

Abstract
The enzyme encoded by the ALOX15B gene has been linked to the development of atherosclerotic plaques in humans and in a mouse model of hypercholesterolemia. In vitro, these enzymes, which share 78% sequence identity, generate distinct products from their substrate arachidonic acid: the human enzyme, a 15-S-hydroperoxy product; and the murine enzyme, an 8-S-product. We probed the activities of these enzymes with nanodiscs as membrane mimics to determine whether they can access substrate esterified in a bilayer and characterized their activities at the membrane interface. We observed that both enzymes transform phospholipid-esterified arachidonic acid to a 15-S-product. Moreover, when expressed in transfected HEK cells, both enzymes result in significant increases in the amounts of 15-hydroxyderivatives of eicosanoids detected. In addition, we show that 15-LOX-2 is distributed at the plasma membrane when the HEK293 cells are stimulated by the addition Ca2+ ionophore and that cellular localization is dependent upon the presence of a putative membrane insertion loop. We also report that sequence differences between the human and mouse enzymes in this loop appear to confer distinct mechanisms of enzyme-membrane interaction for the homologues.
Keywords: arachidonic acid (AA) (ARA), eicosanoid, lipid signaling, lipoxygenase pathway, phospholipid
Introduction
A macrophage 15-lipoxygenase (15-LOX) 2 activity has been linked to elevated levels of oxidized lipids through several experimental approaches that include the heterologous expression of human 15-LOX in a mouse model of hyperlipidemia (1) and pharmacological inhibition of 15-LOX activity (2, 3). These lipid oxidation products can enter the extracellular pool of cholesterol esters transported by LDL (4) and promote the pathological consequences of elevated LDL cholesterol levels (5). Macrophages that take up LDL laden with oxidized lipids are transformed to foam cells, an event that leads to further inflammation and apoptosis and results in the formation of atherosclerotic plaques. Recently, Magnusson et al. (6) demonstrated that silencing production of 15-LOX-2 (the product of the ALOX15B gene) in human macrophages decreased cellular lipid accumulation, the precipitating factor in foam cell formation. These results suggest that inhibition of 15-LOX-2 is a strategy for mitigating the development of cardiovascular disease.
Lipoxygenases oxygenate arachidonic acid (AA) to form a stereo- and regiospecific isomer of hydroperoxyeicosatetraenoic acid (HpETE), and isoforms are named according to which carbon atom is oxygenated (for review see Refs. 7 and 8). In general, within a species, lipoxygenases that differ in regiospecificity share ∼40% sequence identity. In contrast, between-species LOX homologues are expected to share ∼75% or better sequence identity and generate the same HpETE isomer. The mouse homologue of human 15-LOX-2, however, is an 8-LOX (m8S-LOX) with free AA as the substrate, and this altered regiospecificity appears to be unique to the mouse enzyme (9). We asked whether at the membrane and in a cellular context the mouse enzyme might also generate a 15-HpETE product. This information might provide insight into interpreting atherosclerosis studies that employ a murine model of hypercholesterolemia.
We recently reported the crystal structure of 15-LOX-2 and described a possible membrane insertion loop and open active site on one face of the elongated protein that would appear to make it feasible for the enzyme to access AA esterified in a membrane phospholipid (PL) (10). We show that in vitro both m8S-LOX and 15-LOX-2 generate the 15-isomer of the product with PL-esterified substrate in a nanodisc membrane bilayer mimic. However, we observed that the homologues differ in their ability to fully process the discreet pools of substrate in separate nanodiscs. In addition, we report that HEK293 cells transfected to express the mouse-8-S enzyme show elevated levels of 15-peroxidation products of polyunsaturated fatty acids (PUFA), whereas these products are not observed with enzymes incubated with free PUFA in vitro. Moreover, we demonstrate with immunofluorescence data that 15-LOX-2 localizes to the plasma membrane upon Ca2+ stimulation.
Results
Enzyme Activity with a Bilayer Mimic
Binding of 15-LOX-2 to nanodiscs was previously demonstrated by analytical size exclusion chromatography (10), and these results established the conditions to investigate membrane-associated enzyme activity with nanodiscs as substrates. The activities of 15-LOX-2, m8S-LOX, and their loop mutants were monitored with AA covalently attached to phospholipids in nanodiscs containing 25% 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (SAPC) as substrate. The product HpETE (or the corresponding alcohol generated by its non-enzymatic reduction, a hydroxyeicosatetraenoic acid (HETE)) covalently bound to the PL was first detected by electrospray ionization TOF MS. As shown in Fig. 1A, products with molecular weights of m/z 826.6 and 842.6, consistent with the addition of -OH and -OOH, respectively, to SAPC were detected when 15-LOX-2 or m8S-LOX were present in the reaction mixture. The hydroperoxy products are often rapidly reduced to their corresponding alcohols with the extraction procedure employed. Only the peak corresponding to unmodified SAPC (m/z 810.6) was observed in the negative control reactions without 15-LOX-2 or m8S-LOX. In the absence of Ca2+, less product was detected for 15-LOX-2.
FIGURE 1.

Enzyme activity in a nanodisc assay. A, electrospray ionization MS analysis of the 15-LOX-2 and m8S-LOX reactions with nanodiscs. Nanodiscs (25% SAPC, 75% POPC, 5 μm in 50 mm potassium phosphate (pH 7.4), 0.1 mm EDTA, 100 mm NaCl, and 2 mm CaCl2) were incubated with 2.2 μm 15-LOX-2 or 6 μm m8S-LOX for 5 h at room temperature, after which the PL fraction was extracted with the Bligh-Dyer method. Mass spectrometry analysis was performed in positive ion mode. Products with molecular weights of m/z 826.6 and 842.6 are consistent with oxygenation of SAPC. These peaks were not observed in the absence of enzyme, only the peak corresponding to unmodified SAPC (m/z 810.6). In the absence of Ca2+, oxygenated SAPC was observed to a lesser extent for 15-LOX-2. B, HPLC-based assay to determine the regiospecificity of 15-LOX-2 (top panel) and m8S-LOX (bottom panel) with nanodiscs. 15-LOX-2 (2 μm) or m8S-LOX (6 μm) was incubated with 5 μm of nanodiscs (25% SAPC, 75% POPC) for 5 h at room temperature in a 400-μl reaction volume. Subsequently, the sample was incubated at 85 °C to inactivate the LOX and then incubated with 1 mg/ml PLA2 (37 °C, 2.5 h) to cleave the PL. Free fatty acids for HPLC analysis were isolated by solid phase extraction with C18 columns, and the peroxides were reduced to the corresponding alcohols with the addition of TPP. Only the 15-HETE isomer (solid line) is detected. The dashed lines provide the elution times for HETE standards.
The regiospecificity of the reaction was established by reverse phase HPLC of the phospholipase A2-cleaved products (Fig. 1B). (Prior to HPLC analysis oxygenated fatty acids are reduced; thus detected products are HETEs rather than HpETES.) When either 15-LOX-2 or m8S-LOX were incubated with PL-esterified-AA containing nanodiscs, only 15-HETE was formed as product, as opposed to 8-HETE or a di-HETE, the products observed with m8S-LOX and free AA (9, 11).
Fatty Acid and Head Group Preferences
Inositol-PL are enriched with esterified PUFAs in cells; thus nanodiscs containing PL-esterified polyunsaturated fatty acids carrying an inositol head group were used to generate time course data and measure rate constants for the reaction. HPLC-based time course assays were performed with 15-LOX-2 and m8S-LOX on nanodiscs composed of 40% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 25% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), and 35% l-α-phosphatidylinositol (Avanti Polar Lipids). This commercial phosphatidylinositol contains 17% AA and 13% dihomo-γ-linolenic acid (DGLA) according to the fatty acid distribution data from Avanti Polar Lipids, Inc. Thus both AA (20:4) and DGLA (20:3) substrates were available in these nanodiscs, creating an opportunity to compare enzyme activities with two substrates. The time course of product formation (both 15-HETE from AA and 15-hydroxyeicosatrienoic acid (15-HETrE) from DGLA) by m8S-LOX and 15-LOX-2 with these “mixed PI” containing nanodiscs is shown in Fig. 2A. The data were fit to a one-phase association equation using GraphPad Prism. The amount of product formed by 15-LOX-2 was 6–10 times higher than by m8S-LOX, and m8S-LOX generated more product from DGLA even though there was less of this substrate in the nanodiscs. The observed rates of the reactions were 0.0263 ± 0.0017 min−1 for m8S-LOX/15-HETE, 0.0373 ± 0.0035 min−1 for m8S-LOX/15-HETrE, 0.0573 ± 0.0122 min−1 for 15-LOX-2/15-HETE, and 0.0582 ± 0.0107 min−1 for 15-LOX-2/15-HETrE.
FIGURE 2.

15-LOX-2 and 8-LOX activities with inositol phospholipids. A, HPLC-based time course assay with nanodiscs composed of 40% POPC, 25% POPS, and 35% l-α-phosphatidylinositol. The enzyme concentration was 3 μm in both sets of reactions; nanodisc concentrations were 10 and 15 μm for m8S-LOX and 15-LOX-2 reactions, respectively. Aliquots taken at the different time points were incubated at 85 °C to inactivate the LOX and subsequently cleaved with PLA2 (37 °C, 2.5 h) to liberate free fatty acids. After solid phase extraction and the addition of TPP, oxidized fatty acids were quantitated by HPLC. The data were fit to a one-phase association equation with GraphPad Prism. The amount of product formed by 15-LOX-2 is 6–10 times higher than m8S-LOX. B, comparison of 15-LOX-2 and m8S activities and their loopless variants (LM). Reactions (2 mm Ca2+, 3 μm enzyme, and 15 μm nanodiscs) were performed in 40 μl. Nanodiscs contained 40% POPC, 25% POPS, and 35% l-α-phosphatidylinositol from bovine liver. The l-α-phosphatidylinositol is composed of 17% AA and 14% DGLA, which give 15-HETE (dark gray bars) and 15-HETrE (light gray bars) as products, respectively. Incubations were performed at 37 °C for 40 min and enzymes inactivated at 82.5 °C for 50 min before treatment with PLA2 to liberate the oxidized fatty acids for HPLC analysis.
The l-α-phosphatidylinositol containing nanodiscs were also utilized to compare the wild-type activities of the homologues with those of their loopless counterparts (Δ73–79:15-LOX-2 and Δ74–81:m8S-LOX). In these experiments, the reactions were stopped at 40 min for analysis (Fig. 2B). Consistent with the time course data in Fig. 2A, 15-LOX-2 generated more product (>15-fold) than m8S-LOX for the same amount of substrate in nanodiscs. A possible mechanistic reason for this substantial difference in product formation is discussed below.
These data suggest a preference for reaction with PL-DGLA over PL-AA for the mouse enzyme, whereas the activity at the membrane of the human enzyme was highly dependent on the presence of the putative membrane insertion loop. The absence of the putative membrane insertion loop in the human enzyme resulted in a ∼50% decrease in product. As much of a dramatic decrease in product generated by the mouse counterpart was not observed.
As a means to probe a head group preference for 15-LOX-2, the amount of product formed from AA-esterified nanodiscs of various head group compositions was evaluated. The 15-LOX-2 products of choline, ethanolamine, and inositol PL that carry stearate and arachidonate acyl groups (SAPC, SAPE, and SAPI, respectively) were measured at two time points of 15 min (Fig. 3A) and 90 min (Fig. 3B). All of the reactions were conducted in the presence and absence of Ca2+. At 15 min, there was a significantly higher amount of product from SAPC. At the later time point, the product from SAPI achieved a very similar level to that from SAPC; however the product from SAPE was still significantly lower. For all head groups, the presence of Ca2+ increased product yields significantly. These results suggested a selectivity, rather than specificity, for head group, which might be explained by enzyme-head group interactions.
FIGURE 3.
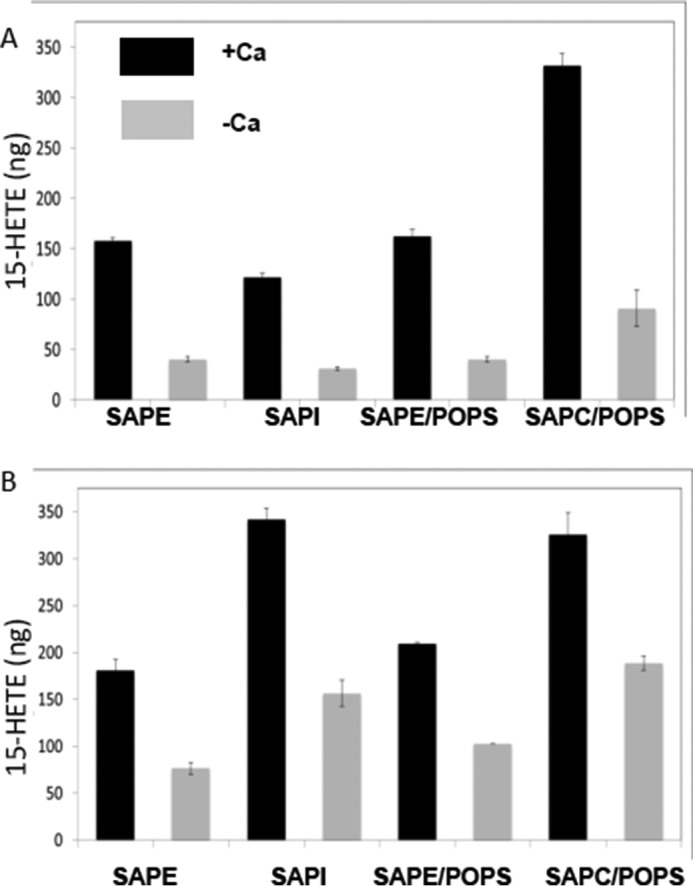
Head group preference and nanodisc composition for 15-LOX-2. Reaction of 15-LOX-2 with nanodiscs composed of AA-bearing PL of distinct head groups, with and without 2 mm Ca2+. Reaction volumes were 40 μl. Enzyme and nanodisc concentrations were 3 and 15 μm, respectively. For each experiment, the substrate PL made up 30% of the bilayer lipids, enough to generate 9.2 μg of product. SAPE and SAPC were also evaluated in nanodiscs, which contained 30% PS:40%PC in the non-substrate PL to minimize any impact of a high concentration of positively charged head groups at the bilayer. With shorter incubation times, SAPC product yields were by far the best, and they were achieved in the presence of PS. However, note that PS has little effect on the activity measured with SAPE as the substrate. However, upon extended incubation, product yields from SAPI and SAPC are comparable. Reactions were performed at 37 °C for 15 min (A) or 90 min (B), after which the enzyme was heat-inactivated, and PL was cleaved with PLA2.
Access to Membrane-embedded Substrates
Enzymes that must access substrate in the bilayer may do so in a processive manner where they are able to move along the membrane to process multiple substrate molecules with a single binding event (scooting mode) or in single turnover mode (hopping mode), in which an enzyme membrane collision is required for each turnover (12, 13). These modes are readily distinguished in a nanodisc assay by the addition of fresh enzyme or substrate to reactions that have been allowed to go to completion. The addition of enzyme to a completed reaction should result in an increase in product only if enzyme functions in scooting mode and is not able to “hop” between nanodiscs to consume all substrate molecules. In contrast, increased product yields because of nanodisc addition is an indication that the enzyme functions in hopping mode because it can readily translocate to the new nanodiscs provided, and the reaction has gone to completion simply because all substrate has been consumed. Fig. 4 shows the results of such experiments for m8S-LOX and 15-LOX-2. The reactions in the top panel were performed with 3 μm m8S-LOX and 10 μm nanodiscs containing 40% POPC, 25% POPS, and 35% l-α-phosphatidylinositol from bovine liver. After 80 min, when the reaction was almost complete, either more m8S-LOX or nanodiscs were added to the reaction for an additional 80 min. There was more product formed only after addition of more enzyme, which suggested the m8S-LOX remains “stuck” on the nanodisc after substrate was consumed. As a control, EDTA (to promote enzyme release from the bilayer) and free linoleic acid (LA) were added at the end of 80 min to establish that the enzyme was still active, and the corresponding reduced hydroxyl product was observed (Fig. 4, top panel, inset). In comparable reactions with 15-LOX-2 (Fig. 4, bottom panel), only the addition of fresh nanodiscs resulted in further product generated, an indication that the entire available substrate pool had been consumed by 15-LOX-2. Thus 15-LOX-2, even in the presence of Ca2+ to promote membrane binding, was able to hop among nanodiscs. This observation is consistent with the fact that 15-LOX-2 generates more product than 8-S-LOX in assay conditions that require access to discrete pools of substrate (Fig. 2).
FIGURE 4.
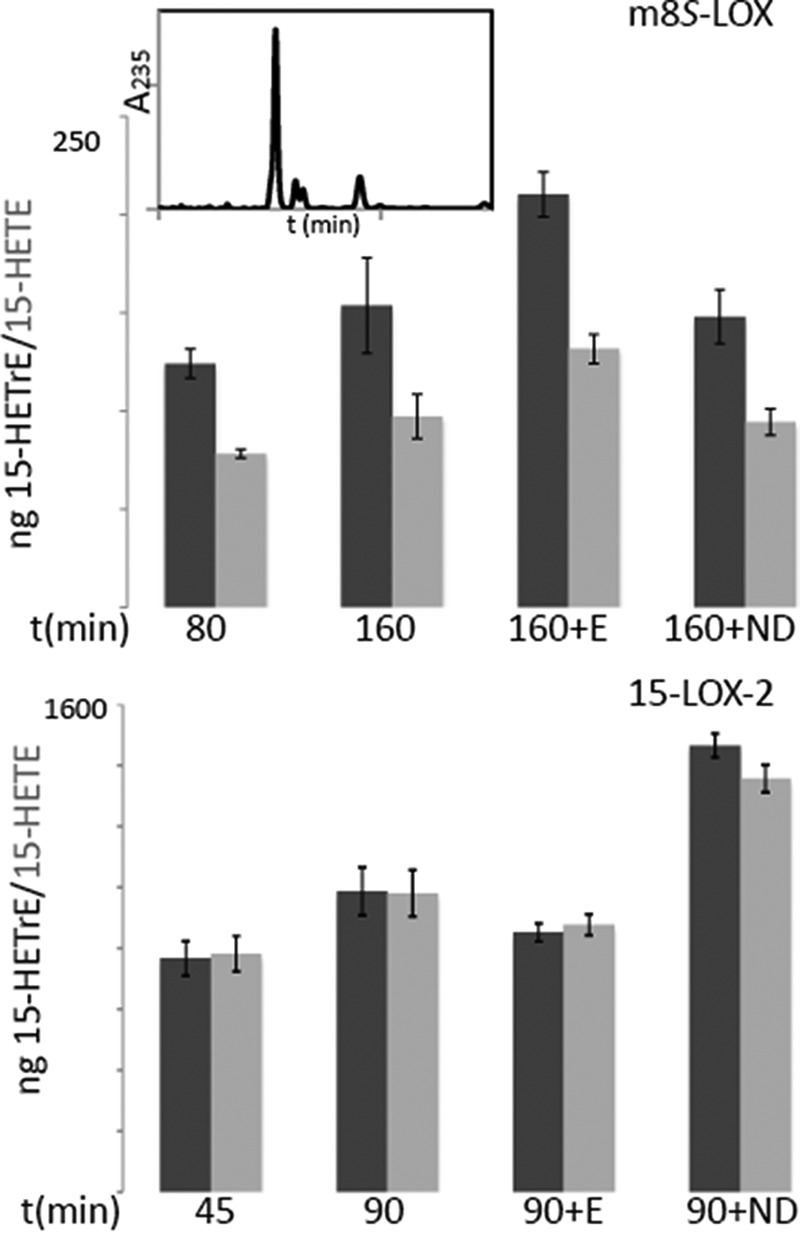
Do m8S-LOX or 15-LOX-2 hop between nanodiscs? Top panel, reactions mixtures of 3 μm m8S-LOX and 10 μm nanodiscs containing 40% POPC, 25% POPS, and 35% l-α-phosphatidylinositol from bovine liver. After 80 min, when the reaction almost reached completion, either fresh m8S-LOX (+E) or nanodiscs (+ND) were added for an additional 80 min. In another set of reactions, EDTA and free linoleic acid were added to verify the enzyme is active, and a HODE product was confirmed (the inset is an HPLC trace). Bottom panel, reactions of 3 μm 15-LOX-2 with 15 μm nanodiscs were incubated for 45 min to reach completion, and either fresh 15-LOX-2 (+E) or nanodiscs (+ND) were added to the samples for a second 45-min incubation. Both 15-HETE (dark gray bars bars) and 15-HETrE (light gray bars) were quantitated.
Activity with Esterified versus Free Substrates
Because the nanodisc assays indicated that the mouse enzyme transforms PL-esterified DGLA more readily than the AA counterpart, we asked whether free DGLA or its ethyl ester derivative were substrates (Fig. 5 and Table 1). 15-LOX-2 with AA displayed substrate inhibition, whereas no substrate inhibition was observed with DGLA (Fig. 5A); m8S-LOX showed substrate inhibition with both AA and DGLA, and DGLA was a poorer substrate than AA as the free fatty acid. The kcat/Km values for the two substrates for 15-LOX-2 are 0.264 ± 0.121 and 0.0895 ± 0.0075 s−1 μm−1 for AA and DGLA, respectively, which suggested a slight preference for AA (Table 1). A preference was not observed for the ethyl esters of the substrates because the kcat/Km values were 0.0464 ± 0.0084 s−1 μm−1 versus 0.0335 ± 0.0056 s−1 μm−1. The free substrates with m8S-LOX displayed considerable substrate inhibition, and enzymatic parameters suggested that both kcat and catalytic efficiency were less for DGLA than for AA, which was the opposite of that observed with nanodisc products for m8S-LOX. The ethyl ester substrates with m8S-LOX did not yield enough activity to be analyzed.
FIGURE 5.

Reactions of 15-LOX-2 (A) and m8S-LOX (B) with free fatty acids: AA and DGLA. Reactions (in 50 mm Tris, pH 7.5, 150 mm NaCl, 0.5 mm EDTA, with the addition of 5% glycerol for m8S-LOX) were monitored in UV spectrophotometer (237 nm). The enzyme concentration was 0.2 μm, and substrate varied from 1 to 40 μm. For assays with ethyl-esters, 20 mm cholate was included. The Michaelis-Menten equation with substrate inhibition was fitted to the curves using least squares fitting in GraphPad Prism. Substrate inhibition is more pronounced for m8S-LOX, and there is no substrate inhibition for 15-LOX-2 with DGLA as substrate. For m8S-LOX, free DGLA is a poorer substrate than AA. The kinetic parameters of the two substrates with 15-LOX-2 are similar. Kinetic parameters are summarized in Table 1.
TABLE 1.
Kinetic parameter
| kcat | Km | kcat/Km | Ki | |
|---|---|---|---|---|
| s−1 | μm | s−1 μm−1 | μm | |
| AA15-LOX-2 | 0.46 ± 0.09 | 1.74 ± 0.72 | 0.264 ± 0.121 | 32.6 ± 9.6 |
| DGLA15-LOX-2 | 0.31 ± 0.01 | 3.46 ± 0.27 | 0.0895 ± 0.0075 | N/A |
| AAm8S-LOX | 0.22 ± 0.07 | 2.86 ± 1.51 | 0.0758 ± 0.0467 | 9.77 ± 2.20 |
| DGLAm8S-LOX | 0.045 ± 0.004 | 0.964 ± 0.186 | 0.0467 ± 0.0099 | 6.28 ± 0.49 |
| AA-EE15-LOX-2 | 0.13 ± 0.01 | 2.80 ± 0.46 | 0.0464 ± 0.0084 | N/A |
| DGLA-EE15-LOX-2 | 0.211 ± 0.014 | 6.30 ± 0.97 | 0.0335 ± 0.0056 | N/A |
Products Observed in Transfected HEK293 Cells
Supplementation conditions to enhance the PUFA content of the HEK cell membranes were established (Fig. 6A). Exogenous 15-LOX-2 was added to crude cell lysates to transform AA and LA to the corresponding hydroperoxides. The products that were PL-esterified were liberated by the addition of PLA2. Note that lysates from cell cultures grown without supplementation had minor amounts of product in the free fatty acid pool, and this amount was increased when PLA2 was added to release additional free fatty acids. Lysates from cells cultured in supplemented media yielded at least 5-fold more free and PLA2-released 15-HETE. Moreover, most of the 15-HETE was only detected after treatment with PLA2, likely an indication that product was generated as the esterified substrate.
FIGURE 6.

Oxidized fatty acids in HEK cells supplemented with AA and LA. A, non-transfected HEK cells supplemented with LA and AA provide substrates for exogenous 15-LOX-2. Treatment of the 15-LOX-2/lysate incubations with PLA2 releases additional 15-HETE and 13-HODE. B, oxidized free fatty acids in HEK293 cells transfected to express 15-LOX-2 (black) or m8S-LOX (striped) are increased over levels observed in non-transfected cells (stippled) similarly enriched with LA and AA. The free fatty acid pool in 15-LOX-2 cells is enriched in 15-HETE and 13-HODE, those products observed in vitro with unesterified substrates. In contrast, elevated levels of various HETEs are observed in those cells transfected with the m8S-LOX, whereas only the 8-HETE product is observed with the unesterified substrate in vitro. The 9-HODE is the product of the reaction with free LA, and 13-HODE is generated from PL-esterified LA.
The results of MS lipidomics analyses of the oxidized fatty acids found in HEK293 cells transfected with pCDNA3.1(+) encoding either the 15-LOX-2 or m8S-LOX are shown in Fig. 6B. We asked whether unesterified LOX products indicative of activity at the bilayer might be identified in the cellular pools of free fatty acids. Because the mouse enzyme only generates 15-HETE from AA as the PL-ester, the presence of 15-HETE in cell lysates of cells expressing m8S-LOX likely reflects enzyme activity at the membrane. The presence of 15-HETrE in cell lysates is also consistent with m8S-LOX activity at the membrane, given the fact that the free DGLA is a poor substrate. For analysis, cells were first washed to remove extracellular free fatty acids and then stimulated with Ca2+ ionophore to induce membrane binding of the LOX. Application of ionophore was also expected to activate PLA2. For cell expressing 15-LOX-2, we could not differentiate between products generated as phospholipid esters or free fatty acids, but these cells clearly yielded elevated levels of 15-HETE and 15-HETrE. For the m8S-LOX-expressing cells, both products were detected at levels comparable with those observed in the 15-LOX-2 transfected cells. The high levels of 8-HETE and 9-HODE in these same extracts reflected activity with free AA and LA, respectively.
However, for HEK cells expressing m8S-LOX, additional products to those predicted from the in vitro nanodisc and free AA data are observed. An apparent loss of specificity has been reported for lipoxygenases when the reaction is slow because the enzyme must work on a membrane embedded PL (14) or at low O2 concentrations (15) (see below). These same off target products have been detected in mouse lipidomics studies and are observed in 5-LOX and 12/15-LOX knock-out mice (16).
Cellular Localization of 15-LOX-2
Earlier experiments with 15-LOX-2 have demonstrated a Ca2+-dependent association with the membrane fraction in cell lysates (17). In addition, 15-LOX-1 is found in both cytosolic and membrane fractions upon Ca2+ stimulation (18). 5-LOX has been shown to localize to the nuclear membrane where the helper protein 5-lipoxygenase-activating protein is located (19), although nuclear membrane localization does not require the presence of the its transmembrane partner (20). Immunofluorescence detection of 15-LOX-2 expressed in transfected HEK293 cells was performed to reveal the cellular location of 15-LOX-2 upon stimulation with the Ca2+ ionophore. Under these conditions, wild-type 15-LOX-2 was detected with a distribution similar to that of a pan-cadherin antibody, a marker for the plasma membrane. In addition, neither the loopless 15-LOX-2 or 15-LOX-2 in which the Ca2+ binding amino acids had been mutated to Ala revealed this same pattern of distribution upon stimulation (Fig. 7); both variants have wild-type enzymatic properties with free AA as the substrate (10). Only the wild-type enzyme displays an antibody distribution pattern consistent with that of the plasma membrane marker.
FIGURE 7.
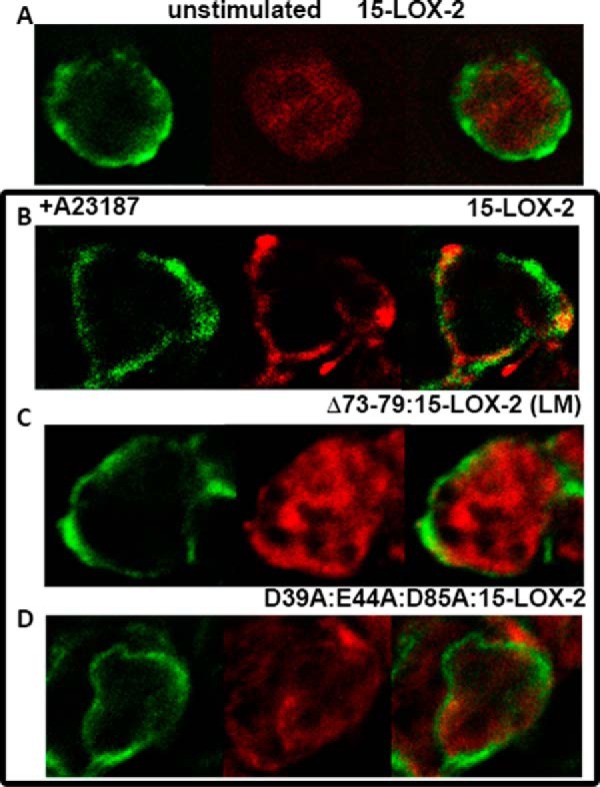
Cellular localization of 15-LOX-2 expressed in HEK293 cells. Upon stimulation by addition of Ca2+ ionophore A23187, only the distribution of wild-type 15-LOX-2 conforms to that of the plasma membrane marker Cadherin. Green, cadherin; red, 15-LOX-2. A, HEK cells expressing wild-type 15-LOX-2, without Ca2+ stimulation. B–D, in the box, wild type (B), Δ73–79:15-LOX-2 (LM) (C), and D39A:E44A:D85A:15-LOX-2 (D), all after Ca2+ simulation.
Discussion
The crystal structure of human 15-LOX-2 revealed an open active site and a Ca2+-stabilized putative membrane insertion loop along one face of the enzyme, raising the possibility that the enzyme can access the PL-esterified AA groups of membrane phospholipids and thus not be dependent on a phospholipase activity to liberate its fatty acid substrates (Fig. 8). A distinct mechanism for substrate acquisition by 15-LOX-2 could provide a context for development of isozyme-specific inhibitors in this enzyme family and circumvent the fact that the core active site and catalytic machinery are so highly conserved among the various lipoxygenases (10, 21–24). 15-LOX-2 activity with solubilized phospholipids was previously reported (25). In this work, we asked whether 15-LOX-2 might process esterified AA in bilayer phospholipids encompassed in nanodiscs. In addition, we looked at the same reaction with the mouse homologue of 15-LOX-2, which generates exclusively the 8-S-isomer of the hydroperoxy product from free AA, rather than the 15-S, and eventually a di-HETE product. Although the activity of the mouse enzyme at the bilayer was reduced relative to that of the human enzyme, the enzyme was able to process the membrane-embedded substrate and yield only the 15-S-isomer of HETE; the 8-S-isomer was not detected. In addition, immunofluorescence studies indicated that 15-LOX-2 translocated to the plasma membrane upon Ca2+ stimulation, and this translocation required the putative membrane insertion loop that projected from the amino-terminal β-barrel domain. In contrast, the same β-barrel domain in 5-LOX targets it to the nuclear membrane (26–28). Finally, HEK293 cells transformed to express the 15-LOX-2 and m8S-LOX enzymes yielded increased pools of both 15-HETE and 15-HETrE (as well as 8-HETE for the 8-enzyme). In vitro, only the action of the mouse enzyme on PL-esterified AA or DGLA (the precursor to HETrEs) generates 15-HETE or 15-HETrE, respectively. Neither free DGLA nor its ethyl ester were transformed to the HETrEs by the 8S-enzyme at an appreciable rate. We might infer from these results that in a cellular context the m8S-enzyme can process PL-esterified PUFAs and lead to the production of 15-HETrEs and 15-HETEs. From the experiments with 15-LOX-2-expressing HEK293 cells, we were unable to conclude that the enzyme generated 15-HETE as the PL-ester in a cellular context. However, our data from incubations of exogenous 15-LOX-2 incubated with lysates of PUFA-enriched untransfected HEK293 cells strongly hinted at a significant membrane-bound activity for this enzyme as well (Fig. 6A). Exogenous PLA2 was added after heat inactivation of 15-LOX-2, and the free 15-HETE levels increased over 5-fold once the incubations were treated with the phospholipase.
FIGURE 8.

Human 15-LOX-2. A, cartoon rendering of the 15-LOX-2 structure. The putative membrane insertion loop (residues 70–85) is colored in black. Fe2+ and Ca2+ are represented by rust and purple spheres, respectively. A competitive inhibitor, the detergent C8E4, is shown in red. Amino acids Asp-85 (Ca2+ ligand) and Val-603 are shown in stick rendering. B, schematic showing how direction of substrate entry determines product stereochemistry. The LOX reaction involves the abstraction of a hydrogen from the central carbon of a pentadiene followed by the oxygenation of the incipient free radical on the opposite side of the substrate. In this drawing, the iron (red sphere) sits below the plane of the figure and the O2 (blue spheres) accesses the free radical from above. In the top panel, (for 15-S product), the tail slides in to position C13 for attack, whereas in the bottom panel (for 8-S product), the AA slides in carboxyl first, presumably into a slightly deeper cavity, such that C10 is positioned for attack. C, a slab of the corresponding surface rendering is shown such that the contours of the fatty acids binding site are apparent. D, a close-up of the binding cavity. Superimposed on the 15-LOX-2 plus inhibitor structure is a phospholipid as observed in the LOX from Pseudomonas aeruginosa (4G32 and 4G33 (49)). Note that the funnel-shaped cavity of 15-LOX is of sufficient size to accommodate a PL head group.
The mouse and human enzymes encoded by the ALOX15B genes share 78% sequence identity at the amino acid level, yet the products of these enzymes differ with respect to regiospecificity with AA as the common substrate. This difference in positional specificity is conferred by the substitution of Asp-602 and Val-603 deep in the 15-LOX-2 binding pocket with Tyr-His in the mouse enzyme. Site-directed mutagenesis of these two amino acids to their 15-LOX-2 counterparts converts the 8-S enzyme to a 15-S enzyme (29). As one can see from Fig. 8 (A–C), Val-603 sits at the deepest part of the AA binding site, positioned to define the depth of the binding pocket and abut C20 of the substrate so that C15 can be positioned for oxygenation. The altered specificity of the mouse 8-S-enzyme is explained by the fact that a His at this position could favor the reverse orientation of substrate: the carboxyl end entering deepest to interact with the His. This inverse entry model is consistent with the stereochemistry of the product as well, because it can position C8 of the substrate for oxygenation to a product with S-stereochemistry (Fig. 8D).
A sequence database search for m8S-LOX homologues retrieved 80 unique sequences with as little as 72% overall sequence identity to the mouse enzyme. Although all but one of these sequences maintain the insertion in the amino-terminal β-barrel domain that appears to serve as a membrane binding loop, only the mouse enzyme has a His rather than a Val, Ile, or Met at the position corresponding to Val-603 in 15-LOX-2 (Fig. 9). This suggests that only the mouse ALOX15B enzyme directs carboxyl-first binding of free AA into the active site pocket. However, if the cellular substrate is the PL-esterified PUFA, the bulky PL head group cannot be accommodated innermost in the cavity, and the PUFA must enter methyl end first, making the mouse enzyme a de facto 15-S-LOX. This possible in vivo 15-activity rather than 8-activity has important implications for the use of mouse model systems (ApoE−/− or LDLR−/−) to study atherosclerosis (30).
FIGURE 9.
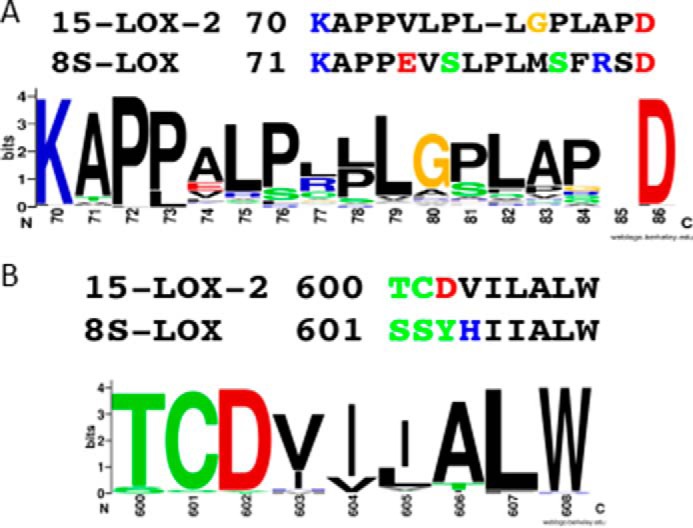
Sequence conservation in the membrane insertion loop and cavity regions. A, the hydrophobicity and rigidity of the membrane insertion loop of 15-LOX-2 is well conserved in 15-LOX-2 homologues. The sequence of this region in the m8S-LOX deviates from this rule. B, 15-LOX-2 homologues have a hydrophobic amino acid at the deepest part of the binding cavity. The mouse sequence is an outlier in this region with a His at the corresponding position. This figure was generated with WebLogo (50, 51) from a ClustalW (52, 53) multisequence lineup of 80 homologous retrieved with the mouse 8-S-LOX sequence.
Rao and co-workers (31) have observed that 15-HETE (generated by the non-enzymatic reduction of the product of a 15-LOX to the corresponding alcohol) contributes to atherosclerosis in the ApoE−/− murine model. Mice express a 12/15-lipoxygenase as well as the 8S-enzyme, and it has been linked to the production of 15-HETE (32). However, in a lipidomics study Demetz et al. (16) performed with ALOX12/15-LOX−/− (the 15-LOX-1 or ALOX15A, homologue) knock-out mice, the level of 12-HETEs detected fell to less than 50% of that of the wild-type mouse, whereas a comparable, or significant, drop in 15-HETE levels was not detected. These data support our suggestion that m8S-LOX may generate 15-HETE in a cellular context, because these knock-out mice generate 15-HETE in the absence of the 15-LOX-1 counterpart (∼40% sequence identity with 15-LOX-2). Moreover, Magnusson et al. (6) have demonstrated that ALOX15B gene-silencing experiments in human primary macrophages decrease cellular lipid accumulation and that the equivalent knockdown in a murine model (LDLR−/−) leads to a reduction in markers of atherosclerosis.
The Role of the Membrane Insertion Loop
The crystal structure of 15-LOX-2 revealed two Ca2+ binding sites at the base of a hairpin-like loop that emanates from the membrane binding amino-terminal domain. Phospholipase A2, which hydrolyzes esterified AA from the phospholipid bilayer upon Ca2+-stimulated membrane binding, also harbors a separate Ca2+-dependent membrane-binding domain (33). Kinetic studies with PLA2 indicate that it can process multiple substrates before falling off the membrane and reassociating for further action, i.e. it scoots along the membrane to acquire more substrate as the off rate for membrane binding is slow (34, 35). An alternative mechanism for interfacial enzymes that function at the bilayer where off-rates for binding are fast relative to the catalytic turnover is referred to as a hopping mode. In this mode of action, the enzyme hops from one contact point to the next. The use of nanodisc assays allows one to readily distinguish between these modes of action, because an enzyme that is free to hop among the nanodiscs can reach all available substrate. In a cellular context, a hopping enzyme might be able to more easily access distinct pools of substrate. On the other hand, a scooting enzyme may have prolonged access to membrane-embedded substrate.
We found that the membrane insertion loop of 15-LOX-2 was required for full enzyme activity at the membrane, because deletion of the loop lowers both the rate and amount of product detected. In contrast, with its putative membrane insertion loop, m8S-LOX cannot hop between nanodiscs, and deletion of the loop has little impact on the amount of product formed. Note that the loop is a second region of sequence divergence between the mouse and human enzyme where the mouse sequence is an outlier in a line-up (Fig. 9).
Influence of the Phospholipid Head Group
Comparison of 15-LOX-2 activity with PL bearing different head groups reveals that it may discriminate among the phospholipids. Choline appears to be the preferred head group, over ethanolamine or inositol, because more product is accumulated at the earlier time point (15 min) with PC. Plasma membranes are generally composed of 39% PC, 23% PE, 9% PS, and 8% PI (36). Although zwitterionic PC and PE are abundant in biological membranes, PI phospholipids carry 20 times more AA than PS-containing PL in lipidomic analysis of the human plasma (37). Thus it was important to evaluate the effect of the PI head group as well. A more thorough investigation of head group preference with a facile continuous assay suited to high throughput format is warranted, but at this point we can conclude that PC-, PI-, and PE-bearing PL are all substrates for 15-LOX-2. There is, however, a preference for PC and PI, which, interestingly, are both bulkier than PE. As one can see in Fig. 8, there is sufficient room in the large crevice that narrows like a funnel to the substrate binding site to accommodate a PL head group and confers some sort of selection mechanism. It is possible that the less bulky PE head group permits the fatty acid to enter further into the substrate tunnel and adopt a non-productive conformation.
PUFA Preference Is Context-dependent
Using the nanodisc assay, we were able to directly observe a substrate preference among esterified PUFA not observed for the free fatty acid substrates. Moreover, analysis of the cell extracts indicated that this preference may reflect an activity in a cellular context. The mouse enzyme preferentially processes PI-esterified DGLA over its AA counterpart in the nanodisc assays. Cell extracts from HEK cells that have been transfected to express the 8S-enzyme show increases in 15-HETrE over non-transfected cells. Free DGLA or its ethyl ester are poor substrates for the mouse enzyme; the enzyme is ∼5-fold more active with free AA than free DGLA, and free DGLA displays potent substrate inhibition. This apparent inconsistency for the mouse enzyme (i.e. that when DGLA is PL-esterified it is the preferred substrate, but when free it is a poor substrate compared with AA) can be understood in terms of the steric constraints a PL-esterified substrate might impose on whether or how the LOX can align the substrate pentadiene for attack. The same constraints that force the bulky phospholipid head group to be on the surface of the enzyme and set the direction of entry of the fatty acid tail first into the active site may also mitigate substrate inhibition. Substrate inhibition by the free PUFA suggests it can bind in the active site in non-productive modes. The presence of the PL head group might preclude non-productive substrate-enzyme interactions and obviate substrate inhibition by simply restricting its modes of binding.
Product Specificity Is Context-dependent
One must also consider the effect of experimental context on product specificity. Although overexpression of 15-LOX-2 in HEK cells leads to an increase in only 15-HETE and 15-HETrE products in cell extracts, the cell extracts from HEK cells expressing m8S-LOX contain isomers not observed in the nanodisc assays (or with free AA). The explanation for this apparent discrepancy could be simply that the nanodisc assays do not incorporate the variety of PL-esterified AA the enzyme can encounter in HEK cells. However, there is precedent for compromised product specificity in conditions in which the enzymatic reaction is slowed, as it is for m8S-LOX at the bilayer. It has been reported that soybean LOX generates a unique regio- and stereospecific product from cholate-solubilized PL substrate (38). However, when the same substrate was incorporated into liposomes, a random product mixture was observed. A random product mixture is not observed when a free fatty acid substrate is presented in liposomes (14). This loss of specificity can be understood in terms of the LOX enzymatic mechanism. The reaction is initiated by hydrogen abstraction from the central carbon of the pentadiene (Fig. 8). An O2 pocket positions molecular oxygen for attack on the incipient radical. The membrane-embedded substrate is constrained and cannot assume optimal alignment in the enzyme active site; this distortion leads to a significantly slowed reaction and allows exposure of the free radical intermediate to O2 in an alternate conformation. A similarly compromised specificity was observed at low O2 concentrations for rabbit 15-LOX-1 (15). The overall interpretation is that if the lifetime of the free radical intermediate is prolonged, alternative positioning in, or even escape from, the active site can lead to a less specific pattern of oxygenation.
Our in vitro observations with the m8S-LOX can be understood in this context. The enzymatic reaction is significantly slowed when the enzyme works at the bilayer. This slowed enzymatic rate may be the source of the additional HETEs detected. However, despite this slower reaction rate, one can see from Fig. 6B that the total amount of HETEs produced by m8S-LOX in HEK cells may even be greater than that produced by 15-LOX-2 under equivalent conditions. Yet this result too is consistent with the in vitro experiments. Recall that m8S-LOX is not readily released from the nanodisc: although 15-LOX-2 effectively hops among nanodiscs, m8S-LOX remains stuck and cannot access all available substrate. This same property—that m8S-LOX scoots rather than hops—may allow m8S-LOX to process more substrate than 15-LOX-2 in a cellular context, even though it means less substrate with the limited small size of a nanodisc.
The combined results led us to ask which experimental system, nanodiscs or HEK cell expression, accurately emulates the m8S-LOX in vivo activity? Both have their shortcomings. As mentioned above, the PL composition of the ND is not as diverse as that in HEK cells, and all possible substrates are not evaluated. On the other hand, constitutive overexpression of the enzyme in HEK cells cannot incorporate any regulatory events that may occur in a biological context. However, what we can clearly conclude is that m8S-LOX is active at the bilayer, because in both nanodiscs and HEK cells, it generates products that are not seen when the substrate is free AA and that are consistent with tail end entry. The data indicate that that m8S-LOX can produce other HETE isomers in addition to 8-HETE and should not be ignored as a possible source of 15-HETE in mouse models of atherosclerosis. When mouse models are used, it should be considered that the potentially disease causing enzyme differs in significant aspects from the human homologue, and small molecule inhibitors used to target membrane activity of the mouse enzyme may not be effective against the human enzyme.
Concluding Remarks
Both 15-LOX-2 and its murine counterpart (78% sequence identity), which generates 8-HpETE from free AA, produce a PL-esterified 15-HpETE when the substrate is incorporated in a bilayer. Experiments with stably transfected HEK cells suggest that these activities can contribute to cellular pools of free 15-HpETE/15-HETE. Despite the fact that the homologous enzymes generate the same product when they function at the bilayer, they differ significantly in their interactions with the membrane. The interaction of 15-LOX-2 with the bilayer is reversible despite the presence of Ca2+, whereas that for m8S-LOX is not.
Materials and Methods
LOX Cloning, Overexpression, and Purification
15-LOX-2, m8S-LOX, and their loop mutants were overexpressed and purified essentially as previously described for 15-LOX-2 (10). For all Escherichia coli overexpression, the pET Duet-1 vector with the E. coli yjgD gene after promoter 2 was used. Wild-type and mutant 15-LOX-2 plasmids were previously generated (10); the m8S-LOX plasmid was created as follows from a pet3C plasmid obtained as a gift from the laboratory of Alan Brash. The m8S-LOX gene was first cloned from pET3C to pET28a using the restriction sites NdeI and HindIII. The m8S gene was amplified from pET28a using primers with BspHI and HindIII restriction sites, and the PCR product was cut with BspHI and HindIII and inserted into the pET-Duet-1 vector cut with NcoI and HindIII. BspHI and NcoI restriction sites have the same sticky ends and can be annealed to each other. The BspHI site was used at the 5′ end of the m8S gene instead of NcoI because of the internal NcoI site in m8S. Cloning was confirmed by sequencing the final pET-Duet-1 vector. The pET-Duet-1 vectors with desired genes were transformed into Rosetta 2 (DE3) cells, and the overexpression was performed as described previously (10).
Enzymes were purified on a Talon Co2+ affinity column using gravity flow and a step gradient. After protein binding, the resin was washed with two column volumes of 20 mm Tris (pH 8.0), 500 mm NaCl, 20 mm imidazole. The enzymes were eluted with 20 mm Tris (pH 8.0), 500 mm NaCl, 200 mm imidazole. After the Talon affinity column the 15-LOX-2 samples were highly purified. For m8S-LOX, 20% glycerol was added to the purification and storage buffers as done by Kawajiri et al. (11); otherwise protein activity was rapidly lost. The m8S-LOX was further purified by size exclusion chromatography on a Superdex 200 column mounted on an AKTA FPLC (GE Healthcare) and eluted with 20 mm Tris-HCl (pH 8.0), 250 mm NaCl.
Nanodisc Preparation
Nanodiscs were prepared according to previously published protocols (10, 39). During the preparation of the phospholipid pellets, the desired ratios of the various sn2 fatty acids and lipid head groups were adjusted. Nanodisc concentration was calculated from the extinction coefficient of the MSPE3D1 protein: 26,600 m−1 cm−1, taking into consideration the two proteins per nanodiscs.
Extraction for Mass Spectrometry of Nanodiscs
Phospholipids were extracted from the enzyme-nanodisc incubations using the Bligh/Dyer method (40). To 100 μl of reaction volume, 700 μl of distilled water was added, after which 2 ml of methanol and 1 ml of CHCl3 were added using Pasteur pipettes. The mixture was vortexed and incubated on the nutator for 20 min at 22 °C. Subsequently, 1 ml of chloroform and 1.8 ml of distilled water were added, and the mixture was incubated again on the nutator for 20 min. The mixture was centrifuged at room temperature at 4000 rpm for 2 min. The lower lipid-containing layer containing was removed with a Pasteur pipette, transferred to a glass tube, dried under a stream of N2, and resuspended in a solution of 2:1 CHCl3:methanol. Analysis by mass spectrometry was performed in the positive ion mode for phospholipids with choline head groups (e.g. SAPC).
Enzyme Assays with Nanodiscs
Assays were set up using 3 μm enzyme (either 15-LOX-2 or m8S-LOX) and 15 μm nanodiscs in 50 mm potassium phosphate buffer (pH 7.4), 100 mm NaCl, 0.5 mm EDTA, and 2 mm CaCl2, at 37 °C. For m8S-LOX, 10% glycerol was added to the reaction buffer. For studying the time course of product formation, 40-μl aliquots were removed and flash frozen in liquid N2. The aliquots were then incubated at 85 °C for 50 min to inactivate the LOX, and 0.1 mg/ml final concentration of phospholipase A2 (PLA2, bee venom; Sigma) was added to cleave the fatty acids (at 37 °C, 2 h) for HPLC analysis, after reduction to their corresponding HETEs with triphenylphosphine (TPP). The reactions were stopped with equal volumes of methanol and TPP (TPP, 25 mg/100 ml). Prior to solid phase extraction with C18 cartridges (UCT CLEAN-UP C18 CEC1811Z), 3 μl of 1 m HCl, 5 μl of 50 ng/μl prostaglandin B1 (Cayman) standard, and 100 μl of PBS were added to a 100-μl stopped reaction mixture. The 100% methanol elutions from the C18 cartridges were dried under a N2 stream and then resuspended in the mobile phase (60% acetonitrile, 0.1% formic acid) for isocratic reverse phase HPLC with a Supelco Discovery HSC18 column monitored at 235 nm. The extinction coefficients used for the products 15-HETE and 15-hydroxy-eicosatrienoic acid (15-HETrE) were 27,000 and 23,000 m−1 cm−1, respectively (41–44). The amount of product was calculated from comparing HPLC peak areas to the peak area of prostaglandin B1 standard of known concentration, correcting for the difference in extinction coefficients.
Enzyme Kinetics with Free Substrates
The assays of 15-LOX-2 and m8S-LOX with the free substrates were performed in 50 mm Tris (pH 7.5), 150 mm NaCl, 0.5 mm EDTA, with the addition of 5% glycerol to m8S-LOX buffers. The enzyme concentration was 200 nm, and the substrate concentrations varied between 1 μm and 40 μm. When ethyl ester derivatives of substrates were used, 20 mm cholic acid was included in reaction buffer. When there was no substrate inhibition, classical Michaelis-Menten equation was used to fit the data. When there was substrate inhibition, the following equation was used in GraphPad Prism least square regression fitting (45–47) as shown in Equation 1.
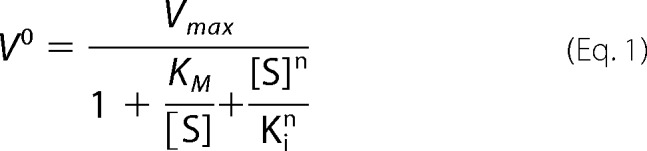 |
Kinetic Analysis of Reactions on Nanodiscs
The protocol for analyzing kinetics of enzymes on lipid surfaces differs from enzyme kinetics in solution (12, 48). Instead of analyzing the data using the integrated Michaelis-Menten equation adapted to interfacial reactions, which requires much higher precision reaction progress curves, the curves created from the intensities of the product peaks from the HPLC assay were fit with the one-phase association equation in GraphPad Prism software.
This equation describes a first order rise to a maximum for product formation. The assumption is that it is a first order reaction, where the rate is dependent only on the concentration of one reactant, which is the phospholipid substrate concentration in the nanodisc. Rate constants observed with different enzymes and PL substrates are compared under equivalent conditions (e.g. same Ca2+ concentrations, buffers, and reaction conditions). In other reactions, the amounts of product formed at a given time point were compared for different head groups, different variants of the enzymes, and in different conditions, to assess the effects of these variables on the rate of product formation.
Supplementation for PUFA Enrichment of HEK Cell Membranes
The membranes of HEK293 cells are generally a poor source of AA. The PUFA content of the cells was increased by supplementation of the media with BSA, AA, and LA over multiple growth cycles, as described below in the details of the transfection procedure. To evaluate the effectiveness of the supplementation, exogenous 15-LOX-2 and PLA2 were utilized to detect the presence of 15-LOX-2 substrates in the membrane fraction. To this end, HEK293 cells were resuspended in 20 ml of lysis buffer (PBS, 1.5 mm CaCl2, 0.5 mm EDTA, protease inhibitors, and sodium orthovanadate) to a final cell concentration of 2 million cells/ml. Lysates were passed through a 23-gauge needle 20 times to rupture cell membranes. Lysate reaction volumes (10 ml) were maintained on ice. The 15-LOX-2 reactions were initiated at 37 °C by addition of enzyme (final concentration, 0.8 μm) to the lysate and incubated for 1 h at 37 °C. The reactions were either stopped with methanol or processed further with PLA2 to release the fatty acids in the PL sn2 position. Prior to the addition of PLA2 (450 μl of 1 mg/ml, 2 h, 37 °C), 15-LOX-2 was heat-inactivated. The lysates were processed for HPLC analysis as above, after clarification by centrifugation.
HEK293 Transfection and Mass Spectrometry Analysis of Oxidized Fatty Acids
The 15-LOX-2 and m8S-LOX genes were cloned into a pCDNA 3.1(+) vector using the NheI and HindIII restriction sites. The constructs were transfected into HEK293 cells using Lipofectamine 3000 (Invitrogen) per manufacturer's instructions. The cells that picked up the plasmid were grown using G418 selection. The untransfected cells were treated in parallel as negative control in experiments. The unsaturated fatty acid content of HEK293 cells was boosted by supplementing the growth medium (DMEM, 10% FBS (Atlanta Biologicals), penicillin/streptomycin, and G418 (Sigma)) with 30 μm (final concentration) AA, 30 μm LA in solution with 15 μm BSA (Sigma) for four cell passages (90% confluence to 40% confluence every 3 days). The cells were harvested at 90% confluence with trypsin-EDTA and washed once with DMEM + BSA (2 mg/ml), once with PBS + BSA, and once with PBS + BSA + glucose (0.1%), and finally resuspended in PBS + glucose at ∼0.5 million cells/ml. After additions of 0.5 mm CaCl2 and 5 μm Ca ionophore A23187, the cells were incubated at 37 °C for 1 h and subsequently cooled on ice for 10 min. Pelleted cells were processed for lipidomic analysis of oxidized fatty acids (see below).
Oxidized Fatty Acid Analysis
Just before analysis, supernatants containing (50% methanol) from 3 × 106 cells were diluted in water to a final methanol concentration of less than 15% and deuterated internal standards (see below for transitions) added. The methanolic solution was extracted using a solid phase extraction cartridge (Strata Polymeric Reversed Phase, 60 mg/ml; Phenomenex, Torrance, CA). The eluant (1 ml of methanol) was reconstituted in HPLC solvent A (8.3 mm acetic acid, buffered to pH 5.7 with NH4OH). Solvent B was acetonitrile/methanol (65/35, v/v). An aliquot of 20 μl was injected into an HPLC system, and separation of the different metabolites was conducted using a C18 column (Kinetex 5 μm EVO C18 100A; Phenomenex) eluted at a flow rate of 300 μl/min with the initial conditions, 35% B, held for 1 min. The gradient was then increased from 35% to 75% solvent B in 8 min; subsequently B was increased to 98% in 1 min and held for 5 min. The HPLC system was directly interfaced into the electrospray source of a triple quadrupole mass spectrometer (AB SCIEX Q-Trap 5500; PE-Sciex, Thornhill, Canada) where mass spectrometric analyses were performed in the negative ion mode using multiple reaction monitoring of the specific transitions: d4 9-HODE m/z 299 → 172, d4 13-HODE m/z 299 → 198, d8 5(S)-HETE m/z 327 → 116, 9-HETE m/z 319 → 123, 8-HETE m/z 319 → 163, 11-HETE m/z 319 → 167, 8-HETrE m/z 321 → 157, 5-HETrE m/z 321 → 205, 15-HETrE m/z 321 → 221, 9-HODE m/z 295→171, 13-HODE m/z 295 → 195, 5-HETE m/z 319 → 115, 15-HETE m/z 319 → 219, and 12-HETE m/z 319 → 179. Quantitation was performed using a standard isotope dilution curve for each of the lipid targets for which a stable isotope standard was added (13-HODE, 9-HODE, and 5-HETE) and reference standard curves determined for each of the other oxidized polyunsaturated fatty acids (unlabeled analytes obtained from Cayman Chemical, Ann Arbor, MI), using d8 5(S)-HETE as the internal standard. Representative MS data are presented in supplementary Figs. S1 and S2.
Immunofluorescence Labeling of 15-LOX-2 in Transfected HEK293 Cells
Transfected HEK293 cells expressing 15-LOX-2, wild-type and variants, were seeded out at 200 cells/ml in each well of an Ibdi 12-well slide treated with 0.01% poly-l-lysine. After 24–48 h at 37 °C and 5.0% CO2, 2.5 μm A23187 Ca2+ ionophore was added to stimulate cells, which were then incubated for 5 min at 37 °C, 5.0% CO2. The cells were fixed with 4% paraformaldye and then washed three times with PBS. The samples were then incubated with a 50 mm NH4Cl solution, followed by a wash cycle. The cells were permeabilized with ice-cold acetone followed by an additional wash cycle, blocked for 1 h with 10% donkey serum, 0.1% Tween 20 in PBS, and subsequently incubated overnight at 4 °C with a primary antibody solution consisting of a 1:1000 dilution of rabbit anti-15-LOX-2 and mouse anti-pan-cadherin (both AbCam) antibodies in 0.1% Tween 20 PBS. After a wash cycle with 0.1% Tween 20 PBS, the cells were incubated with a secondary antibody solution consisting a 1:10000 dilution of goat anti-rabbit antibody Alexa Fluor 647 and donkey anti-mouse Alexa Flour 488 (both Invitrogen) in a 0.1% Tween 20 PBS solution. After another wash cycle, the cells were incubated with 2 μg/μl DAPI solution for 3 min followed by another wash cycle. Prolong Gold antifade reagent and a coverslip were then added to the slide. The slide was imaged with a Leica DM RXA2 upright microscope with a SensiCam QE 12-bit CCD camera using DAPI, FITC (pan-cadherin) and Cy5 (15-LOX-2) filter channels followed by no neighbors deconvolution image analysis.
Author Contributions
G. B. designed, performed, and interpreted the experiments and wrote the paper. E. E. S. performed the immunofluorescence studies. C. U. performed the oxidized fatty acid mass spectrometry. R. C. M. and M. E. N. conceived the studies and contributed to data interpretation and manuscript preparation.
Supplementary Material
This work was funded in part by National Institutes of Health Grants HL107887 (to M. E. N.) and HL117798 (to R. C. M.) and American Heart Association Grant 16GRNT31000010 (to M. E. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1 and S2.
- LOX
- lipoxygenase
- AA
- arachidonic acid
- DGLA
- dihomo-γ-linolenic acid
- HETE
- hydroxyeicosatetraenoic acid
- 15-HpETE
- 15-hydroperxyeicosatetraenoic
- 15-HETrE
- 15-hydroxyeicosatrienoic acid
- LA
- linoleic Acid
- m8S-LOX
- murine 8-S-lipoxygenase
- PUFA
- polyunsaturated fatty acid
- PC
- phosphatidylcholine
- PE
- phosphatidylethanol-amine
- PS
- phosphatidylserine
- PI
- phosphatidylinositol
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine
- PL
- phospholipid
- SAPC
- 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- SAPE
- 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- SAPI
- 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoinositol
- TPP
- triphenylphosphine.
References
- 1. Harats D., Shaish A., George J., Mulkins M., Kurihara H., Levkovitz H., and Sigal E. (2000) Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 20, 2100–2105 [DOI] [PubMed] [Google Scholar]
- 2. Sendobry S. M., Cornicelli J. A., Welch K., Bocan T., Tait B., Trivedi B. K., Colbry N., Dyer R. D., Feinmark S. J., and Daugherty A. (1997) Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 120, 1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bocan T. M., Rosebury W. S., Mueller S. B., Kuchera S., Welch K., Daugherty A., and Cornicelli J. A. (1998) A specific 15-lipoxygenase inhibitor limits the progression and monocyte-macrophage enrichment of hypercholesterolemia-induced atherosclerosis in the rabbit. Atherosclerosis 136, 203–216 [DOI] [PubMed] [Google Scholar]
- 4. Hutchins P. M., and Murphy R. C. (2012) Cholesteryl ester acyl oxidation and remodeling in murine macrophages: formation of oxidized phosphatidylcholine. J. Lipid Res. 53, 1588–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldstein J. L., and Brown M. S. (2001) Molecular medicine: The cholesterol quartet. Science 292, 1310–1312 [DOI] [PubMed] [Google Scholar]
- 6. Magnusson L. U., Lundqvist A., Karlsson M. N., Skålén K., Levin M., Wiklund O., Borén J., and Hultén L. M. (2012) Arachidonate 15-lipoxygenase type B knockdown leads to reduced lipid accumulation and inflammation in atherosclerosis. PLoS One 7, e43142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brash A. R. (1999) Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 274, 23679–23682 [DOI] [PubMed] [Google Scholar]
- 8. Thomas C. P., Morgan L. T., Maskrey B. H., Murphy R. C., Kühn H., Hazen S. L., Goodall A. H., Hamali H. A., Collins P. W., and O'Donnell V. B. (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 285, 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jisaka M., Kim R. B., Boeglin W. E., Nanney L. B., and Brash A. R. (1997) Molecular cloning and functional expression of a phorbol ester-inducible 8S-lipoxygenase from mouse skin. J. Biol. Chem. 272, 24410–24416 [DOI] [PubMed] [Google Scholar]
- 10. Kobe M. J., Neau D. B., Mitchell C. E., Bartlett S. G., and Newcomer M. E. (2014) The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 289, 8562–8569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawajiri H., Piao Y., Takahashi Y., Murakami T., Hamanaka N., and Yoshimoto T. (2005) Synthesis of 8,9-leukotriene A4 by murine 8-lipoxygenase. Biochem. Biophys. Res. Commun. 338, 144–148 [DOI] [PubMed] [Google Scholar]
- 12. Berg O. G., Gelb M. H., Tsai M. D., and Jain M. K. (2001) Interfacial enzymology: the secreted phospholipase A2-paradigm. Chem. Rev. 101, 2613–2654 [DOI] [PubMed] [Google Scholar]
- 13. Gelb M. H., Min J. H., and Jain M. K. (2000) Do membrane-bound enzymes access their substrates from the membrane or aqueous phase: interfacial versus non-interfacial enzymes. Biochim. Biophys. Acta 1488, 20–27 [DOI] [PubMed] [Google Scholar]
- 14. Noguchi N., Yamashita H., Hamahara J., Nakamura A., Kühn H., and Niki E. (2002) The specificity of lipoxygenase-catalyzed lipid peroxidation and the effects of radical-scavenging antioxidants. Biol. Chem. 383, 619–626 [DOI] [PubMed] [Google Scholar]
- 15. Ludwig P., Holzhütter H. G., Colosimo A., Silvestrini M. C., Schewe T., and Rapoport S. M. (1987) A kinetic model for lipoxygenases based on experimental data with the lipoxygenase of reticulocytes. Eur. J. Biochem. 168, 325–337 [DOI] [PubMed] [Google Scholar]
- 16. Demetz E., Schroll A., Auer K., Heim C., Patsch J. R., Eller P., Theurl M., Theurl I., Theurl M., Seifert M., Lener D., Stanzl U., Haschka D., Asshoff M., Dichtl S., et al. (2014) The arachidonic acid metabolome serves as a conserved regulator of cholesterol metabolism. Cell Metab. 20, 787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kilty I., Logan A., and Vickers P. J. (1999) Differential characteristics of human 15-lipoxygenase isozymes and a novel splice variant of 15S-lipoxygenase. Eur. J. Biochem. 266, 83–93 [DOI] [PubMed] [Google Scholar]
- 18. Brinckmann R., Schnurr K., Heydeck D., Rosenbach T., Kolde G., and Kühn H. (1998) Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 91, 64–74 [PubMed] [Google Scholar]
- 19. Pouliot M., McDonald P. P., Krump E., Mancini J. A., McColl S. R., Weech P. K., and Borgeat P. (1996) Colocalization of cytosolic phospholipase A2, 5-lipoxygenase, and 5-lipoxygenase-activating protein at the nuclear membrane of A23187-stimulated human neutrophils. Eur. J. Biochem. 238, 250–258 [DOI] [PubMed] [Google Scholar]
- 20. Gerstmeier J., Weinigel C., Barz D., Werz O., and Garscha U. (2014) An experimental cell-based model for studying the cell biology and molecular pharmacology of 5-lipoxygenase-activating protein in leukotriene biosynthesis. Biochim. Biophys. Acta 1840, 2961–2969 [DOI] [PubMed] [Google Scholar]
- 21. Gillmor S. A., Villaseñor A., Fletterick R., Sigal E., and Browner M. F. (1997) The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol. 4, 1003–1009; Correction (1998) Nat. Struct. Biol.5, 242 [DOI] [PubMed] [Google Scholar]
- 22. Xu S., Mueser T. C., Marnett L. J., and Funk M. O. Jr. (2012) Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure 20, 1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neau D. B., Bender G., Boeglin W. E., Bartlett S. G., Brash A. R., and Newcomer M. E. (2014) Crystal structure of a lipoxygenase in complex with substrate: the arachidonic acid-binding site of 8R-lipoxygenase. J. Biol. Chem. 289, 31905–31913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Newcomer M. E., and Brash A. R. (2015) The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 24, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coffa G., and Brash A. R. (2004) A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc. Natl. Acad. Sci. U.S.A. 101, 15579–15584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen X. S., and Funk C. D. (2001) The N-terminal “β-barrel” domain of 5-lipoxygenase is essential for nuclear membrane translocation. J. Biol. Chem. 276, 811–818 [DOI] [PubMed] [Google Scholar]
- 27. Kulkarni S., Das S., Funk C. D., Murray D., and Cho W. (2002) Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J. Biol. Chem. 277, 13167–13174 [DOI] [PubMed] [Google Scholar]
- 28. Hammarberg T., Provost P., Persson B., and Rådmark O. (2000) The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J. Biol. Chem. 275, 38787–38793 [DOI] [PubMed] [Google Scholar]
- 29. Jisaka M., Kim R. B., Boeglin W. E., and Brash A. R. (2000) Identification of amino acid determinants of the positional specificity of mouse 8S-lipoxygenase and human 15S-lipoxygenase-2. J. Biol. Chem. 275, 1287–1293 [DOI] [PubMed] [Google Scholar]
- 30. Getz G. S., and Reardon C. A. (2012) Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kotla S., Singh N. K., Heckle M. R., Tigyi G. J., and Rao G. N. (2013) The transcription factor CREB enhances interleukin-17A production and inflammation in a mouse model of atherosclerosis. Sci. Signal. 6, ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kotla S., Singh N. K., Traylor J. G. Jr, Orr A. W., and Rao G. N. (2014) ROS-dependent Syk and Pyk2-mediated STAT1 activation is required for 15(S)-hydroxyeicosatetraenoic acid-induced CD36 expression and foam cell formation. Free Radic. Biol. Med. 76, 147–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Six D. A., and Dennis E. A. (2003) Essential Ca(2+)-independent role of the group IVA cytosolic phospholipase A2 C2 domain for interfacial activity. J. Biol. Chem. 278, 23842–23850 [DOI] [PubMed] [Google Scholar]
- 34. Jain M. K., Ranadive G., Yu B. Z., and Verheij H. M. (1991) Interfacial catalysis by phospholipase-A2: monomeric enzyme is fully catalytically active at the bilayer interface. Biochemistry 30, 7330–7340 [DOI] [PubMed] [Google Scholar]
- 35. Jain M. K., and Gelb M. H. (1991) Phospholipase A2-catalyzed hydrolysis of vesicles: uses of interfacial catalysis in the scooting mode. Methods Enzymol. 197, 112–125 [DOI] [PubMed] [Google Scholar]
- 36. Hauser H., and Poupart G. (2004) Lipid Structure in The Structure of Biological Membranes (Yeagle P., ed) 2nd ed., pp. 1–52, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 37. Quehenberger O., Armando A. M., Brown A. H., Milne S. B., Myers D. S., Merrill A. H., Bandyopadhyay S., Jones K. N., Kelly S., Shaner R. L., Sullards C. M., Wang E., Murphy R. C., Barkley R. M., Leiker T. J., et al. (2010) Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brash A. R., Ingram C. D., and Harris T. M. (1987) Analysis of a specific oxygenation reaction of soybean lipoxygenase-1 with fatty acids esterified in phospholipids. Biochemistry 26, 5465–5471 [DOI] [PubMed] [Google Scholar]
- 39. Boldog T., Li M., and Hazelbauer G. L. (2007) Using Nanodiscs to create water-soluble transmembrane chemoreceptors inserted in lipid bilayers. Methods Enzymol. 423, 317–335 [DOI] [PubMed] [Google Scholar]
- 40. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 41. Ramis I., Roselló-Catafau J., Bulbena O., Picado C., and Gelpi E. (1989) 15-Hydroxyeicosatetraenoic acid as a major eicosanoid in nasal secretions: assay by high-performance liquid chromatographic-radioimmunoassay and gas chromatographic-mass spectrometric procedures. J. Chromatogr. 496, 416–422 [DOI] [PubMed] [Google Scholar]
- 42. Lecomte M., Laneuville O., Ji C., DeWitt D. L., and Smith W. L. (1994) Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 269, 13207–13215 [PubMed] [Google Scholar]
- 43. Petrich K., Ludwig P., Kühn H., and Schewe T. (1996) The suppression of 5-lipoxygenation of arachidonic acid in human polymorphonuclear leucocytes by the 15-lipoxygenase product (15S)-hydroxy-(5Z,8Z,11Z,13E)-eicosatetraenoic acid: structure-activity relationship and mechanism of action. Biochem. J. 314, 911–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haviv F., Ratajczyk J. D., DeNet R. W., Martin Y. C., Dyer R. D., and Carter G. W. (1987) Structural requirements for the inhibition of 5-lipoxygenase by 15-hydroxyeicosa-5,8,11,13-tetraenoic acid analogues. J. Med. Chem. 30, 254–263 [DOI] [PubMed] [Google Scholar]
- 45. Dewal M. B., and Firestine S. M. (2013) Site-directed mutagenesis of catalytic residues in N5-carboxyaminoimidazole ribonucleotide synthetase. Biochemistry 52, 6559–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. LiCata V. J., and Allewell N. M. (1997) Is substrate inhibition a consequence of allostery in aspartate transcarbamylase? Biophys. Chem. 64, 225–234 [DOI] [PubMed] [Google Scholar]
- 47. Willemoës M., and Larsen S. (2003) Substrate inhibition of Lactococcus lactis cytidine 5′-triphosphate synthase by ammonium chloride is enhanced by salt-dependent tetramer dissociation. Arch. Biochem. Biophys. 413, 17–22 [DOI] [PubMed] [Google Scholar]
- 48. Jain M. K., and Berg O. G. (1989) The kinetics of interfacial catalysis by phospholipase A2 and regulation of interfacial activation: hopping versus scooting. Biochim. Biophys. Acta 1002, 127–156 [DOI] [PubMed] [Google Scholar]
- 49. Garreta A., Val-Moraes S. P., García-Fernández Q., Busquets M., Juan C., Oliver A., Ortiz A., Gaffney B. J., Fita I., Manresa À., and Carpena X. (2013) Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa. Faseb J. 27, 4811–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schneider T. D., and Stephens R. M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., and Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.