

Abstract
Death domain superfamily members typically act as adaptors mediating in the assembly of supramolecular complexes with critical apoptosis and inflammation functions. These modular proteins consist of death domains, death effector domains, caspase recruitment domains, and pyrin domains (PYD). Despite the high structural similarity among them, only homotypic interactions participate in complex formation, suggesting that subtle factors differentiate each interaction type. It is thus critical to identify these factors as an essential step toward the understanding of the molecular basis of apoptosis and inflammation. The proteins apoptosis-associated speck-like protein containing a CARD (ASC) and NLRP3 play key roles in the regulation of apoptosis and inflammation through self-association and protein-protein interactions mediated by their PYDs. To better understand the molecular basis of their function, we have characterized ASC and NLRP3 PYD self-association and their intermolecular interaction by solution NMR spectroscopy and analytical ultracentrifugation. We found that ASC self-associates and binds NLRP3 PYD through equivalent protein regions, with higher binding affinity for the latter. These regions are located at opposite sides of the protein allowing multimeric complex formation previously shown in ASC PYD fibril assemblies. We show that NLRP3 PYD coexists in solution as a monomer and highly populated large-order oligomerized species. Despite this, we determined its monomeric three-dimensional solution structure by NMR and characterized its binding to ASC PYD. Using our novel structural data, we propose molecular models of ASC·ASC and ASC·NLRP3 PYD early supramolecular complexes, providing new insights into the molecular mechanisms of inflammasome and apoptosis signaling.
Keywords: apoptosis, inflammasome, inflammation, NLRP3, nuclear magnetic resonance (NMR), ASC, pyrin
Introduction
Proteins of the death domain superfamily mediate in apoptotic, innate immunity, and inflammatory responses mainly through homotypic interactions (1). Specifically, they are responsible for the assembly of large signaling complexes in which kinases and caspases are activated (1–4). Despite the lack of sequence convergence within the superfamily, its members are characterized by adopting the so-called death fold, consisting of a globular fold with six α-helices arranged in a greek-key topology (1, 3). However, some structural differences between the subfamilies are observed within this common fold, including different charge distributions throughout the protein surface, diverse locations of hydrophobic patches, and the different lengths of certain α-helices. These differences are thought to lead to homotypic binding specificity (5), albeit certain heterotypic interactions are also known with proposed inhibitory roles in apoptotic and inflammatory pathways (1, 6).
Detailed structural information on complexes formed by several members of the death domain superfamily is available. Some examples are the death domain interactions in the PIDDosome (7), the MyDDosome (8), the Fas·FADD (9, 10), and the Pelle·Tube death domain complexes (11), among others. In addition, structural studies on CARD·CARD 3 interactions have been reported for the Caenorhabditis elegans apoptosome (12), the procaspase-9·Apaf-1 (13), and CED-4·CED-9 complexes (14), together with the death effector domain-containing protein of the MC159 tandem complex (15). In contrast, little information is currently available on the interactions established by PYD domains (6, 16–19). This kind of information is key for designing strategies targeted to modulate complex formation and thus develop therapeutic applications in the context of inflammatory and apoptosis-related disorders (2, 20, 21).
Only three different types of death domain interactions responsible for the formation of multimeric complexes have been characterized thus far (18, 22). The type I interaction defines contacts between helices 1 and 4 of one death domain and helices 2 and 3 of the other; the type II interaction occurs when helix 4 and the loop connecting helices 4 and 5 of one domain interact with the loop between helices 5 and 6 of the other; and finally, the type III interaction includes contacts between helix 3 of one domain and the loops linking helices 1 and 2 and helices 3 and 4 of the other domain (3). It is noteworthy that these binding interfaces do not overlap, and thus it is possible for a single death domain to establish multiple interactions (7, 18). It is assumed that all death domain subfamilies are capable of forming these three interaction types. However, important differences present in each type are believed to be responsible for the homotypic specificity (18).
The apoptosis-associated speck-like protein containing a CARD (ASC) and NLRP3 (also known as cryopyrin) are two members of the PYD subfamily that play crucial roles in inflammasome activation. Upon the onset of danger signals, ASC and NLRP3 activate the inflammasome leading to procaspase-1 activation and further proteolytic activation of the pleiotropic inflammatory cytokines IL-1β and IL-18 (23). This process will ultimately prompt NF-κB activation thus inducing proinflammatory responses (1, 2, 24–27). ASC contains a PYD and a CARD domain (28, 29), whereas NLRP3 carries a PYD, a nucleotide-binding oligomerization domain, and a leucine-rich repeat domain (1). It is assumed that both proteins interact through their PYD domains, whereas ASC and caspase-1 binding would be mediated by a CARD·CARD interaction, subsequently leading to a highly multimeric complex formation, which is essential for the proteolytic activation of the initiator caspases (30, 31).
The PYD domain is characterized at the structural level by a large loop connecting helices 2 and 3, with the latter being particularly short (6, 17, 32–38). Despite the abundant structural information reported for PYD domains (the three-dimensional structures of 10 different PYD domains have been deposited in the PDB) (6, 17, 29, 32–38), our understanding on the ASC·NLRP3 PYD homotypic interaction is very limited. Several models for this interaction have been proposed based on the NLRP3 crystallographic structure and mutational data (4, 16), in addition to the recent semi-atomic structure characterization of ASC PYD mature fibril assemblies by cryo-EM and solid-state NMR (18, 39).
We report here the three-dimensional solution structure of NLRP3 PYD and a detailed study of ASC PYD self-association and its interaction with NLRP3 by NMR and analytical ultracentrifugation (AUC) using different conditions of pH and salinity. All in all, our data indicate that the ASC·ASC and ASC·NLRP3 complexes share the same binding interface. However, we observed higher affinity for the heteromeric interaction. Furthermore, using the available atomic structures and our structural results on the interactions, we have built models of the supramolecular complexes for both ASC·ASC an ASC·NLRP3 PYDs, which provide molecular insights into the first possible stages of inflammasome formation. In addition, our data could be used to target ASC and NLRP3 proteins in the design of therapeutic agents for the treatment of diseases associated with inflammasome disjunction.
Results and Discussion
ASC PYD Self-associates through Two Opposing Binding Interfaces with Similar Affinities
ASC self-association is an essential step in the formation of macromolecular assemblies like the pyroptosome and the inflammasome (40), which have critical roles in cell death and inflammation. NMR or x-ray crystallographic studies are necessary to characterize these interactions at the atomic level. However, oligomerization poses significant challenges for both techniques. It is well known that ASC PYD solubility is too low at neutral pH for conducting NMR experiments (29, 32). Therefore, previous NMR attempts to characterize ASC PYD interactions used an L25A mutant that is soluble at the typical NMR concentrations (27, 37). Unfortunately, this mutation has been shown to diminish the binding capabilities of the protein (see below). Remarkably, recent advances in cryo-EM imaging and solid-state NMR enabled the reconstitution at quasi-atomic resolution of ASC PYD inside a fibril, providing novel structural details into the homo-oligomerization mechanism of ASC (18, 39). However, these groundbreaking structural reconstitutions only provide detailed characterization on mature fibrils, leaving the initial stages of aggregation still unrevealed.
To improve our limited understanding on PYD death domain interaction, we have monitored ASC PYD self-association by NMR under conditions that help increase protein solubility. The chemical shift of NMR signals in 1H-15N HMQC spectra of native 15N-labeled ASC PYD varies upon the addition of unlabeled ASC PYD at different concentrations (Fig. 1A) until reaching a plateau (Fig. 2A), indicating that the protein is self-associating under these conditions. Once the titration reaches an end, the resulting spectrum overlies that recorded at high 15N-labeled ASC PYD concentration (Fig. 1A). The presence of a single set of signals during titration indicates that ASC PYD self-association is fast on the NMR chemical shift time scale. Residues with the largest chemical shift changes are shown in Fig. 1B and appear highlighted in the structure of ASC PYD (PDB code 2KN6 (29)) in Fig. 1D. Our results indicate that self-association is mediated by two interacting surfaces, one involving helices H1, H4, and H5 N terminus and the other composed of helices H2, H3, and H5 C terminus (Fig. 1D).
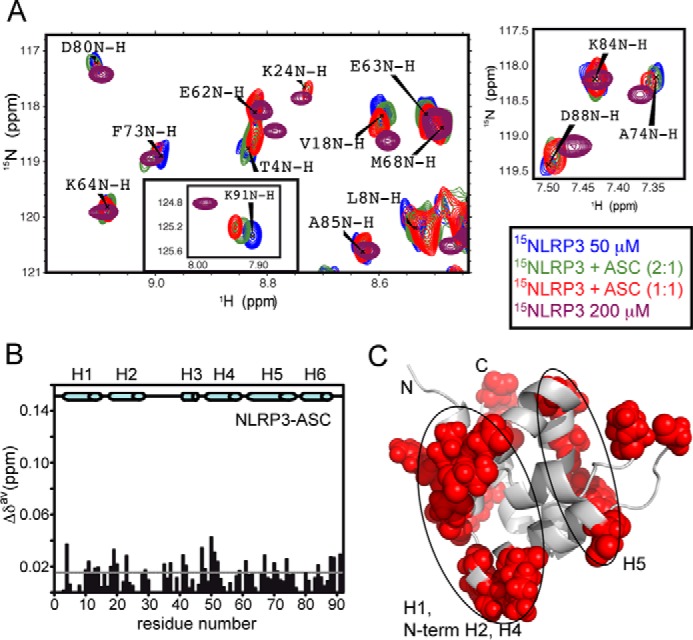
FIGURE 1.

ASC PYD interacts with other PYDs through two equivalent interfaces. A, superimposed SOFAST-HMQC spectra resulting from the titration of 15N-labeled (constant concentration) ASC PYD with unlabeled ASC PYD (increasing concentration, left). Some signals (e.g. Leu-12 NH) remain unmodified, and other show clear changes in δ upon ASC·ASC binding (e.g. Arg-5, Thr-16, or Asp-54 NHs). Similar shifts were observed upon titration with unlabeled NLRP3 PYD (right). For comparison, the inset shows a representative region of the spectra in ASC·NLRP3 titrations where the observed shifts upon binding are equivalent to those found in ASC·ASC. B and C, chemical shift changes upon binding versus residue sequence for ASC·ASC (B) and ASC·NLRP3 (C) interactions. Dashed lines represent the threshold value calculated as 1.5 times the S.D. of the average chemical shift change obtained as explained under “Experimental Procedures.” D and E, mapping on ASC PYD structure of exposed residues with δ changes above the threshold value upon ligand binding for ASC·ASC (D, in red) and ASC·NLRP3 (E, in blue).
FIGURE 2.
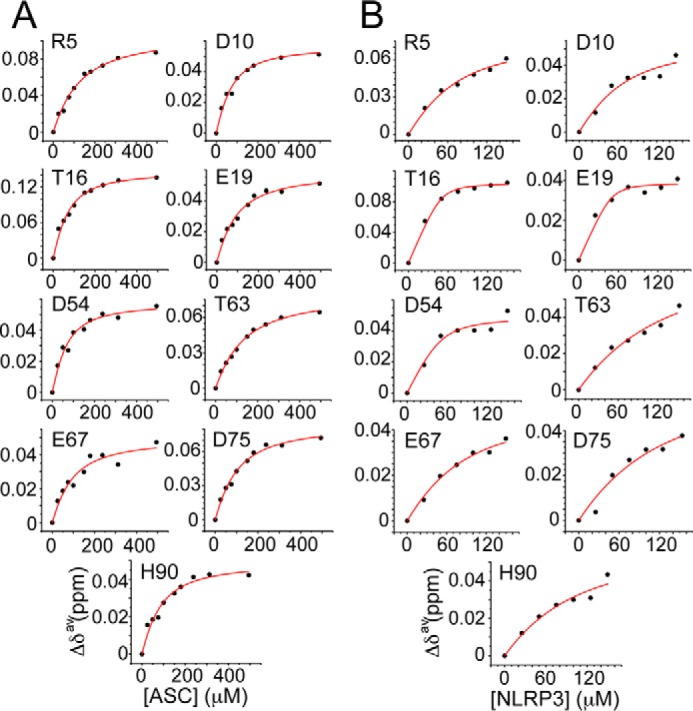
Dissociation constant (KD) values indicate preference of ASC PYD for NLRP3 versus self-association. Changes in chemical shift versus protein concentration for the ASC·ASC (A) and the ASC·NLRP3 (B) interaction. KD values for the ASC·ASC interaction are as follows: Arg-5 (98 ± 14 μm (r = 0.99)); Asp-10 (43 ± 8 μm (r = 0.98)); Thr-16 (41 ± 8 μm (r = 0.98)); Glu-19 (63 ± 12 μm (r = 0.98)); Asp-54 (38 ± 10 μm (r = 0.97)); Thr-63 (106 ± 11 μm (r = 0.99)); Glu-67 (65 ± 25 μm (r = 0.93)); Asp-75 (74 ± 11 μm (r = 0.99)); and His-90 (56 ± 15 μm (r = 0.96)). KD values for the ASC·NLRP3 interaction are as follows: Arg-5 (42 ± 17 μm (r = 0.98)); Asp-10 (37 ± 36 μm (r = 0.92)); Thr-16 (3 ± 1 μm (r = 0.99)); Glu-19 (2 ± 3 μm (r = 0.96)); Asp-54 (8 ± 7 μm (r = 0.95)); Thr-63 (101 ± 58 μm (r = 0.97)); Glu-67 (55 ± 20 μm (r = 0.99)); Asp-75 (95 ± 85 μm (r = 0.94)); and His-90 (71 ± 47 μm (r = 0.96)). Note that the fittings show poor quality for ASC·NLRP3 because of the lower ratio reached (1:3).
In general terms, this finding agrees with previous mutational studies on ASC PYD oligomerization (16, 41). However, several mutations of solvent-exposed residues that abolished ASC PYD self-association do not show, according to our data, the largest variations in chemical shift upon interaction (like Glu-13, Lys-21, and Asp-48, Fig. 1B). This discrepancy could be explained if the absence of the interaction results from an overall destabilization of the protein structure caused by the mutations, rather than from a perturbation of the interaction interface in the complex. Moreover, in one mutational study, the Thr-63A mutant does not seem to significantly affect ASC self-association (16). However, given that Thr-63 is located at the boundaries of the H1, H4, and N-terminal H5 interface (Fig. 1D), it is likely that its contribution to the interaction is minor.
Electrostatic interactions have been traditionally considered as carrying most of the weight with PYD·PYD complex formation (16, 18, 37, 41). As a result, information on the importance of apolar or hydrophobic interactions is scarce. Our NMR titration data show several hydrophobic residues with some of the largest chemical shift perturbations, including Ala-4, Leu-25, Val-30, Leu-44, Leu-45, and Leu-68. Among them, Leu-25, Val-30, and Leu-45 are sufficiently exposed to participate in side chain-side chain interactions. Interestingly, these residues are all located in the interface formed by H2 and H3. Our results thus indicate that hydrophobic interactions play a critical role in one of the two binding surfaces, in agreement with mutational studies pointing to an important function for Leu-25 and Leu-45 in ASC PYD filament formation (41). On the contrary, the V30A mutant can still form filaments, probably because Val-30 is located at the boundaries of H2-H3 interface, whereas Leu-25 and Leu-45 show a central position in this interface (Fig. 1D). Besides, a significant number of the residues showing large chemical shift perturbations have some degree of polarity (Thr-16, Ser-46, Thr-63, Tyr-64, Met-76, and His-90), which indicates that polar interactions are playing an important role in self-association.
However, binding affinity data on ASC PYD self-association is so far unknown, probably due to the previously mentioned aggregation propensity of ASC PYD at neutral pH (29). NMR is an optimum technique to obtain binding affinity constants that fall within the micromolar to millimolar range. We have selected some of the residues with the largest chemical shift variations to obtain apparent values of the ASC PYD·PYD dissociation constant (Fig. 2A). Our results indicate that both surfaces in ASC PYD (H1, H4, N-H5 and H2, H3, C-H5) self-associate with similar affinity showing KD values in the micromolar range (∼40 to ∼100 μm).
However, because the observed chemical shift perturbations are an average of the species resulting from the self-association process, they do not provide the association state of ASC. To gain insight into ASC PYD aggregation state, AUC experiments were performed. The protein appears mainly monomeric in the range of concentrations tested (50–750 μm, Fig. 3A); however, AUC data indicate a tendency of the protein to oligomerize upon increasing concentrations, in agreement with the NMR observations. Nevertheless, the increase in the association state is very small precluding the determination of the oligomeric state formed.
FIGURE 3.
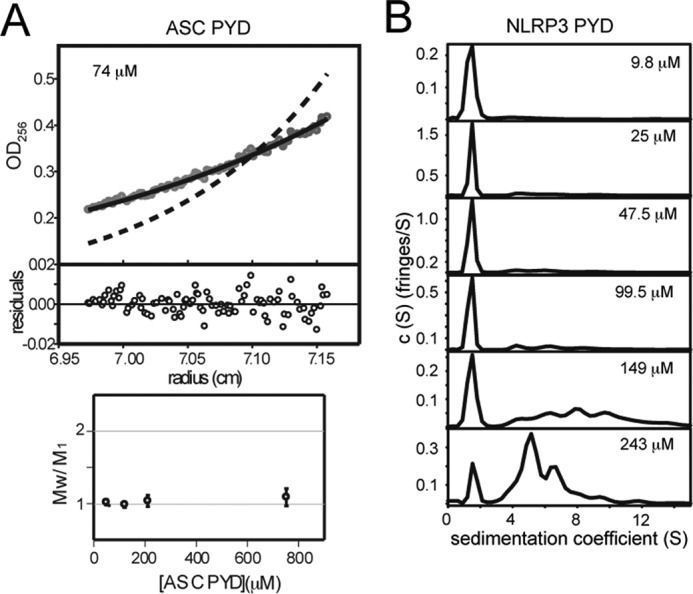
AUC proves oligomerization of PYDs. A, sedimentation equilibrium analysis of ASC PYD at 74 μm in 20 mm glycine, 1 mm TCEP (pH 3.7). Experimental data fit well to a model of the protein being monomeric (solid line) but not to a dimer model (dashed line). The residuals of the fitting of the experimental data to the monomeric model are shown. Plot at the bottom shows the variation of the association state (Mw/M1) with protein concentration (50–745 μm). B, sedimentation velocity plots for NLRP3 at different protein concentrations in 1 mm TCEP (pH 3.6). The percentage of monomeric species in the samples (peak at ∼1.5 S) decreases as follows (from top to bottom): 84.4, 77.3, 75.1, 69.3, 29.1, and 9.8%. Samples were polydisperse, and the aggregation number of the oligomerized species could not be determined.
Solution Structure and Oligomerization Properties of NLRP3 PYD
The conformational behavior and self-associating capabilities of NLRP3 PYD are not very well understood. Previous work using multiangle light scattering coupled to size exclusion chromatography indicates that NLRP3 PYD, although mainly monomeric, can form dimers in solution at pH 5 and 150 mm NaCl (36). However, the effect of protein concentration was not analyzed. Here, we have used sedimentation velocity experiments to study the effect of salt and protein concentration on NLRP3 PYD aggregation (Figs. 3B and 4). We found that in the absence of NaCl, NLRP3 PYD is mainly monomeric at low concentrations (peak at ∼1.5 S; Fig. 3B). At higher protein concentrations, the population of monomeric species decreases, and several higher order oligomerized species are observed. In the presence of NaCl, the population of monomeric species drastically decreases even at the lower protein concentration (Fig. 4), which indicates that the presence of salt favors self-association and could point to an important role of hydrophobic interactions in the NLRP3 PYD aggregation process.
FIGURE 4.
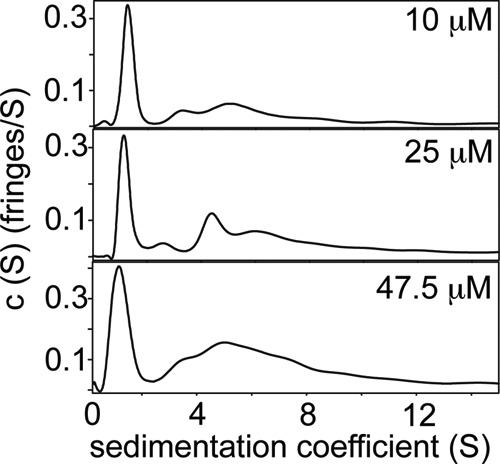
NLRP3 aggregation is significantly enhanced in the presence of NaCl. Sedimentation velocity plots for NLRP3 at different protein concentrations in 1 mm TCEP, 100 mm NaCl (pH 3.6). Monomeric species (peak at ∼1.5 S) decreases from top to bottom as follows: 33.9, 24.3, and 25%.
Results from the sedimentation velocity experiments agree with the solution behavior of NLRP3 PYD observed by NMR. At 0.2 mm protein concentration (pH 3.6, 303 K) in the absence of salt, the NMR 1H-15N HSQC spectra of NLRP3 PYD shows at least two sets of signals (Fig. 5A) as follows: one set clearly corresponds to the monomer according to the NMR signal linewidth, and the other set results from soluble aggregated species. The presence of different sets of signals indicates that monomer-oligomer formation is slow on the NMR chemical shift time scale. It is noteworthy that on the basis of the sedimentation velocity experiments (Fig. 3B), less than 20% of monomeric species are present at 0.2 mm protein concentration, rendering the NMR study with significant challenges. Moreover, the intensity of monomeric NMR signals gradually decreases with time (Fig. 5B), and signals disappear at higher concentration (Fig. 5C). Temperature increase (Fig. 5D) and the presence of NaCl (Fig. 5E) also promote aggregation, in line with sedimentation data (Fig. 4).
FIGURE 5.

Factors affecting NLRP3 aggregation: sample incubation time, protein concentration, temperature, and salt concentration. 1H-15N HSQC spectra of NLRP3 at pH 3.6. A, 200 μm protein concentration and 303 K (conditions used for structural analysis). Sharp monomeric signals are observed together with a central blurb coming from resonances of higher order species. B, same sample used in A after overnight incubation at 303 K. C, protein concentration of 650 μm, 303 K. D, protein concentration of 200 μm, 310 K. E, protein concentration of 200 μm, 303 K, and 100 mm NaCl.
Despite the mentioned challenges, the solution structure of NLRP3 PYD was calculated (Fig. 6). It consists of the typical six-helix bundle motif, with a long loop connecting helices 2 and 3, with the latter being the shortest out of the six helices. The backbone r.m.s.d. of the solution ensemble of conformers, excluding and including loops, is 0.49 and 0.71 Å, respectively. These results indicate the high convergence of the NMR data. MolProbity structural check indicates a 65th percentile of clash score and a 5.13% of poor rotamers. PROCHECK-NMR program (42) shows that the NMR ensemble has 91.9% of the residues in the most favored regions of the Ramachandran plot, 7.8% in additionally allowed regions, 0.3% in generously allowed regions, and 0% in disallowed regions (supplemental Table S1).
FIGURE 6.
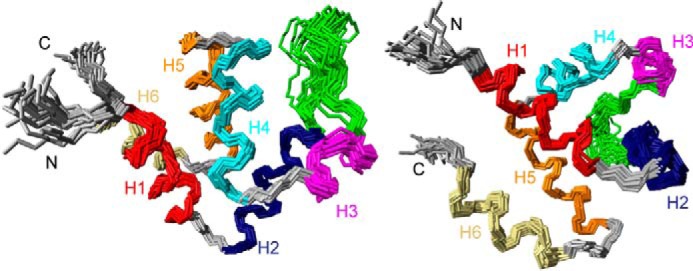
NMR structure of NLRP3 PYD. Two different views of the superposition of the 20 NMR conformers of NLRP3 PYD with the lowest energy, showing a six-helix bundle (PDB code 2NAQ). Helices are color-coded (red, helix 1; blue, helix 2; magenta, helix 3; cyan, helix 4; orange, helix 5; ivory, helix 6). The loop between helices 2 and 3 is colored in green. Superimposed residues (4–28 and 41–88) include helices 1–6. The backbone r.m.s.d. is 0.49 Å. The image was displayed with MOLMOL (65).
Overall, the NLRP3 PYD structure is similar to other PYD domains (Fig. 7A), although by superimposing the different structures, we found that it is more similar to NLRP4 and NLRP10 than to NLRP1, NLRP7, or NLRP12 (6, 32, 35, 37, 38). Strikingly, those NLRPs showing higher structural similarities to NLRP3 (namely NLRP4 and NLRP10) contain an opposing inhibitory role of the inflammatory response, whereas those showing higher structural divergence contain a similar regulatory role (43). This points to the relevance of slight structural divergences determining the specific functions of NRLPs.
FIGURE 7.
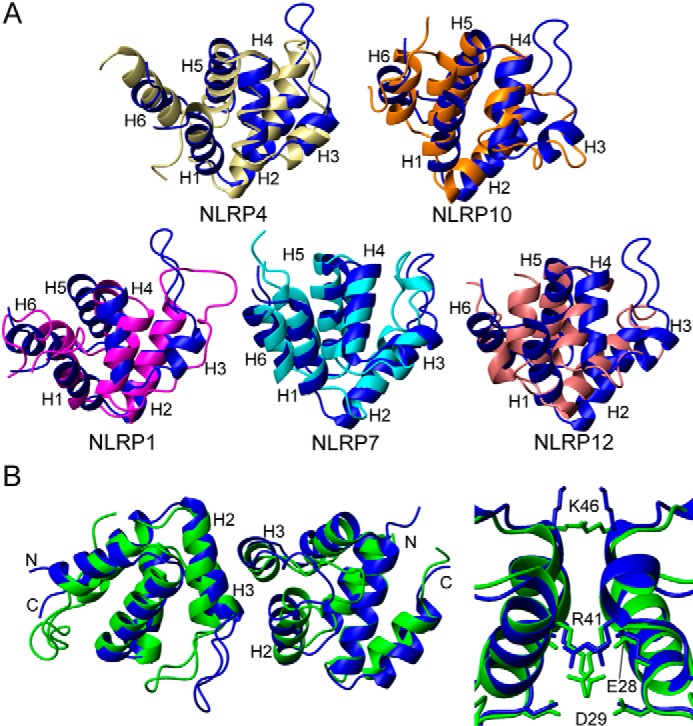
Comparison of NLRP3 PYD with other pyrin domains. A, superposition of the known three-dimensional solution structures of NLRP pyrin domains to NLRP3 PYD represented as ribbon diagrams (PDB codes: NLRP3, 2NAQ; NLRP1, 1PN5; NLRP4, 4EWI; NLRP7, 2KM6; NLRP10, 2DO9; and NLRP12, 2l6A). Blue (NLRP3), magenta (NLRP1), ivory (NLRP4), cyan (NLRP7), orange (NLRP10), and pink (NLRP12) are shown. Helices are numbered. Superimposed residues belong to helices 1, 2, and 4–6. Helix 3 (absent in NLRP1, NLRP10, and NLRP12) and the loop connecting helices 2 and 3 were excluded because of significant conformational variability. The r.m.s.d. values between NLRP3 and the other PYDs are as follows: NLRP4, 3.9 Å; NLRP10, 3.4 Å; NLRP1, 4.9 Å; NLRP7, 5.2 Å; and NLRP12, 5.9 Å. B, superposition of the crystallographic dimeric structure of NLRP3 PYD (green, PDB code, 3QF2; Ref. 36) onto the monomeric NMR structure (blue). The image at the right side shows a detailed view of the dimeric interface, including side chain orientation. The image was displayed with MOLMOL (65).
Moreover, the crystallographic (36) and solution structures of NLRP3 PYD share a high degree of resemblance, with a backbone r.m.s.d. value of 1.66 Å (Fig. 7B). The arrangement of helices 2 and 3 (both involved in the dimer interface of the crystallographic structure) and helix 4 shows the largest differences between both crystal and solution structures. In contrast, side chain orientation of residues in close contact within the dimer interface of the crystallographic structure is very similar both in the NMR monomeric structure and the crystallographic dimer (Fig. 7B). This is a striking result as side chain intermolecular interactions likely contribute to the formation of the dimer, and thus side chain orientation is expected to be different in the monomeric solution structure. Besides, several confronted residues in the dimeric crystallographic interface are charged and therefore should induce repulsive interactions (Fig. 7B). In addition, as mentioned above, NLRP3 PYD is mainly monomeric at the concentrations used for crystallization (36). Therefore, the evidence indicates that the dimeric structure could result from a crystal-packing artifact, as suggested previously (36, 37).
ASC PYD Interacts with NLRP3 PYD through the Same Binding Sites Involved in Its Self-association Albeit with Higher Affinity
After studying the oligomerization capabilities and conformational behavior of NLRP3 in solution, we attempted to analyze a possible interaction between ASC PYD and NLRP3 PYD in solution. For this purpose, 1H-15N HMQC spectra of a 15N-enriched ASC PYD sample were recorded upon the addition of increasing amounts of unlabeled NLRP3 PYD. Chemical shift changes of ASC NMR signals show binding of both PYDs (Fig. 1, A, C, and E), indicating that ASC PYD self-association and its interaction with NLRP3 PYD are mediated by almost identical binding regions. This evidence agrees with a previous study showing that upon mutation of critical residues in the ASC PYD binding interfaces described, both ASC·ASC and ASC·NLRP3 PYD interactions were identically diminished (16). Moreover, the influence of hydrophobic interactions in ASC·NLRP3 PYD binding is again highlighted in residues Leu-25, Val-30, and Leu-45 (Fig. 1C).
In this case, it was also possible to measure the dissociation constants from the NMR titration experiments, which fall within the micromolar range (Fig. 2B). However, the determination of these dissociation constants is somewhat less accurate than for the ASC·ASC PYD binding because NLRP3 increased the tendency for aggregation, which precluded reaching the plateau. Nevertheless, several accurate values were obtained from the change in chemical shifts of Arg-5, Thr-16, Glu-19, Asp-54, and Glu-67 (Fig. 2B). The average value for these residues is 22 μm, which indicates that the affinity of the ASC·NLRP3 PYD complex is slightly higher. A comparable dissociation constant was reported for the ASC·POP1 PYD interaction (∼ 4 μm; 27). However, no analysis on the oligomeric state of ASC PYD was shown in that study.
ASC PYD Self-association and Interaction with NLRP3 Are Enhanced by Increasing the pH and NaCl Concentration of the Milieu
Because we have already shown both by NMR and AUC that the presence of salt enhances NLRP3 PYD oligomerization, we also analyzed its potential effect on ASC PYD self-association and its interaction with NLRP3 PYD. In the presence of 100 mm NaCl, the 1H-15N HMQC spectrum of ASC shows characteristic patterns corresponding to the ASC·ASC interaction, as if the protein had already oligomerized (Fig. 8A). Analogously, the first spectra obtained during the titrations in the present of salt are more similar to the spectra obtained at the end of the titration with no salt, indicating that the complex is formed even at lower protein concentrations under these conditions.
FIGURE 8.
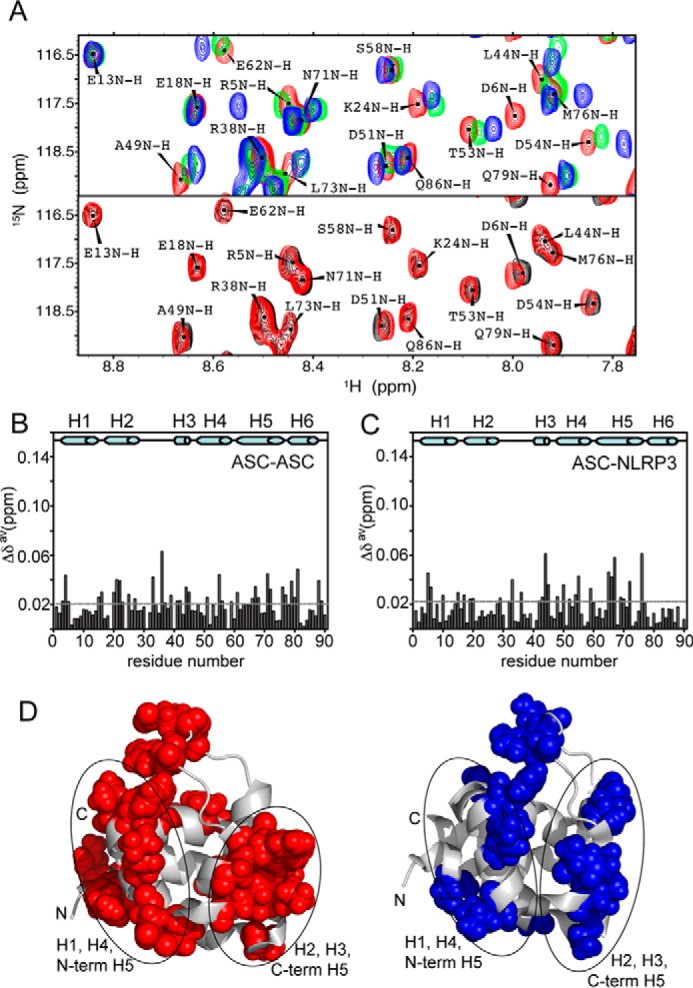
ASC PYD mode of interaction in different conditions. A, top, regions of SOFAST-HMQC spectra of ASC at different concentrations showing typical shifts upon ASC·ASC binding in the absence and presence of NaCl. Top, blue, 50 μm 15N ASC; green, 50 μm 15N ASC + 500 μm ASC; red, 50 μm 15N ASC, 100 mm NaCl. Bottom, red, 50 μm 15N ASC, 100 mm NaCl (same as in top); black, 50 μm 15N ASC + 250 μm ASC, 100 mm NaCl. B and C, chemical shift differences versus residue number of ASC PYD upon binding for the ASC·ASC and ASC·NLRP3 interaction at pH 5.8 (both at a ratio of 1:5 protein/protein concentration). D, residues with the largest chemical shift differences upon binding are mapped in the three-dimensional structure (red and blue for the ASC·ASC and ASC·NLRP3 interaction, respectively).
The effect of pH in the ASC·ASC and ASC·NLRP3 PYD interactions was also studied. NMR 1H-15N HMQC spectra of labeled ASC PYD at pH 5.8 were acquired upon the addition of unlabeled ASC and NLRP3 PYD. Even though significant precipitation occurred, changes in chemical shift values were observed (Fig. 8, B–D). The obtained data indicate that the binding regions at higher pH are very similar to those found at more acidic pH.
ASC PYD L25A Mutation Hinders ASC Interaction through the H2-H3 Face
The L25A mutant has been typically used in ASC PYD interaction studies because of its enhanced solubility (27, 37). In agreement with previously reported data (16), the KD values that we obtained from the NMR titration studies of ASC L25A PYD with itself indicate a reduced tendency to self-associate (Fig. 9). The average KD value for self-association is 165 μm compared with the average value obtained for the wild type of 65 μm (Fig. 2A). However, the binding interfaces remain H1, H4, and N-terminal H5 and H2, H3, and C-terminal H5 (where the L25A substitution is located). Interestingly, the NMR analysis for the L25A mutant shows that the mutation induces structural changes in key residues located in the interface formed by helices H2, H3, and H5 C terminus (particularly in the vicinity of Leu-25 and Leu-45, Fig. 10) and around residue Thr-16, also positioned between the two interfaces. Remarkably, it was proposed that residue Lys-24 is critical for stabilizing H3, and therefore L25A could show a destabilized H3 and hence reduced binding capabilities (17).
FIGURE 9.
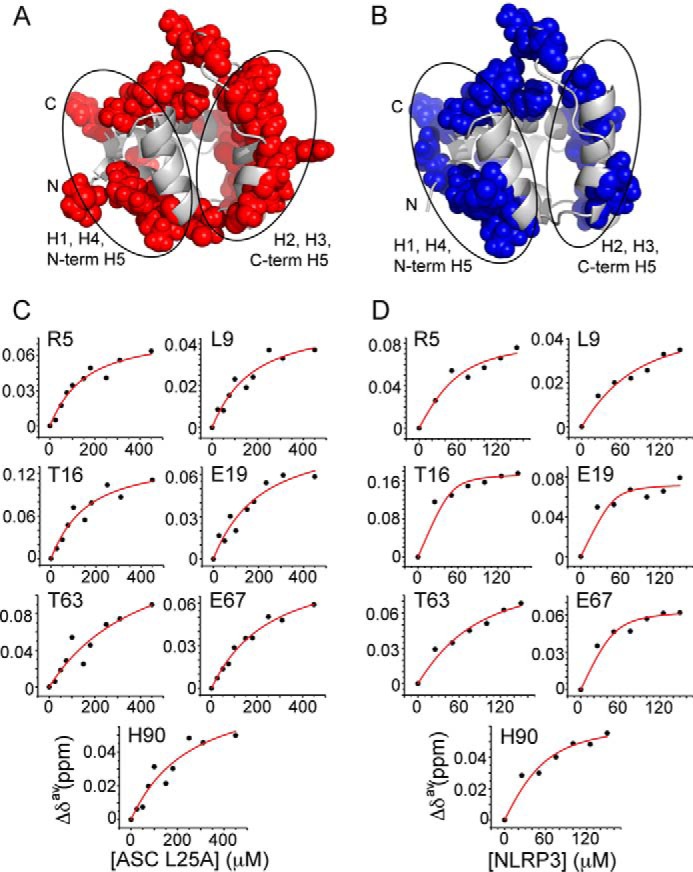
ASC L25A mutant shows reduced binding affinity. A and B, mapping of the exposed residues undergoing above-threshold changes in δ for ASC L25A upon self-binding (A) and NLRP3-binding (B). C, KD values for the ASC L25A·ASC L25A interaction are as follows: Arg-5 (120 ± 42 μm (r = 0.95)); Leu-9 (143 ± 61 μm (r = 0.92)); Thr-16 (126 ± 55 μm (r = 0.92)); Glu-19 (174 ± 76 μm (r = 0.92)); Thr-63 (375 ± 264 μm (r = 0.87)); Glu-67 (216 ± 49 μm (r = 0.98)); His-90 (214 ± 115 μm (r = 0.90)). D, KD values for the ASC L25A-NLRP3 PYD interaction are as follows: Arg-5 (23 ± 20 μm (r = 0.93)); Leu-9 (49 ± 32 μm (r = 0.96)); Thr-16 (3 ± 5 μm (r = 0.91)); Glu-19 (4 ± 7 μm (r = 0.85)); Thr-63 (51 ± 32 μm (r = 0.96)); Glu-67 (7 ± 6 μm (r = 0.94)); His-90 (21 ± 17 μm (r = 0.94)).
FIGURE 10.
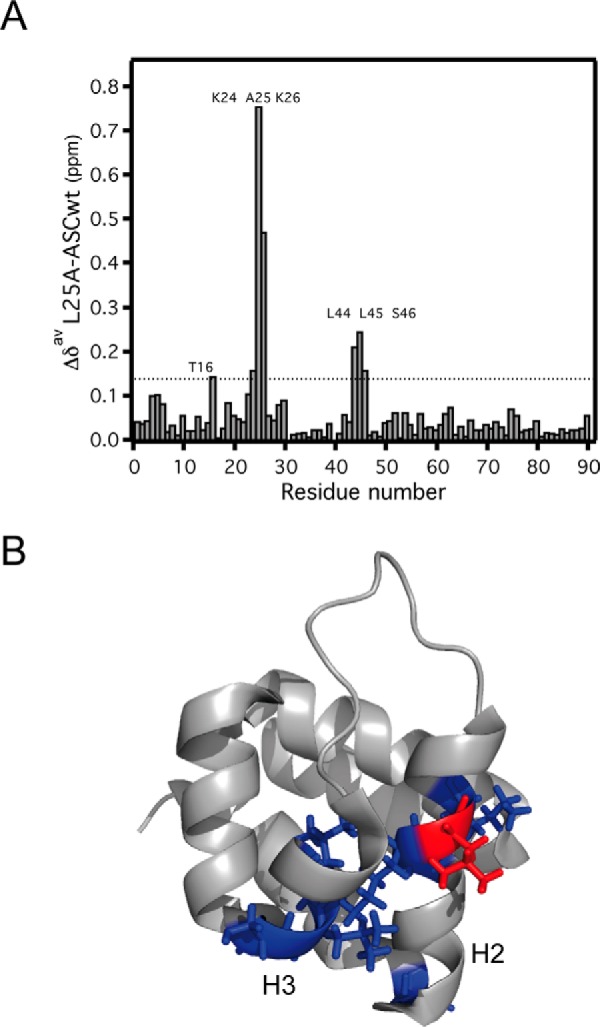
Structural perturbations caused by the L25A mutation. A, chemical shift differences between wild type and mutant (L25A) ASC PYD versus residue number. B, mapping of the largest chemical shift variations onto the ASC PYD structure. Leu-25 is shown in red. The strongest structural effects (shown in “blue”) involve H2-H3.
In addition, these structural effects on the H2-H3 interface provide a possible explanation for previous studies reporting that the L25A mutant interacts with the PYD-only protein POP1 exclusively through the H1-H4 interface of ASC PYD (27). Interestingly, the relevance of this interface for complex formation was discussed recently regarding ASC·NLRP10 PYD interaction (38). Moreover the L25A mutant was also used to test the interaction between ASC and NLRP4 PYDs with negative results (37). More importantly, the L25A mutant was shown unable to form fibrils (41), likely because the interaction through H2-H3 interface is diminished and therefore only dimers interacting through H1-H4 interface can be formed with no possibility of further oligomerization. Besides, the area where Leu-25 is located was shown to be critical for the stability of H3 in the protein AIM2 PYD, which likely has an effect in the protein interaction capabilities (17). All the evidence supports the importance of using WT ASC PYD rather than solubility-enhanced variants for interaction studies.
NLRP3 Binding to ASC
In spite of the high tendency of NLRP3 PYD to aggregate, we were able to perform NMR titrations adding unlabeled ASC and to monitor the regions of the protein affected by the interaction. NLRP3 1H-15N HMQC spectra show severe signal overlap in the amide central region (Fig. 5), precluding the analysis of residues with chemical shift values that fall within this area. Nevertheless, we could monitor the changes in δ upon titration of up to 76% of the total number of signals (Fig. 11A). We observed the following two opposing interfaces on NLRP3 that would be directly involved in association with ASC (Fig. 11, B and C): one interface involves α-helix H1, the N terminus of H2 and H4 (residues Arg-9, Tyr-10, Glu-13, Asp-14, Val-18, Asp-19, Leu-20, Ala-47, Asp-48, Val-50, Asp-51, Lys-84, and Asp-88). and the other is formed by H5 α-helix (residues Thr-4, Gly-35, Ile-37, Phe-59, Gly-61, Glu-63, Trp-66, Ala-67, Val-70, Trp-71, Ala-74, Glu-89, and Lys-91). Interestingly, signal shifts observed upon titration with ASC coincide in direction with those observed when a higher concentration of NLRP3 is used (Fig. 11A), indicating that NLRP3 self-associates using identical interfaces as those used for its interaction with ASC PYD (as it occurs in ASC PYD). In general, these interfaces coincide with those in pyrin involved in binding to ASC PYD (44). As in the case of PYD ASC, the presence of opposing interfaces for the interaction enables the formation of oligomeric ASC·NLRP3 PYD heteromolecular complexes, which is the basis for inflammasome molecular activation (Fig. 12) (1–4, 18).
FIGURE 11.
NLRP3 PYD interacts with ASC PYD through similar interfaces. A, superimposed SOFAST-HMQC spectra acquired in the titration of 15N-labeled NLRP3 with ASC. NLRP3 severe aggregation precluded titration data at molar ratios higher than 1:1. Peak shifts upon titration with ASC reproduce those observed at high NLRP3 concentration, indicating that NLRP3 PYD self-association and its interaction with ASC PYD employ identical interfaces. B and C, mapping of the NLRP3 residues undergoing above-threshold δ upon ASC binding shows similar interfaces (H1, N-terminal H2, H4, and H5) to those found in ASC (Fig. 1) and in pyrin (44). Note that the magnitude of δ in this case is significantly lower than for ASC PYD (Fig. 1, B and C; the scale on the y axis is maintained for comparison), and those residues whose resonances appeared in the central blurb were discarded from this mapping (Cys-6, Lys-7, Leu-8, Ala-9, Lys-21, Leu-27, Lys-34, Leu-55, Ile-57, Ala-65, Arg-78, Arg-79, and Tyr-82).
FIGURE 12.

Summary of the binding interfaces of the PYDs studied. A, overlay of the solution structures of ASC PYD (dark gray, PDB code 2KN6) and NLRP3 PYD (light gray, PDB code 2NAQ) showing a good alignment (backbone r.m.s.d. 3.6 Å). B, mapping of the binding interfaces observed in ASC PYD. This orientation clearly shows that both interfaces are found in opposing sides of the structure. C, in NLRP3 PYD, those interfaces do not seem to follow the same orientation as in ASC.
Supramolecular Complex Models of ASC·ASC and ASC·NLRP3 Oligomers
Using the NMR data on the specific residues critical for interaction, we generated several models of the dimeric complexes formed by docking the interfaces observed. Specifically, for ASC·ASC PYD pair we only obtained continuous oligomeric structures using as unity the complex formed by ASC PYD H1-H4 with ASC PYD H2-H3 (Fig. 13). In turn, this second PYD domain would still contain an accessible H1-H4 interface, which we used as a template for the structural alignment of a second ASC·ASC pair. Given the distribution of charged residues in the domain, we can clearly observe differently charged interfaces that could promote polymerization (Fig. 14A).
FIGURE 13.

ASC PYD oligomeric fibril formation based on the monomer NMR data. Using the ASC·ASC PYD dimeric complex formed by the type I interaction (H1–H4 helices of one domain binding to H2–H3 of the other domain) as unit template, we generate a double-helical fibril that could polymerize based on charge complementarity (Fig. 14). The ribbon diagram represents the best dimer obtained by Haddock (one ASC PYD unit is colored in green and the other in light blue). The selected residues that drive the docking calculation are represented in sticks. The colored circles represent the type I interaction taking place for fibril formation. Each replicate is axially rotated ∼38°. The structures were aligned and represented with PyMOL (73).
FIGURE 14.
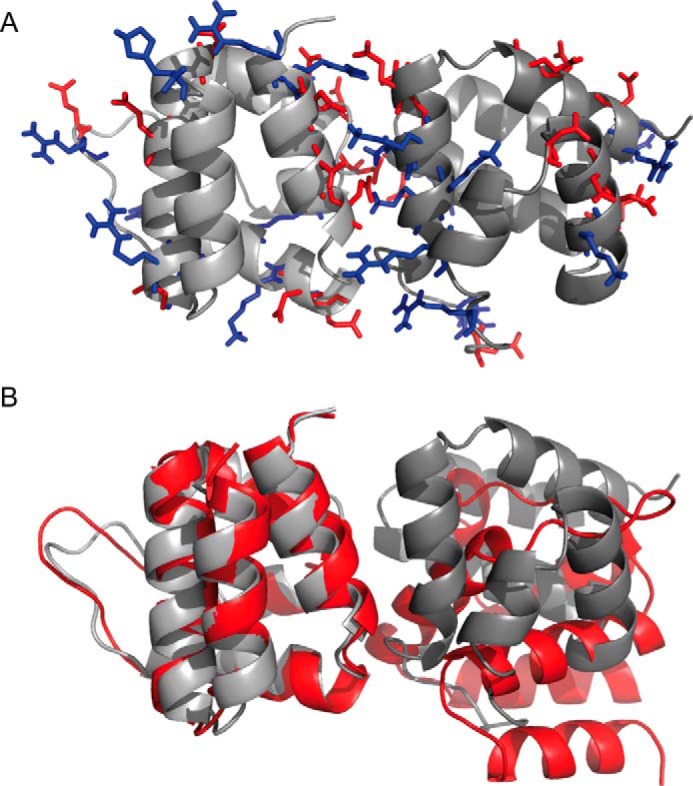
Electrostatic complementarity of ASC PYD dimer and comparison with a previously proposed dimer. A, structural model of the ASC PYD dimer template used to create the fibrils shown in Fig. 10. Side chains of residues involved in the interaction are shown in color to differentiate the positive (blue) and negative (red) net charge. Both the interaction interface and the interfaces remaining at the other end of the domains show complementary charges, allowing for the polymerization of the fibril. B, comparison of our ASC PYD dimer (gray) with one proposed previously (red) (16). The first protomer of each dimer are aligned for clarity showing the different orientation of the remaining protomer in both dimer models.
In this way, the structure obtained is an intertwined helical fibril, which somehow resembles the mature ASC PYD fibril described recently by cryo-electron microscopy (18). However, these mature fibrils contain a helicoidal arrangement of PYD domains with six repeats per turn, with much larger dimensions than the resulting fibril obtained based on our NMR data. Interestingly, the authors also show that the fibrils grow significantly fast from protofibrils with small dimensions to larger assemblies. The mature fibrils are stabilized by three types of interactions between the PYD subunits with the type I as dominant and the only one found within each strand, whereas type II and III are highly variable and are found between the strands stabilizing the mature fibril. In our experimental conditions, we only observed the type I of interaction (Figs. 1, B and D) (13). Besides, the structure of the ASC PYD domain inside the mature fibrils is perturbed (18), with significant conformational changes on H3, which the authors attribute to conformational rearrangements. Along these lines, the type I interaction characterized here is slightly different from the one described in the mature fibril. We observed the following three critical electrostatic clusters in our dimer: Glu-18 and Glu-19 from H2 contact with Arg-5 from H1; Lys-21 from H2 interacts with Asp-6 and Asp-10 from H1; and Lys-22 and Lys-26 from H2 interact with Asp-47 from H4 (Fig. 14A). In contrast, the electrostatic clusters found in the type I interaction observed in the mature fibril were as follows: Lys-21 from H2 interacts with Asp-48 and Asp-51 from H3-H4; Lys-22 and Arg-41 from H2-H3 interact with Asp-54 from H4; and Lys-26 from H2 contacts with Arg-3 and Arg-5 from H1 (18). In general lines, these differences result from the rotation of one protomer relative to the other. Remarkably, in the protofibril proposed here, the residues responsible for the type II and III interactions found in the mature fibril remain accessible, and therefore, it would still be possible to pack protofibrils laterally to form the mature fibril.
Nonetheless, whether the so-called protofibril we describe here represents relevant initial steps of fibrillization is still to be investigated. However, it is worth mentioning that among the 12 residues mutated in the study with effects in fibril formation (18), seven participate in the dimer interface shown here (Glu-13, Lys-21, Lys-22, Lys-26, Arg-41, Leu-50, and Asp-51). Another ASC·ASC PYD homodimer model has been proposed (16). In this case, the arrangement of the protomers is significantly different from what our NMR data indicate (Fig. 14B). The differences might arise from the low number of restraints used in the mentioned study to drive the docking calculations.
The interaction between ASC and NLRP3 PYDs has remained elusive in in vitro studies despite much evidence of its occurrence in vivo (27). However, NMR data clearly indicate the presence of such interaction under our experimental conditions. Based on the titration results, we modeled by docking four different types of binding between ASC and NLRP3 PYDs that agree with the interaction interfaces characterized on monomeric PYDs (Figs. 1 and 11) as follows: ASC H1-H4/NLRP3 H1-H4; ASC H1-H4/NLRP3 H5; ASC H2-H3/NLRP3 H1-H4; and ASC H2-H3/NLRP3 H5 (Fig. 15). By structural alignment of consecutive pair units we built the oligomeric structure shown in Fig. 15.
FIGURE 15.

Proposed ASC·NLRP3 PYD oligomeric complex. A, four different types of interaction between ASC and NLRP3 PYDs are possible based on our NMR data as follows: ASC H1-H4/NLRP3 H5; ASC H1-H4/NLRP3 H1-H4; ASC H2-H3/NLRP3 H5; and ASC H2-H3/NLRP3 H1-H4. ASC PYD and NLRP3 PYD are shown as ribbon diagrams and colored in green and purple, respectively. The residues highlighted as stick representation in the structures were used to drive the specific dockings. In each pair, a green circle represents ASC PYD, and a purple square represents NLRP3 PYD, and the numbers indicate the corresponding type of interaction. B, hexameric ring modeled using as template the resulting docking pairs of the different dimeric units shown in A. ASC PYD, ASC CARD, and ASC linker and NLRP3 PYD are colored in green, blue, gray, and purple, respectively. The C termini of NLRP3 PYDs placed in the ring point to the bottom of the image shown on the right. The structures were structurally aligned and represented with PyMOL (73).
Strikingly, by using only restrictions based on our NMR data, we could build a hexameric ring that reproduces the classical platform architecture formed by other death domain superfamily members (45). The rotational symmetry in this model is of the 6th order, whereas different apoptosomes from C. elegans, human, and Drosophila melanogaster display 4-, 7-, and 8-fold symmetry, respectively (45). Double-ring structures were also described for NLRP1 with 5- and 7-fold symmetry (46), and the NLRC4 inflammasome was shown as a ring structure with 11-fold symmetry (47). NLRP3 self-oligomerization via its NACHT domain is also proposed to form hexameric/heptameric rings (2). Importantly, the PYD-only hexameric ring described here shows the same symmetry as the PYD-only fibrils described recently (18), with similar dimensions of the inner hub (24.6 Å versus 20 Å in the fibril). These dimensions are also similar to those from other death domain oligomeric protein rings described elsewhere (reviewed in Ref. 48 and references therein). Furthermore, several residues from NLRP3 that were previously found to be involved in its interaction with ASC PYD (18) were also found to be critical for the interactions stabilizing the ASC·NLRP3 ring (namely Glu-15, Lys-23, Glu-64, and Asp-82).
Similarly to what was previously found in PYD fibrils (18, 39), the topology of the ring (Fig. 15B) makes the CARD domains of ASC molecules accessible for further interaction given the flexibility of the linker between ASC PYD and CARD domains (29). This flexibility could in turn serve as a mechanism for oligomerization into ring structures (45, 48). Interestingly, several mutations associated with hereditary periodic auto-inflammatory syndromes were found in the PYD of pyrin protein (49, 50). The residues involved in some of these disease-related mutations (corresponding to Tyr-11, Val-18, Lys-24, Arg-41, and Asp-90 of NLRP3) are located in the ASC·NLRP3 interaction interfaces described here. Thus, it is reasonable to speculate that those mutations could hamper the formation of the ring shown here.
In our study we also show that the PYD·PYD dimeric interaction is low affinity. This feature, typical within the death domain superfamily (3, 45), might allow the formation of flexible oligomeric structures that would recruit more components to form robust multimeric ensembles. Although ASC·NLRP3 PYD interaction is of higher affinity, we cannot discard simultaneous self-association and heteromeric interaction. Thus, different combinations would result in different supramolecular complexes formed. Indeed, an auto-inhibition mechanism is proposed to occur in NLRP3 through oligomerization by its leucine-rich repeat domain (18), and thus the oligomerization properties might be different from the ones described here. Therefore, it is of paramount importance to describe in detail the supramolecular structure of the oligomeric rings triggering NLRP3 inflammasome activation.
All in all, the study reported herein expands our understanding of the role of PYD domains in NLRP3 inflammasome formation. Our results likely represent the initial stages of the oligomerization process in the activation cascade and provide insight into the molecular basis underlying this labyrinthine mechanism. Such insight is in fact of critical importance to target specific oligomeric structures or oligomerization steps in NLRP3 inflammasome formation in the therapeutic treatment of a wide range of diseases (23, 25, 51). Along this line, our results anticipate that NMR spectroscopy can be used as a powerful, robust technique for the detailed analysis of antagonists of inflammasome complex formation.
Experimental Procedures
Protein Cloning, Expression, and Purification
Human ASC PYD (residues 1–90; UniProtKB/Swiss-Prot accession number Q9ULZ3), ASC PYD L25A mutant, and NLRP3 PYD cDNAs (residues 3–93; UniProtKB/Swiss-Prot accession number Q96P20) were cloned into pET15b expression vectors (Novagen). ASC PYDs contained an N-terminal six-histidine tag and a thrombin cleavage site, whereas NLRP3 did not contain tags to avoid any possible perturbation in its solution structure.
Proteins were expressed in C41(DE3) Escherichia coli strain (52). For unlabeled protein production, cells were grown in LB media, whereas isotopic labeling was achieved in M9 minimal media supplemented with 15NH4Cl or [13C]glucose as the sole nitrogen and carbon sources, respectively. Overexpression of the recombinant proteins was induced during 4 h at 37 °C by the addition of 1 mm isopropyl β-d-1-thiogalactopyranoside to the respective cultures after an A600 ∼0.8 was reached. Bacterial pellets were resuspended in 50 mm Tris-HCl (pH 8.0), and cells were lysed by sonication. The soluble fraction was removed by centrifugation at 48,000 × g for 30 min. Inclusion bodies were then washed twice in the aforementioned buffer. The resulting pellet was then dissolved in the presence of 6 m GdnHCl and centrifuged at 48,000 × g for 1 h.
ASC PYD was purified by Ni2+-affinity chromatography. Specifically, pellets were dissolved in 20 mm Tris-HCl, 500 mm NaCl, 5 mm imidazole, 6 m GdnHCl (pH 7.9) (binding buffer). His Bind resin (Novagen) was charged according to the manufacturer's instructions. Protein was attached to the resin using binding buffer and washed in the presence of 20 mm imidazole. Elution was achieved by using binding buffer in the presence of 1 m imidazole. Immediately after elution, the protein was subjected to several dialysis steps against H2O (pH 4.0) to slow protein self-association processes. The sample was subsequently purified by reverse phase chromatography using a pre-packed C8 column (Higgins Analytical). Binding buffer consisted of 5% acetonitrile, 0.1% trifluoroacetic acid (TFA), and elution buffer was 95% acetonitrile, 0.1% TFA. As described previously and contrary to WT ASC PYD (35, 37), ASC PYD L25A mutant remains in the soluble fraction and thus could be purified in non-denaturing conditions. A single Ni2+-affinity chromatography purification step was performed for this protein.
NLRP3 PYD was purified using a double-reverse phase chromatography, the first one performed in denaturing conditions. C4 resin (Applied Separations) was equilibrated in 20 mm Tris-HCl, 500 mm NaCl, 6 m GdnHCl, 0.1% TFA. Inclusion bodies extract (resuspended in the same buffer used for C4 resin equilibration) was slowly bound to the resin in several cycles. Once the protein was bound, the column was re-equilibrated in 5% acetonitrile, 0.1% TFA using a very slight gradient, and protein was eluted in 60% isopropyl alcohol, 40% acetonitrile, 0.1% TFA. Different fractions of the chromatogram (with different purity) were pooled, lyophilized, and resuspended in 5% acetonitrile, 0.1% TFA prior to their subsequent purification in regular reverse phase chromatography using a C8 pre-packed column as described above.
After purification, all three proteins were >95% pure, as monitored by SDS-PAGE, and the correct molecular weight was calculated from MALDI-TOF mass spectrometry data. Proteins were lyophilized and stored for subsequent studies.
NMR Spectroscopy for Binding Studies
Unless otherwise stated, NMR experiments were acquired on a Bruker Avance 600 MHz spectrometer equipped with a triple-resonance cryogenic probe and on a Bruker Avance III 600 MHz spectrometer with a triple-resonance, three-axis gradient probe in 20 mm glycine, 5 mm d15-TCEP, 0.1 mm NaN3, 10% D2O (pH 3.7) buffer at 303 K. Fast two-dimensional 1H-15N correlation spectra (SOFAST-HMQC, 53) were acquired to monitor chemical shift (δ) changes upon protein binding because of the propensity of ASC PYD and NLRP3 PYD to self-associate even at low pH. Self-association is particularly severe for NLRP3 PYD according to NMR and AUC data (see above). A concentration of 50 μm of the 15N-labeled protein was used in the titration experiments to minimize δ changes resulting from intrinsic self-association. The unlabeled ligand protein in lyophilized form was added directly to the protein solution followed by pH adjustment. Protein concentration in the mixtures was randomly checked by UV absorbance resulting in errors smaller than 10%. Higher molar ratio in the ASC·ASC titration relative to the ASC·NLRP3 titration was achieved as severe NLRP3 protein aggregation beyond 150 μm concentration occurs (see above). Amide 1H-15N chemical shift values for ASC PYD were obtained from previous assignment of full-length ASC (54) and confirmed by classical three-dimensional 15N-1H NOESY-HSQC (110-ms mixing time) and 15N-1H TOCSY-HSQC (60-ms mixing time) experiments. ASC PYD L25A mutant showed significant perturbations in δ relative to WT ASC PYD, and thus a three-dimensional 15N-1H TOCSY-HSQC (60-ms mixing time) experiment was acquired to get full backbone assignment. NLRP3 PYD full assignment is described below. NMR spectra were processed with NMRPipe (55) and analyzed with SPARKY (56).
Calculation of Dissociation Constants
The observed δ values in the NMR titration experiments are weighted average values of the free and bound forms of the protein because the exchange rate between both forms is fast on the NMR chemical shift time scale. The difference in δ (Δdav) was obtained as follows: ((δ1Hbound − δ1Hfree)2 + (δ15Nbound − δ15Nfree/5)2)1/2. Those residues showing Δdav values higher than 1.5 S.D. upon ligand binding and with a total solvent exposure index larger than 30% as calculated by GETAREA (57) were defined as critical for the corresponding interaction. To obtain dissociation constants (KD), the Δdav versus ligand protein concentration plots were fit to the equation as follows: Δdav = Δdmax(KD + L + P0) − ((KD + L + P0) − 4P0L)1/2/2P0, where P0 is protein concentration (50 μm), and L is total ligand concentration (58). Data fitting was done with OriginPro 8 (OriginLab).
NMR Spectroscopy for NLRP3 Structural Studies
NMR samples were prepared at 0.1–0.2 mm NLRP3 PYD in 5 mm d15-TCEP, 0.1 mm NaN3, 5% D2O (pH 3.6). NMR experiments were acquired at 303 K in a Bruker Avance 600 MHz spectrometer equipped with a triple-resonance cryogenic probe. Sequence backbone assignments were obtained from the following experiments: 1H-15N HSQC, three-dimensional HNCACB, and three-dimensional CBCA(CO)NH. Side chain assignments were obtained from three-dimensional HBHA(CO)NH, three-dimensional H(CCCO)NH TOCSY (22.5-ms mixing time), and three-dimensional HCCH TOCSY (15-ms mixing time). NOE data were obtained from three-dimensional 15N-1H-1H NOESY (110-ms mixing time) and three-dimensional 13C-1H-1H NOESY (120-ms mixing time). Information on NMR experiments for protein structure determination can be found elsewhere (59, 60). All experiments were processed with NMRPipe (55) and analyzed with PIPP (61).
NLRP3 Structure Calculation
Peak intensities from NOESY experiments were translated into a continuous distribution of interproton distances. Distances involving methyl groups, aromatic ring protons, and non-stereospecifically assigned methylene protons were represented as a summation averaging, (∑r−6)−1/6 (62). Errors of 25 and 40% of the distances were applied to obtain lower and upper distance limits. The lower limit of some distance restraints that appeared smaller in the resulting structures for protons belonging to sequential amino acids was increased to 50%. A total of 32 hydrogen bond distance restraints (rNH-O = 1.9–2.5 Å and rN-O = 2.8–3.4 Å) was defined according to the experimentally determined secondary structure of the protein. The TALOS+ program was used to obtain 81 φ and ψ restraints for those residues with statistically significant predictions, and the associated predicted errors were used. Structures were calculated with the program X-PLOR-NIH 2.16.0 (63). The starting structure was heated to 3000 K and cooled in 10,000 steps of 0.002 ps during simulated annealing. The final ensemble of 20 NMR structures was selected based on lowest energy and no restraint-violation criteria. The 20 lowest energy conformers have no distance restraint violations and no dihedral angle violations greater than 0.5 Å and 5°, respectively. Structure quality was assessed with PROCHECK-NMR (42) and MolProbity (64). Structures were analyzed with MOLMOL (65). Atomic coordinates were deposited in the Protein Data Bank with accession code 2NAQ.
Analytical Ultracentrifugation
AUC analysis of ASC PYD and NLRP3 PYD was performed in an XL-A analytical ultracentrifuge (Beckman-Coulter Inc.) with UV-visible optics detection system, using an An50Ti rotor and 3- or 12-mm double-sector centerpieces.
Sedimentation equilibrium experiments were performed on ASC PYD samples (50–745 μm) resuspended in 20 mm glycine, 1 mm TCEP (pH 3.7) at several speeds (16,000 and 20,000 rpm) and the adequate wavelength (256–292 nm), using short columns (23 or 90 μl, depending on the pathlength, 3 or 12 mm, respectively). After the equilibrium scans, a high speed centrifugation run (48,000 rpm) was performed to estimate the corresponding baseline offsets. Weight-average buoyant molecular mass of ASC PYD samples was determined by fitting data to the single species model using the Heteroanalysis program (66). The molecular masses of the proteins were determined from the experimental buoyant values using 0.74 ml/g as the partial specific volume for ASC PYD (calculated from the amino acid composition using the SEDNTERP program (67)).
Non-ideality correction due to very high concentrations of protein was estimated according to Ref. 68. The actual weight-average molecular weight is related to the apparent weight-average molecular weight obtained experimentally by the simple Equation 1 (69),
where Mw, a is the experimentally measured apparent weight-average molecular weight, and ν is the experimentally measured partial specific volume of the protein (we used the calculated ν from the amino acid composition with SEDNTERP (67)).
Sedimentation velocity experiments were carried out at 48,000 rpm and 20 or 30 °C. Samples of ASC PYD ranged from 50 to 745 μm. Samples of NLRP3 PYD (9.8–243 μm) were resuspended in H2O, 1 mm TCEP (pH 3.6). Sedimentation profiles were registered every 5–8 min at the adequate wavelength. The sedimentation coefficient distributions were calculated by least squares boundary modeling of sedimentation velocity data using the c(s) method (70), as implemented in the SEDFIT program. These s values were corrected to standard conditions (water, 20 °C, and infinite dilution (71)) using the SEDNTERP program (67) to obtain the corresponding standard s values (s20, w).
Molecular Modeling
Molecular docking was performed with the program HADDOCK (72) using both the ASC PYD domain of full-length ASC solution structure (PDB code 2KN6 (29)) and the NLRP3 PYD solution structure described in this work (PDB code 2NAQ) as templates. Those residues showing a Δdav value of >1.5 S.D. upon ligand binding and with a total solvent exposure index of >30% as calculated by GETAREA (57) were selected as “active” residues to create a list of ambiguous interaction restraints used in the docking calculation. Specifically, the interfaces used for the docking calculations were those formed by α-helices H1, H4, and the N-terminal end of H5 (residues Gly-2, Arg-3, Asp-6, Asp-10, Thr-16, Leu-50, Asp-54, Glu-62, Thr-63, Glu-67, and His-90) and by helices H2, H3, and the C-terminal end of H5 (residues Glu-19, Lys-22, Lys-24, Leu-25, Ser-29, Val-30, Leu-45, and Asp-75) from ASC PYD, whereas the interfaces formed by α-helices H1 and H4 (residues Arg-10, Glu-13, Asp-14, Glu-16, Val-18, Lys-21, Asp-48, Val-50, and Asp-51) and by residues from H5 (Gly-61, Glu-63, Ala-67, and Trp-71) were selected from NLRP3 PYD. As a result, three different combinations of docking models were used for the ASC·ASC complex (ASC [H1,H4]-ASC [H2,H3]; ASC [H1,H4]-ASC [H1,H4]; and ASC [H2,H3]-ASC [H2,H3]) and four for the ASC·NLRP3 complex (ASC [H1,H4]-NLRP3 [H1,H4]; ASC [H2,H3]-NLRP3 [H1,H4]; ASC [H1,H4]-NLRP3 [H5]; ASC [H2,H3]-NLRP3[H5]). HADDOCK default parameters were accepted for the docking protocol. No other restraints were incorporated.
The top seven HADDOCK clusters of structures out of 1,000 calculated were inspected to build the models. For the ASC·ASC interaction, only the supramolecular complexes resulting from the ASC [Η1,H4]-ASC [H2,H3] pair are shown because they are the only ones conforming to a continuous oligomeric structure inducing no steric clashes, whereas for ASC·NLRP3, we show all the possible combinations of pairs following our docking models. The complexes were built by structural alignment on PyMOL using as templates consecutive units of the corresponding pair.
Author Contributions
J. O. produced and purified all proteins, conducted the great majority of NMR experiments, performed data analysis, and built all supramolecular models. J. O. and E. dA. conducted and analyzed NMR experiments, discussed results, and wrote the manuscript. E. dA. conceived the project, analyzed NMR data on NLRP3, and determined the NLRP3 three-dimensional structure. S. B.-V. produced and purified NLRP3 and analyzed preliminary NMR data on NLRP3. C. A. and G. R. conducted and analyzed the AUC experiments.
Supplementary Material
Acknowledgments
We thank Ángel Pey and José Manuel Sánchez Ruíz for helpful discussions.
This work was supported in part by Spanish Ministry of Science and Innovation Plan Nacional I+D+i Program, Grants BFU2008-03278 and BFU2011-23797, and Programa Ingenio-2010-CONSOLIDER Grant CSD2009-00088 (to E. dA.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (code 2NAQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NLRP3 chemical shifts were deposited in the BMRB under the accession number 25943.
- CARD
- caspase recruitment domain
- PYD
- pyrin domain
- ASC
- apoptosis-associated speck-like protein containing a CARD
- AUC
- analytical ultracentrifugation
- HMQC
- heteronuclear multiple-Quantum correlation
- NOESY
- nuclear Overhauser effect spectroscopy
- TOCSY
- total correlation spectroscopy
- r.m.s.d.
- root-mean-square deviation
- PDB
- Protein Data Bank
- GdnHCl
- guanidine HCl
- TCEP
- tris(2-carboxyethyl)phosphine
- NLRP3
- NACHT, LRR, and PYD domains-containing protein 3.
References
- 1. Park H. H., Lo Y.-C., Lin S.-C., Wang L., Yang J. K., and Wu H. (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25, 561–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schroder K., and Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 3. Kersse K., Verspurten J., Vanden Berghe T., and Vandenabeele P. (2011) The death-fold superfamily of homotypic interaction motifs. Trends Biochem. Sci. 36, 541–552 [DOI] [PubMed] [Google Scholar]
- 4. Park H. H. (2012) PYRIN domains and their interactions in the apoptosis and inflammation signaling pathway. Apoptosis 17, 1247–1257 [DOI] [PubMed] [Google Scholar]
- 5. Chu L. H., Gangopadhyay A., Dorfleutner A., and Stehlik C. (2015) An updated view on the structure and function of PYRIN domains. Apoptosis 20, 157–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pinheiro A. S., Eibl C., Ekman-Vural Z., Schwarzenbacher R., and Peti W. (2011) The NLRP12 pyrin domain: structure, dynamics, and functional insights. J. Mol. Biol. 413, 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park H. H., Logette E., Raunser S., Cuenin S., Walz T., Tschopp J., and Wu H. (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin S. C., Lo Y. C., and Wu H. (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scott F. L., Stec B., Pop C., Dobaczewska M. K., Lee J. J., Monosov E., Robinson H., Salvesen G. S., Schwarzenbacher R., and Riedl S. J. (2009) The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457, 1019–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L., Yang J. K., Kabaleeswaran V., Rice A. J., Cruz A. C., Park A. Y., Yin Q., Damko E., Jang S. B., Raunser S., Robinson C. V., Siegel R. M., Walz T., and Wu H. (2010) The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat. Struct. Mol. Biol. 17, 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao T., Towb P., Wasserman S. A., and Sprang S. R. (1999) Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell 99, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qi S., Pang Y., Hu Q., Liu Q., Li H., Zhou Y., He T., Liang Q., Liu Y., Yuan X., Luo G., Li H., Wang J., Yan N., and Shi Y. (2010) Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell 141, 446–457 [DOI] [PubMed] [Google Scholar]
- 13. Qin H., Srinivasula S. M., Wu G., Fernandes-Alnemri T., Alnemri E. S., and Shi Y. (1999) Structural basis of the procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399, 549–557 [DOI] [PubMed] [Google Scholar]
- 14. Yan N., Chai J., Lee E. S., Gu L., Liu Q., He J., Wu J. W., Kokel D., Li H., Hao Q., Xue D., and Shi Y. (2005) Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature 437, 831–837 [DOI] [PubMed] [Google Scholar]
- 15. Yang J. K., Wang L., Zheng L., Wan F., Ahmed M., Lenardo M. J., and Wu H. (2005) Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol. Cell 20, 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vajjhala P. R., Mirams R. E., and Hill J. M. (2012) Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 287, 41732–41743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin T., Perry A., Smith P., Jiang J., and Xiao T. S. (2013) Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 auto-inhibition and inflammasome assembly. J. Biol. Chem. 288, 13225–13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu A., Magupalli V. G., Ruan J., Yin Q., Atianand M. K., Vos M. R., Schröder G. F., Fitzgerald K. A., Wu H., and Egelman E. H. (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwon D., Yoon J. H., Shin S.-Y., Jang T.-H., Kim H.-G., So I., Jeon J.-H., and Park H. H. (2012) A comprehensive manually curated protein-protein interaction database for the death domain superfamily. Nucleic Acids Res. 40, D331–D336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis B. K., Wen H., and Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elliott E. I., and Sutterwala F. S. (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265, 35–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weber C. H., and Vincenz C. (2001) The death domain superfamily: a tale of two interfaces? Trends Biochem. Sci. 26, 475–481 [DOI] [PubMed] [Google Scholar]
- 23. Rathinam V. A., Vanaja S. K., and Fitzgerald K. A. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stehlik C., Hayashi H., Pio F., Godzik A., and Reed J. C. (2003) CARD6 is a modulator of NF-κB activation by Nod1- and Cardiak-mediated pathways. J. Biol. Chem. 278, 31941–31949 [DOI] [PubMed] [Google Scholar]
- 25. Franchi L., Eigenbrod T., Muñoz-Planillo R., and Nuñez G. (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinon F. (2008) Detection of immune danger signals by NALP3. J. Leukocyte Biol. 83, 507–511 [DOI] [PubMed] [Google Scholar]
- 27. Srimathi T., Robbins S. L., Dubas R. L., Chang H., Cheng H., Roder H., and Park Y. C. (2008) Mapping of POP1-binding site on pyrin domain of ASC. J. Biol. Chem. 283, 15390–15398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Masumoto J., Taniguchi S., Ayukawa K., Sarvotham H., Kishino T., Niikawa N., Hidaka E., Katsuyama T., Higuchi T., and Sagara J. (1999) ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 274, 33835–33838 [DOI] [PubMed] [Google Scholar]
- 29. de Alba E. (2009) Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 284, 32932–32941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salvesen G. S., and Dixit V. M. (1999) Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. U.S.A. 96, 10964–10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kubota T., and Koike R. (2010) Cryopyrin-associated periodic syndromes: background and therapeutics. Mod. Rheumatol. 20, 213–221 [DOI] [PubMed] [Google Scholar]
- 32. Hiller S., Kohl A., Fiorito F., Herrmann T., Wider G., Tschopp J., Grütter M. G., and Wüthrich K. (2003) NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure 11, 1199–1205 [DOI] [PubMed] [Google Scholar]
- 33. Liepinsh E., Barbals R., Dahl E., Sharipo A., Staub E., and Otting G. (2003) The death-fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 332, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 34. Natarajan A., Ghose R., and Hill J. M. (2006) Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. J. Biol. Chem. 281, 31863–31875 [DOI] [PubMed] [Google Scholar]
- 35. Pinheiro A. S., Proell M., Eibl C., Page R., Schwarzenbacher R., and Peti W. (2010) Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 285, 27402–27410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bae J. Y., and Park H. H. (2011) Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eibl C., Grigoriu S., Hessenberger M., Wenger J., Puehringer S., Pinheiro A. S., Wagner R. N., Proell M., Reed J. C., Page R., Diederichs K., and Peti W. (2012) Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51, 7330–7341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su M.-Y., Kuo C.-I., Chang C.-F., and Chang C.-I. (2013) Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS ONE 8, e67843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sborgi L., Ravotti F., Dandey V. P., Dick M. S., Mazur A., Reckel S., Chami M., Scherer S., Huber M., Böckmann A., Egelman E. H., Stahlberg H., Broz P., Meier B. H., and Hiller S. (2015) Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 13237–13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu A., and Wu H. (2015) Structural mechanisms of inflammasome assembly. FEBS J. 282, 435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moriya M., Taniguchi S., Wu P., Liepinsh E., Otting G., and Sagara J. (2005) Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry 44, 575–583 [DOI] [PubMed] [Google Scholar]
- 42. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 43. Tian X., Pascal G., and Monget P. (2009) Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol. Biol. 9, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vajjhala P. R., Kaiser S., Smith S. J., Ong Q.-R., Soh S. L., Stacey K. J., and Hill J. M. (2014) Identification of multifaceted binding modes for pyrin and ASC pyrin domains gives insights into pyrin inflammasome assembly. J. Biol. Chem. 289, 23504–23519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yuan S., and Akey C. W. (2013) Apoptosome structure, assembly, and procaspase activation. Structure 21, 501–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faustin B., Lartigue L., Bruey J. M., Luciano F., Sergienko E., Bailly-Maitre B., Volkmann N., Hanein D., Rouiller I., and Reed J. C. (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 [DOI] [PubMed] [Google Scholar]
- 47. Halff E. F., Diebolder C. A., Versteeg M., Schouten A., Brondijk T. H., and Huizinga E. G. (2012) Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Santiveri C. M., Oroz J., and de Alba E. (2014) A ring-like model for ASC self-association via the CARD domain. Inflammasome 1, 44–54 [Google Scholar]
- 49. Hull K. M., Shoham N., Chae J. J., Aksentijevich I., and Kastner D. L. (2003) The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr. Opin. Rheumatol. 15, 61–69 [DOI] [PubMed] [Google Scholar]
- 50. Touitou I., Lesage S., McDermott M., Cuisset L., Hoffman H., Dode C., Shoham N., Aganna E., Hugot J. P., Wise C., Waterham H., Pugnere D., Demaille J., and Sarrauste de Menthiere C. (2004) Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 24, 194–198 [DOI] [PubMed] [Google Scholar]
- 51. Cook G. P., Savic S., Wittmann M., and McDermott M. F. (2010) The NLRP3 inflammasome, a target for therapy in diverse disease states. Eur. J. Immunol. 40, 631–634 [DOI] [PubMed] [Google Scholar]
- 52. Miroux B., and Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 53. Schanda P., and Brutscher B. (2005) Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Sci. 127, 8014–8015 [DOI] [PubMed] [Google Scholar]
- 54. de Alba E. (2007) 1H, 15N and 13C backbone and side chain chemical shifts of human ASC (apoptosis-associated speck-like protein containing a CARD domain). Biomol. NMR Assign. 1, 135–137 [DOI] [PubMed] [Google Scholar]
- 55. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 56. Goddard T. D., and Kneller D. G. (2004) Sparky. University of California, San Francisco [Google Scholar]
- 57. Fraczkiewicz R., and Braun W. (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 19, 319–333 [Google Scholar]
- 58. Fielding L. (2007) NMR methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magnet. Reson. Spec. 51, 219–242 [DOI] [PubMed] [Google Scholar]
- 59. Bax A., and Grzesiek S. (1993) Methodological advances in protein NMR. Acc. Chem. Res. 26, 131–138 [Google Scholar]
- 60. Cavanagh J., Fairbrother W. J., Palmer A. G., Rance M., and Skelton N. J. (2006) Protein NMR Spectroscopy: Principles and Practice, Academic Press, Inc., San Diego [Google Scholar]
- 61. Garret D. S., Powers R., Gronenborn A., and Clore G. M. (1991) A common sense approach to peak spiking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95, 214–220 [DOI] [PubMed] [Google Scholar]
- 62. Nilges M. (1993) A calculation strategy for the structure determination of symmetric dimers by 1H NMR. Proteins 17, 297–309 [DOI] [PubMed] [Google Scholar]
- 63. Schwieters C. D., Kuszewski J. J., and Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 64. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd., Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Koradi R., Billeter M., and Wütrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 29–32 [DOI] [PubMed] [Google Scholar]
- 66. Cole J. L. (2004) Analysis of heterogeneous interactions. Methods Enzymol. 384, 212–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., and Horton J. C., eds) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 68. Chatelier R. C., and Minton A. P. (1987) Sedimentation equilibrium in macromolecular solutions of arbitrary concentration. I. Self-associating proteins. Biopolymers 26, 507–524 [DOI] [PubMed] [Google Scholar]
- 69. Muramatsu N., and Minton A. P. (1989) Hidden self-association of proteins. J. Mol. Recognit. 1, 166–171 [DOI] [PubMed] [Google Scholar]
- 70. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van Holde K. E. (1986) in Physical Biochemistry (Carlson G., ed) 2nd Ed., pp. 110–136, Prentice-Hall, Englewood Cliffs, NJ [Google Scholar]
- 72. de Vries S. J., van Dijk M., and Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 73. DeLano W. L. (2014) The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.