

Abstract
DNA methylation is a fundamental epigenetic mark that plays a critical role in differentiation and is mediated by the actions of DNA methyltransferases (DNMTs). TGF-β1 is one of the most potent inducers of fibroblast differentiation, and although many of its actions on fibroblasts are well described, the ability of TGF-β1 to modulate DNA methylation in mesenchymal cells is less clear. Here, we examine the ability of TGF-β1 to modulate the expression of various DNMTs in primary lung fibroblasts (CCL210). TGF-β1 increased the protein expression, but not RNA levels, of both DNMT1 and DNMT3a. The increases in DNMT1 and DNMT3a were dependent on TGF-β1 activation of focal adhesion kinase and PI3K/Akt. Activation of mammalian target of rapamycin complex 1 by Akt resulted in increased protein translation of DNMT3a. In contrast, the increase in DNMT1 by TGF-β1 was not dependent on new protein synthesis and instead was due to decreased protein degradation. TGF-β1 treatment led to the phosphorylation and inactivation of glycogen synthase kinase-3β, which resulted in inhibition of DNMT1 ubiquitination and proteosomal degradation. The phosphorylation and inactivation of glycogen synthase kinase-3β was dependent on mammalian target of rapamycin complex 1. These results demonstrate that TGF-β1 increases expression of DNMT1 and DNMT3a through different post-transcriptional mechanisms. Because DNA methylation is critical to many processes including development and differentiation, for which TGF-β1 is known to be crucial, the ability of TGF-β1 to increase expression of both DNMT1 and DNMT3a demonstrates a novel means by which TGF-β1 may regulate DNA methylation in these cells.
Keywords: Akt PKB, DNA methylation, DNA methyltransferase, fibroblast, glycogen synthase kinase 3 (GSK-3), mammalian target of rapamycin (mTOR), transforming growth factor beta (TGF-B), ubiquitylation (ubiquitination), DNMT1, DNMT3a
Introduction
DNA methylation is a key epigenetic process involved in controlling gene expression and cellular phenotype. Specific DNA methylation patterns are established and maintained during cell differentiation and are critical to normal development (1, 2). Because the establishment of DNA methylation is highly coordinated, understanding how DNA methylation machinery is regulated may provide insight into how DNA methylation patterns are altered during development and disease.
DNA methyltransferases (DNMTs) 2 are a family of enzymes responsible for the addition of methyl groups to DNA. Mammals possess three catalytically active DNMT isoforms: DNMT1, DNMT3a, and DNMT3b, each with independent, nonredundant functions (3). Deletion of any of these enzymes in mice results in embryonic lethality (1, 4). Alterations in specific DNMT isoform expression and activity have been identified in cancer (5–7), aging (8), autoimmune disease (9, 10), and fibrotic disorders (11–13) and have been associated with changes in the global DNA methylation patterns of cells. Expression of various DNMT isoforms has been demonstrated to be regulated by both transcriptional and post-transcriptional mechanisms (14, 15). Although DNMTs play an important role in establishing and maintaining DNA methylation patterns, how DNMT expression and activity are regulated is still incompletely understood.
TGF-β1 is a soluble mediator with pleiotropic actions that depend on cell type. TGF-β1 is one of the most well recognized mediators of wound repair and fibrosis (16), on top of its recognized ability to modulate adaptive immunity (17) and tumor growth (18). Acting in a paracrine or autocrine fashion, TGF-β1 is one of the most potent activators of fibroblasts, stimulating the increased production of extracellular matrix proteins and differentiating fibroblasts into contractile-capable myofibroblasts (16, 19). The changes in fibroblasts induced by TGF-β1 are persistent and are believed to contribute to the pathogenesis of many fibrotic disorders (16, 20, 21) in which activated myofibroblasts are associated with the dysregulated expression of a myriad of genes (22). Although TGF-β1 is well recognized to induce gene expression changes through the actions of several transcription factors (including the family of Smad proteins) (23), the persistence of these gene expression changes suggest that epigenetic changes may also be involved, and the extent to which TGF-β1 alters epigenetic machinery in fibroblasts is unclear. TGF-β1 has been reported to induce global DNA methylomic changes in hepatocellular tumors (24) and increase expression of multiple DNMT isoforms in prostate cancer cells (25). TGF-β1 has recently been shown to increase expression of DNMT1 in lung fibroblasts (26) but conversely inhibit DNMT1 and DNMT3a expression in cardiac fibroblasts (27). These studies suggest that the actions of TGF-β1 on DNA methylation machinery are context specific, and the mechanism(s) by which TGF-β1 affects DNMT expression are not known.
We previously demonstrated that prostaglandin E2, a lipid mediator that inhibits fibroblast activity and myofibroblast differentiation, induces global DNA methylation changes in fibroblasts that are due, in part, to increased expression of DNMT3a (28). Here, we investigate the effects of TGF-β1 on the expression of various DNMT isoforms in lung fibroblasts. We observed that TGF-β1 increased the expression of both DNMT1 and DNMT3a. Interestingly, the increase in expression of these isoforms occurred through different post-transcriptional mechanisms, and we delineate the signaling pathways responsible for these changes.
Results
Treatment with TGF-β1 Increased Expression of DNMT1 and DNMT3a in Normal Lung Fibroblasts
To determine whether TGF-β1 affects the expression of various DNMT isoforms, adult normal lung fibroblasts (CCL210 cells) were treated with TGF-β1 (2 ng/ml) for varying time periods, and expression of DNMT1 and DNMT3a were assayed by immunoblot. A dose of 2 ng/ml of TGF-β1 was chosen because we previously showed that it was sufficient to induce myofibroblast differentiation at the low end of the dose range. TGF-β1 increased the expression of both DNMT1 (Fig. 1A) and DNMT3a (Fig. 1B). Expression of DNMT1 and DNMT3a increased as early as 4 h and remained elevated for 48 h after TGF-β1 treatment. The increase in DNMT1 and DNMT3a expression by TGF-β1 was supported by immunofluorescence microscopy (Fig. 1, C and D), which additionally demonstrate that TGF-β1 treatment increased localization of both DNMT1 and DNMT3a to the cell nucleus.
FIGURE 1.

Treatment with TGF-β1 increased DNMT1 and DNMT3a protein expression. A and B, CCL210 cells were treated with 2 ng/ml of TGF-β1 for 4, 8, 24, and 48 h, and DNMT1 (A) and DNMT3a (B) protein expression was assayed by immunoblot. A representative immunoblot is shown with the mean fold change in expression by densitometry, relative to no-TGF-β1 control, shown below (n = 4). The data are shown as means ± S.E. * p < 0.05 relative to no-TGF-β1 treatment. C and D, cells were treated with or without TGF-β1 (2 ng/ml) for 8 h and immunostained for DNMT1 (C) and DNMT3a (D) with a TRITC-conjugated secondary antibody. Nuclei were counterstained with DAPI.
The Increase in DNMT1 and DNMT3a by TGF-β1 Was Dependent on focal adhesion kinase (FAK) and PI3K/Akt Pathways
TGF-β1 is capable of activating both the canonical Smad and non-Smad signaling pathways (29). Activation of FAK is a downstream consequence common to both pathways and has been shown to be important in TGF-β1-mediated increase in fibroblast differentiation and matrix adherence (29–31). FAK has been shown to activate the PI3K/Akt pathway (29, 32), which has also been demonstrated to be important to fibroblast activation and differentiation. To determine whether FAK and PI3K/Akt activation are required for the increase in DNMT1 and DNMT3a by TGF-β1, cells were treated with the FAK inhibitor PF62271 or the PI3K inhibitor LY294002 in the presence or absence of TGF-β1, and expression of DNMT1 and DNMT3a was evaluated by immunoblot. Inhibition of either FAK or PI3K abolished the ability of TGF-β1 to increase both DNMT1 (Fig. 2A) and DNMT3a (Fig. 2B).
FIGURE 2.

The increase in DNMT1 and DNMT3a expression by TGF-β1 was inhibited by FAK and PI3K inhibitors. CCL210 cells were treated with 10 μm of the FAK inhibitor PF62271 or 10 μm of the PI3K inhibitor LY294002 in the presence or absence of TGF-β1 (2 ng/ml) for 24 h, and expression of DNMT1 (A) and DNMT3a (B) was assayed by immunoblot. The mean densitometric values relative to untreated control of four independent experiments are shown below a representative immunoblot. *, p < 0.05. inh, inhibitor.
TGF-β1 Increased DNMT3a Expression via New Protein Synthesis
We next examined whether the increase in DNMT3a expression by TGF-β1 was due to an increase in DNMT3a mRNA. CCL210 cells were treated with TGF-β1 for 4, 8, 24, and 48 h. Levels of DNMT3a mRNA were unaffected by TGF-β1 treatment at all of the time points assayed (Fig. 3A). Because DNMT3a mRNA levels were unchanged in the presence of TGF-β1, we sought to determine whether the increase in DNMT3a protein was due to either an increase in protein translation or decrease in DNMT3a degradation. We first treated cells with the protein synthesis inhibitor cycloheximide and examined expression of DNMT3a protein over time. Levels of DNMT3a protein decreased over time, becoming barely detectable after 16 h, in the presence of cycloheximide (Fig. 3B). The cells were then treated for 2, 4, 8, and16 h with cyclohexidime in the presence or absence of TGF-β1. DNMT3a expression continued to decline with cycloheximide over time, even in the presence of TGF-β1 treatment (Fig. 3C). Cycloheximide inhibited the ability of TGF-β1 to increase expression of DNMT3a, especially at the 8- and 16-h time points (Fig. 3D). These data indicate that new protein synthesis is indeed necessary for the increase in DNMT3a by TGF-β1.
FIGURE 3.

The increase in DNMT3a by TGF-β1 was inhibited by the protein synthesis inhibitor cycloheximide (CHX). A, CCL210 cells were treated with TGF-β1 (2 ng/ml) for 4, 8, 24, and 48 h, and DNMT3a mRNA levels were assayed by real time RT-PCR (n = 4). B, CCL210 cells were treated with CHX (15 μm) for the indicated times, and DNMT3a protein was assayed by immunoblot. A representative immunoblot is shown, with the mean densitometric values of five independent experiments shown below. *, p < 0.05 relative to the 2-h time point with no CHX. C, cells were treated for varying times with CHX (15 μm) ± TGF-β1 (2 ng/ml), and expression of DNMT3a was compared relative to untreated cells at the 2-h time point (n = 3). *, p < 0.05. D, cells were treated for 8 or 16 h in the presence or absence of CHX (15 μm) and TGF-β1 (2 ng/ml), and expression of DNMT3a was assayed by immunoblot. The mean densitometric values relative to untreated control of seven independent experiments are shown below a representative immunoblot. *, p < 0.05. CHX, cycloheximide.
TGF-β1 Increases DNMT3a via Activation of mTORC1
Akt activates multiple downstream targets including mTOR, which is best recognized for its role in stimulating protein translation machinery and inducing protein synthesis (33, 34). Given that the increase in DNMT3a by TGF-β1 is dependent on new protein synthesis, we sought to determine whether mTOR activation is required for the increase in DNMT3a by TGF-β1. mTOR participates in two complexes: mTORC1 and mTORC2. Treatment of fibroblasts with rapamycin, an mTORC1 inhibitor, inhibited the increase in DNMT3a (Fig. 4A). Likewise, silencing expression of Raptor, an adaptor molecule specific for mTORC1, also inhibited the increase in DNMT3a by TGF-β1 (Fig. 4B). These data demonstrate that TGF-β1 signals through the Akt/mTORC1 pathway to result in new protein synthesis of DNMT3a.
FIGURE 4.
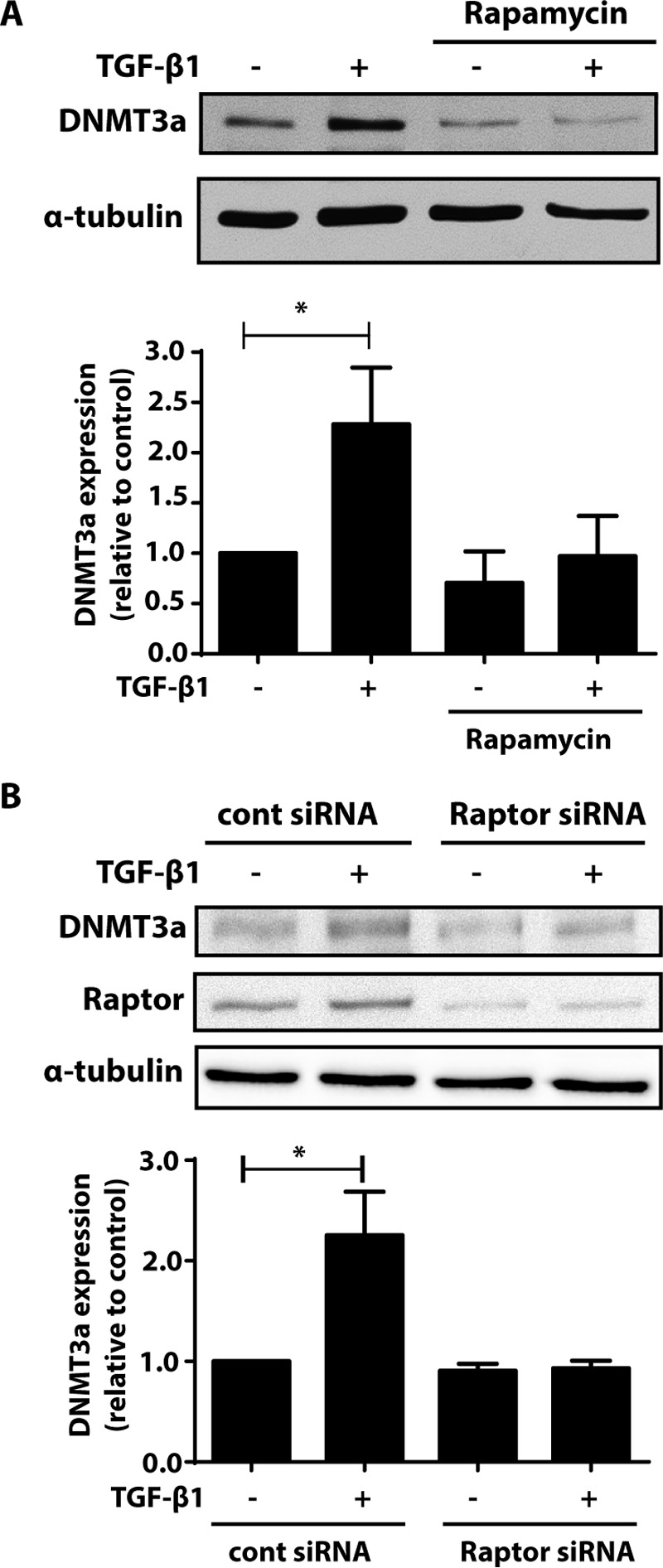
Inhibition of mTORC1 inhibited the ability of TGF-β1 to increase DNMT3a. A, CCL210 cells were treated with TGF-β1 (2 ng/ml) in the presence or absence of rapamycin (1 μm), an mTORC1 inhibitor, and DNMT3a protein was assayed by immunoblot. The mean densitometric values of three independent experiments are shown below a representative immunoblot. *, p < 0.05. B, CCL210 cells were treated with either control siRNA or siRNA targeting Raptor. The cells were then treated in the presence or absence of TGF-β1 (2 ng/ml), and expression of DNMT3a and Raptor protein was assayed by immunoblot. Mean densitometric values of DNMT3a protein from four independent experiments are shown below a representative immunoblot. *, p < 0.05.
TGF-β1 Increased DNMT1 Expression by Inhibiting DNMT1 Ubiquitination and Degradation
To determine whether an increase in DNMT1 mRNA levels was responsible for the increase in DNMT1 expression by TGF-β1, CCL210 cells were treated with TGF-β1 for varying times, and levels of DNMT1 mRNA were assayed by RT-PCR. As with DNMT3a, we did not observe a significant change in DNMT1 mRNA levels with TGF-β1 treatment at any of the time points assayed (Fig. 5A). To determine whether the increase in DNMT1 protein was dependent on new protein synthesis, we treated cells with the protein synthesis inhibitor cycloheximide. Treatment with cycloheximide resulted in a rapid decrease in DNMT1 expression as early as 2 h after treatment, with levels continuing to decline over time (Fig. 5B). We next treated cells with TGF-β1 in the presence or absence of cycloheximide for varying times. In contrast to that observed with DNMT3a, the relative half-life of DNMT1 in the presence of cycloheximide was markedly different when cells were treated with TGF-β1 (Fig. 5C). DNMT1 expression increased with TGF-β1 treatment, even in the presence of cycloheximide at both 8 and 16 h of treatment (Fig. 5C). These data indicate that the increase in DNMT1 by TGF-β1 does not depend on an increase in either DNMT1 gene transcription or protein synthesis.
FIGURE 5.

The increase in DNMT1 by TGF-β1 was not inhibited by the protein synthesis inhibitor CHX. A, CCL210 cells were treated with TGF-β1 (2 ng/ml) for 4, 8, 24, and 48 h, and DNMT1 mRNA levels were assayed by real time RT-PCR (n = 4). B, CCL210 cells were treated with CHX (15 μm) for the indicated times, and DNMT1 protein was assayed by immunoblot. A representative immunoblot is shown, with the mean densitometric values of four independent experiments shown below. *, p < 0.05 relative to the 2-h time point with no CHX. C, cells were treated for varying times with CHX (15 μm) ± TGF-β1 (2 ng/ml), and expression of DNMT1 was compared relative to untreated cells at the 2-h time point (n = 5). *, p < 0.05. D, cells were treated for 8 or 16 h in the presence or absence of CHX (15 μm) and TGF-β1 (2 ng/ml), and expression of DNMT1 was assayed by immunoblot. The mean densitometric values relative to untreated control of three independent experiments are shown below a representative immunoblot. *, p < 0.05.
DNMT1 undergoes ubiquitination prior to degradation (35, 36), and studies have shown that the increase in DNMT1 protein expression in cancers is a result of increased DNMT1 protein stability rather than abundance of mRNA (37, 38). To determine whether TGF-β1 alters DNMT1 ubiquitination and protein stability, lysates from fibroblasts treated with or without TGF-β1 were immunoprecipitated for DNMT1 and immunoblotted for ubiquitin. Because DNMT1 expression was observed to increase as early as 4 h after TGF-β1 treatment, we examined DNMT1 ubiquitination at 4 and 8 h. TGF-β1-treated cells exhibited less DNMT1 ubiquitination compared with control (Fig. 6A). In comparison, ubiquitination of DNMT3a was unaffected by TGF-β1 (Fig. 6B). The cells were then treated with MG-132, a proteosomal inhibitor, in the presence or absence of TGF-β1. Expression of DNMT1 was increased in the presence of MG-132, with no further increase in the presence of TGF-β1 (Fig. 6C). These data demonstrate that TGF-β1 inhibits DNMT1 ubiquitination and that TGF-β1 was unable to increase expression of DNMT1 any further than that achieved by inhibiting proteosomal degradation alone.
FIGURE 6.
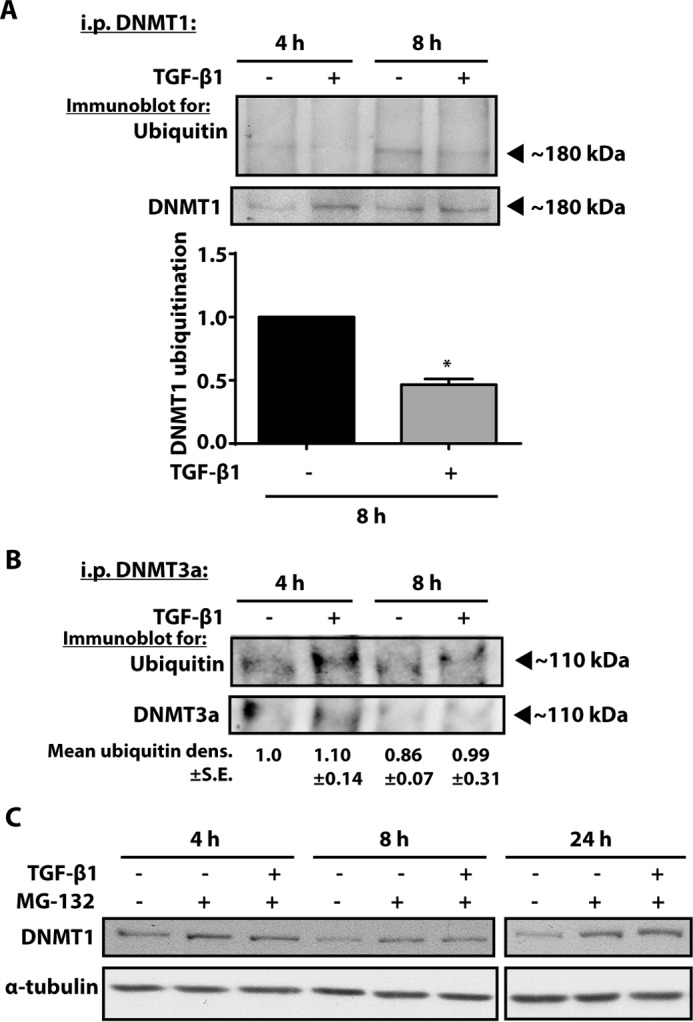
Treatment with a proteosomal inhibitor increased expression of DNMT1, and TGF-β1 treatment decreased DNMT1 ubiquitination. CCL210 cells were treated with TGF-β1 (2 ng/ml) for 4 or 8 h in the presence of the proteosomal inhibitor MG-132 (10 μm). Cell lysates were immunoprecipitated for DNMT1 or DNMT3a, resolved by SDS-PAGE, and probed with antibodies against ubiquitin and DNMT1. A, representative blot of ubiquitinylated DNMT1 is shown. Mean densitometry of ubiquitinylated DNMT1 from three independent experiments assayed at the 8-h time point is shown below. * p < 0.05. B, a representative blot of ubiquitinylated DNMT3a in cells treated with or without TGF-β1 is shown. Relative mean densitometry ± S.E. from three independent experiments is indicated by the numbers below. C, CCL210 cells were treated with MG-132 (10 μm) in the presence or absence of TGF-β1 (2 ng/ml), and expression of DNMT1 was assayed by immunoblot. A representative blot of two independent experiments is shown. i.p., immunoprecipitation.
TGF-β1 Inhibited DNMT1 Ubiquitination by Phosphorylating and Inactivating Glycogen Synthase Kinase (GSK)-3β
Several proteins have been demonstrated to interact with DNMT1 and regulate its ubiquitination (35, 36, 39, 40). In particular, the ubiquitin protein β-TrCP (ligase β-transducin repeat protein) has been shown to ubiquitinate DNMT1 through increased GSK-3β activity (41). GSK-3β has been shown to be inactivated by TGF-β1 through phosphorylation of serine residue 9 (42). We first confirmed in CCL210 fibroblasts that GSK-3β was phosphorylated after TGF-β1 treatment (Fig. 7A). This was inhibited in the presence of FAK and PI3K inhibitors, indicating that phosphorylation of GSK-3β occurs downstream of these signaling molecules. The cells were next transfected with a plasmid that overexpressed a constitutively active form of GSK-3β, in which serine 9 is mutated to adenine (S9A). Cells transfected with GSK-3β S9A did not exhibit an increase in DNMT1 expression with TGF-β1 treatment, as compared with control plasmid (Fig. 7B). By comparison, DNMT3a expression increased with TGF-β1 treatment even in the presence of GSK-3β S9A overexpression. Although TGF-β1 treatment resulted in decreased DNMT1 ubiquitination of cells treated with the control plasmid, TGF-β1 treatment did not affect DNMT1 ubiquitination in cells treated with GSK-3β S9A (Fig. 7C). These results indicate that TGF-β1 phosphorylates and inactivates GSK-3β, resulting in decreased DNMT1 ubiquitination and degradation.
FIGURE 7.

TGF-β1 phosphorylated and inactivated GSK-3β, inhibiting DNMT1 ubiquitination. A, cells treated with 10 μm of the FAK inhibitor PF62271 or 10 μm of the PI3K inhibitor LY294002 in the presence or absence of TGF-β1 (2 ng/ml) were assayed by immunoblot for phosphorylation of GSK-3β at serine 9. The mean densitometric values of phosphorylated GSK-3β from seven independent experiments are shown below a representative immunoblot. *, p < 0.05 relative to untreated control; #, p < 0.05 relative to TGF-β1 treatment without inhibitors. B, CCL210 cells transfected with either control plasmid (pcDNA 3.1) or plasmid that overexpressed a constitutively active form of GSK-3β S9A were treated with or without TGF-β1 (2 ng/ml), and expression of DNMT1 and DNMT3a was assayed by immunoblot. The mean densitometric values relative to untreated pcDNA 3.1 control from three independent experiments are shown below the representative immunoblots. *, p < 0.05. C, cells transfected with either control or GSK-3β S9A plasmid were treated with MG-132 (1 μm) in the presence or absence of TGF-β1 (2 ng/ml). Cell lysates were immunoprecipitated for DNMT1 and immunoblotted for ubiquitin and DNMT1 (n = 4). *, p < 0.05. i.p., immunoprecipitation; inh, inhibitor.
mTORC1 Is Necessary for TGF-β1-mediated GSK-3β Phosphorylation and Increase in DNMT1
GSK-3β can be either directly phosphorylated by Akt, or its phosphorylation can occur downstream of mTORC1 signaling. Because mTORC1 was an important downstream signaling molecule for the increase in DNMT3a by TGF-β1, we sought to determine whether mTORC1 was also necessary for TGF-β1-mediated increase in DNMT1. Phosphorylation of GSK-3β by TGF-β1 was completely inhibited by treatment with the mTORC1 inhibitor rapamycin (Fig. 8A). Cells treated with rapamycin did not exhibit an increase in DNMT1 expression after TGF-β1 treatment (Fig. 8B). Likewise, silencing Raptor, an adaptor for mTORC1, resulted in an inability for TGF-β1 to increase DNMT1 expression (Fig. 8C). These data demonstrate that mTORC1 is necessary for TGF-β1-mediated increase in both DNMT3a and DNMT1.
FIGURE 8.

mTORC1 is necessary for GSK-3β phosphorylation and TGF-β1-mediated increase in DNMT1. A and B, CCL210 cells were treated with TGF-β1 (2 ng/ml) in the presence or absence of rapamycin (1 μm), phosphorylated GSK3β (P-GSK3β) (A), and DNMT1 protein (B) were assayed by immunoblot. The mean densitometric values of three independent experiments are shown below a representative immunoblot. *, p < 0.05. C, CCL210 cells were treated with either control siRNA or siRNA targeting Raptor. The cells were then treated in the presence or absence of TGF-β1 (2 ng/ml), and expression of DNMT1 and Raptor protein were assayed by immunoblot. Mean densitometric values of DNMT1 protein from three independent experiments are shown below a representative immunoblot. cont, control.
Treatment with TGF-β1 Did Not Alter DNA Methylation of Long Interspersed Nuclear Elements (LINE)-1
Previous studies have demonstrated that TGF-β1 treatment increases the DNA methylation of THY1 in fibroblasts through the actions of DNMT1 (26). Thy-1 is a cell surface glycoprotein that inhibits myofibroblast differentiation and survival, and its hypermethylation by TGF-β1 leads to its decreased expression in differentiated myofibroblasts. TGF-β1 has also been shown to increase the expression of collagen type I by inducing DNA methylation changes in cardiac fibroblasts (27). To determine whether global DNA methylation levels were affected by TGF-β1 treatment, we assayed the DNA methylation of the retrotransposon LINE-1, which is frequently used to estimate global methylation (43). Bisulfite sequencing revealed no change in LINE-1 methylation after TGF-β1 treatment (Fig. 9).
FIGURE 9.
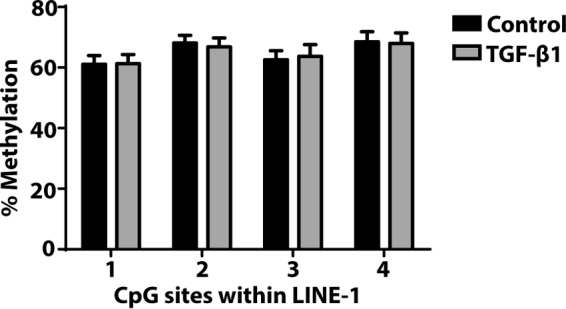
Effect of TGF-β1 treatment on LINE-1. CCL210 cells were treated with or without TGF-β1 (2 ng/ml) for 24 h, and the DNA methylation level of LINE-1 was assayed by bisulfite pyrosequencing. The percentage of methylation refers to the degree of methylation identified at each CpG site (n = 6).
Discussion
TGF-β1 plays a pivotal role during development and is one of the most potent inducers of fibroblast differentiation (16). Here, we demonstrate that TGF-β1 up-regulates the expression of DNMT1 and DNMT3a, two enzymes that critically maintain and establish DNA methylation, by distinct post-transcriptional mechanisms. Expression of DNMT3a was increased by TGF-β1 via an increase in its protein synthesis and translation. By contrast, TGF-β1 increased DNMT1 expression by inhibiting its ubiquitination and protein degradation. Although the increase in both DNMT1 and DNMT3a expression by TGF-β1 involved the activation of FAK, PI3K/Akt, and mTORC1, different mechanisms downstream of mTORC1 were responsible for the up-regulation of these two DNMTs. Activation of protein translational machinery by mTORC1 was sufficient to increase expression of DNMT3a, whereas the increase in DNMT1 expression by TGF-β1 was not dependent on protein translation. Instead, TGF-β1 treatment resulted in phosphorylation and inhibition of GSK-3β, which was associated with a decrease in DNMT1 ubiquitination and degradation (Fig 10). These data thus illustrate the various ways in which expression of different DNMT isoforms are regulated and provide insight into how TGF-β1, a potent mediator of myofibroblast differentiation, may also modulate the DNA methylation pattern of these cells.
FIGURE 10.
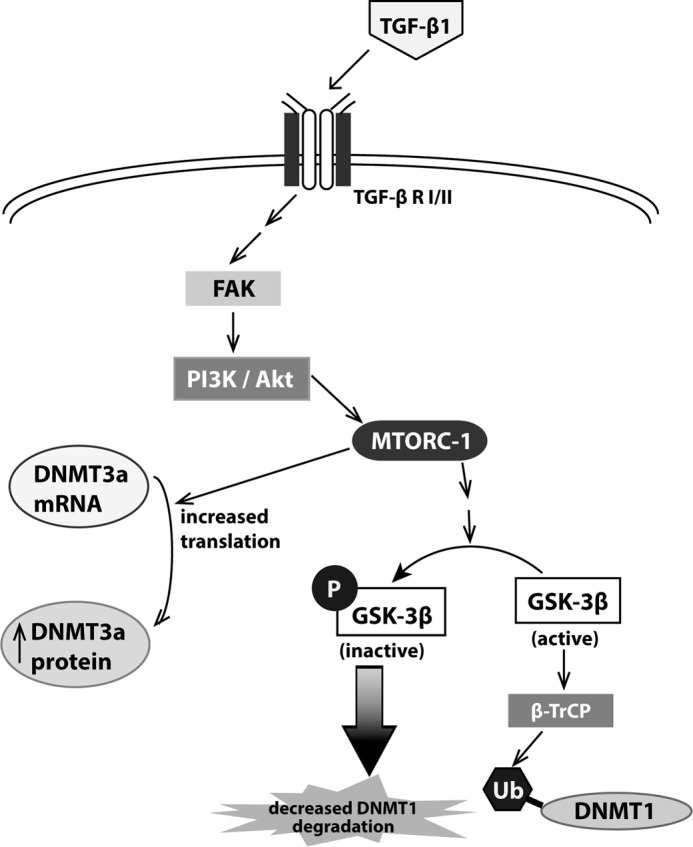
Schematic diagram of the signaling pathway by which TGF-β1 increases DNMT1 and DNMT3a. TGF-β1 signals through FAK, PI3K/Akt, and mTORC1 to increase protein translation of DNMT3a. Through the same signaling pathway, TGF-β1 phosphorylates and inactivates GSK-3β, which results in decreased DNMT1 ubiquitination and degradation. This results in an overall increase in DNMT1 expression.
DNMTs maintain and establish de novo DNA methylation patterns, and regulation of their expression is tightly regulated during cell differentiation and development (1–3). DNMT1 can be regulated at the transcriptional level, where transcription factors Sp1, SP3 (44), and E2F (45) are recognized to up-regulate expression of DNMT1. E2F is recognized to play a major role in cell cycle and is responsible for the increased expression of DNMT1 during DNA replication and cell proliferation (46). There is emerging evidence, however, that post-translational regulation of DNMT1 stability is also crucial in establishing abundance of total DNMT1 protein (37, 38). Our studies showed that the increase in DNMT1 by TGF-β1 is exclusively driven by post-translational mechanisms. Several ubiquitinases, such as the E3 ligase ubiquitin-like with plant homeodomain and ring finger domains 1 (UHRF1) (35) and β-TrCP (41), have been shown to ubiquitinate DNMT1. Our data show that DNMT1 ubiquitination is inhibited by TGF-β1 treatment. TGF-β1 is recognized to inactivate GSK-3β through phosphorylation (42), and previous studies have shown that GSK-3β enhances DNMT1 ubiquitination through recruitment of β-TrCP (41). Overexpression of a constitutively active form of GSK-3β led to an increase in DNMT1 ubiquitination and attenuated the ability of TGF-β1 to increase DNMT1 expression, confirming that the inactivation of GSK-3β by TGF-β1 was indeed necessary for the increase in DNMT1 expression.
DNMT1 has been described to undergo other post-translational modifications, including acetylation (35), lysine methylation (39, 47), and phosphorylation (41, 48) that also affect its stability. Direct interactions with other proteins including histone deacetylases (49) and the deubiquitinase herpesvirus-associated ubiquitin-specific protease (HAUSP) (35) also affect DNMT1 protein turnover. Whether TGF-β1 induces other DNMT1 post-translational modifications or alters the expression of interacting proteins is not known. We showed that PI3K/Akt signaling is necessary for the increase in DNMT1 expression by TGF-β1. Our data also demonstrate that treatment with TGF-β1 enhanced DNMT1 (and DNMT3a) localization to the cell nucleus. PI3K/Akt signaling has not only been shown to affect DNMT1 stability (41, 48) but also can directly phosphorylate DNMT1 at the nuclear localization signal (50), and this may be a mechanism by which TGF-β1 increases DNMT1 nuclear localization.
DNMT3a expression can be regulated at the transcriptional level through the increase of transcription factors such as Sp1 and Sp3 (51), but we found that TGF-β1, like DNMT1, increased DNMT3a expression in a post-transcriptional manner. As with DNMT1, TGF-β1signaled through FAK, PI3K/Akt, and mTORC1 to increase DNMT3a. mTORC1 is well recognized to increase total cellular protein synthesis and protein translation machinery (33, 34), and activation of mTORC1 by TGF-β1 was necessary to increase DNMT3a protein translation. Although the increase in DNMT1 by TGF-β1 was not dependent on new protein synthesis, mTORC1 was, interestingly, also necessary in mediating the ability of TGF-β1 to increase DNMT1. These results emphasize the pleiotropic actions of mTORC1 in stimulating new protein translation and also in modulating the stability and degradation of other proteins. Although phosphorylation of GSK-3β can be a direct consequence of Akt actions, our data indicate that mTORC1 is necessary in this process and are consistent with studies that have shown that mTORC1 can indirectly phosphorylate GSK-3β through activation of S6 kinase 1 (52).
The three catalytically active mammalian DNMTs, DNMT1, DNMT3a, and DNMT3b, all have distinct, nonredundant functions, and increased expression of each results in different effects on the overall global DNA methylation pattern (3, 14). Global deletion of any one isoform results in embryonic lethality (1, 4). DNMT1 is classically described as the “maintenance” methyltransferase, whereas DNMT3a and DNMT3b are responsible for de novo DNA methylation, but increasing evidence over the past several years has challenged this distinction and subverted this simplistic model (53). We did not observe a difference in LINE-1 methylation, which is often used to approximate global methylation (43), after TGF-β1 treatment. DNA methylation of LINE-1 and other retrotransposon elements, however, do not encapsulate the methylation changes that might occur in other regions of the genome. We hypothesize that the increase in DNMT1 and DNMT3a by TGF-β1 may result in methylation changes that are locus-specific. Whole genome methylation studies would be required to identify these sites of methylation change. Future studies would also be needed to determine whether identified changes in methylation are attributable to increases specific to either DNMT1 or DNMT3a. At least one study showed that Thy-1, whose expression is lost during myofibroblast differentiation, becomes hypermethylated in fibroblasts by TGF-β1 because of an increase in DNMT1 (26). This study confirms that increased DNMT1 expression by TGF-β1 leads to increased DNA methylation and decreased expression of at least one gene that is critical to the function of fibroblasts and their differentiation to myofibroblasts.
TGF-β1 is an important driver of fibroproliferative diseases, and the activation and differentiation of fibroblasts into myofibroblasts by TGF-β1 is accompanied by a change in the expression of many genes (22). Although some of these gene expression changes are driven by transcription factors, many of these changes persist beyond the half-life of these proteins, indicating that epigenetic regulation may be responsible for some of these changes. The finding that TGF-β1 increases expression of DNMT1 and DNMT3a provides insight into how this may occur. DNA methylation is often associated with suppression of gene expression, and in microarray studies of fibroblasts, nearly equal numbers of genes are down-regulated as they are up-regulated by TGF-β1 (22). In addition, these up-regulated genes may also occur as a consequence of DNA methylation because hypermethylation of gene bodies has been shown to increase gene expression (54). Because TGF-β1 is one of the most potent mediators of differentiation, the ability of TGF-β1 to affect DNA methylation may indicate a broader role for DNA methylation in modulating myofibroblast differentiation. Of note, prostaglandin E2 (PGE2), a potent anti-fibrotic mediator that inhibits and reverses myofibroblast differentiation, was also shown to induce global changes in DNA methylation (28). TGF-β1 has been shown to down-regulate PGE2 synthesis (55–57). Whether the methylation changes induced by PGE2 are opposite to that of TGF-β1 is unknown. PGE2 was also shown to increase expression of DNMT3a but not DNMT1, so DNA methylation differences that occur between PGE2 and TGF-β1 may be a result of the actions of DNMT1 or other factors independent of increases in DNMT3a.
DNA methylation changes have been identified in several fibrotic disorders, including renal fibrosis (58) and idiopathic pulmonary fibrosis (IPF) (12, 13). Fibroblasts, in particular, have been demonstrated to exhibit gene-specific and global DNA methylomic changes in IPF (11, 59) and are felt to contribute to the profibrotic phenotype of these cells. How alterations in DNA methylation arise in IPF and other fibrotic disorders is unknown. IPF fibroblasts are noted to have increased expression of DNMT1 and DNMT3a (11, 13), which in the context of our findings, may be due to the increased levels of TGF-β1 in fibrotic lung tissue. The ability of TGF-β1 to increase DNMT1 and DNMT3a in fibroblasts provides a potential mechanism by which DNA methylation changes may arise in these diseases.
TGF-β1 possesses pleiotropic activity, and how TGF-β1 affects the expression of DNMT isoforms in other cell types varies. TGF-β1 is an important modulator of T cell differentiation (17) and has been shown to antagonize DNMT1 accumulation in T cells (60), where DNMT1 plays a critical role in T cell development (61). In alveolar macrophages, we previously showed that TGF-β1 increased the expression of DNMT1 by inhibiting expression of microRNA-29b (62). microRNA-148b and microRNA-152 have also been shown to inhibit DNMT1 expression (63). We did not observe a change in DNMT1 mRNA levels to suggest a role for miRs in the ability of TGF-β1 to increase DNMT1 expression in fibroblasts. Studies in liver tumors and prostate cancer also demonstrate that TGF-β1 increases DNMT expression and global DNA methylation (24, 25). Expression of DNMT3b is low at baseline in lung fibroblasts, and further studies would be needed to determine whether TGF-β1 also regulates DNMT3b expression in fibroblasts and other cell types. Our findings, together with these other studies, demonstrate that the effects of TGF-β1 on DNMT expression and DNA methylation vary depending on cell and tissue type.
DNA methylation is often considered an important link that explains how environmental exposure contributes to disease (64), but the mechanisms by which the environment induces DNA methylation changes are diverse and not well understood. Exposure to air pollutants (65) and cigarette smoke (66) are associated with changes in DNA methylation patterns and are also associated with increased expression and activity of TGF-β1 (67, 68). Identifying TGF-β1 as a mediator capable of altering expression of DNMTs thus provides a possible indication of how environmental exposures induce DNA methylation changes.
In conclusion, our study demonstrates that TGF-β1 increases expression of DNMT1 and DNMT3a through different post-transcriptional mechanisms in lung fibroblasts. The increase in both DNMT1 and DNMT3a by TGF-β1 occurs through FAK, PI3K/Akt, and mTORC1 signaling. mTORC1 directly increases protein translation of DNMT3a, whereas phosphorylation and inactivation of GSK-3β by mTORC1 was necessary to prevent DNMT1 ubiquitination and degradation. These findings demonstrate the diverse mechanisms by which different DNMT isoforms can be regulated in a post-transcriptional manner to establish and maintain DNA methylation in development and disease.
Experimental Procedures
Cell Culture
Normal primary human lung fibroblasts (CCL210) were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and 1% penicillin/streptomycin. Prior to treatment, the cells were plated at 0.25 × 106 cells/well in 6-well plates overnight. Medium was subsequently removed, and the cells were incubated between 24 and 48 h in serum-free DMEM. The cells were then treated with the following reagents for 24 h unless otherwise indicated: TGF-β1 (2 ng/ml; R&D Systems, Minneapolis, MN), the protein synthesis inhibitor cycloheximide (15 μm; EMD Millipore, Billerica, MA), the FAK inhibitor PF62271 (10 μm; Tocris, Minneapolis, MN), the PI3K inhibitor LY294002 (10 μm; Cell Signaling, Danvers, MA), the mTOR inhibitor rapamycin (1 μm; Enzo, Farmingdale, NY), and the proteosomal inhibitor MG-132 (10 μm; Cayman Chemicals, Ann Arbor, MI). All inhibitors except for MG-132 were added 30 min prior to the addition of TGF-β1. MG-132 was added 1 h prior to the addition of TGF-β1. For siRNA experiments, the cells were treated with 50 nm of either control siRNA or siRNA against Raptor (GE Dharmacon, Lafayette, CO) using RNAiMAX (Thermo Fisher Scientific) for 24 h in OptiMEM (Thermo Fisher Scientific) and then incubated in serum-free DMEM for another 24 h prior to the addition of TGF-β1 for 24 h.
In some experiments, the cells were transfected with either pcDNA3.1 control plasmid or PCIneo plasmid expressing constitutively active GSK-3β in which serine 9 is mutated to adenine (S9A). GSK-3β S9A was a gift from Scott Friedman (69) (Addgene plasmid 49492). Following transfection, the cells were incubated in OptiMEM for 24 h and then in serum-free DMEM for another 24 h. For immunoprecipitation experiments, cells were pretreated with MG-132 (10 μm) for 1 h before the addition of TGF-β1 for 24 h.
Immunoprecipitation and Immunoblotting
Protein lysates were collected in lysis buffer (PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor (Roche) and phosphatase inhibitor I and II (EMD Millipore) cocktails. For immunoprecipitation, the cell lysates were sonicated and centrifuged for 10 min at 14,000 × g, and the supernatants were normalized by total protein concentration. Lysates were incubated with monoclonal mouse antibody against DNMT1 (1:80; antibody 13537; Abcam, Cambridge, MA) or with monoclonal rabbit antibody against DNMT3a (1:50; antibody 3598; Cell Signaling) overnight at 4 ºC before pulldown with protein G or A magnetic beads (Cell Signaling). Magnetic beads were washed three times with lysis buffer before elution in SDS buffer. For nonimmunoprecipitation immunoblotting, the lysates were normalized by total protein concentration. All samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were immunoblotted using the following antibodies (all monoclonal mouse unless otherwise stated): DNMT1 (1:1000, ab13537 Abcam), DNMT3a (1:1000, ab13888 Abcam), GSK-3β phosphorylated at Ser9 (1:500, monoclonal rabbit, ab75814; Abcam), ubiquitin (1:1000 MA1-10035; Thermo Scientific) or α-tubulin (1:1000; Sigma). Membranes were then incubated with the appropriate horseradish-peroxide-conjugated secondary antibody (Cell Signaling) and visualized with enhanced chemiluminescence reagent (GE Healthcare). For all protein bands, densitometry was analyzed by ImageJ Software (National Institutes of Health) and normalized to α-tubulin.
Immunofluorescence Staining
25,000 cells/well were plated on four-chamber glass slides, serum-starved for 24 h, and then treated for 8 h with TGF-β1 (2 ng/ml). The slides were washed and fixed with 4% paraformaldehyde for 30 min. The slides were blocked for 1 h in 3% BSA in PBS with 0.5% Triton X-100 and incubated overnight at 4 ºC with antibody against DNMT1 (1:50, IMG-261A; Imgenex, San Diego, CA) or DNMT3a (1:50, ab13888; Abcam). The slides were then washed and incubated with mouse secondary antibody (1:200) conjugated to TRITC and mounted with fluorescent mounting medium containing DAPI. The cells were visualized under fluorescent microscopy.
Real Time RT-PCR
RNA was isolated from cell using TRIzol, and quantitative mRNA levels were assayed by real time RT-PCR using the StepOnePlus real time PCR system (Applied Biosystems, South San Francisco, CA). Primers for human DNMT1, DNMT3a, and DNMT3b were obtained from Applied Biosystems, and primers for β-actin were used as previously reported (70).
LINE-1 Methylation
Genomic DNA was isolated from 1 × 106 cells treated with or without TGF-β1 (2 ng/ml) for 24 h using the DNeasy kit (Qiagen). 1 μg of genomic DNA was subject to bisulfite conversion using the EZ DNA methylation kit from Zymo Research (Irvine, CA). Biotin-labeled primers (Qiagen) were used to PCR amplify LINE-1. Amplicons were isolated using Sepharose beads and sequenced on the Pyromark Q24 pyrosequencer (Qiagen).
Statistical Analysis
The data were analyzed on GraphPad Prism 6.0 (GraphPad Prism Software, San Diego, CA) using analysis of variance or Student's t test, as appropriate, with p < 0.05 defined as statistically significant. All data are shown as means ± S.E.
Author Contributions
S. K. H. conceived and coordinated the study and wrote the paper. All authors designed, performed, and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
This work was funded by Grants HL119289 and HL127203 from the NHLBI, National Institutes of Health (to S. K. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DNMT
- DNA methyltransferase
- PG
- prostaglandin
- GSK
- glycogen synthase kinase
- IPF
- idiopathic pulmonary fibrosis
- FAK
- focal adhesion kinase
- mTOR
- mammalian target of rapamycin
- TRITC
- tetramethylrhodamine isothiocyanate
- CHX
- cycloheximide
- LINE
- long interspersed nuclear elements.
References
- 1. Okano M., Bell D. W., Haber D. A., and Li E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 [DOI] [PubMed] [Google Scholar]
- 2. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 3. Jurkowska R. Z., Jurkowski T. P., and Jeltsch A. (2011) Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–222 [DOI] [PubMed] [Google Scholar]
- 4. Li E., Bestor T. H., and Jaenisch R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 [DOI] [PubMed] [Google Scholar]
- 5. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C., Payton J. E., Baty J., Welch J., Harris C. C., Lichti C. F., Townsend R. R., Fulton R. S., Dooling D. J., et al. (2010) DNMT3A mutations in acute myeloid leukemia. The New Engl. J. Med. 363, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pathania R., Ramachandran S., Elangovan S., Padia R., Yang P., Cinghu S., Veeranan-Karmegam R., Arjunan P., Gnana-Prakasam J. P., Sadanand F., Pei L., Chang C. S., Choi J. H., Shi H., Manicassamy S., et al. (2015) DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 6, 6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin R. K., Wu C. Y., Chang J. W., Juan L. J., Hsu H. S., Chen C. Y., Lu Y. Y., Tang Y. A., Yang Y. C., Yang P. C., and Wang Y. C. (2010) Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 70, 5807–5817 [DOI] [PubMed] [Google Scholar]
- 8. Li Y., Liu Y., Strickland F. M., and Richardson B. (2010) Age-dependent decreases in DNA methyltransferase levels and low transmethylation micronutrient levels synergize to promote overexpression of genes implicated in autoimmunity and acute coronary syndromes. Exp. Gerontol. 45, 312–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gorelik G., Fang J. Y., Wu A., Sawalha A. H., and Richardson B. (2007) Impaired T cell protein kinase C delta activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J. Immunol. 179, 5553–5563 [DOI] [PubMed] [Google Scholar]
- 10. Quddus J., Johnson K. J., Gavalchin J., Amento E. P., Chrisp C. E., Yung R. L., and Richardson B. C. (1993) Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Invest. 92, 38–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang S. K., Fisher A. S., Scruggs A. M., White E. S., Hogaboam C. M., Richardson B. C., and Peters-Golden M. (2010) Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am. J. Pathol. 177, 2245–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rabinovich E. I., Kapetanaki M. G., Steinfeld I., Gibson K. F., Pandit K. V., Yu G., Yakhini Z., and Kaminski N. (2012) Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One 7, e33770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanders Y. Y., Ambalavanan N., Halloran B., Zhang X., Liu H., Crossman D. K., Bray M., Zhang K., Thannickal V. J., and Hagood J. S. (2012) Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 186, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hermann A., Gowher H., and Jeltsch A. (2004) Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 61, 2571–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin R. K., and Wang Y. C. (2014) Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 4, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Border W. A., and Noble N. A. (1994) Transforming growth factor β in tissue fibrosis. New Engl. J. Med. 331, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 17. Li M. O., Sanjabi S., and Flavell R. A. (2006) Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471 [DOI] [PubMed] [Google Scholar]
- 18. Pardali K., and Moustakas A. (2007) Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 1775, 21–62 [DOI] [PubMed] [Google Scholar]
- 19. Thannickal V. J., Lee D. Y., White E. S., Cui Z., Larios J. M., Chacon R., Horowitz J. C., Day R. M., and Thomas P. E. (2003) Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 278, 12384–12389 [DOI] [PubMed] [Google Scholar]
- 20. Li M., Krishnaveni M. S., Li C., Zhou B., Xing Y., Banfalvi A., Li A., Lombardi V., Akbari O., Borok Z., and Minoo P. (2011) Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Invest. 121, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aschner Y., and Downey G. P. (2016) Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 54, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wettlaufer S. H., Scott J. P., McEachin R. C., Peters-Golden M., and Huang S. K. (2016) Reversal of the transcriptome by prostaglandin E2 during myofibroblast dedifferentiation. Am. J. Respir. Cell Mol. Biol. 54, 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi Y., and Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 24. Martin M., Ancey P. B., Cros M. P., Durand G., Le Calvez-Kelm F., Hernandez-Vargas H., and Herceg Z. (2014) Dynamic imbalance between cancer cell subpopulations induced by transforming growth factor β (TGF-β) is associated with a DNA methylome switch. BMC Genomics 15, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Q., Chen L., Helfand B. T., Jang T. L., Sharma V., Kozlowski J., Kuzel T. M., Zhu L. J., Yang X. J., Javonovic B., Guo Y., Lonning S., Harper J., Teicher B. A., Brendler C., et al. (2011) TGF-β regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS One 6, e25168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neveu W. A., Mills S. T., Staitieh B. S., and Sueblinvong V. (2015) TGF-β1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am. J. Physiol. Cell Physiol. 309, C616–C626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pan X., Chen Z., Huang R., Yao Y., and Ma G. (2013) Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS One 8, e60335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang S. K., Scruggs A. M., Donaghy J., McEachin R. C., Fisher A. S., Richardson B. C., and Peters-Golden M. (2012) Prostaglandin E2 increases fibroblast gene-specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J. 26, 3703–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horowitz J. C., Rogers D. S., Sharma V., Vittal R., White E. S., Cui Z., and Thannickal V. J. (2007) Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cell. Signal. 19, 761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garrison G., Huang S. K., Okunishi K., Scott J. P., Kumar Penke L. R., Scruggs A. M., and Peters-Golden M. (2013) Reversal of myofibroblast differentiation by prostaglandin E2. Am. J. Respir. Cell Mol. Biol. 48, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas P. E., Peters-Golden M., White E. S., Thannickal V. J., and Moore B. B. (2007) PGE2 inhibition of TGF-β1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L417–L428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia H., Nho R. S., Kahm J., Kleidon J., and Henke C. A. (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a β1 integrin viability signaling pathway. J. Biol. Chem. 279, 33024–33034 [DOI] [PubMed] [Google Scholar]
- 33. Inoki K., Li Y., Zhu T., Wu J., and Guan K. L. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 [DOI] [PubMed] [Google Scholar]
- 34. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Du Z., Song J., Wang Y., Zhao Y., Guda K., Yang S., Kao H. Y., Xu Y., Willis J., Markowitz S. D., Sedwick D., Ewing R. M., and Wang Z. (2010) DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 3, ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shamma A., Suzuki M., Hayashi N., Kobayashi M., Sasaki N., Nishiuchi T., Doki Y., Okamoto T., Kohno S., Muranaka H., Kitajima S., Yamamoto K., and Takahashi C. (2013) ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol. Cell. Biol. 33, 3113–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Agoston A. T., Argani P., Yegnasubramanian S., De Marzo A. M., Ansari-Lari M. A., Hicks J. L., Davidson N. E., and Nelson W. G. (2005) Increased protein stability causes DNA methyltransferase 1 dysregulation in breast cancer. J. Biol. Chem. 280, 18302–18310 [DOI] [PubMed] [Google Scholar]
- 38. De Marzo A. M., Marchi V. L., Yang E. S., Veeraswamy R., Lin X., and Nelson W. G. (1999) Abnormal regulation of DNA methyltransferase expression during colorectal carcinogenesis. Cancer Res. 59, 3855–3860 [PubMed] [Google Scholar]
- 39. Estève P. O., Chin H. G., Benner J., Feehery G. R., Samaranayake M., Horwitz G. A., Jacobsen S. E., and Pradhan S. (2009) Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 106, 5076–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Q., Agoston A. T., Atadja P., Nelson W. G., and Davidson N. E. (2008) Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res. 6, 873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin R. K., Hsieh Y. S., Lin P., Hsu H. S., Chen C. Y., Tang Y. A., Lee C. F., and Wang Y. C. (2010) The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Invest. 120, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caraci F., Gili E., Calafiore M., Failla M., La Rosa C., Crimi N., Sortino M. A., Nicoletti F., Copani A., and Vancheri C. (2008) TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res. 57, 274–282 [DOI] [PubMed] [Google Scholar]
- 43. Tabish A. M., Baccarelli A. A., Godderis L., Barrow T. M., Hoet P., and Byun H. M. (2015) Assessment of changes in global DNA methylation levels by pyrosequencing(R) of repetitive elements. Methods Mol. Biol. 1315, 201–207 [DOI] [PubMed] [Google Scholar]
- 44. Kishikawa S., Murata T., Kimura H., Shiota K., and Yokoyama K. K. (2002) Regulation of transcription of the Dnmt1 gene by Sp1 and Sp3 zinc finger proteins. Eur. J. Biochem. 269, 2961–2970 [DOI] [PubMed] [Google Scholar]
- 45. Kimura H., Nakamura T., Ogawa T., Tanaka S., and Shiota K. (2003) Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res. 31, 3101–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szyf M., Bozovic V., and Tanigawa G. (1991) Growth regulation of mouse DNA methyltransferase gene expression. J. Biol. Chem. 266, 10027–10030 [PubMed] [Google Scholar]
- 47. Estève P. O., Chang Y., Samaranayake M., Upadhyay A. K., Horton J. R., Feehery G. R., Cheng X., and Pradhan S. (2011) A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 18, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun L., Zhao H., Xu Z., Liu Q., Liang Y., Wang L., Cai X., Zhang L., Hu L., Wang G., and Zha X. (2007) Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell. Signal. 19, 2255–2263 [DOI] [PubMed] [Google Scholar]
- 49. Peng L., Yuan Z., Ling H., Fukasawa K., Robertson K., Olashaw N., Koomen J., Chen J., Lane W. S., and Seto E. (2011) SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell. Biol. 31, 4720–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hodge D. R., Cho E., Copeland T. D., Guszczynski T., Yang E., Seth A. K., and Farrar W. L. (2007) IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics 4, 387–398 [PubMed] [Google Scholar]
- 51. Jinawath A., Miyake S., Yanagisawa Y., Akiyama Y., and Yuasa Y. (2005) Transcriptional regulation of the human DNA methyltransferase 3A and 3B genes by Sp3 and Sp1 zinc finger proteins. Biochem. J. 385, 557–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang H., Brown J., Gu Z., Garcia C. A., Liang R., Alard P., Beurel E., Jope R. S., Greenway T., and Martin M. (2011) Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-β-signaling pathways regulates the innate inflammatory response. J. Immunol. 186, 5217–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jeltsch A., and Jurkowska R. Z. (2014) New concepts in DNA methylation. Trends Biochem. Sci. 39, 310–318 [DOI] [PubMed] [Google Scholar]
- 54. Maunakea A. K., Nagarajan R. P., Bilenky M., Ballinger T. J., D'Souza C., Fouse S. D., Johnson B. E., Hong C., Nielsen C., Zhao Y., Turecki G., Delaney A., Varhol R., Thiessen N., Shchors K., et al. (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Diaz A., Chepenik K. P., Korn J. H., Reginato A. M., and Jimenez S. A. (1998) Differential regulation of cyclooxygenases 1 and 2 by interleukin-1β, tumor necrosis factor-α, and transforming growth factor-β1 in human lung fibroblasts. Exp. Cell Res. 241, 222–229 [DOI] [PubMed] [Google Scholar]
- 56. Harding P., Balasubramanian L., Swegan J., Stevens A., and Glass W. F. 2nd (2006) Transforming growth factor β regulates cyclooxygenase-2 in glomerular mesangial cells. Kidney Int. 69, 1578–1585 [DOI] [PubMed] [Google Scholar]
- 57. Keerthisingam C. B., Jenkins R. G., Harrison N. K., Hernandez-Rodriguez N. A., Booth H., Laurent G. J., Hart S. L., Foster M. L., and McAnulty R. J. (2001) Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-β in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 158, 1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bechtel W., McGoohan S., Zeisberg E. M., Müller G. A., Kalbacher H., Salant D. J., Müller C. A., Kalluri R., and Zeisberg M. (2010) Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 16, 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang S. K., Scruggs A. M., McEachin R. C., White E. S., and Peters-Golden M. (2014) Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS One 9, e107055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li C., Ebert P. J., and Li Q. J. (2013) T cell receptor (TCR) and transforming growth factor β (TGF-β) signaling converge on DNA (cytosine-5)-methyltransferase to control forkhead box protein 3 (foxp3) locus methylation and inducible regulatory T cell differentiation. J. Biol. Chem. 288, 19127–19139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee P. P., Fitzpatrick D. R., Beard C., Jessup H. K., Lehar S., Makar K. W., Pérez-Melgosa M., Sweetser M. T., Schlissel M. S., Nguyen S., Cherry S. R., Tsai J. H., Tucker S. M., Weaver W. M., Kelso A., et al. (2001) A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 [DOI] [PubMed] [Google Scholar]
- 62. Domingo-Gonzalez R., Wilke C. A., Huang S. K., Laouar Y., Brown J. P., Freeman C. M., Curtis J. L., Yanik G. A., and Moore B. B. (2015) Transforming growth factor-β induces microRNA-29b to promote murine alveolar macrophage dysfunction after bone marrow transplantation. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L86–L95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Azizi M., Teimoori-Toolabi L., Arzanani M. K., Azadmanesh K., Fard-Esfahani P., and Zeinali S. (2014) MicroRNA-148b and microRNA-152 reactivate tumor suppressor genes through suppression of DNA methyltransferase-1 gene in pancreatic cancer cell lines. Cancer Biol. Ther. 15, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fraga M. F., Ballestar E., Paz M. F., Ropero S., Setien F., Ballestar M. L., Heine-Suñer D., Cigudosa J. C., Urioste M., Benitez J., Boix-Chornet M., Sanchez-Aguilera A., Ling C., Carlsson E., et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Baccarelli A., Wright R. O., Bollati V., Tarantini L., Litonjua A. A., Suh H. H., Zanobetti A., Sparrow D., Vokonas P. S., and Schwartz J. (2009) Rapid DNA methylation changes after exposure to traffic particles. Am. J. Respir. Crit. Care Med. 179, 572–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Breton C. V., Byun H. M., Wenten M., Pan F., Yang A., and Gilliland F. D. (2009) Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 180, 462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen T. L., Liao J. W., Chan W. H., Hsu C. Y., Yang J. D., and Ueng T. H. (2013) Induction of cardiac fibrosis and transforming growth factor-β1 by motorcycle exhaust in rats. Inhal. Toxicol. 25, 525–535 [DOI] [PubMed] [Google Scholar]
- 68. Soltani A., Sohal S. S., Reid D., Weston S., Wood-Baker R., and Walters E. H. (2012) Vessel-associated transforming growth factor-β1 (TGF-β1) is increased in the bronchial reticular basement membrane in COPD and normal smokers. PLoS One 7, e39736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lang U. E., Kocabayoglu P., Cheng G. Z., Ghiassi-Nejad Z., Muñoz U., Vetter D., Eckstein D. A., Hannivoort R. A., Walsh M. J., and Friedman S. L. (2013) GSK3β phosphorylation of the KLF6 tumor suppressor promotes its transactivation of p21. Oncogene 32, 4557–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huang S., Wettlaufer S. H., Hogaboam C., Aronoff D. M., and Peters-Golden M. (2007) Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L405–L413 [DOI] [PubMed] [Google Scholar]