

Abstract
A subset of thyroid carcinomas contains a t(2;3)(q13;p25) chromosomal translocation that fuses paired box gene 8 (PAX8) with the peroxisome proliferator-activated receptor γ gene (PPARG), resulting in expression of a PAX8-PPARγ fusion protein, PPFP. We previously generated a transgenic mouse model of PPFP thyroid carcinoma and showed that feeding the PPARγ agonist pioglitazone greatly decreased the size of the primary tumor and prevented metastatic disease in vivo. The antitumor effect correlates with the fact that pioglitazone turns PPFP into a strongly PPARγ-like molecule, resulting in trans-differentiation of the thyroid cancer cells into adipocyte-like cells that lose malignant character as they become more differentiated. To further study this process, we performed cell culture experiments with thyrocytes from the PPFP mouse thyroid cancers. Our data show that pioglitazone induced cellular lipid accumulation and the expression of adipocyte marker genes in the cultured cells, and shRNA knockdown of PPFP eliminated this pioglitazone effect. In addition, we found that PPFP and thyroid transcription factor 1 (TTF-1) physically interact, and that these transcription factors bind near each other on numerous target genes. TTF-1 knockdown and overexpression studies showed that TTF-1 inhibits PPFP target gene expression and impairs adipogenic trans-differentiation. Surprisingly, pioglitazone repressed TTF-1 expression in PPFP-expressing thyrocytes. Our data indicate that TTF-1 interacts with PPFP to inhibit the pro-adipogenic response to pioglitazone, and that the ability of pioglitazone to decrease TTF-1 expression contributes to its pro-adipogenic action.
Keywords: cancer, differentiation, fusion protein, gene expression, thyroid
Introduction
Approximately 30% of follicular thyroid carcinomas, as well as small subsets of follicular variant papillary carcinomas and follicular adenomas, harbor a t(2,3)(q13;p25) chromosomal translocation that fuses paired box gene 8 (PAX8) 2 with the peroxisome proliferator-activated receptor γ gene (PPARG), resulting in the production of a PAX8-PPARγ fusion protein (PPFP) (1). This fusion protein is composed of all but the very carboxyl-terminal segment of PAX8 followed by fully intact PPARγ1, and its expression is driven by the strong PAX8 promoter. Both the PAX8 and the PPARγ DNA binding domains within PPFP are functional, as determined by chromatin immunoprecipitation deep sequencing (ChIP-seq) (2). The transcription factor PAX8 is a master regulator of thyroid development and function. PAX8 mutations in humans (3) and mice (4) result in failure of thyroid gland development. In the mature thyroid gland, PAX8 induces the expression of thyroid-specific genes such as those encoding thyroglobulin, thyroid peroxidase, and the sodium iodide symporter (5–7). PPARγ is a member of the nuclear receptor superfamily of ligand-activated transcription factors (8, 9) and among its functions, it is essential for adipogenesis (10). Synthetic agonist ligands for PPARγ such as pioglitazone are insulin sensitizers and hence are used to treat type 2 diabetes mellitus. PPARγ ligands also are ligands for PPFP. PPARγ has no identified role in the normal thyroid and is expressed at extremely low levels in that organ.
We have recently developed a transgenic mouse model of PPFP thyroid carcinoma (11). In this model, pioglitazone was highly therapeutic, greatly shrinking thyroid size and preventing metastatic disease. The most remarkable aspect of this therapeutic response is that pioglitazone converted PPFP into a strongly PPARγ-like transcription factor, resulting in the induction of numerous adipocyte PPARγ target genes and the accumulation of large amounts of intracellular lipid. This pro-differentiation effect likely underlies the therapeutic efficacy of pioglitazone, but the factors that regulate the adipogenic response to ligand-bound PPFP have not been investigated.
Thyroid transcription factor 1 (TTF-1), formally denoted Nkx2–1, is a homeobox transcription factor essential for the development of the thyroid, lung and brain (12). In the mature thyroid, TTF-1 interacts physically with PAX8 to induce the expression of thyroglobulin and thyroid peroxidase (13, 14). TTF-1 appears to have a complex role in cancer biology. In lung adenocarcinoma, both pro-oncogenic and anti-oncogenic effects have been described (reviewed in Ref. 15). A germline missense mutation (A339V) of TTF-1 has been identified in families with multinodular goiter and papillary thyroid carcinoma (16). A genome-wide association study of thyroid cancer cases revealed the variant SNP rs944289 located on chromosome 14q13.3, which maps close to TTF-1, was associated with an increased risk of thyroid cancer (17). Concurrent overexpression of RET/PTC1 and TTF-1 confers tumorigenicity to thyrocytes in nude mice (18).
In the present study, we established a PPFP thyroid cell culture model system that replicates the pioglitazone-dependent trans-differentiation of thyroid carcinoma cells into adipocyte-like cells, and we show this effect requires the expression of PPFP. We show that TTF-1 physically interacts with PPFP, that these transcription factors bind near each other on numerous target genes, and that TTF-1 has an inhibitory effect on PPFP/pioglitazone-mediated adipogenic function. Importantly, PPFP down-regulates endogenous TTF-1 expression in a pioglitazone-dependent manner, suggesting that this effect contributes to the pro-adipogenic trans-differentiation and anti-tumor activities of pioglitazone in PPFP thyroid carcinomas.
Results
The Pioglitazone-dependent Adipogenic Differentiation of Thyroid Cancer Cells Is Mediated through PPFP
Thyroid-specific expression of PPFP combined with thyroid-specific deletion of Pten is an established murine model of PPFP thyroid carcinoma (PPFPThy;PtenThy−/− mice) (11). The PPARγ agonist pioglitazone has a profound therapeutic effect in these mice, and results in broad up-regulation of adipocyte PPARγ target genes as the thyroid cancer cells adopt an adipocyte-like phenotype. To elucidate the molecular mechanisms underlying this process, we created a thyroid cell line from the PPFPThy;PtenThy−/− mice. These cells highly express PPFP (Fig. 1A, lane 1) but there is no detectable expression of PPARγ protein. Nine days of treatment with pioglitazone resulted in the accumulation of intracellular lipid as assessed by Oil Red O staining and quantification (Fig. 1B), as well as the induction of adipocyte PPARγ target genes such as Fabp4, Plin1, Cd36, and Lpl at the RNA level (Fig. 1C). The induction of Fabp4, Plin1, and Cd36 was confirmed at the protein level, but there was still no detectable PPARγ protein (Fig. 1D). These data support the hypothesis that pioglitazone-induced adipogenic differentiation reflects the PPARγ-like function of PPFP.
FIGURE 1.

Pioglitazone promotes adipogenic differentiation of PPFP thyrocytes. A, immunoblot using a PPARγ antibody of whole cell lysates from mouse PPFP thyrocytes, 3T3-L1 preadipocytes, and fully differentiated 3T3-L1 adipocytes. N.S., nonspecific band. B, left panel: mouse PPFP thyrocytes were induced to adipogenic differentiation by pioglitazone for 9 days or treated with vehicle (DMSO), and then neutral lipid was detected by Oil Red O (ORO) staining. B, right panel: the lipid content stained by ORO was quantified. C, relative mRNA expression of adipocyte genes was determined by RT-qPCR. Data were normalized to the expression of cyclophilin with triplicated samples. D, immunoblot using an antibody to PPARγ, Fabp4, Plin1, or CD36 of whole cell lysates from PPFP thyrocytes induced by pioglitazone or DMSO for 9 days (left two lanes), or cell lysates from 3T3-L1 preadipocytes or adipocytes as indicated in A (right two lanes). A–D, statistical significance was evaluated with Student's t test, **, p < 0.01; data are representative of three independent experiments.
To further demonstrate the central role of PPFP in the pioglitazone proadipogenic effect, we performed a series of PPFP knockdown experiments. Two shRNAs targeting the PPARγ portion of PPFP effectively knocked down PPFP expression in this cell line (Fig. 2A). As expected, silencing of PPFP inhibited the pro-adipogenic effect of pioglitazone as assessed by Oil Red O staining (Fig. 2B) and quantification (Fig. 2C), as well as by decreased expression of adipocyte genes (Fig. 2, D and E).
FIGURE 2.

Depletion of PPFP impairs pioglitazone-induced adipogenic trans-differentiation of mouse PPFP thyrocytes. A, left panel, knockdown of PPFP by two shRNAs targeting its PPARγ sequence (shPPARγ-1 and PPARγ-2) was confirmed by immunoblot using an anti-PPARγ antibody, N.S. nonspecific band. A, right panel, knockdown of PPFP was further confirmed by RT-qPCR using PPFP specific primers. B, ORO staining of PPFP thyrocytes following shControl or shPPARγ knockdown of PPFP and treatment with pioglitazone for 9 days shows that knockdown of PPFP impairs adipogenic differentiation. C, quantification of ORO staining in B. D, RT-qPCR analysis of adipocyte gene expression 9 days postadipogenic differentiation with pioglitazone. Data were normalized to the expression of cyclophilin with triplicated samples. E, immunoblot against Plin1 for whole cell lysates after PPFP knockdown by shPPARγ and 9 days of treatment with pioglitazone. F, knockdown of PPFP protein using an shRNA targeting its PAX8 sequence (shPax8). PPFP was detected using a PPARγ antibody. N.S., nonspecific band. G, ORO staining shows loss of lipid accumulation following knockdown of PPFP by shPax8 and treatment with pioglitazone for 9 days. H, quantification of ORO staining in G. I, RT-qPCR analysis of adipocyte gene expression in the cells of G. A–I, data are representative of three independent experiments. Each qPCR analysis was performed in triplicate samples, and data were normalized to the expression of cyclophilin. Statistical significance was evaluated with Student's t test, or ANOVA followed by Scheffe's test, *, p < 0.05 and **, p < 0.01.
Although there is no detectable PPARγ protein expression in these cells, the possibility remains that a very low level of PPARγ protein is playing a role and that the shRNA is acting by depletion of this putative low level PPARγ. To eliminate this possibility, we also knocked down PPFP using an shRNA against the PAX8 potion of the protein (Fig. 2F). This also impaired the ability of pioglitazone to induce an adipogenic response as assessed by Oil Red O staining (Fig. 2G) and quantification (Fig. 2H), as well as by decreased expression of adipocyte genes (Fig. 2I). These data indicate that PPFP is responsible for the adipogenic differentiation of PPFP thyroid cells when treated with pioglitazone.
Thyroid Transcription Factor 1 (TTF-1) Interacts with PPFP and Inhibits PPFP Target Gene Expression in a Pioglitazone-dependent Manner
We recently identified PPFP genomic binding sites by ChIP-seq and characterized PPFP-dependent gene expression by mRNA deep sequencing (RNA-seq) in PCCL3-PPFP rat thyroid cells (2). We tested for transcription factor motifs overrepresented in the proximal regions surrounding the PPFP peaks from the ChIP-seq data using Genomatix Genome Analyzer, to identify candidate transcription factors that interact with PPFP. As shown in supplemental Table S1, Nkx2 family proteins were prominently over-represented, with the motif matrix for TTF-1 (Nkx2–1) having the fifth highest Z-score in promoters and the ninth highest in the genome. We decided to focus our attention on TTF-1, since as noted above, it interacts physically and functionally with PAX8 and plays a role in carcinogenesis.
We hypothesized that PPFP binds to TTF-1 and that this interaction modulates PPFP target gene expression. We cotransfected Myc-PPFP and HA-TTF1 into JEG3 cells, and found that they co-immunoprecipitate (Fig. 3A). Furthermore, Myc-PPFP also pulled down endogenous TTF-1 in the mouse PPFP thyroid cell line (Fig. 3B). We next investigated the functional importance of the PPFP-TTF-1 interaction by knockdown of endogenous TTF-1 in PCCL3-PPFP cells or PCCL3-EV (empty vector) control cells. Immunoblotting indicated that two different targeting shRNAs (shTTF-1-1 and shTTF-1-2) knocked down endogenous TTF-1 protein expression (Fig. 3C). The effectiveness of this knockdown was confirmed by showing that both shRNAs down-regulated the expression of Tg, a known TTF-1 target gene (Fig. 3D). We then tested several genes that are induced by PPFP, and found that neither shRNA affected expression of Duoxa2, whereas both increased the PPFP/pioglitazone-dependent expression of Plin1 and Crat (Fig. 3D). To simplify the experiments, all subsequent experiments were performed using shTTF-1–2 (denoted as shTTF-1).
FIGURE 3.

TTF-1 binds to PPFP and depletion of TTF-1 up-regulates expression of a subset of PPFP target genes in a pioglitazone-dependent manner. A, co-immunoprecipitation of HA-TTF-1 and Myc-PPFP following co-transfection into JEG3 cells. Left, whole cell lysates were immunoprecipitated with anti-Myc tag or normal IgG, followed by SDS-PAGE and immunoblotting with anti-HA tag. Right, immunoprecipitation was with anti-HA tag or normal IgG, followed by SDS-PAGE and immunoblotting with anti-Myc tag. B, whole cell lysates of mouse PPFP thyrocytes were immunoprecipitated with anti-Myc tag or normal IgG, followed by SDS-PAGE and immunoblotting with anti-TTF-1. C, knockdown of endogenous TTF-1 by two shRNAs targeting rat TTF-1 (shTTF-1-1 and shTTF-1-2) in both PCCL3-EV (left) and PCCL3-PPFP (right) rat thyroid cells. The cells were treated with vehicle DMSO or 1 μm pioglitazone for 24 h prior to harvest, and immunoblotting was with an anti-TTF-1 antibody. N.S., nonspecific band. D, effect of depletion of endogenous TTF-1 on mRNA expression of the TTF-1 target gene Tg, and the PPFP target genes Duoxa2, Plin1, and Crat. Analysis was by RT-qPCR for cells indicated in C. Each qPCR analysis was performed in triplicate, and the data were normalized to the expression of Pgk1. Statistical significances comparing shTTF-1-1 and shTTF-1-2 with shControl in PPFP cells in the presence of pioglitazone were evaluated with ANOVA followed by Scheffe's test, *, p < 0.05 and **, p < 0.01. A–D, data are representative of three independent experiments.
We tested the effect of TTF-1 knockdown on the expression of a series of genes that are induced by PPFP and that contain a PPFP ChIP-seq peak within 5 kb of the transcription start site which also has a TTF-1 motif within 150 bp of the PPFP peak center predicted by FIMO (19). Ten such genes were tested, and all 10 demonstrated increased pioglitazone-dependent expression upon TTF-1 knockdown (Fig. 4A). We also tested 8 PPFP target genes that contain a PPFP ChIP-seq peak within 5 kb of the transcription start site but without a predicted TTF-1 motif, and found the expression of 6 of these genes is unaffected by TTF-1 knockdown (Fig. 4B). However, the other two genes, Plin1 and Crat, were up-regulated by TTF-1 knockdown (see Fig. 3D).
FIGURE 4.

Depletion of TTF-1 up-regulates PPFP target gene expression in a pioglitazone-dependent manner. A, RT-qPCR analysis of gene expression in PCCL3-EV cells and PCCL3-PPFP cells, with either shControl or shTTF-1 knockdown, and following treatment with DMSO or pioglitazone for 24 h. The genes analyzed all have a ChIP-seq PPFP peak that is predicted by FIMO to contain a TTF-1 motif within 150 bp of the PPFP peak center. B, similar to A, except the PPFP peaks of these genes are predicted by FIMO not to contain a TTF-1 motif. A–B, data are representative of three independent experiments. Each qPCR analysis was performed in triplicate, and data were normalized to the expression of Pgk1. Statistical significance was evaluated with Student's t test, **, p < 0.01.
Because a computational tool such as FIMO may give false positive or false negative identification of transcription factor binding sites, we also used ChIP-qPCR to evaluate TTF-1 binding to a subset of the above genes (Fig. 5A). We tested 4 of the PPFP peaks that were predicted to contain TTF-1 binding sites (Ces2g, Cidec, Grepl2, Slc16a6), and all 4 gave strong TTF-1 ChIP-qPCR signals compared with sites in 4 negative control genes that do not contain PPFP peaks and that are not predicted to encompass TTF-1 motifs (Hprt1, Ash1L, Chfr, Ireb2).
FIGURE 5.

Recruitment of TTF-1 to PPFP ChIP-seq peaks. A, ChIP against TTF-1 (or with normal IgG) was performed in PCCL3-PPFP cells that had been treated with pioglitazone for 24 h, followed by qPCR of the immunoprecipitates to determine the recruitment of TTF-1 to potential target sites. The genes Hprt1, Ash1L, Chfr, and Ireb2 are negative controls. These genes do not contain PPFP ChIP-seq peaks and the qPCR amplicons are predicted by FIMO not to contain TTF-1 motifs. The qPCR amplicons for Ces2g, Cidec, Grepl2, and Slc16a6 lie within PPFP ChIP-seq peaks that are predicted by FIMO to contain TTF-1 motifs within 150 bp of the peak center. The qPCR amplicons for Duoxa2, Ddx52, Ech1, Kpna2, and Lgals lie within PPFP ChIP-seq peaks that are predicted by FIMO not to contain TTF-1 motifs. The amplicons for Plin1 and Crat lie outside PPFP-ChIP-seq peaks but encompass predicted TTF-1 motifs. B, PCCL3-PPFP cells were transfected with either HA-TTF-1 or HA-TTF-1HDD. Forty-eight hours post-transfection, the cells were treated with pioglitazone for 24 h followed by ChIP with an anti-HA tag antibody and qPCR for the amplicons described in A. C, PCCL3-PPFP cells were transfected with either HA-TTF-1 or HA-TTF-1HDD. Cytoplasmic and nuclear fractions were isolated and analyzed by immunoblot using an anti-HA tag antibody. The blot also was probed for PPFP (using anti-PPARγ) as a protein expected to be primarily nuclear, and for GAPDH as a protein expected to be primarily cytoplasmic. D, JEG3 cells were cotransfected with HA-TTF-1 plus Myc-PPFP (lane 1), HA-TTF-1HDD plus Myc-PPFP (lane 2), or HA-TTF-1 plus Myc empty vector (lane 3). PPFP was immunoprecipitated using an anti-Myc tag antibody, and the material was analyzed for co-precipitated HA-TTF-1 or HA-TTF-1HDD by immunoblot with an anti-HA tag antibody. A–D, data are representative of two independent experiments. Each qPCR analysis was performed with triplicate samples.
We then tested the genes predicted by FIMO to lack TTF-1 binding sites in their PPFP peaks, initially focusing on Plin1 and Crat since these were unexpectedly induced by TTF-1 knockdown. Although the Plin1 PPFP peak gave only a weak signal with TTF-1 ChIP-qPCR (data not shown), FIMO predicted the presence of three TTF-1 binding sites within a 543 bp segment of the Plin1 first intron, ∼3.7 kb 3′ to the PPFP peak. TTF-1 ChIP-qPCR using primers centered within this 543 bp sequence gave a strong signal (Fig. 5A), suggesting that TTF-1 bound to this region may functionally interact with PPFP. For Crat, FIMO predicted a TTF-1 binding site 351 bp 3′ to the PPFP peak. This region gave a modest ChIP-qPCR signal (Fig. 5A), suggesting that it may or may not account for the effect of TTF-1 knockdown on gene expression.
We also performed TTF-1 ChIP-qPCR on the PPFP peaks in 5 genes that were unaffected by TTF-1 knockdown and that were predicted to lack TTF-1 binding sites. For these sites we expected a low ChIP-qPCR signal. However, for Ech1, Kpna2, and Lgals, a moderate signal was found, about halfway between that in the negative control genes and that in the genes with TTF-1 binding sites (Fig. 5A). For two of the genes, Duoxa2 and Ddx52, a strong signal was found.
All of the genes that showed moderate or high TTF-1 ChIP-qPCR signals have nearby PPFP binding sites, whereas the 4 negative control genes lack PPFP binding sites and have very low TTF-1 ChIP-qPCR signals. Therefore, it is possible that at least part of the moderate to high TTF-1 ChIP-qPCR signal is due to a physical interaction with PPFP (or perhaps endogenous PAX8), rather than a direct binding of TTF-1 to DNA. If this hypothesis is correct, TTF-1 with its DNA binding domain deleted might still give a moderately strong ChIP-qPCR signal on PPFP peaks that lack a TTF-1 motif. To explore this hypothesis, PCCL3-PPFP cells were transfected with either HA-TTF-1 or HA-TTF-1HDD (deletion of the DNA binding domain), and then ChIP was performed with an anti-HA tag antibody followed by qPCR for a negative control gene (Hprt1) and the PPFP peaks in Cidec, Ech1, and Kpna2 (Fig. 5B). These were chosen because TTF-1 binds strongly to the Cidec PPFP peak and TTF-1 knockdown induces this gene, whereas the Ech1 and Kpna2 PPFP peaks only show moderate TTF-1 binding and the expression of these genes is unaffected by TTF-1 knockdown. The ChIP-qPCR signals for HA-TTF-1 (Fig. 5B) closely parallel those for endogenous TTF-1 (Fig. 5A), whereas HA-TTF-1HDD has essentially no binding to any of these sites. The data suggest that direct DNA binding of TTF-1 is important for generating the TTF-1 ChIP-qPCR signal.
We performed control experiments to support the validity of the HA-TTF-1HDD ChIP-qPCR. First, cell fractionation studies were performed to determine whether HA-TTF-1HDD has access to the nucleus. These data show that HA-TTF-1 is confined to the nucleus, whereas HA-TTF-1HDD is found in both the nucleus and the cytoplasm (Fig. 5C). This indicates that HA-TTF-1HDD does have access to the nucleus, although its inability to bind DNA may facilitate its partial extranuclear localization. Second, co-immunoprecipitation experiments showed that deletion of the DNA binding domain (HA-TTF-1HDD) does not impair the physical interaction of HA-TTF-1 with PPFP (Fig. 5D).
The TTF-1 DNA Binding Domain Is Essential for TTF-1 to Regulate the Transcriptional Activity of PPFP
To further examine the importance of TTF-1 DNA binding in its ability to regulate the function of PPFP, we tested the ability of human TTF-1, with or without an intact DNA binding domain, to replace endogenous TTF-1 in rat PCCL3 cells. We treated PCCL3 cells with shControl or shTTF-1 to knock down endogenous TTF-1, and then transiently transfected the cells with HA-TTF-1 or HA-TTF-1HDD, plus a human AQP7 promoter: firefly luciferase vector and a Renilla luciferase internal control vector (Fig. 6A). AQP7 is one of the most highly induced genes in human PPFP thyroid carcinomas, and we have previously shown this reporter plasmid is induced by PPFP ± pioglitazone (20). As shown in Fig. 6A, left panel, PPFP induced luciferase and pioglitazone induced it further (bars 3 versus 1). Knockdown of TTF-1 increased the inductions by PPFP ± pioglitazone (bars 4 versus 3) but had no effect on expression in the absence of PPFP (bars 2 versus 1), indicating that TTF-1 inhibits the PPFP/pioglitazone response. Fig. 6A, middle panel, shows that forced over expression of human TTF-1 represses luciferase expression and overcomes the stimulatory effect of endogenous TTF-1 knockdown (bars 8 versus 4). In contrast, forced overexpression of human TTF-1 with its DNA binding domain deleted (TTF-1HDD) does not (bars 12 versus 4). Control experiments indicate that TTF-1HDD is expressed at least as well as intact TTF-1 (Fig. 6B, lanes 5–8 versus 1–4, and 13–16 versus 9–12) and does not alter the expression of PPFP (lanes 13–16 versus 9–12). In aggregate the data suggest that DNA binding of TTF-1 is necessary but not sufficient to inhibit PPFP/pioglitazone-dependent target gene expression.
FIGURE 6.

The TTF-1 DNA binding domain is required for TTF-1 to inhibit PPFP transcriptional activity. A, PCCL3-EV or PCCL3-PPFP rat thyroid cells expressing either shControl or shTTF-1 to knock down endogenous TTF-1 were transiently co-transfected with a PPFP-responsive human AQP7 promoter- firefly luciferase plasmid, a pRL-TATA Renilla luciferase internal control plasmid, and either pCagen-EV, pCagen-HA-TTF-1 or pCagen-HA-TTF-1HDD. The cells were treated for 24 h with DMSO or pioglitazone followed by assay of luciferases. The y axis represents firefly luciferase normalized to Renilla luciferase. Statistical significance was evaluated with Student's t test, or ANOVA followed Scheffe's test, **, p < 0.01. B, immunoblots against PPARγ (to detect PPFP) and HA tag (to detect HA-TTF-1 and HA-TTF-1HDD) were performed 48 h post cotransfection of the reporter gene assay described in A. Immunoblotting for β-actin was used as a loading control. A–B, data are representative of two independent experiments.
TTF-1 Does Not Impair the Interaction of PPFP with DNA
To further explore the mechanism underlying the inhibitory effect of TTF-1 on PPFP transcriptional activity, we performed ChIP-qPCR for PPFP recruitment to several target genes. Knockdown of TTF-1 tended to decrease, rather than increase, PPFP recruitment to target genes (Fig. 7), even though PPFP/pioglitazone-dependent gene expression increased (Fig. 4A). This indicates that the inhibitory effect of TTF-1 on PPFP function is not mediated by a decreased interaction of PPFP with DNA. Presumably the PPFP-TTF-1 interaction alters the balance of coactivators and corepressors brought to the target gene, but the precise mechanisms involved are unknown.
FIGURE 7.
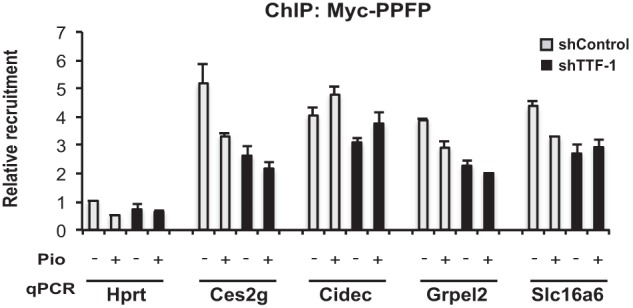
TTF-1 does not impair the binding of PPFP to DNA. PCCL3-PPFP cells expressing either shControl or shTTF-1 to knock down endogenous TTF-1 were treated with DMSO or pioglitazone for 24 h and then subjected to ChIP for PPFP using anti-Myc tag antibody. The precipitates were analyzed by qPCR for binding to Hprt1 as a negative control, and to the previously identified PPFP ChIP-seq peaks in Ces2g, Cidec, Grpel2, and Slc16a6. Data are representative of three independent experiments. Each qPCR analysis was performed with triplicate samples.
TTF-1 Inhibits PPFP-dependent Adipogenic Differentiation in PCCL3 Rat Thyrocytes
The above studies indicate that TTF-1 inhibits at least some gene inductions by PPFP/pioglitazone. Since the major phenotypic response to pioglitazone in PPFP-expressing thyroid cells is pro-adipogenic, we hypothesized that TTF-1 is inhibitory to this response. As predicted, we found that shTTF-1 knockdown enhanced the proadipogenic effect of pioglitazone in PCCL3-PPFP cells, assessed by Oil Red O staining (Fig. 8A) and quantification (Fig. 8B). Knockdown of TTF-1 also induced the expression of numerous adipocyte marker genes in a PPFP and pioglitazone-dependent manner at the RNA level, including Fabp4, Plin2, Cidec, Aqp7, and Dgat2 (Fig. 8C). The inductions of Fabp4 and Plin2 were confirmed at the protein level (Fig. 8D, upper panel, lane 8 versus 6). Immunoblotting for TTF-1 confirmed successful knockdown by shTTF-1 (Fig. 8D, lanes 3–4 versus 1–2, and 7–8 versus 5–6), and also unexpectedly showed that PPFP inhibited endogenous TTF-1 protein expression (upper panel, lanes 5–6 versus 1–2), and pioglitazone further decreased TTF-1 protein expression in PPFP cells (upper panel, lane 6 versus 5, and 8 versus 7), but not in EV cells (upper panel, lane 2 versus 1, and 4 versus 3). These same changes also were seen at the mRNA level (Fig. 8D, lower panel, same lane comparisons as with the upper panel).
FIGURE 8.

Depletion of TTF-1 promotes adipogenic differentiation of PPFP thyroid cells. A, PCCL3-EV and PCCL3-PPFP thyroid cells expressing either shControl or shTTF-1 were treated with DMSO or pioglitazone for 9 days followed by ORO staining of lipid droplets. B, quantification of ORO staining in A. C, relative mRNA expression of adipocyte genes at the conditions described in A was determined by RT-qPCR. D, upper panel, immunoblots for Fabp4, Plin2, and TTF-1 proteins were performed at the end of the differentiation described in A, and β-actin was used as a loading control; Lower panel, RT-qPCR was performed for mouse TTF-1 mRNA expression at the conditions indicated. A–D, data are representative of three independent experiments. Each qPCR analysis was performed in triplicate, and data were normalized to the expression of Pgk1. Statistical significance was evaluated with Student's t test, or ANOVA followed by Scheffe's test, *, p < 0.05 and **, p < 0.01
To test whether TTF-1 overexpression exerts an effect opposite that of TTF-1 depletion on proadipogenic differentiation, we stably overexpressed TTF-1 protein ∼2-fold through retroviral infection of PCCL3-PPFP cells (Fig. 9A). As expected, overexpression of TTF-1 inhibited the proadipogenic effect of pioglitazone, indicated by decreased Oil Red O staining and quantification (Fig. 9, B and C). In agreement with this, TTF-1 overexpression also inhibited the pioglitazone induction of adipocyte marker genes Fabp4, Plin2, Cidec, and Dgat2 at the RNA level (Fig. 9D); and the inhibition Fabp4 was confirmed at the protein level (Fig. 9E, lane 4 versus 2). Importantly, similar to the data in Fig. 8D, pioglitazone inhibited the expression of endogenous TTF-1 in these PCCL3-PPFP cells (Fig. 9E, lane 2 versus 1). However, the expression of exogenous HA-TTF-1, which is driven by a non-native promoter, was not repressed by pioglitazone (Fig. 9E, lane 4 versus 3), suggesting that the effect of PPFP/pioglitazone on inhibiting endogenous TTF-1 is at the level of transcription.
FIGURE 9.

Stable overexpression of TTF-1 inhibits adipogenic differentiation of PPFP thyroid cells. A, immunoblots against TTF-1 or HA tag (to specifically detect exogenous HA-TTF-1) were performed following the stable overexpression of pMSCV-EV or pMSCV-HA-TTF-1 in PCCL3-PPFP rat thyroid cells. B, ORO staining of lipid droplets in pMSCV-EV or pMSCV-HA-TTF-1 expressing PCCL3-PPFP thyroid cells after 9 days of treatment with DMSO or pioglitazone. C, quantification of ORO staining in B. D, relative mRNA expression of adipocyte genes at the conditions described in B, determined by RT-qPCR. Each qPCR analysis was performed in triplicate, and data were normalized to the expression of Pgk1. E, immunoblots against Fabp4, TTF-1, or HA tag were performed at the end of the 9-day proadipogenic differentiation described above. A–D, data are representative of three independent experiments. C and D, statistical significance was evaluated with Student's t test, *, p < 0.05 and **, p < 0.01.
Discussion
A chromosomal translocation fuses the genes encoding PAX8 and PPARγ in ∼30% of follicular thyroid carcinomas, resulting in expression of a PAX8-PPARγ fusion protein, PPFP (1). Since PPFP contains the full sequence of PPARγ1, ligands for PPARγ also are ligands for PPFP. In a transgenic mouse model of PPFP thyroid carcinoma, we previously showed that the PPARγ agonist pioglitazone is highly therapeutic, greatly reducing thyroid tumor size and preventing metastatic disease (11). Histological examination indicated that pioglitazone induced trans-differentiation of the thyroid carcinoma cells into adipocyte-like cells in vivo. Although the tumor cells continue to express thyroid marker genes, they also strongly express a large number of adipocyte PPARγ target genes and accumulate large amounts of intracellular lipid. Since PPARγ is the master regulator of adipogenesis, the in vivo data imply that PPFP is strongly PPARγ-like in the presence of pioglitazone.
To study this further, we created a cell line from the PPFP carcinoma mouse thyrocytes and studied it in culture. We found that these thyrocytes have no detectable endogenous PPARγ protein and that shRNA knockdown of PPFP inhibits pioglitazone-induced adipogenic differentiation in culture (Figs. 1 and 2). These data indicate that pioglitazone is indeed acting via PPFP to induce a PPARγ-like adipogenic response in the thyroid carcinoma cells.
We wanted to identify factors that interact with PPFP/pioglitazone to regulate the pro-adipogenic response, and potentially the anti-tumor response. This led us to study TTF-1 (Nkx2-1). TTF-1 is a homeodomain-containing transcription factor expressed in the thyroid, lung and central nervous system. TTF-1 is essential for thyroid development, and in the mature gland, it regulates the expression of various thyroid-specific genes. TTF-1 also appears to play both pro- and anti-oncogenic roles in the progression of lung carcinoma (15, 21, 22), and it may play a role in thyroid carcinogenesis as well (18). Here, we found that TTF-1 interacts both physically and functionally with PPFP. For unclear reasons knockdown of TTF-1 in PPFP mouse thyroid carcinoma cells led to cell death (data not shown), so we focused on rat PCCL3 thyroid cells with stable expression of PPFP that we have previously studied (23). We found that depletion of TTF-1 up-regulates the expression of a subset of PPFP target genes including adipocyte markers (Fig. 4) and promotes the trans-differentiation of PCCL3-PPFP cells into adipocyte-like cells (Fig. 8). Furthermore, forced overexpression of TTF-1 inhibits the pro-adipogenic action of PPFP/pioglitazone (Fig. 9). Thus, endogenous TTF-1 clearly plays a negative role in regulating the function of PPFP/pioglitazone.
Interestingly, we also found that PPFP inhibits endogenous TTF-1 mRNA and protein expression and that this effect is enhanced by pioglitazone (Figs. 8D and 9E). However, pioglitazone has no effect in the absence of PPFP. The down-regulation of TTF-1 by PPFP±pioglitazone is important for two reasons. First, since TTF-1 inhibits the adipogenic effect of pioglitazone, repression of TTF-1 is an important component of the pro-adipogenic mechanism of PPFP/pioglitazone. Second, since the pro-adipogenic (pro-differentiation) action of pioglitazone is important to its therapeutic anti-tumor effect (11), this implies that down-regulation of TTF-1 is an important component of the anti-tumor effect of this drug. Further, this suggests that an anti-TTF-1 drug could be synergistic with pioglitazone.
The precise molecular mechanism by which TTF-1 inhibits PPFP/pioglitazone target gene expression and adipogenic differentiation is uncertain. The DNA binding domain of TTF-1 is important for this activity (Fig. 6A) and the genes that TTF-1 represses generally have TTF-1 binding sites within 150 bp of the center of PPFP ChIP-seq peaks (Figs. 5 and 7), both of which implicate a role for direct binding of TTF-1 to the target gene. Although the simplest model would be that TTF-1 interferes with binding of PPFP to shared target genes in response to pioglitazone, we found no evidence for this by ChIP-qPCR (Fig. 7). Therefore, it seems more likely that the balance of co-activator and co-repressor complexes shifts more toward the latter when PPFP and TTF-1 bind to target genes. However, this remains to be confirmed and the putative co-regulatory proteins remain to be identified. In fact, the co-regulatory proteins that interact with PPFP are unknown. We performed a co-immunoprecipitation experiment for the co-activator p300 to test whether it might be implicated, but found no interaction of p300 with PPFP in PCCL3-PPFP cells in the absence or presence of shTTF-1 and pioglitazone (data not shown).
In Fig. 10, we present a model that summarizes our data. The model shows PPFP and TTF-1 physically interact in PPFP thyroid cancer cells. Pioglitazone binds to PPFP and inhibits TTF-1 expression. TTF-1 inhibits the ability of pioglitazone-bound PPFP to induce adipocyte target genes and to trans-differentiate the thyroid cancer cells into adipocyte-like thyroid cells.
FIGURE 10.
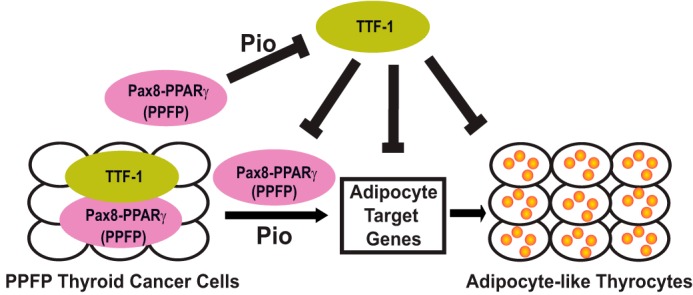
Model of the TTF-1 inhibitory effect on PPFP in thyroid cancer cells. In the presence of pioglitazone, PPFP has PPARγ-like function to promote the induction of adipocyte genes and the trans-differentiation of PPFP thyroid cancer cells into adipocyte-like thyrocytes. PPFP, especially in the presence of pioglitazone, inhibits the expression of TTF-1. However, TTF-1 inhibits the ability of PPFP+pioglitazone to induce adipocyte genes and the trans-differentiation into adipocyte-like cells.
It may seem surprising that TTF-1, which has no known role in adipocyte biology, regulates the pro-adipogenic action of PPFP/pioglitazone. However, an analysis of genome-wide epigenetic changes during adipogenesis identified the motif for TTF-1 as being modified by histone H3 lysine 27 acetylation (24). Since this epigenetic change marks active enhancer elements, it suggests that a transcription factor binds to this motif and contributes to normal adipogenesis. We found that, in thyroid cancer cells, TTF-1 has the opposite effect, inhibiting PPFP/pioglitazone-mediated adipogenic activity. This difference could be explained if the endogenous adipocyte transcription factor is not TTF-1 itself but a related protein. Additional contributing factors could relate to functional differences between PPARγ in adipocytes and PPFP in thyrocytes, or aspects of the timing of expression of the putative true adipocyte transcription factor. Other explanations are possible as well. In any case the data suggest the potential importance of TTF-1 family members in normal adipogenesis.
Experimental Procedures
Cell Culture, Reagents, and Retrovirus Production
Mouse thyroid cells was isolated from thyroid glands of transgenic FVB/N mice with combined thyroid-specific expression of PPFP and thyroid-specific deletion of Pten (denoted PPFPThy;PtenThy−/−), which we have previously established as a mouse model of PPFP thyroid carcinoma (11). For simplicity we use the term “cell line,” but these cells are derived non-clonally from a pool of thyrocytes. The cells have been maintained in culture for over a year with stable expression of PPFP. Cells were cultured at 5% CO2 in Coon's F-12 media (Sigma-Aldrich) with l-glutamine, 5% fetal bovine serum (FBS), penicillin-streptomycin (Cat# 10378, Thermo Scientific, Waltham, MA), 1 mIU/ml bovine thyroid stimulating hormone (Cat. T8931, Sigma-Aldrich),), 10 μg/ml bovine insulin (Cat. I0516, Sigma-Aldrich),), 5 μg/ml bovine apo-transferrin (Cat. T1428, Sigma-Aldrich),), 10 nm hydrococortisone (Cat. H0888, Sigma-Aldrich), 10 nm mouse epidermal growth factor (EGF) (Cat. SRP3196, Sigma-Aldrich), and anti-mycoplasma reagent plasmocin (Cat. ant-mpt, Invivogen, San Diego, CA). We also cultured the PCCL3 rat thyroid cell line that stably expresses PPFP (PCCL3-PPFP cells) or that contains the empty vector (PCCL3-EV), both of which have been described previously (23). PCCL3 cells were cultured as described above except without EGF. JEG3 cells were cultured in Eagles minimum essential medium supplemented with 10% FBS with penicillin-streptomycin at 37 °C in 5% CO2. Differentiation of mouse PPFP thyroid cells or PCCL3-PPFP cells into adipocyte-like cells was achieved by treatment of confluent cells every other day with 1 μm pioglitazone for 7 to 9 days.
Antibodies against the following proteins were obtained as indicated: PPARγ (H-100, sc-7196), TTF-1 (H-190, sc-13040) and GAPDH (sc-32233) from Santa Cruz Biotechnology (Santa Cruz, CA); Fabp4 (D25B3, Cat. 3544), CD36 (DL9T, Cat. 14347), Myc-Tag (9B11, Cat. 2276), HA-Tag (C29F4, Cat. 3724), and β-actin (Cat. 4967) from Cell Signaling Technology (Danvers, MA); Fabp4 (Cat. MAB1443) from R&D Systems, Inc. (Minneapolis, MN); Perilipin A (Plin1) (Cat. PA1-1051) from Thermo Scientific (Waltham, MA); and ADFP (Plin2) (ab52355) from Abcam Inc., (Cambridge, MA).
The pCagen empty vector (pCagen-EV) and pCagen expressing PPFP with three Myc epitopes at the N terminus (pCagen-PPFP) were described previously (23). Human TTF-1 variant 2 (the dominant isoform in the thyroid) with a single HA tag at the N terminus also was expressed from pCagen (pCagen-HA-TTF-1). Human TTF-1 variant 2 with deletion of its homeodomain (DNA binding domain) was purchased from Addgene (Cambridge, MA) (plasmid 52063, pcDNA3-TTF-1HDD). In this construct, TTF-1 amino acids 171–221 are deleted and are replaced by GDL. The PCR was used to add an HA tag to the N terminus of TTF-1HDD as well as a 5′ KpnI site and a 3′ NheI site to facilitate cloning into pCagen. To create the retroviral expression vector pMSCV-HA-TTF-1 (denoted as pMSCV-TTF-1), PCR was used to add BglII and EcoRI sites at the ends of HA-TTF-1 followed by ligation to the same sites in pMSCV (25) using the In-Fusion Dry-Down PCR cloning Kit (Clontech, Mountain View, CA). All constructs were confirmed by sequencing.
For retroviral infection, 293T cells were transfected by calcium phosphate coprecipitation with pMSCV empty vector (pMSCV-EV) and pMSCV-TTF-1 retroviral expression vectors and viral packing vectors as described previously (25). The virus-containing media were collected and applied to sub-confluent PCCL3-PPFP cells followed by 8 μg/ml puromycin selection 72 h postinfection.
shRNA Knockdown of PPFP and TTF-1
Knockdown of PPFP in mouse PPFP thyroid cells was performed by infection with a lentivirus expressing shRNA targeting either its PPARγ or PAX8 portion. Two different Mission Lentivirus-based plasmids of shRNAs (clone numbers: TRCN0000001674 and TRCN0000355926) against human PPARγ, and the shControl using TRC2 pLKO.5-puro non-mammalian shRNA (SHC202) were obtained from Sigma-Aldrich. 293T cells were cotransfected with the shRNA and packaging plasmids psPAX2 and pMD2 by the calcium phosphate method to produce the lentivirus as described previously (25). Similarly, knockdown of PPFP by targeting the PAX8 portion of the protein was performed using Mission Lentivirus-based plasmid shRNA clone number TRCN0000359747. Two inducible shRNA plasmids in the pLKO-puro-IPTG-3xLaco vector against rat TTF-1 were obtained from Sigma-Aldrich, and the targeting sequences were GTTCTCAGTGTCTGACATCTT (shTTF-1–1, clone TRCN0000086267) and CGCCATGTCTTGCTCTACTTT (shTTF-1–2, customer designed). Mission 3× Laco inducible Non-Target shRNA (SHC332 from Sigma-Aldrich) was used as the shControl. After infection of either PCCL3-EV or PCCL3-PPFP cells, selection was achieved with 8 μg/ml puromycin and the shRNAs were induced with 10 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3–5 days.
Oil Red O Staining and Lipid Quantification
Cells were stained for neutral lipid with Oil Red O and photographed as described (25). Oil Red O staining was quantified using the method by Remirez-Zacarias et al. (26).
Cell Lysis, Immunoblotting, and Protein Immunoprecipitation
Cells were harvested in buffer containing 40 mm HEPES, 120 mm sodium chloride, 10 mm sodium pyrophosphate, 10 mm sodium glycerophosphate, 1 mm EDTA, 50 mm sodium fluoride, 0.5 mm sodium orthovanadate, and 1% Triton X-100, and frozen overnight at −80 °C. Cell lysates were resuspended by pipetting and incubated at 4 °C with gentle rocking for 40 min to 1 h, followed by microcentrifugation for 10 min at 4 °C. The supernatants were transferred to new tubes and protein concentrations were determined by Bradford Assay using Bio-Rad Protein Assay Dye Reagent (Cat. 500-0006, Bio-Rad). Nuclear and cytoplasmic extracts were prepared using NE-PER (Cat. 78833) from PCCL3-PPFP cells transiently transfected with pCagen-HA-TTF-1 or pCagen-HA-TTF-1HDD via FuGENE 6 Reagent (Cat. E2691, Promega). Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes, and immunoblotting was performed using the antibodies described above. The primary antibodies were diluted 1:1000 with Signal enhancer HIKARI Solution 1, and the secondary antibodies were diluted 1:10,000 with Signal enhancer HIKARI Solution 2 (Cat. UN00102, Nacalai USA, San Diego, CA). Detection by enhanced chemiluminescence was with a SuperSignal West Dura kit (Cat. 34075, Thermo Scientific, Rockford, IL) and a Bio-Rad Fluor-S Max Multi-Imager.
To test whether PPFP binds to TTF-1 in mammalian cells, Myc-PPFP and HA-TTF-1 were transiently transfected in JEG3 cell using Lipofectamine Plus (Invitrogen). Forty-eight hours post-transfection, cells were lysed in M-PER (Pierce). Immunoprecipitations were performed by incubating cell lysates with either Myc tag or HA tag antibody overnight at 4°C, followed by incubation with Dynabeads Protein G (Cat. 10004D, Thermo Scientific) for 2 h at 4°C. After extensive washing with M-PER, the coimmunoprecipitated proteins were separated by SDS-PAGE and detected by immunoblotting with HA tag or Myc tag antibodies.
RNA Isolation and RT Real-time Quantitative PCR (qPCR)
Total RNA was isolated with an RNeasy Mini Kit (Qiagen, Cat. 74104) according to the manufacturer's instructions. 4 μg of total RNA were reverse transcribed using SuperScript III First Strand Synthesis System (Cat. 18080051, Thermo Scientific) and real-time qPCR was performed as described previously (25). Primer sequences used for qPCR are provided in supplemental Table S2.
Reporter Gene Assays
The plasmid pGL3-AQP7-Luc, in which the human aquaporin 7 (AQP7) gene promoter (bp −2359 to +90) drives firefly luciferase expression, has been described previously (20). The internal control Renilla luciferase plasmid pRL-TATA was constructed by ligation of an E1B TATA box sequence GATCAGGGTATATAATGA (top strand) into the BglII site of pRL-null (Promega). For transfection, shControl/shTTF-1 PCCL3-EV and PCCL3-PPFP cells were induced by IPTG to knock down endogenous TTF-1 followed by splitting the cells into 24-well plates. Afterward, the cells were cotransfected with 150 ng of pGL3-AQP7-Luc, 50 ng of pRL-TATA, and either 50 ng of pCagen-EV, pCagen-HA-TTF-1 or pCagen-HA-TTF-1HDD using Lipofectamine Plus (Invitrogen) followed by incubation of cells for 48 h. The cells were then treated with DMSO (vehicle) or 1 μm pioglitazone for 24 h. The cells were lysed and analyzed for firefly and Renilla luciferase activities using the Promega dual luciferase reporter assay system.
Chromatin Immunoprecipitation (ChIP) Assay
Chromatin immunoprecipitation assays were performed with modifications following the protocol from Chromatin Immunoprecipitation Assay Kit (Upstate, Lake Placid, NY) and as described previously (27). In brief, the cells were cross-linked with 1% formaldehyde, quenched with 0.125 m glycine, and resuspended in SDS lysis buffer (50 mm HEPES, 1%SDS, 10 mm EDTA) containing protease inhibitor mixture (Cat. 78410, Pierce Biotechnology). The cells were sheared twice with a 27-gauge needle and sonicated using three 11 min cycles of 30 s on and 30 s off in a Bioruptor XL (Model UCD310, Diagenode Inc., Denville, NJ) at the high setting. Following centrifugation, the supernatants were diluted 1:10 in ChIP dilution buffer containing 0.05% bovine serum albumin (BSA), 5 μg of sheared salmon sperm DNA (Cat. AM9680, Thermo Fisher Scientific) and protease inhibitor. To decrease nonspecific binding, the samples were precleared by incubation with Protein G or A Dynabeads with sheared salmon sperm DNA/BSA/TE for 2 h at 4 °C. Immunoprecipitation was then performed overnight at 4 °C with anti-TTF-1, HA tag, or Myc tag antibody. After immunoprecipitation, 40 μl of magnetic Protein G or A Dynabeads equilibrated with sheared salmon sperm DNA/BSA/TE was added and the incubation was continued for another 2 h at 4 °C. Precipitates were then washed twice with Low Salt Buffer and once with High Salt Buffer, LiCl Buffer, and Tris-EDTA buffer, pH 8.0. The immunoprecipitated DNAs eluted from the beads with 0.1 m NaHCO3 containing 1% SDS and the 1% input samples were de-crosslinked overnight at 65 °C, and purified using a PCR purification kit (Qiagen, Cat. 28104). Finally, the ChIP-immunoprecipitated DNA fragments were quantified by real-time PCR using primers to PPFP ChIP-seq peaks (2) and TTF-1 binding motifs identified by FIMO (supplemental Table S1). Primer sequences used for ChIP-qPCR are provided in supplemental Table S3.
Over-represented Motif Analysis
We used Genomatix Genome Analyzer to identify overrepresented known motifs in PPFP ChIP-seq peaks (2), which we used to identify candidate interaction partners with PPFP. The input was confined to PPFP peaks that contain a PPARγ or PAX8 motif (65% of all peaks), since peaks with neither motif are more likely to be false positives. Then we masked the nucleotides (converting them to Ns) that were within either a PPARγ or PAX8 motif, to eliminate potential false positive matches to proteins whose motifs are similar to PPARγ or PAX8. We submitted these sequences to Genomatix Region Miner to identify over-represented transcription factor binding sites or modules (using default parameters). TTF-1 and other Nkx2 family members were identified as some of the most over-represented motifs in promoter regions and the genome using Genomatix (supplemental Table S1). We downloaded the TTF-1 motif position weight matrix (PWM) from Genomatix, and utilized FIMO to predict individual TTF-1 motif occurrences across the genome.
Statistical Analysis
Results are presented as the mean ± S.D. When comparing two groups, significance was determined using Student's t test. When more than two groups were compared, an analysis of variance (ANOVA) was followed by Scheffe's test, and the significance is indicated as *, p < 0.05 and **, p < 0.01.
Author Contributions
B. X., Y. Z, M. A. S., and R. J. K. designed the studies and wrote the manuscript. B. X., M. O., J. O., and J. Y. performed the experiments and analyzed the data. Y. Z. and M. A. S. performed the motif analysis.
Supplementary Material
This work was supported by National Institutes of Health (NIH) Grants R01CA151842 and R01CA166033. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S3.
- PAX8
- paired box gene 8
- PPARγ
- peroxisome proliferator activated receptor γ
- PPFP
- Pax8-PPARγ fusion protein
- TTF-1
- thyroid transcription factor 1
- ChIP
- chromatin immunoprecipitation
- ORO
- Oil Red O.
References
- 1. Kroll T. G., Sarraf P., Pecciarini L., Chen C. J., Mueller E., Spiegelman B. M., and Fletcher J. A. (2000) PAX8-PPARγ1 fusion oncogene in human thyroid carcinoma [corrected]. Science 289, 1357–1360 [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y., Yu J., Lee C., Xu B., Sartor M. A., and Koenig R. J. (2015) Genomic binding and regulation of gene expression by the thyroid carcinoma-associated PAX8-PPARγ fusion protein. Oncotarget 6, 40418–40432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macchia P. E., Lapi P., Krude H., Pirro M. T., Missero C., Chiovato L., Souabni A., Baserga M., Tassi V., Pinchera A., Fenzi G., Grüters A., Busslinger M., and Di Lauro R. (1998) PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat. Genet. 19, 83–86 [DOI] [PubMed] [Google Scholar]
- 4. Mansouri A., Chowdhury K., and Gruss P. (1998) Follicular cells of the thyroid gland require Pax8 gene function. Nat. Genet. 19, 87–90 [DOI] [PubMed] [Google Scholar]
- 5. Esposito C., Miccadei S., Saiardi A., and Civitareale D. (1998) PAX 8 activates the enhancer of the human thyroperoxidase gene. Biochem. J. 331, 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fabbro D., Pellizzari L., Mercuri F., Tell G., and Damante G. (1998) Pax-8 protein levels regulate thyroglobulin gene expression. J. Mol. Endocrinol. 21, 347–354 [DOI] [PubMed] [Google Scholar]
- 7. Ohno M., Zannini M., Levy O., Carrasco N., and di Lauro R. (1999) The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol. Cell. Biol. 19, 2051–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evans R. M., Barish G. D., and Wang Y. X. (2004) PPARs and the complex journey to obesity. Nat. Med. 10, 355–361 [DOI] [PubMed] [Google Scholar]
- 9. Rosen E. D., and Spiegelman B. M. (2001) PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 276, 37731–37734 [DOI] [PubMed] [Google Scholar]
- 10. Rosen E. D., Hsu C. H., Wang X., Sakai S., Freeman M. W., Gonzalez F. J., and Spiegelman B. M. (2002) C/EBPα induces adipogenesis through PPARγ: a unified pathway. Genes Dev. 16, 22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dobson M. E., Diallo-Krou E., Grachtchouk V., Yu J., Colby L. A., Wilkinson J. E., Giordano T. J., and Koenig R. J. (2011) Pioglitazone induces a proadipogenic antitumor response in mice with PAX8-PPARγ fusion protein thyroid carcinoma. Endocrinology 152, 4455–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stanfel M. N., Moses K. A., Schwartz R. J., and Zimmer W. E. (2005) Regulation of organ development by the NKX-homeodomain factors: an NKX code. Cell Mol. Biol. (Noisy-le-grand) Suppl. 51, OL785–799 [PubMed] [Google Scholar]
- 13. Di Palma T., Nitsch R., Mascia A., Nitsch L., Di Lauro R., and Zannini M. (2003) The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. J. Biol. Chem. 278, 3395–3402 [DOI] [PubMed] [Google Scholar]
- 14. Miccadei S., De Leo R., Zammarchi E., Natali P. G., and Civitareale D. (2002) The synergistic activity of thyroid transcription factor 1 and Pax 8 relies on the promoter/enhancer interplay. Mol. Endocrinol. 16, 837–846 [DOI] [PubMed] [Google Scholar]
- 15. Yamaguchi T., Hosono Y., Yanagisawa K., and Takahashi T. (2013) NKX2–1/TTF-1: an enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell 23, 718–723 [DOI] [PubMed] [Google Scholar]
- 16. Ngan E. S., Lang B. H., Liu T., Shum C. K., So M. T., Lau D. K., Leon T. Y., Cherny S. S., Tsai S. Y., Lo C. Y., Khoo U. S., Tam P. K., and Garcia-Barceló M. M. (2009) A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J. Natl Cancer Inst. 101, 162–175 [DOI] [PubMed] [Google Scholar]
- 17. Gudmundsson J., Sulem P., Gudbjartsson D. F., Jonasson J. G., Sigurdsson A., Bergthorsson J. T., He H., Blondal T., Geller F., Jakobsdottir M., Magnusdottir D. N., Matthiasdottir S., Stacey S. N., Skarphedinsson O. B., Helgadottir H., et al. (2009) Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 41, 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Endo T., and Kobayashi T. (2013) Concurrent overexpression of RET/PTC1 and TTF1 confers tumorigenicity to thyrocytes. Endocr. Relat. Cancer 20, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grant C. E., Bailey T. L., and Noble W. S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giordano T. J., Au A. Y., Kuick R., Thomas D. G., Rhodes D. R., Wilhelm K. G. Jr, Vinco M., Misek D. E., Sanders D., Zhu Z., Ciampi R., Hanash S., Chinnaiyan A., Clifton-Bligh R. J., Robinson B. G., Nikiforov Y. E., and Koenig R. J. (2006) Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clin. Cancer Res. 12, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 21. Winslow M. M., Dayton T. L., Verhaak R. G., Kim-Kiselak C., Snyder E. L., Feldser D. M., Hubbard D. D., DuPage M. J., Whittaker C. A., Hoersch S., Yoon S., Crowley D., Bronson R. T., Chiang D. Y., Meyerson M., and Jacks T. (2011) Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 473, 101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamaguchi T., Yanagisawa K., Sugiyama R., Hosono Y., Shimada Y., Arima C., Kato S., Tomida S., Suzuki M., Osada H., and Takahashi T. (2012) NKX2–1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 21, 348–361 [DOI] [PubMed] [Google Scholar]
- 23. Vu-Phan D., Grachtchouk V., Yu J., Colby L. A., Wicha M. S., and Koenig R. J. (2013) The thyroid cancer PAX8-PPARG fusion protein activates Wnt/TCF-responsive cells that have a transformed phenotype. Endocr. Relat. Cancer 20, 725–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mikkelsen T. S., Xu Z., Zhang X., Wang L., Gimble J. M., Lander E. S., and Rosen E. D. (2010) Comparative epigenomic analysis of murine and human adipogenesis. Cell 143, 156–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu B., Gerin I., Miao H., Vu-Phan D., Johnson C. N., Xu R., Chen X. W., Cawthorn W. P., MacDougald O. A., and Koenig R. J. (2010) Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS ONE 5, e14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramírez-Zacarías J. L., Castro-Muñozledo F., and Kuri-Harcuch W. (1992) Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry 97, 493–497 [DOI] [PubMed] [Google Scholar]
- 27. Shang Y., Hu X., DiRenzo J., Lazar M. A., and Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.