

Abstract
Proteolysis of the amyloid precursor protein (APP) liberates various fragments including the proposed initiator of Alzheimer disease-associated dysfunctions, amyloid-β. However, recent evidence suggests that the accepted view of APP proteolysis by the canonical α-, β-, and γ-secretases is simplistic, with the discovery of a number of novel APP secretases (including δ- and η-secretases, alternative β-secretases) and additional metabolites, some of which may also cause synaptic dysfunction. Furthermore, various proteins have been identified that interact with APP and modulate its cleavage by the secretases. Here, we give an overview of the increasingly complex picture of APP proteolysis.
Keywords: ADAM, Alzheimer disease, amyloid, amyloid precursor protein (APP), amyloid-beta (AB), beta-secretase 1 (BACE1), cathepsin B (CTSB), presenilin, protease, proteolysis, secretase, ADAM10
Introduction
Currently over 46 million people worldwide are living with dementia (see the Alzheimer's Disease International website) with Alzheimer disease (AD) 3 representing the most common form of dementia. In AD, the amyloid cascade hypothesis posits that amyloid-β (Aβ), produced through the sequential proteolytic cleavage of the amyloid precursor protein (APP) by the β- and γ-secretases, is a key molecule in initiating and propagating disease pathology including neurofibrillary tangle formation, neuronal cell loss, aberrant synaptic activity, and brain atrophy that lead to the clinically recognized symptoms of dementia (1). However, identification of the Aβ peptide 25 years ago has not yet led to the advent of a viable therapeutic strategy that can slow or halt the progression of AD. Recent studies have revealed new complexities in the proteolytic processing of APP, including the identification of novel secretases which generate APP metabolites that accumulate in the brains of AD patients and may contribute to the synaptic dysfunction observed in the disease. In addition, numerous proteins are being identified that interact with APP, modulating its proteolysis and Aβ production. These new APP secretases and metabolites, along with the APP interactors, may present novel therapeutic targets that are independent of direct modulation of the canonical secretases and that will need to be considered when evaluating the results from current Aβ-directed therapies. In this Minireview, we summarize the recent developments in APP proteolysis focusing on the novel secretases, APP interactors, and APP metabolites that are impacting on our understanding of both APP biology and the neurodegenerative disease process.
The Canonical α-, β-, and γ-Secretases and APP Fragments
The generally accepted model of APP proteolysis is that APP is processed by one of two distinct proteolytic pathways (Fig. 1A). In the amyloidogenic pathway, β-secretase, the β-site APP-cleaving enzyme 1 (BACE1), cleaves APP within the ectodomain and liberates a soluble proteolytic fragment, termed soluble APPβ (sAPPβ), primarily in the endosomal system from the transmembrane APP holoprotein (2). The remaining C-terminal membrane-bound APP fragment, CTFβ, is subsequently cleaved by the presenilin (PS)-containing γ-secretase multisubunit complex to liberate the Aβ peptide and the APP intracellular domain (AICD) (Fig. 1A). γ-Secretase cleaves CTFβ at several sites in the transmembrane domain, “trimming” the Aβ peptide from the initial ϵ-cleavage sites to produce shorter, and relatively benign, Aβ species (Fig. 2) (3). However, inefficient trimming of the Aβ peptide at its C terminus results in the release of longer aggregation-prone Aβ species such as Aβ42, which are central to the production of the neurotoxic oligomeric Aβ assemblies (4, 5). In the non-amyloidogenic pathway, α-secretase, members of the “a disintegrin and metalloprotease” (ADAM) family, mainly ADAM10, cleave APP at the cell surface within the Aβ domain, liberating sAPPα and leaving an alternative membrane-bound C-terminal fragment, CTFα. CTFα can also be subsequently proteolytically processed by γ-secretase to liberate an N-terminally truncated version of the Aβ peptide referred to as p3 and the AICD (Fig. 1A). The AICD has a role as a nuclear transcription factor regulating the expression of various genes, including that of the Aβ-degrading neprilysin (6–8). Although the AICD produced via either the amyloidogenic or non-amyloidogenic pathways have the same peptide sequence, they appear to be functionally distinct. The AICD produced following β-secretase cleavage is transported to the nucleus and binds to the neprilysin gene promoter, whereas that produced following α-secretase cleavage is rapidly degraded in the cytosol by insulin-degrading enzyme (6).
FIGURE 1.

The proteolytic processing of APP. A, the traditional model of APP proteolysis involves APP processing either in the non-amyloidogenic pathway, where sequential cleavage by α-secretase (ADAM10; pink) and the γ-secretase multisubunit complex (here shown for simplicity as a single entity; blue) liberates sAPPα and p3, or in the amyloidogenic pathway, where sequential cleavage by β-secretase (BACE1 or possibly cathepsin B; green) and γ-secretase liberates sAPPβ and Aβ. Both pathways produce AICD, which can be proteolytically degraded or translocated to the nucleus, where it has roles in transcriptional regulation. B, APP processing by the δ-secretase (AEP; yellow) causes the release of three soluble fragments of APP (sAPP1–585, sAPP1–373, and sAPP374–585). The remaining CTFδ is then further processed by β- and γ-secretases to release Aβ and AICD. C, η-secretase (suggested to be MT5-MMP; orange) releases the soluble sAPPη fragment and leaves the membrane-bound CTFη. CTFη can be further processed by α- or β-secretase to release Aη-α or Aη-β, respectively. Following α- or β-cleavage, the remaining CTF can be cleaved by γ-secretase to release p3 and AICD, or Aβ and AICD, respectively. sec, secretase. D, meprin β (purple) acts as a β-secretase producing a fragment similar to sAPPβ as well as two shorter, soluble fragments (sAPP1–380/3 and sAPP1–124). The remaining CTF can then be processed by γ-secretase to release Aβ2-X and AICD. See text for further details.
FIGURE 2.
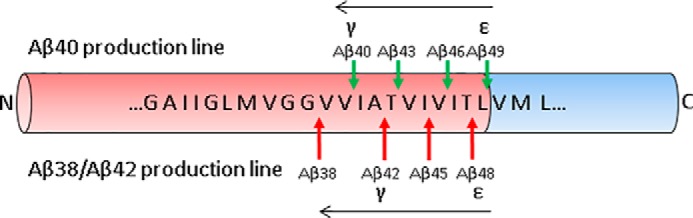
Formation of Aβ by γ-secretase cleavage of APP CTFβ. Cleavage of APP CTFβ by γ-secretase follows a “nibbling” pattern in the direction indicated by the black arrows, where the initial (ϵ) cleavage dictates the final (γ) cleavage. The initial cleavage dictates the C-terminal length of Aβ and thus its amyloidogenic potential. N, N terminus; C, C terminus.
The established view is that these sequential cleavages of APP occur through separate enzyme-substrate interactions that are temporally and spatially separated. However, recent evidence indicates that ADAM10 and γ-secretase physically interact and work in partnership to sequentially cleave APP (9). ADAM10 and γ-secretase co-immunoprecipitated from cells and mouse brain, and were shown to interact by superresolution microscopy. The complex between the two secretases was stabilized by members of the tetraspanin family of scaffold proteins (namely tetraspanin 12 and 17). Limited data also indicated a physical interaction between BACE1 and γ-secretase, albeit in a complex distinct from the ADAM10-γ-secretase complex (9). This comprehensive biochemical study suggests that cells possess large multiprotease complexes capable of sequentially and efficiently processing transmembrane substrates such as APP through a spatially coordinated regulated proteolytic mechanism (9). This raises the interesting question as to whether these multiprotease complexes could be targeted to selectively modulate APP processing in the amyloidogenic pathway.
Whether there is a reciprocal relationship between the amyloidogenic and non-amyloidogenic APP processing pathways has remained a contentious issue, possibly as a result of differences in the model systems being examined (10). In monkeys, BACE1 inhibition decreased sAPPβ and Aβ in cerebrospinal fluid (CSF) and increased sAPPα as measured by ELISA, but there was no change in labeled sAPPα kinetics, suggesting that BACE1 inhibition may not boost α-secretase processing of APP to the same degree (11). In human induced pluripotent stem cell-derived neurons, inhibition of BACE1 reduced sAPPβ and Aβ and reciprocally increased sAPPα (12), whereas inhibition of γ-secretase resulted in an increase in sAPPα and a decrease in sAPPβ (9). This latter result suggests a level of feedback within the ADAM10-γ-secretase complex (9). Activating α-secretase has been suggested as a potential therapeutic approach for reducing Aβ production, although the evidence for this from in vivo studies is somewhat mixed (13, 14). For example, moderate neuronal overexpression of ADAM10 in APPV717I transgenic mice increased the secretion of sAPPα, reduced the formation of Aβ peptides, and prevented their deposition in plaques (15), and in APP/PS-1 transgenic mice the intracerebral injection of the vitamin A analogue acitretin, which stimulates ADAM10 promoter activity, led to a reduction of Aβ40 and Aβ42 (16). However, although a recent clinical trial reported increased CSF sAPPα levels in patients with AD treated with acitretin, there was only a trend to decline in sAPPβ that did not reach significance and there were no changes in the levels of Aβ40 or Aβ42 (17). Moreover, ADAM10 is implicated in the ectodomain shedding of several substrates such as Notch and ErbB2, which promote cancer progression and inflammatory diseases. Thus, further work with more selective α-secretase activators in human neurons and in vivo models is required to clarify whether α-secretase activation is a viable therapeutic approach for AD either by reducing the neurotoxic Aβ and/or by increasing sAPPα, which is reported to have neuroprotective functions (Table 1).
TABLE 1.
Properties of the proteolytic fragments generated from APP
| APP fragment | Function/Characteristics |
|---|---|
| sAPPβ | Induces neural differentiation of stem cells (65) |
| 100-fold less effective than sAPPα at protecting mouse hippocampal neurons from excitotoxic stress (66) | |
| Cleaved to produce an N-terminal fragment that induced axonal pruning (33) but may be independent of β-secretase activity (32) | |
| CTFβ | Evidence for early pathological accumulation in AD mouse models (67) |
| Disruption of LTP (68) | |
| Linked to learning and memory deficits in AD mouse models (69) | |
| Reduction of spine density and induction of synaptotoxicity (70) | |
| Aβ | The major APP metabolic fragment involved in the initiation and propagation of AD; for recent reviews, see Refs. 1 and 5 |
| sAPPα | Induction of Akt cell survival pathway (71) |
| Protection against neuronal damage caused by traumatic brain injury (72) | |
| Protection against synaptic dysfunction (73) and roles in synaptic plasticity (74) | |
| CTFα | Reduction of spine density and induction of synaptotoxicity (70) |
| Possible role as a γ-secretase inhibitor (75) | |
| p3 | Accumulates in amyloid plaques (76) but less aggregation prone than Aβ (77) |
| Forms calcium permeable ion channels resulting in neuronal toxicity (78) | |
| AICD | Transcriptional regulator regulating several proteins linked to AD (8, 79) |
| Contradictory evidence for a pathological role for AICD in AD (80, 81) | |
| sAPPη/sAPP95 | No specific function described but identified in APP transgenic mouse brain lysates with some discrepancy in molecular weight (22, 23) |
| Aη-α | Inhibited LTP in hippocampal brain slices from mice and suppressed activity in hippocampal neurons (23) |
| Aη-β | Has none of the effects reported for Aη-α (23) |
| CTFη | Indirect evidence that CTFη is a better substrate for BACE1 and γ-secretase cleavage (22) |
| Accumulated in dystrophic neurites surrounding amyloid plaques in transgenic mouse and human AD patient brains (23) | |
| CTFδ | The shorter of the two CTFδ forms accumulated in brain lysates from AD patients (20) |
| sAPPδ (1–448, 1–660, 449–660) | sAPP1–448 neurotoxic to neurons |
| No neurotoxicity was observed for other soluble fragments (20) | |
| N-APP fragments | Increased production of 11-kDa fragment during neuronal differentiation (31) |
| N-APP fragment 18–286 bound neurons with higher affinity than sAPPα via an undetermined receptor and increased phosphatidylinositol phosphate lipid levels (82) |
The exact function of several of the APP metabolites (Table 1), including sAPPα, much like the function of the holo-APP molecule, is unclear, in part due to the focus of most APP research being on Aβ. The significantly different, and even opposing, properties of the various APP metabolites adds further to the complexity of the biological functions of APP, and on what the impact will be of inhibiting or activating any particular secretase on the balance of neuroprotective versus neurotoxic outputs.
Although the contribution of the canonical α-, β-, and γ-secretases to APP proteolysis has been studied in depth, the proteolytic cleavage of APP, like many proteins, is more complex than originally envisaged. An increasing number of additional secretases have been identified that also proteolytically process APP in vivo. Here, we review each of these new secretases and, where known, the properties of the APP metabolites produced by their action. A summary of these new secretase cleavage sites within APP is provided in Fig. 1, and the functions and/or characteristics of the metabolites produced through these cleavage events are summarized in Table 1. Although APP can also be cleaved in its intracellular domain (recently reviewed in Ref. 18), this review will focus on cleavages within the APP ectodomain.
δ-Secretase
Asparagine endopeptidase (AEP), previously linked to AD through its capacity to cleave tau, which forms the neurofibrillary tangles (19), was recently shown to cleave APP at two separate sites in the ectodomain (Fig. 1B) (20). AEP is a pH-controlled soluble lysosomal cysteine protease that cleaves after asparagine residues. In an elegant series of experiments, Zhang et al. (20) showed that AEP, or δ-secretase, cleaves APP between Asn373-Glu374 and Asn585-Ile586 (note APP695 numbering used henceforth) in vitro and in vivo, potentially generating three soluble APP fragments and CTFs (Fig. 1B). Cleavage of APP before Thr584 was described previously and referred to as δ-cleavage, although the protease responsible was not identified (21). The AEP cleavage of APP could be blocked in vitro using buffers in which the pH (pH 7.4) inhibited the catalytic activity of AEP, by mutation of critical residues (Cys189 and Asn323) within the catalytic domain of AEP, by mutation of the δ-secretase cleavage sites within APP, or through antibody or peptide inhibition of the enzyme (20). The shorter N-terminal APP fragment, sAPP1–373, produced by AEP cleavage, but not the sAPP1–585 or sAPP374–585 fragments (Fig. 1B), was toxic to primary cultured neurons (Table 1), suggesting that AEP could be a key player in the generation of toxic metabolites within the brain (20). In addition, recombinant APP protein corresponding to the AEP-derived CTF was a better substrate for BACE1 proteolysis in comparison with full-length APP when assayed in vitro, although it is unknown whether this is true for membrane-bound APP in intact cells. Nevertheless, significantly less Aβ was detected in the conditioned medium of AEP knock-out neurons (20). Up-regulation and increased activity of AEP were observed in aged mice, along with increased AEP activity, and AEP produced APP fragments using antibodies specific to the N and C termini of these fragments within the brains of AD patients (20). Furthermore, knock-out of AEP in the 5×FAD mouse model reduced Aβ deposition, synapse and dendritic spine loss, and behavioral deficits (20). AEP gene knock-out similarly protected against memory deficits in another AD mouse model, APP/PS-1 mice (20). Thus, these two studies (19, 20) provide compelling evidence for a role of AEP in AD-like neurodegeneration. However, further work is required to confirm these observations and to clarify the contribution of δ-secretase cleavage of APP to disease pathogenesis and to clarify whether inhibition of AEP may be a therapeutic approach in AD.
η-Secretase
Recent work from two groups revealed that η-secretase contributes to the production of Aβ, while also producing proteolytic fragments with the capacity to induce synaptic dysfunction (22, 23). Both groups identified the matrix metalloproteinase MT5-MMP as contributing to the η-secretase cleavage of APP. Both soluble and membrane-bound matrix metalloproteinases have been of interest to the AD field for some time due to their capacity to proteolytically cleave APP and Aβ in vitro and in vivo (24, 25). In the most recent work, η-secretase was shown to cleave APP between Asn504-Met505, resulting in the shedding of an ∼80–95 kDa soluble fragment, leaving the novel membrane-bound CTF, CTFη (Fig. 1C) (22, 23). Using a suite of antibodies and extensive mass spectrometry analyses, further processing of CTFη by both α-secretases and β-secretases to produce fragments termed Aη-α and Aη-β was elucidated (Fig. 1C) (23). Both Aη-α and Aη-β were detectable in mouse brain homogenates and also in human CSF, where levels were estimated to be 5-fold higher than Aβ (23). Interestingly, Aη-α but not Aη-β inhibited long term potentiation in hippocampal brain slices from mice and suppressed neuronal activity in hippocampal neurons (Table 1) (23). In support of the data obtained in human CSF, η-secretase cleavage of APP exceeded β-secretase cleavage by almost 10-fold in human neurons, and accumulation of ηCTFs was also observed in dystrophic neurites surrounding amyloid plaques in the brains of AD patients (23). In a separate study, and despite MT5-MMP not possessing β-secretase-like activity, MT5-MMP knock-out in 5xFAD mice significantly reduced both Aβ plaque deposition and soluble Aβ and APP CTFs within the brain (22). In addition, a concomitant reduction in markers of gliosis, increases in neuronal integrity as evidenced by increased MAP2 and synaptophysin staining, preserved hippocampal function, and improved performance in spatial memory tasks were all observed in the MT5-MMP knock-out mice (22). MT5-MMP expression in HEK cells expressing APPSWE increased Aβ and CTF production as well as the liberation of the 95-kDa soluble APP N-terminal fragment (sAPP95), presumably that identified by Willem et al. (23) as sAPPη. In the 5xFAD/MT5-MMP knock-out mice, this soluble fragment was significantly reduced in brain homogenates (22). Of concern, both genetic and pharmacological inhibition of BACE1 activity resulted in the accumulation of CTFη and Aη-α (23), highlighting that therapeutic inhibition of BACE1 as currently underway in multiple clinical trials needs to be carefully monitored to ensure that accumulation of alternative neurotoxic APP fragments such as Aη-α does not occur under such therapeutic intervention.
Meprin β
Meprin β is a zinc metalloprotease that has also been proposed as a candidate β-secretase. Meprin β proteolytically cleaves peptide substrates spanning the β-secretase cleavage site in APP at the β-site Met596-Asp597, as well as at the adjacent Asp597-Ala598 and Ala598-Glu599 sites (Fig. 1D) (26). Furthermore, cells expressing meprin β produced significant amounts of Aβ2-X even in the presence of a BACE1 inhibitor, whereas meprin β inhibition significantly reduced its production (26). Further work has suggested that unlike BACE1, meprin β cleaves APP at the cell surface and may directly compete with ADAM10 in vivo, as indicated by increased sAPPα in the soluble fractions derived from meprin β knock-out mice brains (27). Meprin β knock-out also increased sAPPβ in the soluble brain fraction as detected with a neoepitope-specific antibody, whereas conditioned medium from cells expressing APPSWE, which is a better substrate for BACE1 than wild-type APP (12), significantly altered the ratio of Aβ2–40:Aβ1–40 production, indicating direct competition between meprin β and BACE1 for APP (27). Importantly, in the context of AD, Aβ2–40 also showed increased aggregation propensity when compared with Aβ1–40, implicating this species in the preferential production of oligomeric assemblies of Aβ (27). Still, in the brain, Aβ2-X peptides are severalfold lower in abundance relative to Aβ42 species. Thus, the potential contribution of meprin β to AD pathogenesis remains to be established.
In addition to cleavage near the β-secretase site, three further meprin β cleavage sites within APP have been identified (Fig. 1D) (28). Degradomics analysis identified meprin β cleavage sites within APP between Ala124-Asp125, Glu380-Thr381, and Gly383-Asp384, whereas subsequent in vitro analysis identified an N-terminal proteolytic fragment with a molecular mass of 11 kDa, resulting from cleavage at Ala124-Asp125, which directly corresponded to a fragment previously identified in human CSF (28, 29). The site in APP at which meprin β cleaves may depend on its subcellular location, with membrane-bound meprin β cleaving APP at the β-site, whereas soluble meprin β produces the N-terminal APP fragments (30). Other short N-terminal APP fragments have been reported (31–33), although whether they correspond to the same fragments as produced by meprin β or through proteolytic cleavage by alternative proteases remains unknown.
Cathepsin B
The lysosomal cysteine protease cathepsin B has also been proposed as a putative β-secretase (Fig. 1A) (34). The role of cathepsin B as a β-secretase remains controversial, with conflicting evidence indicating that genetic deletion or inhibition of cathepsin B in APP transgenic mice can either enhance (35) or reduce (36) Aβ pathology. Moreover, unlike BACE1 (37), cathepsin B does not accumulate with close spatial proximity to amyloid plaques (38). Cathepsin B cleavage of APP may enhance the production of N-terminally truncated pyroglutamylated forms of Aβ (pGlu-Aβ) in which the N-terminal glutamate in Aβ3-X or Aβ11-X is cyclized by glutaminyl cyclase (34). pGlu-Aβ exhibits increased aggregation propensity and increased cellular toxicity and disrupts long term potentiation to a greater extent than Aβ1-X species (39–41). In addition, recent work has proposed pGlu-Aβ as the predominant Aβ species within the brains of AD patients (42), and efforts to design therapeutics specifically targeting this Aβ species have proved successful in mouse models (43). However, evidence for direct proteolysis of APP between Ala673-Glu674 by cathepsin B remains to be provided. An alternative scenario for the production of N-terminally truncated Aβ species is the truncation of the Aβ1-X peptide by aminopeptidases following BACE1 cleavage.
Mystery Proteases
The recent identification of novel APP secretases indicates that our understanding of APP proteolysis and its contribution to AD is far from complete. Indeed, a series of studies on human CSF has identified numerous APP N-terminal fragments (29) and N-terminally extended Aβ species that may have (patho)physiological functions (44, 45). Evidence also exists for the N-terminal truncation of the Aβ peptide at every one of its first 11 amino acids, although again, the biological relevance of the majority of these species remains uncertain (46). At least one N-terminally truncated species of Aβ (Aβ5-X) has been shown to increase in the CSF of patients treated with a BACE1 inhibitor, suggesting that it is liberated in a BACE1-independent manner and that the protease responsible for its liberation acts in direct competition with BACE1 (47). In addition to the typically monitored fragments (sAPPα, sAPPβ, Aβ, C99 and C83 CTFs, and AICD), a plethora of additional N-terminal and C-terminal APP fragments are likely present in the brain, although it is currently unclear which of these are biologically relevant or functional, how they are regulated, and how the proteolytic pathways interrelate. Until the proteases involved in these novel APP proteolytic pathways are identified, the interrelationship between these distinct pathways is reconciled, and the biological functions of the resulting APP metabolites are fully elucidated, we are a long way from understanding how APP metabolism impacts on normal cellular biology, let alone in AD.
Modulation of APP Proteolysis by Interacting Proteins
A major issue undermining secretases as therapeutic targets in AD is the indiscriminate nature with which their direct inhibition blocks proteolysis of their multiple substrates. Both BACE1 and γ-secretase cleave numerous substrates, producing metabolites that are key in an array of biological roles (48, 49). This has led to a search for alternative mechanisms through which APP proteolysis can be selectively disrupted. Protein-protein interactions have been widely shown to influence APP trafficking and processing, highlighting the APP interactome as a novel source of potential therapeutic targets for the selective disruption of APP proteolysis. A summary of proteins that directly interact with APP and modulate its proteolysis is shown in Table 2. There are likely multiple mechanisms by which interactors modulate APP proteolysis, for example through acting as a physical blockade (e.g. BRI2) to prevent the access of the proteases to APP (50), or by altering APP trafficking (e.g. sortilin-related receptor (SORLA) (51)), and thus either enhancing or reducing the likelihood of APP encountering the secretases.
TABLE 2.
Protein interactors of APP and their effect on APP proteolysis
TGN, trans-Golgi network; GFLD, growth factor-like domain; KPI, Kunitz protease inhibitor; LDLR, low-density lipoprotein receptor; LRP, low-density lipoprotein receptor-related protein; RAP, receptor-associated protein; pThr, phospho-threonine.
| Protein | Interaction details | Effect on proteolysis | Ref. |
|---|---|---|---|
| AP-4 complex | Identified by yeast two-hybrid; μ4 subunit interacts with the YKFFE motif in APP cytoplasmic tail; induces export of APP from the TGN | siRNA knockdown of μ4 subunit ↑Aβ, ↓CTFs | 83 |
| BRI2 | Identified by yeast two-hybrid screen; masks the recognition site for α-secretase and BACE1 | siRNA knockdown ↑sAPPβ, ↑Aβ40, ↑Aβ42 | 50 |
| BRI3 | Identified by a yeast two-hybrid screen; specifically interacts with full-length APP but not CTFs | siRNA knockdown ↑sAPPβ, ↑Aβ40, ↑Aβ42; overexpression ↓sAPPα/β, ↓Aβ40, ↓Aβ42 | 84 |
| CD74 | Identified by yeast two-hybrid screen; binds the ectodomain of APP | Overexpression ↓Aβ40, ↓Aβ42 | 85 |
| Dab2 | Binds the NPXY motif in the cytoplasmic tail of APP; identified due to ability to bind a similar motif in LDLR; mediates endocytosis of APP to endosomes | Overexpression of full-length protein ↑Aβ40, ↑Aβ42 | 86 |
| GRP78 | Binds APP and slows its maturation and secretion | Overexpression ↓Aβ40, ↓Aβ42 | 87 |
| LRP-1 | Identified due to similar ability to bind KPI domain containing tissue factor pathway inhibitor; induces endocytosis of APP | Overexpression ↑Aβ, ↓sAPPα; LRP1 antagonist RAP ↓Aβ | 88, 89 |
| LRP1B | Investigated due to similarities to LRP1; binds full-length APP; increases cell surface levels of APP | Overexpression ↑sAPPα, ↓Aβ40, ↓Aβ42, ↓CTFβ | 90 |
| LRP10 | Investigated due to homology to LDLR family members; interacts with the ectodomain of APP causing retention within the Golgi | Overexpression ↓Aβ40, ↓sAPPα/β | 91 |
| Lingo-1 | Extracellular ligand for APP identified through a brain interactome study | siRNA knockdown ↑CTFα, ↓CTFβ; overexpression ↓sAPPα, ↓sAPPβ | 58, 59 |
| Nogo receptors (NgR) | Investigated due to localization to amyloid plaques in AD brains; interact with APP ectodomain in human brain and cells | Overexpression of NgR1 ↓Aβ; overexpression of NgR2 and NgR3 ↑Aβ40 ↑Aβ42 | 92, 93 |
| PAT1a | Investigated due to prior studies showing APP and PAT interacted directly | siRNA knockdown ↓Aβ, ↓CTFs | 94 |
| Pin1 | Identified due to capacity of Pin1 to bind pThr-Pro motifs; binds APP and alters the cis/trans isomerization of the Pro668/Thr669 bond | Overexpression ↑Aβ40, ↑Aβ42; alternatively, in vivo knockout ↓sAPPα, ↑sAPPβ, ↑Aβ42 | 95, 96 |
| Reelin | Extracellular ligand for APP identified by brain interactome study | Overexpression ↓sAPPα, ↓sAPPβ, ↓CTFs, ↓Aβ40, ↓Aβ42 | 59 |
| SNX17 | Binds the NPXY motif in the cytoplasmic tail of APP; identified due to ability to bind a similar motif in LRP1; proposed to increase APP recycling to cell surface | Dominant negative mutant and siRNA knockdown ↑Aβ40, ↑Aβ42 | 86 |
| SORCS1 | Investigated due to homology to SORLA; binds full-length APP; genetic link to AD | Overexpression ↓Aβ40, ↓Aβ42; siRNA knockdown ↑Aβ40 | 97 |
| SORLA | Investigated due to reduced expression in AD brain; binds to the GFLD and carbohydrate domains of APP; causes retrograde transport of APP to the trans-Golgi network | Overexpression ↓sAPPα, ↓sAPPβ, ↓Aβ40 | 51, 98 |
| Spondin-1 | Identified through a brain interactome study; binds extracellular domain of APP | Overexpression ↓CTFs; independent study showed overexpression had no effect | 59, 99 |
| Syt-1 and Syt-9 | Identified in an ex vivo APP ectodomain interactome study; interact with the APP ectodomain between the E1 and KPI domains | Syt-1 and Syt-9 overexpression ↑sAPPβ, ↑Aβ40, ↑Aβ42; Syt-1 siRNA knockdown ↓sAPPβ, ↓Aβ40, ↓Aβ42 | 55 |
| TRPC6 | Investigated due to previously identified link between presenilin2 and TRPC6 activity; binds directly to APP CTFβ | siRNA knockdown ↑Aβ40, ↑Aβ42; overexpression ↓Aβ40, ↑Aβ42 | 100 |
A wide range of approaches has been taken to identify APP-interacting proteins both in vitro and in vivo. In vitro, yeast two-hybrid studies of the APP interactome have identified several proteins that interact with the intracellular domain of APP, including perhaps the best established APP interactor, Fe65 (Table 2). Fe65 interacts with the cytoplasmic tail of APP (via the YENPTY motif) and influences APP internalization. Despite intensive study, the effect of Fe65 on APP proteolysis has remained controversial (see Ref. 52). In a similar manner, proteins from the Mint family have been shown to interact with the APP YENTPY motif, with conflicting results reported on their effect on APP proteolysis (53). Using a split-ubiquitin version of the yeast two-hybrid approach, several APP interactor proteins were identified, one of which, BRI2, was shown to influence the proteolysis of APP (50) and was also linked to familial forms of dementia in which increased APP proteolysis was observed (54). Gautam et al. (55) used various glutathione S-transferase-tagged APP ectodomain constructs to isolate interacting proteins from the soluble fraction of mouse brains. Novel interactions between the APP ectodomain and three members of the synaptotagmin family of proteins were identified by mass spectrometry, two of which were subsequently shown to influence APP proteolysis (55). Recently, the first study of the APP interactome in a cell culture context was reported using the commonly used, non-neuronal human embryonic kidney cell line, where tagged APP was immunoprecipitated from stable isotope-labeled amino acids in cell culture (SILAC)-labeled cells and interactors were identified by liquid chromatography-tandem mass spectrometry (56). This study identified interactors for wild-type and Swedish mutant APP (and other neurodegeneration-linked proteins), focusing on the interaction between APP and the mitochondrial leucine-rich PPR motif-containing protein (LRPPRC) (56). Although it was not shown to directly influence Aβ generation, the authors suggested that this interaction (which was higher for the Swedish mutant APP) could lead to mitochondrial dysfunction as LRPPRC has a key role in mitochondrial gene regulation (56). We recently employed a similar approach, whereby the interactome of two APP isoforms was studied in the neuron-like SH-SY5Y cells. 4 Key differences in the interactomes of the two APP isoforms, APP695 and APP751, which differ in their ability to be cleaved by β-secretase and to produce Aβ, were identified. We determined the proteins enriched in the interactome of each APP isoform and identified specific enrichment of proteins implicated in mitochondrial function and nuclear transport specifically in the APP695 interactome. Further interrogation of the APP interactome and subsequent experimental validation revealed Fe65 and ataxin-10 as specific modulators of APP695 proteolysis and GAP43 as a specific modulator of APP751 proteolysis, altering Aβ generation.4 GAP43 was identified in a recent study of the γ-secretase interactome (57), with similar results observed on the production of Aβ.
Various studies have investigated the in vivo interactome of APP. One such study investigated the in vivo APP interactome in wild-type mice, comparing the interactome with that of amyloid precursor-like protein (APLP)1 and APLP2 (58). Thirty-four APP interactors were identified, of which one, leucine-rich repeat and immunoglobulin-like domain-containing protein 1 (LINGO-1), was subsequently shown to influence Aβ generation in human embryonic kidney cells (58), although its effect was later contested (59). In a similar in vivo mouse study, Kohli et al. (60) generated a human APP mouse model with a tandem affinity purification tag inserted into the protein in the AICD region. Various proteins involved in synaptic vesicle trafficking were enriched in the APP interactome, along with members of the 14-3-3 family, leading the authors to propose an important role for APP in synaptic signaling (60). A human in vivo APP interactome study identified 21 proteins that interacted with human APP in AD and control brains, although none of the identified proteins interacted specifically with APP in either the AD or control brains (61).
Specifically targeting the interaction between the Vps29 and Vps35 subunit of the retromer complex pharmacologically (although in this case to stabilize, rather than disrupt their interaction) was recently shown to reduce APP proteolysis through alterations in its trafficking (62), indicating that targeted drug screening can identify compounds that modulate APP processing. Indeed, this approach is being taken to identify drugs that disrupt APP dimer formation, which has been shown to increase Aβ production (63). As a protein-protein interaction is likely indicative of a functional relationship, a complete understanding of the implications of disrupting such interactions is required if targeting these interactions is to become a viable and successful therapeutic option in AD.
Concluding Remarks
The recent identification that APP can be proteolytically processed by secretases other than the canonical α-, β-, and γ-secretases adds considerable complexity to the biology of APP and raises important questions. What is the contribution of the recently identified δ- and η-secretases to AD? Why have no mutations around the δ- and η-secretase cleavage sites in APP that give rise to familial AD been identified to date? As noted above, both of these new secretases can increase Aβ production by initially cleaving the full-length APP molecule, making it a better substrate for BACE1 cleavage by reducing the steric hindrance of the large N-terminal ectodomain, as well as generating additional neurotoxic fragments (20, 22). What is the role of APP metabolites other than Aβ in AD and in normal biology? What is the contribution of other proteases, including meprin β and cathepsin B, to APP processing in AD? Given that several other APP metabolites have been identified in the human brain, how many other proteases remain to be identified that influence APP processing? Careful and detailed studies analyzing multiple processing pathways and APP metabolites under appropriate conditions will be required to determine the relative amount of APP processed by each secretase under normal and disease situations. Will inhibition of either the δ-secretases and/or η-secretases be viable alternative therapeutic approaches to alleviate some or all of the symptoms of AD? Will targeting these new secretases be required in addition to targeting β- and or γ-secretase or Aβ immunotherapy to negate the neurotoxicity of other APP metabolites? What is the role of the recently described APP interactors in regulating the production of Aβ or other biologically active APP fragments? Will any of these interactors offer novel targets for therapeutic intervention in AD or in other diseases associated with mis-metabolism of APP such as fragile X syndrome (64)? These recent reports of novel APP secretases and interactors open up the field and underline that further research is needed before we fully understand the complexities of APP proteolytic processing and the roles of its various metabolites in health and disease.
This work was supported by Alzheimer's Research UK (PhD2012-5), the Dr Donald Dean Fund in Dementia Research, and the University of Manchester (to N. M. H.). This work was also supported by National Institutes of Health Grants AG019070 and AG051230 (to G. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
R. J. Andrew, K. Fisher, K. J. Heesom, K. A. B. Kellett, and N. M. Hooper, unpublished data.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- ADAM
- a disintegrin and metalloprotease
- AEP
- asparagine endopeptidase
- APP
- amyloid precursor protein
- AICD
- APP intracellular domain
- BACE1
- β-site APP-cleaving enzyme 1
- CSF
- cerebrospinal fluid
- CTF
- C-terminal fragment
- PS
- presenilin
- sAPP
- soluble amyloid precursor protein
- FAD
- familial AD
- MT5-MMP
- membrane type-5 matrix metalloproteinase
- pGlu-Aβ
- pyroglutamylated form of Aβ
- SORLA
- sortilin-related receptor
- LTP
- long term potentiation.
References
- 1. Musiek E. S., and Holtzman D. M. (2015) Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajendran L., and Annaert W. (2012) Membrane trafficking pathways in Alzheimer's disease. Traffic 13, 759–770 [DOI] [PubMed] [Google Scholar]
- 3. Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., and Ihara Y. (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Strooper B., and Karran E. (2016) The cellular phase of Alzheimer's disease. Cell 164, 603–615 [DOI] [PubMed] [Google Scholar]
- 5. Jarosz-Griffiths H. H., Noble E., Rushworth J. V., and Hooper N. M. (2016) Amyloid-β receptors: the good, the bad, and the prion protein. J. Biol. Chem. 291, 3174–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belyaev N. D., Kellett K. A., Beckett C., Makova N. Z., Revett T. J., Nalivaeva N. N., Hooper N. M., and Turner A. J. (2010) The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J. Biol. Chem. 285, 41443–41454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grimm M. O., Mett J., Stahlmann C. P., Grösgen S., Haupenthal V. J., Blümel T., Hundsdörfer B., Zimmer V. C., Mylonas N. T., Tanila H., Müller U., Grimm H. S., and Hartmann T. (2015) APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 7, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Multhaup G., Huber O., Buée L., and Galas M.-C.(2015) Amyloid precursor protein (APP) metabolites APP intracellular fragment (AICD), Aβ42, and Tau in nuclear roles. J. Biol. Chem. 290, 23515–23522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen A. C., Kim S., Shepardson N., Patel S., Hong S., and Selkoe D. J. (2015) Physical and functional interaction between the α- and γ-secretases: A new model of regulated intramembrane proteolysis. J. Cell Biol. 211, 1157–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Colombo A., Wang H., Kuhn P. H., Page R., Kremmer E., Dempsey P. J., Crawford H. C., and Lichtenthaler S. F. (2013) Constitutive α- and β-secretase cleavages of the amyloid precursor protein are partially coupled in neurons, but not in frequently used cell lines. Neurobiol. Dis. 49, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dobrowolska J. A., Michener M. S., Wu G., Patterson B. W., Chott R., Ovod V., Pyatkivskyy Y., Wildsmith K. R., Kasten T., Mathers P., Dancho M., Lennox C., Smith B. E., Gilberto D., McLoughlin D., et al. (2014) CNS amyloid-β, soluble APP-α and -β kinetics during BACE inhibition. J. Neurosci. 34, 8336–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ben Halima S., Mishra S., Raja K. M., Willem M., Baici A., Simons K., Brüstle O., Koch P., Haass C., Caflisch A., and Rajendran L. (2016) Specific inhibition of β-secretase processing of the Alzheimer disease amyloid precursor protein. Cell Rep. 14, 2127–2141 [DOI] [PubMed] [Google Scholar]
- 13. Lichtenthaler S. F. (2011) α-Secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21 [DOI] [PubMed] [Google Scholar]
- 14. Saftig P., and Reiss K. (2011) The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur. J. Cell Biol. 90, 527–535 [DOI] [PubMed] [Google Scholar]
- 15. Postina R., Schroeder A., Dewachter I., Bohl J., Schmitt U., Kojro E., Prinzen C., Endres K., Hiemke C., Blessing M., Flamez P., Dequenne A., Godaux E., van Leuven F., and Fahrenholz F. (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tippmann F., Hundt J., Schneider A., Endres K., and Fahrenholz F. (2009) Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 23, 1643–1654 [DOI] [PubMed] [Google Scholar]
- 17. Endres K., Fahrenholz F., Lotz J., Hiemke C., Teipel S., Lieb K., Tüscher O., and Fellgiebel A. (2014) Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83, 1930–1935 [DOI] [PubMed] [Google Scholar]
- 18. Nhan H. S., Chiang K., and Koo E. H. (2015) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol. 129, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Z., Song M., Liu X., Kang S. S., Kwon I. S., Duong D. M., Seyfried N. T., Hu W. T., Liu Z., Wang J. Z., Cheng L., Sun Y. E., Yu S. P., Levey A. I., and Ye K. (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat. Med. 20, 1254–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Z., Song M., Liu X., Su Kang S., Duong D. M., Seyfried N. T., Cao X., Cheng L., Sun Y. E., Ping Yu S., Jia J., Levey A. I., and Ye K. (2015) δ-Secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer's disease. Nat. Commun. 6, 8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simons M., de Strooper B., Multhaup G., Tienari P. J., Dotti C. G., and Beyreuther K. (1996) Amyloidogenic processing of the human amyloid precursor protein in primary cultures of rat hippocampal neurons. J. Neurosci. 16, 899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baranger K., Marchalant Y., Bonnet A. E., Crouzin N., Carrete A., Paumier J. M., Py N. A., Bernard A., Bauer C., Charrat E., Moschke K., Seiki M., Vignes M., Lichtenthaler S. F., Checler F., et al. (2016) MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer's disease. Cell. Mol. Life Sci. 73, 217–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willem M., Tahirovic S., Busche M. A., Ovsepian S. V., Chafai M., Kootar S., Hornburg D., Evans L. D., Moore S., Daria A., Hampel H., Müller V., Giudici C., Nuscher B., Wenninger-Weinzierl A., et al. (2015) η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526, 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hernandez-Guillamon M., Mawhirt S., Blais S., Montaner J., Neubert T. A., Rostagno A., and Ghiso J. (2015) Sequential amyloid-β degradation by the matrix metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 290, 15078–15091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fragkouli A., Tsilibary E. C., and Tzinia A. K. (2014) Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer's 5xFAD mice. Neurobiol. Dis. 70, 179–189 [DOI] [PubMed] [Google Scholar]
- 26. Bien J., Jefferson T., Causević M., Jumpertz T., Munter L., Multhaup G., Weggen S., Becker-Pauly C., and Pietrzik C. U. (2012) The metalloprotease meprin β generates amino terminal-truncated amyloid β peptide species. J. Biol. Chem. 287, 33304–33313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schönherr C., Bien J., Isbert S., Wichert R., Prox J., Altmeppen H., Kumar S., Walter J., Lichtenthaler S. F., Weggen S., Glatzel M., Becker-Pauly C., and Pietrzik C. U. (2016) Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site. Mol. Neurodegener. 11, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jefferson T., Čauševič M., auf dem Keller U., Schilling O., Isbert S., Geyer R., Maier W., Tschickardt S., Jumpertz T., Weggen S., Bond J. S., Overall C. M., Pietrzik C. U., and Becker-Pauly C. (2011) Metalloprotease meprin β generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J. Biol. Chem. 286, 27741–27750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Portelius E., Brinkmalm G., Tran A., Andreasson U., Zetterberg H., Westman-Brinkmalm A., Blennow K., and Ohrfelt A. (2010) Identification of novel N-terminal fragments of amyloid precursor protein in cerebrospinal fluid. Exp. Neurol. 223, 351–358 [DOI] [PubMed] [Google Scholar]
- 30. Jäckle F., Schmidt F., Wichert R., Arnold P., Prox J., Mangold M., Ohler A., Pietrzik C. U., Koudelka T., Tholey A., Gütschow M., Stirnberg M., and Becker-Pauly C. (2015) Metalloprotease meprin β is activated by transmembrane serine protease matriptase-2 at the cell surface thereby enhancing APP shedding. Biochem. J. 470, 91–103 [DOI] [PubMed] [Google Scholar]
- 31. Vella L. J., and Cappai R. (2012) Identification of a novel amyloid precursor protein processing pathway that generates secreted N-terminal fragments. FASEB J. 26, 2930–2940 [DOI] [PubMed] [Google Scholar]
- 32. Olsen O., Kallop D. Y., McLaughlin T., Huntwork-Rodriguez S., Wu Z., Duggan C. D., Simon D. J., Lu Y., Easley-Neal C., Takeda K., Hass P. E., Jaworski A., O'Leary D. D., Weimer R. M., and Tessier-Lavigne M. (2014) Genetic analysis reveals that amyloid precursor protein and death receptor 6 function in the same pathway to control axonal pruning independent of β-secretase. J. Neurosci. 34, 6438–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikolaev A., McLaughlin T., O'Leary D. D., and Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Hook G., Yu J., Toneff T., Kindy M., and Hook V. (2014) Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer's disease therapeutic. J. Alzheimers Dis. 41, 129–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun B., Zhou Y., Halabisky B., Lo I., Cho S.-H, Mueller-Steiner S., Devidze N., Wang X., Grubb A., and Gan L. (2008) Cystatin C–cathepsin B axis regulates amyloid β levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 60, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hook V. Y., Kindy M., and Hook G. (2008) Inhibitors of cathepsin B improve memory and reduce β-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, β-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753 [DOI] [PubMed] [Google Scholar]
- 37. Sadleir K. R., Kandalepas P. C., Buggia-Prévot V., Nicholson D. A., Thinakaran G., and Vassar R. (2016) Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer's disease. Acta Neuropathol. 132, 235–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gowrishankar S., Yuan P., Wu Y., Schrag M., Paradise S., Grutzendler J., De Camilli P., and Ferguson S. M. (2015) Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc. Natl. Acad. Sci. U. S. A. 112, E3699–E3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schilling S., Lauber T., Schaupp M., Manhart S., Scheel E., Böhm G., and Demuth H. U. (2006) On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45, 12393–12399 [DOI] [PubMed] [Google Scholar]
- 40. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., et al. (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deng Y., Wang Z., Wang R., Zhang X., Zhang S., Wu Y., Staufenbiel M., Cai F., and Song W. (2013) Amyloid-β protein (Aβ) Glu11 is the major β-secretase site of β-site amyloid-β precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Aβ Asp1 contributes to Alzheimer pathogenesis. Eur. J. Neurosci. 37, 1962–1969 [DOI] [PubMed] [Google Scholar]
- 42. Portelius E., Lashley T., Westerlund A., Persson R., Fox N. C., Blennow K., Revesz T., and Zetterberg H. (2015) Brain amyloid-β fragment signatures in pathological ageing and Alzheimer's disease by hybrid immunoprecipitation mass spectrometry. Neurodegener. Dis. 15, 50–57 [DOI] [PubMed] [Google Scholar]
- 43. Frost J. L., Liu B., Rahfeld J.-U., Kleinschmidt M., O'Nuallain B., Le K. X., Lues I., Caldarone B. J., Schilling S., Demuth H.-U., and Lemere C. A. (2015) An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol. Aging 36, 3187–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Portelius E., Mattsson N., Andreasson U., Blennow K., and Zetterberg H. (2011) Novel Aβ isoforms in Alzheimer's disease: their role in diagnosis and treatment. Curr. Pharm. Des. 17, 2594–2602 [DOI] [PubMed] [Google Scholar]
- 45. Welzel A. T., Maggio J. E., Shankar G. M., Walker D. E., Ostaszewski B. L., Li S., Klyubin I., Rowan M. J., Seubert P., Walsh D. M., and Selkoe D. J. (2014) Secreted amyloid β-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry 53, 3908–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bayer T. A., and Wirths O. (2014) Focusing the amyloid cascade hypothesis on N-truncated Aβ peptides as drug targets against Alzheimer's disease. Acta Neuropathol. 127, 787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Portelius E., Dean R. A., Andreasson U., Mattsson N., Westerlund A., Olsson M., Demattos R. B., Racke M. M., Zetterberg H., May P. C., and Blennow K. (2014) β-Site amyloid precursor protein-cleaving enzyme 1 (BACE1) inhibitor treatment induces Aβ5-X peptides through alternative amyloid precursor protein cleavage. Alzheimers Res. Ther. 6, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haapasalo A., and Kovacs D. M. (2011) The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vassar R. (2014) BACE1 inhibitor drugs in clinical trials for Alzheimer's disease. Alzheimers Res. Ther. 6, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsuda S., Giliberto L., Matsuda Y., McGowan E. M., and D'Adamio L. (2008) BRI2 inhibits amyloid β-peptide precursor protein processing by interfering with the docking of secretases to the substrate. J. Neurosci. 28, 8668–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andersen O. M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C. A., Breiderhoff T., Jansen P., Wu X., Bales K. R., Cappai R., Masters C. L., Gliemann J., Mufson E. J., et al. (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McLoughlin D. M., and Miller C. C. (2008) The FE65 proteins and Alzheimer's disease. J. Neurosci. Res. 86, 744–754 [DOI] [PubMed] [Google Scholar]
- 53. Ho A., Liu X., and Südhof T. C. (2008) Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease. J. Neurosci. 28, 14392–14400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsuda S., Tamayev R., and D'Adamio L. (2011) Increased AβPP processing in familial Danish dementia patients. J. Alzheimers Dis. 27, 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gautam V., D'Avanzo C., Berezovska O., Tanzi R. E., and Kovacs D. M. (2015) Synaptotagmins interact with APP and promote Aβ generation. Mol. Neurodegener. 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hosp F., Vossfeldt H., Heinig M., Vasiljevic D., Arumughan A., Wyler E., Genetic and Environmental Risk for Alzheimer's Disease GERAD1 Consortium, Landthaler M., Hubner N., Wanker E. E., Lannfelt L., Ingelsson M., Lalowski M., Voigt A., and Selbach M. (2015) Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 11, 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Inoue M., Hur J. Y., Kihara T., Teranishi Y., Yamamoto N. G., Ishikawa T., Wiehager B., Winblad B., Tjernberg L. O., and Schedin-Weiss S. (2015) Human brain proteins showing neuron-specific interactions with γ-secretase. FEBS J. 282, 2587–2599 [DOI] [PubMed] [Google Scholar]
- 58. Bai Y., Markham K., Chen F., Weerasekera R., Watts J., Horne P., Wakutani Y., Bagshaw R., Mathews P. M., Fraser P. E., Westaway D., St George-Hyslop P., and Schmitt-Ulms G. (2008) The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteomics 7, 15–34 [DOI] [PubMed] [Google Scholar]
- 59. Rice H. C., Young-Pearse T. L., and Selkoe D. J. (2013) Systematic evaluation of candidate ligands regulating ectodomain shedding of amyloid precursor protein. Biochemistry 52, 3264–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kohli B. M., Pflieger D., Mueller L. N., Carbonetti G., Aebersold R., Nitsch R. M., and Konietzko U. (2012) Interactome of the amyloid precursor protein APP in brain reveals a protein network involved in synaptic vesicle turnover and a close association with Synaptotagmin-1. J. Proteome Res. 11, 4075–4090 [DOI] [PubMed] [Google Scholar]
- 61. Cottrell B. A., Galvan V., Banwait S., Gorostiza O., Lombardo C. R., Williams T., Schilling B., Peel A., Gibson B., Koo E. H., Link C. D., and Bredesen D. E. (2005) A pilot proteomic study of amyloid precursor interactors in Alzheimer's disease. Ann. Neurol. 58, 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mecozzi V. J., Berman D. E., Simoes S., Vetanovetz C., Awal M. R., Patel V. M., Schneider R. T., Petsko G. A., Ringe D., and Small S. A. (2014) Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 10, 443–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. So P. P., Zeldich E., Seyb K. I., Huang M. M., Concannon J. B., King G. D., Chen C. D., Cuny G. D., Glicksman M. A., and Abraham C. R. (2012) Lowering of amyloid β peptide production with a small molecule inhibitor of amyloid-β precursor protein dimerization. Am. J. Neurodegener. Dis. 1, 75–87 [PMC free article] [PubMed] [Google Scholar]
- 64. Pasciuto E., Ahmed T., Wahle T., Gardoni F., D'Andrea L., Pacini L., Jacquemont S., Tassone F., Balschun D., Dotti C. G., Callaerts-Vegh Z., D'Hooge R., Müller U. C., Di Luca M., De Strooper B., and Bagni C. (2015) Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron 87, 382–398 [DOI] [PubMed] [Google Scholar]
- 65. Freude K. K., Penjwini M., Davis J. L., LaFerla F. M., and Blurton-Jones M. (2011) Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J. Biol. Chem. 286, 24264–24274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Furukawa K., Sopher B. L., Rydel R. E., Begley J. G., Pham D. G., Martin G. M., Fox M., and Mattson M. P. (1996) Increased activity-regulating and neuroprotective efficacy of α-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J. Neurochem. 67, 1882–1896 [DOI] [PubMed] [Google Scholar]
- 67. Lauritzen I., Pardossi-Piquard R., Bauer C., Brigham E., Abraham J. D., Ranaldi S., Fraser P., St-George-Hyslop P., Le Thuc O., Espin V., Chami L., Dunys J., and Checler F. (2012) The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 32, 16243–16255a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tamayev R., Matsuda S., Arancio O., and D'Adamio L. (2012) β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol. Med. 4, 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mitani Y., Yarimizu J., Saita K., Uchino H., Akashiba H., Shitaka Y., Ni K., and Matsuoka N. (2012) Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 32, 2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bittner T., Fuhrmann M., Burgold S., Jung C. K., Volbracht C., Steiner H., Mitteregger G., Kretzschmar H. A., Haass C., and Herms J. (2009) γ-Secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J. Neurosci. 29, 10405–10409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Milosch N., Tanriöver G., Kundu A., Rami A., François J. C., Baumkötter F., Weyer S. W., Samanta A., Jäschke A., Brod F., Buchholz C. J., Kins S., Behl C., Müller U. C., and Kögel D. (2014) Holo-APP and G-protein-mediated signaling are required for sAPPα-induced activation of the Akt survival pathway. Cell Death Dis. 5, e1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Corrigan F., Vink R., Blumbergs P. C., Masters C. L., Cappai R., and van den Heuvel C. (2012) sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J. Neurochem. 122, 208–220 [DOI] [PubMed] [Google Scholar]
- 73. Fol R., Braudeau J., Ludewig S., Abel T., Weyer S. W., Roederer J. P., Brod F., Audrain M., Bemelmans A. P., Buchholz C. J., Korte M., Cartier N., and Müller U. C. (2016) Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer's disease mouse model. Acta Neuropathol. 131, 247–266 [DOI] [PubMed] [Google Scholar]
- 74. Hick M., Herrmann U., Weyer S. W., Mallm J. P., Tschäpe J. A., Borgers M., Mercken M., Roth F. C., Draguhn A., Slomianka L., Wolfer D. P., Korte M., and Müller U. C. (2015) Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 129, 21–37 [DOI] [PubMed] [Google Scholar]
- 75. Tian Y., Crump C. J., and Li Y. M. (2010) Dual role of α-secretase cleavage in the regulation of γ-secretase activity for amyloid production. J. Biol. Chem. 285, 32549–32556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gowing E., Roher A. E., Woods A. S., Cotter R. J., Chaney M., Little S. P., and Ball M. J. (1994) Chemical characterization of Aβ17–42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J. Biol. Chem. 269, 10987–10990 [PubMed] [Google Scholar]
- 77. Dulin F., Léveillé F., Ortega J. B., Mornon J. P., Buisson A., Callebaut I., and Colloc'h N. (2008) P3 peptide, a truncated form of Aβ devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS Lett. 582, 1865–1870 [DOI] [PubMed] [Google Scholar]
- 78. Jang H., Arce F. T., Ramachandran S., Capone R., Azimova R., Kagan B. L., Nussinov R., and Lal R. (2010) Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer's disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 6538–6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beckett C., Nalivaeva N. N., Belyaev N. D., and Turner A. J. (2012) Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell. Signal. 24, 402–409 [DOI] [PubMed] [Google Scholar]
- 80. Ghosal K., Vogt D. L., Liang M., Shen Y., Lamb B. T., and Pimplikar S. W. (2009) Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. U. S. A. 106, 18367–18372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Giliberto L., d'Abramo C., Acker C. M., Davies P., and D'Adamio L. (2010) Transgenic expression of the amyloid-β precursor protein-intracellular domain does not induce Alzheimer's Disease-like traits in vivo. PLoS ONE 5, e11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dawkins E., Gasperini R., Hu Y., Cui H., Vincent A. J., Bolós M., Young K. M., Foa L., and Small D. H. (2014) The N-terminal fragment of the β-amyloid precursor protein of Alzheimer's disease (N-APP) binds to phosphoinositide-rich domains on the surface of hippocampal neurons. J. Neurosci. Res. 92, 1478–1489 [DOI] [PubMed] [Google Scholar]
- 83. Burgos P. V., Mardones G. A., Rojas A. L., daSilva L. L., Prabhu Y., Hurley J. H., and Bonifacino J. S. (2010) Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18, 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Matsuda S., Matsuda Y., and D'Adamio L. (2009) BRI3 inhibits amyloid precursor protein processing in a mechanistically distinct manner from its homologue dementia gene BRI2. J. Biol. Chem. 284, 15815–15825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Matsuda S., Matsuda Y., and D'Adamio L. (2009) CD74 interacts with APP and suppresses the production of Aβ. Mol. Neurodegener. 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee J., Retamal C., Cuitiño L., Caruano-Yzermans A., Shin J. E., van Kerkhof P., Marzolo M. P., and Bu G. (2008) Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yang Y., Turner R. S., and Gaut J. R. (1998) The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Aβ40 and Aβ42 secretion. J. Biol. Chem. 273, 25552–25555 [DOI] [PubMed] [Google Scholar]
- 88. Ulery P. G., Beers J., Mikhailenko I., Tanzi R. E., Rebeck G. W., Hyman B. T., and Strickland D. K. (2000) Modulation of β-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP): evidence that LRP contributes to the pathogenesis of Alzheimer's disease. J. Biol. Chem. 275, 7410–7415 [DOI] [PubMed] [Google Scholar]
- 89. Kounnas M. Z., Moir R. D., Rebeck G. W., Bush A. I., Argraves W. S., Tanzi R. E., Hyman B. T., and Strickland D. K. (1995) LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell 82, 331–340 [DOI] [PubMed] [Google Scholar]
- 90. Cam J. A., Zerbinatti C. V., Knisely J. M., Hecimovic S., Li Y., and Bu G. (2004) The low density lipoprotein receptor-related protein 1B retains β-amyloid precursor protein at the cell surface and reduces amyloid-β peptide production. J. Biol. Chem. 279, 29639–29646 [DOI] [PubMed] [Google Scholar]
- 91. Brodeur J., Thériault C., Lessard-Beaudoin M., Marcil A., Dahan S., and Lavoie C. (2012) LDLR-related protein 10 (LRP10) regulates amyloid precursor protein (APP) trafficking and processing: evidence for a role in Alzheimer's disease. Mol. Neurodegener. 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Park J. H., Gimbel D. A., GrandPre T., Lee J. K., Kim J. E., Li W., Lee D. H., and Strittmatter S. M. (2006) Alzheimer precursor protein interaction with the Nogo-66 receptor reduces amyloid-β plaque deposition. J. Neurosci. 26, 1386–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhou X., Hu X., He W., Tang X., Shi Q., Zhang Z., and Yan R. (2011) Interaction between amyloid precursor protein and Nogo receptors regulates amyloid deposition. FASEB J. 25, 3146–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kuan Y.-H., Gruebl T., Soba P., Eggert S., Nesic I., Back S., Kirsch J., Beyreuther K., and Kins S. (2006) PAT1a modulates intracellular transport and processing of amyloid precursor protein (APP), APLP1, and APLP2. J. Biol. Chem. 281, 40114–40123 [DOI] [PubMed] [Google Scholar]
- 95. Akiyama H., Shin R. W., Uchida C., Kitamoto T., and Uchida T. (2005) Pin1 promotes production of Alzheimer's amyloid β from β-cleaved amyloid precursor protein. Biochem. Biophys. Res. Commun. 336, 521–529 [DOI] [PubMed] [Google Scholar]
- 96. Pastorino L., Sun A., Lu P-J, Zhou X. Z., Balastik M., Finn G., Wulf G., Lim J., Li S.-H., Li X., Xia W., Nicholson L. K., and Lu K. P. (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 440, 528–534 [DOI] [PubMed] [Google Scholar]
- 97. Reitz C., Tokuhiro S., Clark L. N., Conrad C., Vonsattel J. P., Hazrati L. N., Palotás A., Lantigua R., Medrano M., Z Jiménez-Velázquez I., Vardarajan B., Simkin I., Haines J. L., Pericak-Vance M. A., Farrer L. A., et al. (2011) SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer's disease risk. Ann. Neurol. 69, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Andersen O. M., Schmidt V., Spoelgen R., Gliemann J., Behlke J., Galatis D., McKinstry W. J., Parker M. W., Masters C. L., Hyman B. T., Cappai R., and Willnow T. E. (2006) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45, 2618–2628 [DOI] [PubMed] [Google Scholar]
- 99. Ho A., and Südhof T. C. (2004) Binding of F-spondin to amyloid-β precursor protein: a candidate amyloid-β precursor protein ligand that modulates amyloid-β precursor protein cleavage. Proc. Natl. Acad. Sci. U.S.A. 101, 2548–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang J., Lu R., Yang J., Li H., He Z., Jing N., Wang X., and Wang Y. (2015) TRPC6 specifically interacts with APP to inhibit its cleavage by γ-secretase and reduce Aβ production. Nat. Commun. 6, 8876. [DOI] [PMC free article] [PubMed] [Google Scholar]