Neill S. M., Dalton J. P., and Cwiklinski K. (2015) Fasciola hepatica vaccine: we may not be there yet but we're on the right road. Vet. Parasitol. 208, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mas-Coma S., Bargues M. D., and Valero M. A. (2005) Fascioliasis and other plant-borne trematode zoonoses. Int. J. Parasitol. 35, 1255–1278 [DOI] [PubMed] [Google Scholar]
- 5. Robinson M. W., Menon R., Donnelly S. M., Dalton J. P., and Ranganathan S. (2009) An integrated transcriptomics and proteomics analysis of the secretome of the helminth pathogen Fasciola hepatica proteins associated with invasion and infection of the mammalian host. Mol. Cell.Proteomics 8, 1891–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cwiklinski K., Dalton J. P., Dufresne P. J., La Course J., Williams D. J., Hodgkinson J., and Paterson S. (2015) The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol. 16, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cwiklinski K., de la Torre-Escudero E., Trelis M., Bernal D., Dufresne P. J., Brennan G. P., O'Neill S., Tort J., Paterson S., Marcilla A., Dalton J. P., and Robinson M. W. (2015) The extracellular vesicles of the helminth pathogen, Fasciola hepatica: biogenesis pathways and cargo molecules involved in parasite pathogenesis. Mol. Cell. Proteomics 14, 3258–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jefferies J. R., Campbell A. M., van Rossum A. J., Barrett J., and Brophy P. M. (2001) Proteomic analysis of Fasciola hepatica excretory-secretory products. Proteomics 1, 1128–1132 [DOI] [PubMed] [Google Scholar]
- 9. Dalton J. P., O'Neill S. O., Stack C., Collins P., Walshe A., Sekiya M., Doyle S., Mulcahy G., Hoyle D., Khaznadji E., Moiré N., Brennan G., Mousley A., Kreshchenko N., Maule A. G., et al. (2003) Fasciola hepatica cathepsin L-like proteases: biology, function, and potential in the development of first generation liver fluke vaccines. Int. J. Parasitol. 33, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 10. Young N. D., Hall R. S., Jex A. R., Cantacessi C., and Gasser R. B. (2010) Elucidating the transcriptome of Fasciola hepatica—a key to fundamental and biotechnological discoveries for a neglected parasite. Biotechnol. Adv. 28, 222–231 [DOI] [PubMed] [Google Scholar]
- 11. Ranasinghe S., and McManus D. P. (2013) Structure and function of invertebrate Kunitz serine protease inhibitors. Dev. Comp. Immunol. 39, 219–227 [DOI] [PubMed] [Google Scholar]
- 12. Eng W. S., Fry B. G., Sunagar K., Takacs Z., Jackson T. N. W., and Guddat L. W. (2015) in Venomous Reptiles and Their Toxins: Evolution, Pathophysiology and Biodiscovery (Fry B., ed) p. 281, Oxford University Press, New York [Google Scholar]
- 13. Laskowski M. Jr., and Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 [DOI] [PubMed] [Google Scholar]
- 14. Krowarsch D., Cierpicki T., Jelen F., and Otlewski J. (2003) Canonical protein inhibitors of serine proteases. Cell. Mol. Life Sci. 60, 2427–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerwinsky E. W., Burck P. J., and Grinnan E. L. (1967) Acidic bovine pancreatic trypsin inhibitor. I. Purification and physical characterization. Biochemistry 6, 3175–3179 [DOI] [PubMed] [Google Scholar]
- 16. Olsen J. V., Ong S.-E., and Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 [DOI] [PubMed] [Google Scholar]
- 17. Farady C. J., and Craik C. S. (2010) Mechanisms of macromolecular protease inhibitors. Chembiochem 11, 2341–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H., Wang L., Zhou M., Yang M., Ma C., Chen T., Zhang Y., Zeller M., Hornshaw M., and Shaw C. (2012) Functional peptidomics of amphibian skin secretion: a novel Kunitz-type chymotrypsin inhibitor from the African hyperoliid frog, Kassina senegalensis. Biochimie 94, 891–899 [DOI] [PubMed] [Google Scholar]
- 19. Abreu P. A., Soares T. S., Buarque D. S., Torquato R. S., and Tanaka A. S. (2014) RmKK, a tissue kallikrein inhibitor from Rhipicephalus microplus eggs. Biochem. Biophys. Res. Commun. 449, 69–73 [DOI] [PubMed] [Google Scholar]
- 20. Macedo-Ribeiro S., Almeida C., Calisto B. M., Friedrich T., Mentele R., Stürzebecher J., Fuentes-Prior P., and Pereira P. J. (2008) Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick. PLoS ONE 3, e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wan H., Lee K. S., Kim B. Y., Zou F. M., Yoon H. J., Je Y. H., Li J., and Jin B. R. (2013) A spider-derived Kunitz-type serine protease inhibitor that acts as a plasmin inhibitor and an elastase inhibitor. PLoS ONE 8, e53343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu D., Bungiro R. D., Ibanez M., Harrison L. M., Campodonico E., Jones B. F., Mieszczanek J., Kuzmic P., and Cappello M. (2004) Molecular characterization of Ancylostoma ceylanicum Kunitz-type serine protease inhibitor–evidence for a role in hookworm-associated growth delay. Infect. Immun. 24, 2214–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milstone A. M., Harrison L. M., Bungiro R. D., Kuzmic P., and Cappello M. (2000) A broad spectrum Kunitz-type serine protease inhibitor secreted by the hookworm Ancylostoma ceylanicum. J. Biol. Chem. 275, 29391–29399 [DOI] [PubMed] [Google Scholar]
- 24. Jin X., Deng L., Li H., Zhang Z., He Q., Yang C., Jiang H., Zhu X.-Q., and Peng L. (2011) Identification and characterization of a serine protease inhibitor with two trypsin inhibitor-like domains from the human hookworm Ancylostoma duodenale. Parasitol. Res. 108, 287–295 [DOI] [PubMed] [Google Scholar]
- 25. Ranasinghe S. L., Fischer K., Zhang W., Gobert G. N., and McManus D. P. (2015) Cloning and characterization of two potent Kunitz type protease inhibitors from Echinococcus granulosus. PLoS Negl. Trop. Dis. 9, e0004268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. González S., Fló M., Margenat M., Durán R., González-Sapienza G., Graña M., Parkinson J., Maizels R. M., Salinas G., Alvarez B., and Fernández C. (2009) A family of diverse Kunitz inhibitors from Echinococcus granulosus potentially involved in host-parasite cross-talk. PLoS One 4, e7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ranasinghe S. L., Fischer K., Gobert G. N., and McManus D. P. (2015) Functional expression of a novel Kunitz type protease inhibitor from the human blood fluke Schistosoma mansoni. Parasites & Vectors 8, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ranasinghe S., Fischer K., Gobert G. N., and McManus D. P. (2015) A novel coagulation inhibitor from Schistosoma japonicum. Parasitology 142, 1663–1672 [DOI] [PubMed] [Google Scholar]
- 29. Tsujimoto H., Kotsyfakis M., Francischetti I. M., Eum J. H., Strand M. R., and Champagne D. E. (2012) Simukunin from the salivary glands of the black fly Simulium vittatum inhibits enzymes that regulate clotting and inflammatory responses. PLoS ONE 7, e29964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soares T. S., Watanabe R. M., Tanaka-Azevedo A. M., Torquato R. J., Lu S., Figueiredo A. C., Pereira P. J., and Tanaka A. S. (2012) Expression and functional characterization of boophilin, a thrombin inhibitor from Rhipicephalus (Boophilus) microplus midgut. Vet. Parasitol. 187, 521–528 [DOI] [PubMed] [Google Scholar]
- 31. Ceraul S. M., Dreher-Lesnick S. M., Mulenga A., Rahman M. S., and Azad A. F. (2008) Functional characterization and novel rickettsiostatic effects of a Kunitz-type serine protease inhibitor from the tick Dermacentor variabilis. Infect. Immun. 76, 5429–5435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Francischetti I. M., Valenzuela J. G., Andersen J. F., Mather T. N., and Ribeiro J. M. (2002) Ixolaris, a novel recombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood 99, 3602–3612 [DOI] [PubMed] [Google Scholar]
- 33. Bozas S. E., Panaccio M., Creaney J., Dosen M., Parsons J. C., Vlasuk G. V., Walker I. D., and Spithill T. W. (1995) Characterisation of a novel Kunitz-type molecule from the trematode Fasciola hepatica. Mol. Biochem. Parasitol. 74, 19–29 [DOI] [PubMed] [Google Scholar]
- 34. Lowther J., Robinson M. W., Donnelly S. M., Xu W., Stack C. M., Matthews J. M., and Dalton J. P. (2009) The importance of pH in regulating the function of the Fasciola hepatica cathepsin L1 cysteine protease. PLoS Negl. Trop. Dis. 3, e369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smooker P. M., Whisstock J. C., Irving J. A., Siyaguna S., Spithill T. W., and Pike R. N. (2000) A single amino acid substitution affects substrate specificity in cysteine proteinases from Fasciola hepatica. Protein Sci. 9, 2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stack C. M., Caffrey C. R., Donnelly S. M., Seshaadri A., Lowther J., Tort J. F., Collins P. R., Robinson M. W., Xu W., McKerrow J. H., Craik C. S., Geiger S. R., Marion R., Brinen L. S., and Dalton J. P. (2008) Structural and functional relationships in the virulence-associated cathepsin L proteases of the parasitic liver fluke, Fasciola hepatica. J. Biol. Chem. 283, 9896–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perona J. J., Tsu C. A., Craik C. S., and Fletterick R. J. (1993) Crystal structure of rat anionic trypsin complexed with the protein inhibitors APPI and BPTI. J. Mol. Biol. 230, 919–933 [DOI] [PubMed] [Google Scholar]
- 38. Illy C., Quraishi O., Wang J., Purisima E., Vernet T., and Mort J. S. (1997) Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 272, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 39. Sasaki S. D., Cotrin S. S., Carmona A. K., and Tanaka A. S. (2006) An unexpected inhibitory activity of Kunitz-type serine proteinase inhibitor derived from Boophilus microplus trypsin inhibitor on cathepsin L. Biochem. Biophys. Res. Commun. 341, 266–272 [DOI] [PubMed] [Google Scholar]
- 40. Gil D. F., García-Fernández R., Alonso-del-Rivero M., Lamazares E., Pérez M., Varas L., Díaz J., Chávez M. A., González-González Y., and Mansur M. (2011) Recombinant expression of ShPI-1A, a non-specific BPTI-Kunitz-type inhibitor, and its protection effect on proteolytic degradation of recombinant human miniproinsulin expressed in Pichia pastoris. FEMS Yeast Res. 11, 575–586 [DOI] [PubMed] [Google Scholar]
- 41. Delfín J., Martínez I., Antuch W., Morera V., González Y., Rodríguez R., Márquez M., Saroyán A., Larionova N., Díaz J., Márquez M., Saroyán A., Larionova N., Díaz J., Padrón G., and Chávez M. (1996) Purification, characterization and immobilization of proteinase inhibitors from Stichodactyla helianthus. Toxicon 34, 1367–1376 [DOI] [PubMed] [Google Scholar]
- 42. Azzolini S. S., Sasaki S. D., Campos I. T., S Torquato R. J., Juliano M. A., and Tanaka A. S. (2005) The role of HiTI, a serine protease inhibitor from Haematobia irritans irritans (Diptera: Muscidae) in the control of fly and bacterial proteases. Exp. Parasitol. 111, 30–36 [DOI] [PubMed] [Google Scholar]
- 43. Threadgold L. T., and Gallagher S. S. (1966) Electron microscope studies of Fasciola hepatica. I. The ultrastructure and interrelationships of the parenchymal cells. Parasitology 56, 299–304 [DOI] [PubMed] [Google Scholar]
- 44. Threadgold L. T., and Arme C. (1974) Electron microscope studies of Fasciola hepatica: XI. Autophagy and parenchymal cell function. Exp. Parasitol. 35, 389–405 [DOI] [PubMed] [Google Scholar]
- 45. Hanna R. (1976) Fasciola hepatica: a light and electron microscope autoradiographic study of incorporation of monosaccharides into glycogen and glycoprotein. Exp. Parasitol. 39, 204–213 [DOI] [PubMed] [Google Scholar]
- 46. Bennett J. L., and Gianutsos G. (1977) Distribution of catecholamines in immature Fasciola hepatica: a histochemical and biochemical study. Int. J. Parasitol. 7, 221–225 [DOI] [PubMed] [Google Scholar]
- 47. Mathews C. K., Van Holde K. E., Appling D. R., and Anthony-Cahill S. J. (2012) in Biochemistry (Matthews C. K., ed) 4th Ed., p. 192, Pearson, Toronto, Canada [Google Scholar]
- 48. McGonigle L., Mousley A., Marks N. J., Brennan G. P., Dalton J. P., Spithill T. W., Day T. A., and Maule A. G. (2008) The silencing of cysteine proteases in Fasciola hepatica newly excysted juveniles using RNA interference reduces gut penetration. Int. J. Parasitol. 38, 149–155 [DOI] [PubMed] [Google Scholar]
- 49. Muiño L., Perteguer M. J., Gárate T., Martínez-Sernández V., Beltrán A., Romarís F., Mezo M., González-Warleta M., and Ubeira F. M. (2011) Molecular and immunological characterization of Fasciola antigens recognized by the MM3 monoclonal antibody. Mol. Biochem. Parasitol. 179, 80–90 [DOI] [PubMed] [Google Scholar]
- 50. Zavasnik-Bergant T. (2008) Cystatin protease inhibitors and immune functions. Front. Biosci. 13, 4625–4637 [DOI] [PubMed] [Google Scholar]
- 51. Hartmann S., and Lucius R. (2003) Modulation of host immune responses by nematode cystatins. Int. J. Parasitol. 33, 1291–1302 [DOI] [PubMed] [Google Scholar]
- 52. Barrett A. J., Woessner J. F., and Rawlings N. D. (eds) (2012) Handbook of Proteolytic Enzymes, pp. 1097–1099, Elsevier, London and San Diego [Google Scholar]
- 53. Ishidoh K., and Kominami E. (1998) Gene regulation and extracellular functions of procathepsin L. Biol. Chem. 379, 131–135 [DOI] [PubMed] [Google Scholar]
- 54. Honey K., and Rudensky A. Y. (2003) Lysosomal cysteine proteases regulate antigen presentation. Nat. Rev. Immunol. 3, 472–482 [DOI] [PubMed] [Google Scholar]
- 55. Robinson M. W., Alvarado R., To J., Hutchinson A. T., Dowdell S. N., Lund M., Turnbull L., Whitchurch C. B., O'Brien B. A., Dalton J. P., and Donnelly S. (2012) A helminth cathelicidin-like protein suppresses antigen processing and presentation in macrophages via inhibition of lysosomal vATPase. FASEB J. 26, 4614–4627 [DOI] [PubMed] [Google Scholar]
- 56. Asagiri M., Hirai T., Kunigami T., Kamano S., Gober H.-J., Okamoto K., Nishikawa K., Latz E., Golenbock D. T., Aoki K., Ohya K., Imai Y., Morishita Y., Miyazono K., Kato S., et al. (2008) Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 319, 624–627 [DOI] [PubMed] [Google Scholar]
- 57. Falcón C. R., Masih D., Gatti G., Sanchez M. C., Motrán C. C., and Cervi L. (2014) Fasciola hepatica Kunitz type molecule decreases dendritic cell activation and their ability to induce inflammatory responses. PLoS One 9, e114505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li B., Cao Y., Zhou L., Liang C., and Sun F. (2013) A novel protein expression system–PichiaPinkTM-and a protocol for fast and efficient recombinant protein expression. African J. Biotechnol. 10, 19464–19472 [Google Scholar]
- 59. Copeland R. A. (ed) (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, pp. 305–317, John Wiley & Sons, Inc., New York [Google Scholar]
- 60. Kawamura K., Yamada T., Kurihara K., Tamada T., Kuroki R., Tanaka I., Takahashi H., and Niimura N. (2011) X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Crystallogr. D Biol. Crystallogr. 67, 140–148 [DOI] [PubMed] [Google Scholar]
- 61. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., and Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 [DOI] [PubMed] [Google Scholar]
- 62. Jacobson M. P., Friesner R. A., Xiang Z., and Honig B. (2002) On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608 [DOI] [PubMed] [Google Scholar]
- 63. Ritchie D. W., and Venkatraman V. (2010) Ultra-fast FFT protein docking on graphics processors. Bioinformatics 26, 2398–2405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.