

Abstract
Protein synthesis inhibition is an immediate response during stress to switch the composition of protein pool in order to adapt to the new environment. It was reported that this response could be either protective or deleterious. However, how cells choose to live or die upon protein synthesis inhibition is largely unknown. Previously, we have shown that elongation factor-2 kinase (eEF2K), a protein kinase that suppresses protein synthesis during elongation phase, is a positive regulator of apoptosis both in vivo and in vitro. Consistently, here we report that knock-out of eEF2K protects mice from a lethal dose of whole-body ionizing radiation at 8 Gy by reducing apoptosis levels in both bone marrow and gastrointestinal tracts. Surprisingly, similar to the loss of p53, eEF2K deficiency results in more severe damage to the gastrointestinal tract at 20 Gy with the increased mitotic cell death in small intestinal stem cells. Furthermore, using epithelial cell lines, we showed that eEF2K is required for G2/M arrest induced by radiation to prevent mitotic catastrophe in a p53-independent manner. Specifically, we observed the elevation of Akt/ERK activity as well as the reduction of p21 expression in Eef2k−/− cells. Therefore, eEF2K also provides a protective strategy to maintain genomic integrity by arresting cell cycle in response to stress. Our results suggest that protective versus pro-apoptotic roles of eEF2K depend on the type of cells: eEF2K is protective in highly proliferative cells, such as small intestinal stem cells and cancer cells, which are more susceptible to mitotic catastrophe.
Keywords: apoptosis, cell cycle, cell death, radiation biology, stem cells, eukaryotic elongation factor-2 kinase (eEF2K), mitotic catastrophe
Introduction
Elongation factor-2 (eEF2) 3 kinase (eEF2K) belongs to the α-kinase family, which has no homology to conventional protein kinases (1). By phosphorylating eukaryotic elongation factor-2 (eEF2), eEF2K suppresses protein synthesis during the elongation stage (2).The activity of eEF2K depends on Ca2+/calmodulin, pH (3), and AMP/ATP ratio (4), and it can be activated during stress (5, 6). A recent study demonstrated that depletion of eEF2K reduces apoptosis in normal cells, such as fibroblasts, granulosa cells, and oocytes, and results in accumulation of defective oocytes (7). In addition, lack of eEF2K in Caenorhabditis elegans results in a similar phenotype in germ cell apoptosis (7). Intriguingly, eEF2K plays paradoxical roles in normal versus cancer cells; although activation of eEF2K leads to apoptosis in normal cells (7), its activity is critical to maintain the viability of oncogene-transformed cells after adaptation to nutrient deprivation (8).
In addition to its role in apoptosis induction, recent studies also suggested that eEF2K could be involved in cell cycle regulation. During checkpoint silencing, the degradation of eEF2K by the SCF(βTrCP) ubiquitin-proteasome system was connected with protein synthesis resumption (9). Moreover, in an APC-deficient colorectal cancer mouse model, the inhibition of eEF2K by mTORC1 hyperactivation was critical for tumor growth. Depletion of eEF2K also resulted in an alteration of some cyclin proteins (10). However, under the normal condition, depletion of eEF2K in mice did not affect cell cycle progression and the growth rate (7).
Here, we employed a classic ionizing radiation model to study the functions of eEF2K under DNA damage stress. Consistent with the role of eEF2K in programmed cell death, knock-out of eEF2K protected mice from 8 Gy of IR by reducing hematopoietic stem cell death. Unexpectedly, we found that when gastrointestinal syndrome was triggered with a higher dose of ionizing radiation, Eef2k−/− mice became more sensitive due to massive intestinal stem cell loss. We found that eEF2K-deficient cells escaped from G2/M arrest and resulted in more severe DNA damage and mitotic catastrophe post-IR. Collectively, our data demonstrated that eEF2K regulates both apoptosis and cell cycle in response to IR to govern stem cell survival.
Results
Knock-out of eEF2K in Mice Increases the Survival after 8 Gy of IR but Not after 20 Gy of IR
To study the role of eEF2K in radiation-induced damage, eEF2K wild type and knock-out mice were exposed to various doses of ionizing radiation: 8, 15, and 20 Gy. At 8 Gy, 50% of wild type mice died, but only 10% of Eef2k−/− mice died within 30 days (Fig. 1A, p = 0.041). When the dose was further increased to 15 Gy, both eEF2K wild type and deficient mice died within 2 weeks, but no significant difference was observed at this dose (Fig. 1B, p = 0.140). Surprisingly, when the dose was even further increased to 20 Gy, Eef2k−/− mice became more sensitive (Fig. 1C, p = 0.003) compared with their wild type littermates. All of the Eef2k−/− mice died in 6 days, whereas wild type mice survived up to 8 days. To further verify the cause of lethality at different doses, autopsy analysis was conducted immediately after death of mice. At 15 Gy, H&E staining showed a massive cell loss in bone marrow, whereas crypts with abnormal structures were still present in the small intestines. However, a complete destruction of small intestine microstructures was observed when mice were dead after exposure to 20 Gy of IR, with most of the bone marrow cells surviving at this time (Fig. 1D). These results suggest that eEF2K regulates cell death in bone marrow cells and leads to bone marrow failure upon IR. However, when gastrointestinal complications became the main cause of lethality induced by escalated dose of IR (20 Gy), eEF2K extended the life span.
FIGURE 1.

Eef2k−/− mice displayed opposite sensitivities at low dose and high dose of ionizing radiation. A, Kaplan-Meier survival analyses of Eef2k+/+ and Eef2k−/− mice after 8-Gy total body γ-irradiation. Each group contained 10 mice. The p value was obtained by the log-rank test. B, Kaplan-Meier survival analyses of Eef2k+/+ and Eef2k−/− mice after 15-Gy total body γ-irradiation. Each group contained 10 mice. The p value was obtained by the log-rank test. C, Kaplan-Meier survival analyses of Eef2k+/+ and Eef2k−/− mice after 20-Gy total body γ-irradiation. Each group contained five mice. The p value was obtained by the log-rank test. D, H&E staining of autopsy samples from mouse bone marrow and small intestine tissues after mice succumbed to death after 0-, 15-, or 20-Gy total body γ-irradiation. Scale bars, 100 μm for 0-Gy tissue and 50 μm for other tissues. E, Western blotting analysis of p-eEF2 in ovary, small intestine, and muscle under normal conditions, after 8- and 20-Gy ionizing radiation. Actin was used as a loading control. Quantification was done by using Quantity One software. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The different sensitivities of Eef2k−/− mice toward various doses of ionizing radiation may indicate that eEF2K functions differently according to tissue and cell types. To answer this question, phosphorylated eEF2 (p-eEF2) was measured by Western blotting in ovary, small intestine, and muscle at 4 and 24 h after 8- and 20-Gy γ-irradiation. In highly proliferative tissues, such as ovary and small intestine, eEF2K activity was quickly suppressed within 24 h independent of the doses. However, in non-proliferative tissues (e.g. muscle), the change of p-eEF2 level was not as robust as in other tissues after radiation treatment (Fig. 1E). These results suggest that the dynamics of eEF2K activity in DNA damage response is distinct in different cell types and could be related to cell proliferating status.
eEF2K Is Pro-apoptotic in Hematopoietic Stem Cells in Response to Ionizing Radiation
To further investigate the role of eEF2K in IR-induced bone marrow failure, both wild type and Eef2k−/− mice were exposed to 8 Gy ionizing radiation. Femurs and tibias were collected at 4 and 24 h post-IR for immunohistochemistry analysis. To monitor the activity of eEF2K, immunostaining of p-eEF2 was conducted. Under normal conditions, p-eEF2 staining was barely observed in bone marrow cells. After IR exposure, the intensity of p-eEF2 staining was dramatically increased in various types of bone marrow cells. Specifically, the most intense staining was detected in the cells with condensed chromosomes, a characteristic of apoptotic cell death (Fig. 2A). Because apoptosis plays an important role in bone marrow cell loss at post-IR, cleaved caspase-3 and TUNEL staining were used to evaluate apoptosis levels in bone marrow cells. Consistent with the role of eEF2K in apoptosis, cleaved caspase-3- and TUNEL-positive cells were reduced in Eef2k−/− mice (Fig. 2A). In addition, the quantification for the number of cleaved caspase-3-positive and TUNEL-positive cells also showed a significant decrease in Eef2k−/− mice (Fig. 2 (B and C), p = 0.001, p < 0.0001).
FIGURE 2.

Knock-out of eEF2K protects bone marrow cells from ionizing radiation. A, representative images of immunohistochemistry staining of p-eEF2 (brown), cleaved caspase-3 (brown), and TUNEL (brown) in bone marrow cells at 4 and 24 h after 8 Gy of total body γ-irradiation. Scale bars, 100 μm. B, quantification of cleaved caspase-3-positive cells in Eef2k+/+ and Eef2k−/− mouse bone marrow cells at 4 h after 8-Gy γ-irradiation. The p value was obtained by the Mann-Whitney test. Results are presented as mean ± S.E. (error bars). C, quantification of TUNEL-positive cells in Eef2k+/+ and Eef2k−/− mouse bone marrow cells at 4 h after 8-Gy γ-irradiation. The p value was obtained by the Mann-Whitney test. Results are presented as mean ± S.E. D, comparison of the number of white blood cells extracted from the femur and tibia of 2-month wild type and Eef2k−/− mice. Results are presented as mean ± S.E. E, comparison of the distribution of lymphocyte, eosinophil, basophil, and neutrophil of 2-month wild type and Eef2k−/− mice. The blood smears were prepared from mouse tails, and a total of 100 white blood cells were counted after Wright-Giemsa staining. Results are presented as mean ± S.E. F, comparison of the number of colonies formed with 2 × 104 bone marrow cells after they were plated onto soft agar plates with complete culture medium for 10 days. Results are presented as mean ± S.E. G, colony formation assay for Eef2k+/+ and Eef2k−/− mouse hematopoietic stem cells after ionizing radiation. Results are presented as mean ± S.E. The p value was obtained by the Mann-Whitney test. H, Western blotting analysis of PUMA protein in Eef2k+/+ and Eef2k−/− mouse bone marrow cells after ionizing radiation. Actin was used as a loading control. Quantification was done by using Quantity One software, and results are presented as mean ± S.E. I, RT-PCR analysis of PUMA mRNA level in Eef2k+/+ and Eef2k−/− MEFs as well as Eef2k−/− MEFs transfected with plxs-eEF2K plasmid. Results are presented as mean ± S.E. The p value was obtained by the Mann-Whitney test. J, Western blotting analysis of PUMA protein in Eef2k+/+ and Eef2k−/− MEFs as well as Eef2k−/− MEFs transfected with plxs-eEF2K plasmid. Quantification was done by using Quantity One software. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To assess the function of eEF2K in hematopoietic stem/progenitor cell survival after exposure to IR, a colony formation assay was performed using freshly isolated hematopoietic stem/progenitor cells from Eef2k+/+ and Eef2k−/− mice. Briefly, hematopoietic stem/progenitor cells were exposed to 0, 1, 2, 3, 4, and 5 Gy of IR, and the survival of bone marrow colony-forming cells was measured by counting the number of colonies in soft agar plates after culture for 10 days. The value of D0, which represents the amounts of radiation to reduce the number of colonies by 63% (11), was calculated by linear regression analysis. At basal level without radiation treatment, the total number of bone marrow cells extracted from Eef2k+/+ and Eef2k−/− femur and tibia was similar, and the cells displayed similar abilities to generate colonies (Fig. 2, D–F) with comparable percentages of colonies exhibiting myeloid and erythroid lineages (data now shown). After radiation, the calculated D0 for Eef2k−/− bone marrow colony-forming cells was significantly higher than the D0 of wild type cells (Fig. 2G, p = 0.02), indicating that depletion of eEF2K results in increased resistance to IR in hematopoietic cells.
The radioresistant phenotype was described previously in PUMA knock-out mice (12). Surprisingly, we found that the expression level of PUMA, a critical player in radiation-induced apoptosis, was reduced in the bone marrow cells of Eef2k−/− mice (Fig. 2H), suggesting that PUMA could be a downstream factor of eEF2K to mediate apoptotic cell death in response to IR. To further investigate the regulatory role of eEF2K in PUMA expression, PUMA mRNA and protein levels were compared between Eef2k+/+ and Eef2k−/− mouse embryonic fibroblasts (MEFs). Consistently, we found that knock-out of eEF2K reduces PUMA expression at both RNA and protein levels. Moreover, re-expression of eEF2K in Eef2k−/− MEFs restored the levels of PUMA expression in both RNA and protein, suggesting that eEF2K could specifically regulate PUMA expression (Fig. 2 (I and J), p = 0.011, p = 0.0043).
eEF2K Regulates Intestinal Stem Cell Death in Response to IR
eEF2K knock-out mice displayed an increased sensitivity to elevated dose of IR, indicating that eEF2K may function in intestinal stem cell death. In addition to bone marrow stem cells, intestinal stem cells represent another pool of adult stem cells sensitive to IR. Previous studies showed that IR as low as 1 Gy can cause massive apoptosis in small intestine crypt epithelium (13). Therefore, small intestine tissues were collected from wild type and Eef2k−/− mice for a TUNEL assay at 4 and 24 h after exposure to 8 Gy of IR. The intestinal crypt has a very well organized structure, and the cell position at the very bottom is called the +1 position (Fig. 3B). In a stem cell niche, there are mainly two types of stem cells residing in the crypts; Lgr5-positive cells are actively proliferating cells between Paneth cells, and the others are label retention cells at the +4 position (14). Once the stem cells lose the contact with the stem cell niche (above +4 position), they quickly migrate upward and differentiate into various lineages of epithelial cells to maintain the high turnover rate within the crypt-villus units (15). To quantify the apoptosis level in stem cells, the percentage of TUNEL staining-positive cells was counted and calculated from +1 to +14 position. At 4 h, when maximum apoptosis occurs, the apoptosis level in the small intestine of Eef2k−/− mice was reduced at the stem cell zone (+1 to +4 position) (Fig. 3, A and C). However, when the apoptosis level began to decrease in the small intestine of wild type mice at 24 h, the number of TUNEL-positive cells was increased in Eef2k−/− mouse small intestine crypts (Fig. 3, A and D). The examination of TUNEL-positive cells showed giant cell appearance with abnormally distributed condensed chromosomes in the small intestine of Eef2k−/− mice at 24 h post-IR (16), suggesting that eEF2K functions to suppress mitotic cell death after ionizing radiation. Moreover, when mice were exposed to 18 Gy radiation, the cell death level became comparable between Eef2k+/+ and Eef2k−/− small intestines at both 4 and 24 h post-IR (Fig. 3E).
FIGURE 3.

Knock-out of eEF2K protects mouse small intestine from first wave of apoptosis but sensitizes them to a second wave of cell death after 8-Gy radiation. A, representative images of TUNEL staining (brown) in Eef2k+/+ and Eef2k−/− mouse small intestine at 4 and 24 h after 8-Gy total body γ-irradiation. Scale bars, 50 μm. B, schematic illustration of small intestine microstructure and cell position in intestinal crypt. C, comparison of TUNEL-positive cells in Eef2k+/+ and Eef2k−/− mouse intestinal crypts at 4 h after 8-Gy total body γ-irradiation. Quantification was done in 100 half-crypts at each position from position 1 to position 14 starting from the very bottom of the crypt. Results are presented as mean ± S.E. (error bars). D, comparison of TUNEL-positive cells in Eef2k+/+ and Eef2k−/− mouse intestinal crypts at 24 h after 8-Gy total body γ-irradiation. Quantification was done in 100 half-crypts at each position from position 1 to position 14 starting from the very bottom of the crypt. Results are presented as mean ± S.E. E, representative images of TUNEL staining (brown) in Eef2k+/+ and Eef2k−/− mouse small intestine at 4 and 24 h after 18-Gy total body γ-irradiation. Scale bars, 50 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To analyze the effects of eEF2K on intestinal stem cell survival after ionizing radiation, a microcolony formation assay was conducted. It is generally thought that a single stem cell could regenerate a whole crypt within 4 days if it survives after IR insults. Therefore, mice were sacrificed at day 4 post-IR, and small intestine tissues were collected from at least three mice for cross-section, followed by H&E staining. The number of regenerated crypts was counted in each section as evidence of surviving intestinal stem cells. The number of regenerated crypts was comparable between Eef2k+/+ and Eef2k−/− mice at 8 and 10 Gy; whereas, the regenerated crypt number was significantly reduced in Eef2k−/− mice at doses of 12 and 14 Gy (Fig. 4 (A and B), p < 0.001). In addition, the loss of regeneration capacity could be represented by reduction of the proliferative index. To analyze the proliferative index in intestinal stem cells, BrdU was injected into mice 2 h before sacrifice at day 4 post-IR. Consistently, the number of BrdU-positive crypts was significantly decreased in Eef2k−/− mice compared with their wild type cohorts (Fig. 4 (C and D), p < 0.001). To exclude the possibility that eEF2K may directly affect cell proliferation under normal conditions, Ki67 staining was used to label proliferative cells in intestinal crypts, and the expression level of Ki67 was similar between wild type and Eef2k−/− mice (Fig. 4E). These results indicate that eEF2K protects intestinal stem cells from an extremely high dose of radiation.
FIGURE 4.

Knock-out of eEF2K sensitizes small intestinal stem cells to ionizing radiation. A, cross-section of wild type and Eef2k−/− mouse small intestines at 4 days after ionizing radiation. Scale bars, 100 μm. B, microcolony formation assay of Eef2k+/+ and Eef2k−/− mouse intestinal stem cells after 0-, 8-, 10-, 12-, or 14-Gy total body γ-irradiation. The number of regenerated crypts was counted on at least 10 different sections for each mouse, and each counted section was at least 1 cm apart. Three mice were used for each genotype at the indicated dose. Results are presented as mean ± S.E. (error bars). The p value was obtained by the Mann-Whitney Test. C, representative images of BrdU immunohistochemical staining in wild type and Eef2k−/− mouse small intestine 4 days after 14-Gy γ-irradiation. A group of 4 mice was irradiated with 14-Gy γ-irradiation, and 100 mg/kg BrdU was injected 2 h before scarification. The positive staining of BrdU was more often observed in small intestine of wild type mice than in Eef2k−/− mice. Scale bars, 50 μm. D, quantification of the number of small intestine crypts with positive BrdU staining. Results are presented as mean ± S.E. The BrdU incorporation was significantly reduced in Eef2k−/− mouse small intestine. E, representative images of Ki67 staining (brown) in wild type and Eef2k−/− mouse small intestine. Scale bars, 50 μm. ***, p < 0.001.
eEF2K Suppresses Intestinal Mitotic Cell Death in Response to γ-Irradiation
Previous studies have shown that mitotic cell death or clonogenic death occurs during the second wave of cell death in the small intestine in response to IR (16, 17). Because we found that the second wave of cell death in the small intestine was increased in Eef2k−/− mice, we wanted to test whether eEF2K regulates the mitotic cell death. We further investigated the cell cycle progression of crypt epithelial cells in Eef2k−/− mice post-IR. As a marker of cell proliferation, Ki67 antigen is present in all cells residing in G1, S, G2, and mitosis phases (18). To visualize actively cycling cells, Ki67 was used for immunohistochemistry staining in the small intestine at 24 h after exposure to 8 Gy of IR. By counting 50 crypts, the number of Ki67-positive cells was higher in Eef2k−/− mice crypts at the stem cell zone compared with wild type mouse crypts (Fig. 5, A and B).
FIGURE 5.

Knock-out of eEF2K increases mitotic catastrophe in small intestinal cells after ionizing radiation. A, representative images of immunohistochemistry staining of Ki67 (brown) in Eef2k+/+ and Eef2k−/− mouse small intestine crypts at 24 h after 8-Gy total body γ-irradiation. Scale bars, 50 μm. B, comparison of Ki67-positive cells in Eef2k+/+ and Eef2k−/− mouse intestinal crypts at 24 h after 8-Gy total body γ-irradiation. Quantification was done in at least 50 half-crypts for each mouse, and the number of mice used for each genotype is indicated in the figure. Results are presented as mean ± S.E. (error bars). C, comparison of the number of mitotic cells in normal small intestine crypts of Eef2k+/+ and Eef2k−/− mice. 100 crypts per mouse were counted. Results are presented as mean ± S.E. D, representative images of intestinal crypts at 24 h after 8-Gy total body γ-irradiation. The cells undergoing mitotic catastrophe are labeled with a red asterisk. Scale bars, 20 μm. E, comparison of mitotic catastrophe cells in Eef2k+/+ and Eef2k−/− mouse small intestine crypts at 24 and 72 h after 8-Gy total body γ-irradiation. Quantification was done in at least 50 half-crypts for each mouse, and at least two mice were used for each genotype at the indicated time. Results are presented as mean ± S.E. The p value was obtained by the Mann-Whitney test. *, p < 0.05.
Moreover, the mitotic cell death was compared between Eef2k+/+ and Eef2k−/− small intestine after IR. Although no difference was observed in the number of mitotic cells before IR (Fig. 5C), the mitotic cell death was significantly higher in the intestinal crypt of Eef2k−/− mice (Fig. 5, D and E). The epithelial cells eliminated by mitotic cell death displayed abnormal mitotic morphology, which could be easily distinguished by simple nuclear staining by hematoxylin (Fig. 5D). The quantification results also indicated significantly greater mitotic cell death in Eef2k−/− mouse small intestine at 24 and 72 h after 8-Gy γ-irradiation (Fig. 5E, p = 0.049, p = 0.028), most of which displayed abnormal mitotic condensed chromosomes and chromosome fragments. Thus, overall, these results suggest that the higher stem cell loss rate in Eef2k−/− small intestinal crypts after IR is at least partially due to the improper cell cycle progression, especially mitosis.
eEF2K Deficiency Disrupts Cell Cycle Arrest and Increases Mitotic Catastrophe upon IR
The same phenotype in response to ionizing radiation was also observed in p53−/− mice, which is due to the dual role of p53 in DNA damage response, including apoptosis and cell cycle arrest (17, 19). We then asked whether eEF2K regulates cell cycle progression upon DNA damage. Further investigations of the functions of eEF2K in DNA damage response were conducted in epithelial cell lines after radiation treatment. The cell lines were derived from baby mouse kidney (BMK) of Eef2k+/+ and Eef2k−/− mice and immortalized with dominant negative p53 to avoid the impact of p53 (20).
Interestingly, when Eef2k+/+ and Eef2k−/− BMK cells were exposed to various doses of IR, more cells were surviving at 5 Gy in Eef2k−/− cells at both 48 and 72 h post-IR, whereas the levels of cell death were higher in Eef2k−/− cells at 10 and 20 Gy at 48 h (Fig. 6A, p < 0.001, p < 0.001, p < 0.001, p < 0.001). We further tested three pairs of Eef2k+/+ and Eef2k−/− cells derived from different mice. Consistently, a significant increase in cell survival was observed in all Eef2k−/− cell lines after 5 Gy of IR (Fig. 6B, p = 0.006). Moreover, the impact of eEF2K in cell cycle arrest was studied by propidium iodide staining after exposure to IR. Before IR, the cell cycle distribution was similar between Eef2k+/+ and Eef2k−/− cells. At 12 h post-IR, when >80% wild type cells were arrested at G2 phase, far fewer Eef2k−/− cells were arrested. At this time point, <5% of G1 cells were observed in Eef2k+/+ cells, but >20% of G1 cells were observed in Eef2k−/− cells. Furthermore, at 24 h post-IR, when most of the wild type cells continually resided in G2 phase, Eef2k−/− cells already displayed a nearly normal cell cycle distribution. Moreover, a similar change pattern was observed after exposure to 10-, 15-, and 20-Gy ionizing radiation (Fig. 6C). To further test whether cells were arrested in G2 phase after DNA damage, BrdU labeling was used to study cell cycle progression. When most of the BrdU-positive Eef2k+/+ cells were arrested at G2 phase at 12 h post-IR, some Eef2k−/− cells escaped from G2/M checkpoint and progressed into G1 phase (Fig. 6D), indicating that depletion of eEF2K renders cells incapable of arresting at G2/M checkpoint.
FIGURE 6.

eEF2K mediates G2/M checkpoint after ionizing radiation treatment. A, BMK cells were exposed to 5, 10, and 20 Gy of IR, and cell survival was analyzed by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Results are presented as mean ± S.E. (error bars). The p value was obtained by the Mann-Whitney test. B, three independent BMK cell lines derived from Eef2k+/+ or Eef2k−/− mice were exposed to 5-Gy γ-irradiation, and cell survival was analyzed at 72 h post-IR by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Results are presented as mean ± S.E. Each bar represents an independent cell line. The p value was obtained by the Mann-Whitney test. C, cell cycle analysis of BMK cells at 6, 12, and 24 h after 5-, 10-, 15-, and 20-Gy γ-irradiation. D, Eef2k−/− BMK cells escape from G2/M checkpoint after 5-Gy ionizing radiation. BMK cells were exposed to 5-Gy γ-irradiation and labeled with BrdU for 1 h. Cell cycle and BrdU-positive cells were analyzed by FACS at 6 h post-IR and 12 h post-IR. More eEF2K-deficient cells progressed into G1 phase at 12 h post-IR. E, Western blotting analysis of p-eEF2, eEF2K, phospho-AKT(Ser-473), AKT, phospho-ERK, ERK, p21, PUMA, and cleaved caspase-3 protein in WT and KO BMK cells after 5-, 10-, 15-, and 20-Gy ionizing radiation. Actin was used as a loading control. Quantification was done by using Quantity One software. *, p < 0.05; ***, p < 0.001.
To explore the underlying mechanism mediating the functions of eEF2K in DNA damage-induced cell cycle arrest and apoptosis, various molecular signaling markers were analyzed by Western blotting. The Akt and ERK pathways play critical roles in promoting cell cycle progression (21, 22). Interestingly, we observed an increase of activities of both pathways in Eef2k−/− cells. Additionally, p21, a potent cell cycle kinase inhibitor, was decreased after the loss of eEF2K, which may also contribute to the insufficient cell cycle arrest in eEF2K-deficient cells. Moreover, consistent with the results obtained from bone marrow cells and MEFs, there was a reduction of PUMA expression level in Eef2k−/− BMK cells (Fig. 6E).
Evidence has shown that deficiency in cell cycle arrest induced by DNA damage leads to large pieces of DNA fragments escaping from the nucleus to form micronuclei and causes cell death due to mitotic catastrophe (23). We further quantified the number of micronuclei in Eef2k+/+ and Eef2k−/− BMK cells after 5 Gy of IR. Consistent with the observations in mice, more cells underwent abnormal mitosis after ionizing radiation (Fig. 7 (A and B), p = 0.030), and more cells exhibited micronuclei in Eef2k−/− cells (Fig. 7 (A and C), p = 0.002), indicating that eEF2K protects cells from mitotic catastrophe induced by IR. γH2AX is a sensitive marker to detect DNA damage. Consistently, more γH2AX was accumulated in Eef2k−/− cells after 5-Gy ionizing radiation (Fig. 7D), indicating a higher level of DNA damage due to insufficient cell cycle arrest with the loss of eEF2K. To study whether the regulation of eEF2K in DNA damage response is dose-dependent, Eef2k+/+ and Eef2k−/− BMK cells were treated with 5-, 10-, 15-, and 20-Gy γ-irradiation, and the percentage of cells with micronuclei was quantified at 24 and 48 h post-treatment. Eef2k−/− cells displayed a higher level of micronuclei after various doses of ionizing radiation (Fig. 7E, p < 0.001 for all doses and time points).
FIGURE 7.

Knock-out of eEF2K renders cells more susceptible to mitotic catastrophe after ionizing radiation. A, representative images of nuclear DAPI staining in WT and KO BMK cells after 5-Gy γ-irradiation. Mitotic cells are marked with a yellow arrow, and cells with micronuclei are labeled with a red asterisk. B, comparison of the percentage of mitotic cells at 0 and 24 h after γ-irradiation. Quantification was done in at least six different fields for each treatment, each field with at least 100 cells. The overall percentage was calculated based on more than 600 cells. The p value was obtained by the Mann-Whitney test. Results are presented as mean ± S.E. (error bars). C, comparison of the percentage of cells with micronuclei at 0 and 24 h after γ-irradiation. Quantification was done in at least six different fields for each treatment, each field with at least 100 cells. The overall percentage was calculated based on more than 600 cells. The p value was obtained by the Mann-Whitney test. Results are presented as mean ± S.E. D, Western blotting analysis of p-eEF2 and γ-H2AX protein in WT and KO BMK cells at various time points after exposure to 5-Gy γ-irradiation. Tubulin was used as a loading control. Quantification was done by using Quantity One software. E, comparison of the percentage of cells with micronuclei at 24 and 48 h after 5-, 10-, 15-, and 20-Gy γ-irradiation. Quantification was done in at least four different fields for each treatment, each field with at least 100 cells. The overall percentage was calculated based on more than 400 cells. The p value was obtained by the Mann-Whitney test. Results are presented as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Here we report that eEF2K plays important roles in IR-induced injuries of bone marrow and small intestine. Knock-out of eEF2K protects mice from hematopoietic syndrome but sensitizes them to gastrointestinal syndrome, which is due to the dual functions of eEF2K in response to DNA damage induced by IR. Our results demonstrate that eEF2K promotes apoptosis; however, it protects cells from mitotic cell death by regulating cell cycle arrest (Fig. 8).
FIGURE 8.
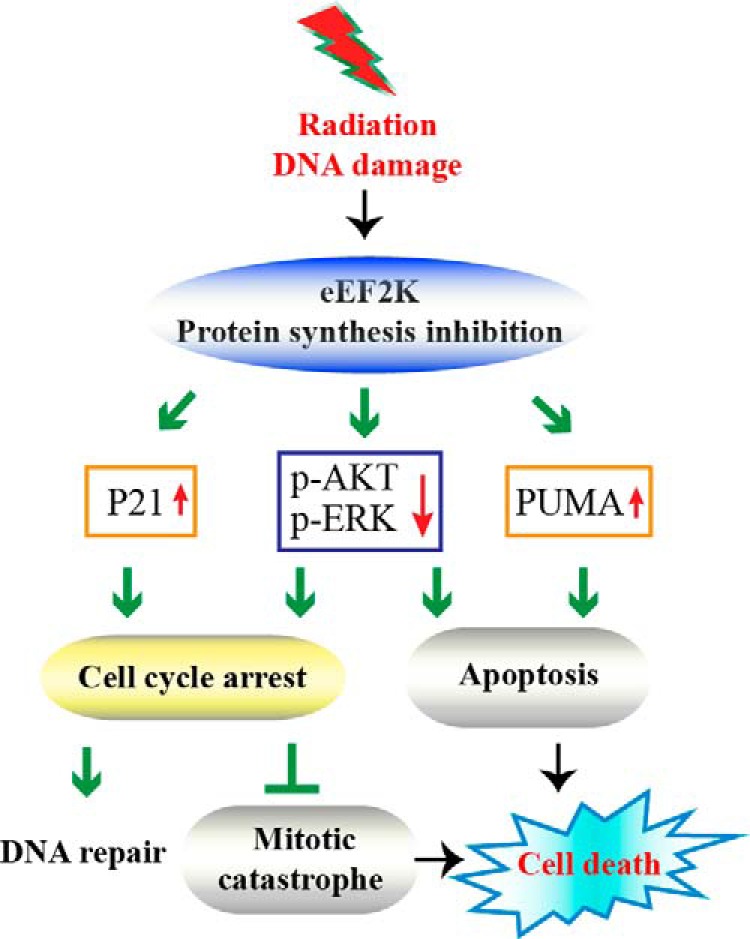
Model of eEF2K medicated cell death machinery in response to DNA damage. eEF2K has two roles in cell death induced by DNA damage; one is to promote apoptosis, and the other is to control cell cycle arrest to prevent mitotic cell death. When mitotic catastrophe takes charge, pro-apoptotic eEF2K will switch to being an anti-death protector by arresting the cell cycle to suppress mitotic catastrophe.
In response to DNA damage, a multilayer surveillance system was evolved to maintain genomic stability, in which programmed cell death collaborates closely with cell cycle progression to determine cell fate. Whereas interphase cells may die from apoptosis or step into senescence, mitotic cells usually die during mitosis or after mitosis due to mitotic catastrophe. Mitotic catastrophe has been described as the major form of cell death after ionizing radiation because of premature and inappropriate entry of cells into mitosis (24, 25) and has also been reported as the major response of cancer cells to several anti-cancer drugs (26, 27). Therefore, the functions of eEF2K in cell cycle arrest and mitotic catastrophe may provide a clue to explain its paradoxical roles in normal versus cancer cell death under stress conditions. The elevated proliferating rate of cancer cells renders them more susceptible to mitotic catastrophe in stress environment, whereas normal cells are prone to arrest in the cycle or die from apoptosis. The inactivation of eEF2K could potentially increase mitotic catastrophe in cancer cells but prevent normal cells from apoptosis induced by DNA damage.
Our study uncovers the role of eEF2K in both apoptosis and cell cycle arrest, two major aspects of DNA damage response. The previous study has reported that the level of eEF2K was regulated during DNA damage response in vitro (9). During DNA damage checkpoint silencing, a process required to allow cell cycle reentry, eEF2K was degraded by the ubiquitin-proteasome system through the ubiquitin ligase SCF(βTrCP) to enable rapid resumption of translation elongation. Here we demonstrated the functions of eEF2K in response to DNA damage in vivo. In addition, recent studies suggested that the role of eEF2K in apoptosis was due to its function in protein synthesis arrest and down-regulation of the short lived anti-apoptotic protein cFLIP(L) and XIAP (7), but its regulation in cell cycle arrest was unknown. Similarly, a number of cell cycle regulatory proteins were also subjected to quick regulation of synthesis and degradation during DNA damage checkpoints, including various cyclins, polo-like kinase 1 (Plk1) (28), Cdc25A, and Wee1 kinase (29). Specifically, the short lived protein survivin has dual roles in apoptosis and cell cycle arrest, which may contribute to the functions of eEF2K in DNA damage response (30). Moreover, it has been shown that Cdc2-cyclin B suppresses eEF2K activity by phosphorylation (31), whereas CDK2-cyclin A phosphorylates eEF2 to facilitate eEF2K function (32). Also, the studies from APC-deficient mice have suggested that eEF2K could affect the expression of cyclin D3 to modulate cell cycle progression and cell proliferation (10). Therefore, all of these lines of evidence suggest that translational regulation by eEF2K controls cell cycle progression in response to DNA damage.
Previously, multiple survival pathways have been shown to suppress eEF2K activity, including mTORC1 (33), p70S6K1, and p90RSK1 (34). Moreover, knockdown of eEF2, the substrate of eEF2K, inactivates the PKB/Akt pathway in gastrointestinal cancer cell lines (35). Working in the same direction, here we show that eEF2K-deficient BMK cells displayed a higher activity of Akt. In addition, we also found an activation of ERK in Eef2k−/− cells. The alteration of activities of survival pathways may underlie the pro-death role of eEF2K in response to various stresses. Even more importantly, as both Akt and ERK pathways regulate cell survival and cell cycle progression (21, 22), they may contribute to the functions of eEF2K in DNA damage response. However, the questions regarding how eEF2K modulates Akt and ERK pathway activity remain elusive.
The activity of eEF2K was shown to be stimulated by intracellular free Ca2+, acidic pH (3), oxidative stress, and increased AMP/ATP ratios (4). However, after irradiation, the signaling input for eEF2K activation remains elusive. Recently, studies of AMPK have revealed its role in DNA damage response, including apoptosis and cell cycle checkpoint (36). Therefore, the activity of eEF2K could be regulated by AMPK in response to ionizing radiation. In addition to DNA damage, ionizing radiation produces oxidative stress, which also can regulate the activity of eEF2K post-IR.
The dual sensitivities toward ionizing radiation were also described previously in p53−/− mice (17). In our study, whether there is a cross-talk between eEF2K and p53 signaling in DNA damage response remains to be uncovered. By Western blotting analysis, we found that expression of PUMA was down-regulated in Eef2k−/− bone marrow cells under both normal conditions and ionizing radiation treatment (Fig. 2C). PUMA is a downstream target for p53 to mediate apoptosis (37). These results suggest that eEF2K may alter the p53 signaling pathway via regulation of the levels of PUMA. On the other hand, blocking the functions of p53 using dominant negative p53 does not compromise the responses of Eef2k−/− cells to IR. Thus, the regulation of eEF2K in DNA damage could be independent of p53, or eEF2K is a downstream mediator in the p53 signaling pathway.
In addition to p53, the alterations in the immune system post-IR also play an important role in damage response. The modulation of immune response by IL-12 also results in similar phenotypes in mice (38). Therefore, the question of whether knock-out of eEF2K could affect the immune system, especially the expression of IL-12, would be interesting to investigate in follow-up experiments.
Upon radiation insults, the cause of intestinal stem cell depletion remains controversial. Early studies indicate that endothelial cell apoptosis is the primary lesion to initiate stem cell death (39). However, genetic studies by gene knock-out mice support the idea that elimination of intestinal stem cells is due to the death of epithelial lineage cells (19). Studies in p21−/− mice have suggested that the cell death is caused by misregulation of cell cycle arrest rather than apoptosis (40, 41). Although it seems controversial that knock-out of PUMA could protect intestinal stem cells from high dose ionizing radiation (42), recent studies have suggested that this may be due to the up-regulation of p21 in Puma−/− mice with an unknown mechanism (40). In contrast with Puma−/− mice, we found a decrease of p21 in eEF2K-deficient cells, which may underlie the defects in cell cycle arrest and mitotic cell death after the loss of eEF2K in intestinal stem cells, instead of apoptosis. Overall, our data provide more supportive evidence for the critical role of clonogenic death of epithelial cells in intestinal stem cell lethality.
Overall, in addition to its role in apoptosis, we unveiled the function of eEF2K in G2/M checkpoint and mitotic catastrophe. Previously, most clinical and experimental anti-cancer approaches were designed to trigger apoptosis in cancer cells; the therapeutic potential of mitotic catastrophe was largely neglected. Nowadays, the increased resistance of more and more cancer cells to pro-apoptotic agents and the side effects mediated by apoptosis in normal tissues largely hinder the improvement of anti-cancer therapies. Conversely, cancer cells seem to be intrinsically more sensitive to mitotic catastrophe than normal cells (43, 44). Therefore, our finding indicates that eEF2K could be a potential target for drug development to improve cancer-killing efficacy and simultaneously protect normal cells from apoptosis.
Experimental Procedures
Mice and Ionizing Radiation
The procedures of all animal experiments were approved by the institutional animal care and use committee at the University of Medicine and Dentistry of New Jersey. The Eef2k−/− mice were generated in our laboratory by back-crossing to C57BL/6 for 10 generations. The targeting vector was constructed by using a 1.2-kb DNA fragment as the short arm, which was a PCR fragment from the end of exon 8 to exon 10 (primer pairs: SA2 (5′-TGGAGATGGTAACCTTG-3′) and SA4 (5′-TCAAGATGGTCTTGGCTGATTG-3′)). The long arm was the BamHI fragment, which contains exon 6. In this knock-out strategy, the entire exon 7 and the majority of exon 8 have been replaced by the neo gene cassette. After electroporation of embryonic stem cells, surviving colonies in G418 were expanded, and PCR analysis was performed to identify clones that had undergone homologous recombination. PCR was done using primer pairs SA8 (5′-GGCCGGCTGCTAGAGAGTGTC-3′) and Neo1 (5′-TGCGAGGCCAGAGGCCACTTGTGTAGC-3′). The correctly targeted ES cell lines were microinjected into C57BL/6J host blastocysts. The chimeric mice were generated, and they gave germ line transmission of the disrupted eEF2K gene. The genotyping of Eef2k−/− mice was performed using PCR with two pairs of primers (Neo1/SA8, SA8/SA5; sequence of SA5: 5′-CATCAGCTGATTGTAGTGGACATC-3′). Mice were irradiated at doses ranging from 0 to 20 Gy using a 173Cs irradiator.
Generation of Immortalized BMK Cells
Primary epithelial cells are isolated from wild type and Eef2k−/− BMK (ages 4–8 days) and immortalized with adenovirus E1A and dominant-negative p53 (p53DD) as described previously (45, 46). Briefly, the isolated primary epithelia were transfected by electroporation with expression vectors for E1A and p53DD. Following 1 week of in vitro culture, untransfected epithelial cells died off, leaving behind immortal epithelial colonies that emerged in the next 2–3 weeks without the requirement for drug selection.
Immunohistochemistry; 3,3′-Diaminobenzidine (DAB) Reaction
Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Tissues were sectioned (4 μm), mounted on glass slides, and subjected to immunohistochemical staining for the presence and distribution of p-eEF2 (Cell Signaling, catalog no. 2331) and BrdU incorporation (Invitrogen). Antigen unmasking was performed in 10 mm sodium citrate, pH 6.0, with 0.1% Tween 20. Sections were then washed and blocked with TBST buffer (100 mm Tris-HCl (pH 7.5), 9% NaCl, 0.025% Triton X-100) supplemented with 1% bovine serum albumin and 10% normal goat serum and then incubated with primary antibody diluted 1:200 in 1% BSA in TBS. The secondary antibody was prepared from the biotinylated antibody stock of the Vectastain Elite ABC kit (Vector Laboratories Inc., catalog nos. PK-6101, PK-6102, and PK-6105). After incubation, sections were incubated in 3% hydrogen peroxide, followed by Vectastain Elite ABC, and finally ImmPACT DAB peroxidase substrate (Vector Laboratories, catalog no. SK4105). Sections were counterstained with hematoxylin.
TUNEL and BrdU Staining
A group of at least three mice was injected with 100 mg/kg BrdU 2 h before ionizing radiation or sacrifice, depending on experimental designs. Sections (4 μm) from paraffin-embedded intestinal bundles were subjected to TUNEL staining and BrdU staining. TUNEL staining was performed using an ApopTag kit (Chemicon International, Temecula, CA). To score the apoptotic index, the crypts with at least 17 cells in full longitudinal sections were selected. The frequency of apoptosis for each cell position from the base of crypts was recorded in 100 half-crypt sections. Following BrdU staining, the number of crypts with positive BrdU staining was recorded in more than 10 sections from each mouse. The data were reported as mean ± S.E. For Fig. 2B, the TUNEL analysis was obtained from six different locations in each mouse, and two mice of each genotype were utilized.
Colony Formation Assay
Hematopoietic stem cell survival was quantified by colony formation assay as described before with modifications (11). Adult femur bone marrow cells were obtained from eEF2K knock-out male mice or their wild type littermates (8–12 weeks of age). Five doses of 137Cs γ-irradiation were used to generate the linear regression function for stem/progenitor cell sensitivity. For 0 and 1 Gy, bone marrow cells were suspended at a concentration of 5 × 104 cells/ml into 2.5 ml of methylcellulose-based complete medium (R&D Systems); for 2 and 3 Gy, a concentration of 1 × 105 cells/ml was used; and for 4 and 5 Gy, a concentration of 2.5 × 105 cells/ml was used. The irradiated cell mixture was equally plated into two 35-mm culture dishes and incubated at 37 °C under 5% CO2. Colony number was counted 7 days after incubation by scoring the colony with more than 50 cells. The average colony number of each dose was used to calculate the survival (S) as the fraction of the nonirradiated culture. D0 was determined from the line of best fit, expressed as the dose when ln S = −1.
Crypt Microcolony Assay
The stem cell survival was quantified by scoring the number of surviving/regenerated crypts in H&E-stained cross-sections 4 days after various doses of radiation. A surviving crypt was counted as containing at least five adjacent chromophilic non-Paneth cells, one Paneth cell, and a lumen. Three mice were used in each group. The crypt survival difference was subjected to the Mann-Whitney test.
Western Blotting
Antibodies against phosphorylated eEF2, cleaved caspase-3 (5A1) (Cell Signaling), α-tubulin (B-5–1-2) (Sigma), PUMA (Novus Biologicals, Abzoom), eEF2K (Abcam), p21 (Proteintech), phospho-AKT(Ser-473) (Cell Signaling), phospho-ERK (Cell Signaling), AKT (Abzoom), ERK (Proteintech), β-actin (Transgene), and γH2AX (JBW301) (Millipore) were used. For Western blotting, cells were lysed in SDS lysis buffer (20 mm HEPES, pH 7.5, 50 mm NaCl, 25 mm KCl, 10 mm DTT, 3 mm benzamidine, 1% SDS, 1 mm sodium orthovanadate, 20 mm sodium pyrophosphate, 1 tablet of cysteine protease inhibitors/10 ml) to block the phosphorylation reaction in vitro. Western blotting was performed according to the manufacturer's instructions (Cell Signaling Inc.).
Statistics
Survival data were analyzed by the log-rank test. For other quantification results, the non-parametric two-tailed Mann-Whitney U test was used for comparison.
Study Approval
The institutional animal care and use committee at the Rutgers University Robert Wood Johnson Medical School approved the animal and surgical procedures performed in this study.
Author Contributions
This study was designed by Y. L., H.-P. C., and A. G. R. Mouse experiments were performed and analyzed by Y. L., H.-P. C., and Z. H. The manuscript was written and prepared by Y. L., H.-P. C., and A. G. R. J. J. M. did some of cell cycle analysis. K. D. and E. W. assisted in generating BMK cells. J. C. and Z. L. provided experimental material.
Acknowledgments
We thank C. W. Lu, C. Gélinas, and L. F. Liu for useful suggestions and Y. P. Chen and Y. X. Chen for technical expertise in sample preparation and immunohistochemistry.
This work was supported by National Institutes of Health Grants R01GM57300, R01CA81102, R01AG19890, RC1AI078513, R03TW008217, and R21AG042870 and the Ministry of Education of the People's Republic of China. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- eEF2
- elongation factor-2
- eEF2K
- elongation factor-2 kinase
- Gy
- gray
- IR
- irradiation
- MEF
- mouse embryonic fibroblast
- BMK
- baby mouse kidney.
References
- 1. Ryazanov A. G., Ward M. D., Mendola C. E., Pavur K. S., Dorovkov M. V., Wiedmann M., Erdjument-Bromage H., Tempst P., Parmer T. G., Prostko C. R., Germino F. J., and Hait W. N. (1997) Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 4884–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryazanov A. G., Shestakova E. A., and Natapov P. G. (1988) Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334, 170–173 [DOI] [PubMed] [Google Scholar]
- 3. Dorovkov M. V., Pavur K. S., Petrov A. N., and Ryazanov A. G. (2002) Regulation of elongation factor-2 kinase by pH. Biochemistry 41, 13444–13450 [DOI] [PubMed] [Google Scholar]
- 4. Browne G. J., Finn S. G., and Proud C. G. (2004) Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J. Biol. Chem. 279, 12220–12231 [DOI] [PubMed] [Google Scholar]
- 5. Patel J., McLeod L. E., Vries R. G. J., Flynn A., Wang X., and Proud C. G. (2002) Cellular stresses profoundly inhibit protein synthesis and modulate the states of phosphorylation of multiple translation factors. Eur. J. Biochem. 269, 3076–3085 [DOI] [PubMed] [Google Scholar]
- 6. Boyce M., Py B. F., Ryazanov A. G., Minden J. S., Long K., Ma D., and Yuan J. (2008) A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 15, 589–599 [DOI] [PubMed] [Google Scholar]
- 7. Chu H. P., Liao Y., Novak J. S., Hu Z., Merkin J. J., Shymkiv Y., Braeckman B. P., Dorovkov M. V., Nguyen A., Clifford P. M., Nagele R. G., Harrison D. E., Ellis R. E., and Ryazanov A. G. (2014) Germline quality control: eEF2K stands guard to eliminate defective oocytes. Dev. Cell 28, 561–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leprivier G., Remke M., Rotblat B., Dubuc A., Mateo A. R., Kool M., Agnihotri S., El-Naggar A., Yu B., Somasekharan S. P., Faubert B., Bridon G., Tognon C. E., Mathers J., Thomas R., et al. (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kruiswijk F., Yuniati L., Magliozzi R., Low T. Y., Lim R., Bolder R., Mohammed S., Proud C. G., Heck A. J. R., Pagano M., and Guardavaccaro D. (2012) Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci. Signal. 5, ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faller W. J., Jackson T. J., Knight J. R., Ridgway R. A., Jamieson T., Karim S. A., Jones C., Radulescu S., Huels D. J., Myant K. B., Dudek K. M., Casey H. A., Scopelliti A., Cordero J. B., Vidal M., Pende M., Ryazanov A. G., Sonenberg N., Meyuhas O., Hall M. N., Bushell M., Willis A. E., and Sansom O. J. (2015) mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee J. M., and Bernstein A. (1993) P53 mutations increase resistance to ionizing radiation. Proc. Natl. Acad. Sci. U.S.A. 90, 5742–5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeffers J. R., Parganas E., Lee Y., Yang C., Wang J., Brennan J., MacLean K. H., Han J., Chittenden T., Ihle J. N., McKinnon P. J., Cleveland J. L., and Zambetti G. P. (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4, 321–328 [DOI] [PubMed] [Google Scholar]
- 13. Potten C. S., and Grant H. K. (1998) The relationship between ionizing radiation-induced apoptosis and stem cells in the small and large intestine. Br. J. Cancer 78, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Potten C. S., Gandara R., Mahida Y. R., Loeffler M., and Wright N. A. (2009) The stem cells of small intestinal crypts: where are they? Cell Prolif. 42, 731–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clevers H. (2013) The intestinal crypt, a prototype stem cell compartment. Cell 154, 274–284 [DOI] [PubMed] [Google Scholar]
- 16. Merritt A. J., Allen T. D., Potten C. S., and Hickman J. A. (1997) Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after γ-irradiation. Oncogene 14, 2759–2766 [DOI] [PubMed] [Google Scholar]
- 17. Komarova E. A., Kondratov R. V., Wang K., Christov K., Golovkina T. V., Goldblum J. R., and Gudkov A. V. (2004) Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23, 3265–3271 [DOI] [PubMed] [Google Scholar]
- 18. Scholzen T., and Gerdes J. (2000) The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 182, 311–322 [DOI] [PubMed] [Google Scholar]
- 19. Kirsch D. G., Santiago P. M., di Tomaso E., Sullivan J. M., Hou W. S., Dayton T., Jeffords L. B., Sodha P., Mercer K. L., Cohen R., Takeuchi O., Korsmeyer S. J., Bronson R. T., Kim C. F., Haigis K. M., Jain R. K., and Jacks T. (2010) p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327, 593–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Degenhardt K., Sundararajan R., Lindsten T., Thompson C., and White E. (2002) Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem. 277, 14127–14134 [DOI] [PubMed] [Google Scholar]
- 21. Chambard J. C., Lefloch R., Pouysségur J., and Lenormand P. (2007) ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 [DOI] [PubMed] [Google Scholar]
- 22. Liang J., and Slingerland J. M. (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2, 339–345 [PubMed] [Google Scholar]
- 23. Roninson I. B., Broude E. V., and Chang B. D. (2001) If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updat. 4, 303–313 [DOI] [PubMed] [Google Scholar]
- 24. Portugal J., Mansilla S., and Bataller M. (2010) Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr. Pharm. Des. 16, 69–78 [DOI] [PubMed] [Google Scholar]
- 25. Jonathan E. C., Bernhard E. J., and McKenna W. G. (1999) How does radiation kill cells? Curr. Opin Chem. Biol. 3, 77–83 [DOI] [PubMed] [Google Scholar]
- 26. Eom Y. W., Kim M. A., Park S. S., Goo M. J., Kwon H. J., Sohn S., Kim W. H., Yoon G., and Choi K. S. (2005) Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 24, 4765–4777 [DOI] [PubMed] [Google Scholar]
- 27. Tao W. (2005) The mitotic checkpoint in cancer therapy. Cell Cycle 4, 1495–1499 [DOI] [PubMed] [Google Scholar]
- 28. van Vugt M. A., and Yaffe M. B. (2010) Cell cycle re-entry mechanisms after DNA damage checkpoints: giving it some gas to shut off the breaks! Cell Cycle 9, 2097–2101 [DOI] [PubMed] [Google Scholar]
- 29. Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., and Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 30. White-Gilbertson S., Kurtz D. T., and Voelkel-Johnson C. (2009) The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 3, 402–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith E. M., and Proud C. G. (2008) cdc2-cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J. 27, 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hizli A. A., Chi Y., Swanger J., Carter J. H., Liao Y., Welcker M., Ryazanov A. G., and Clurman B. E. (2013) Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A-cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol. Cell. Biol. 33, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Browne G. J., and Proud C. G. (2004) A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell. Biol. 24, 2986–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X., Li W., Williams M., Terada N., Alessi D. R., and Proud C. G. (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura J., Aoyagi S., Nanchi I., Nakatsuka S., Hirata E., Shibata S., Fukuda M., Yamamoto Y., Fukuda I., Tatsumi N., Ueda T., Fujiki F., Nomura M., Nishida S., Shirakata T., et al. (2009) Overexpression of eukaryotic elongation factor eEF2 in gastrointestinal cancers and its involvement in G2/M progression in the cell cycle. Int. J. Oncol. 34, 1181–1189 [PubMed] [Google Scholar]
- 36. Sanli T., Steinberg G. R., Singh G., and Tsakiridis T. (2014) AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 15, 156–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Villunger A., Michalak E. M., Coultas L., Müllauer F., Böck G., Ausserlechner M. J., Adams J. M., and Strasser A. (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302, 1036–1038 [DOI] [PubMed] [Google Scholar]
- 38. Neta R., Stiefel S. M., and Ali N. (1995) In lethally irradiated mice interleukin-12 protects bone marrow but sensitizes intestinal tract to damage from ionizing radiation. Ann. N.Y. Acad. Sci. 762, 274–280; discussion 280–271 [DOI] [PubMed] [Google Scholar]
- 39. Paris F., Fuks Z., Kang A., Capodieci P., Juan G., Ehleiter D., Haimovitz-Friedman A., Cordon-Cardo C., and Kolesnick R. (2001) Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 293, 293–297 [DOI] [PubMed] [Google Scholar]
- 40. Leibowitz B. J., Qiu W., Liu H., Cheng T., Zhang L., and Yu J. (2011) Uncoupling p53 functions in radiation-induced intestinal damage via PUMA and p21. Mol. Cancer Res. 9, 616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sullivan J. M., Jeffords L. B., Lee C. L., Rodrigues R., Ma Y., and Kirsch D. G. (2012) p21 protects “Super p53” mice from the radiation-induced gastrointestinal syndrome. Radiat. Res. 177, 307–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qiu W., Carson-Walter E. B., Liu H., Epperly M., Greenberger J. S., Zambetti G. P., Zhang L., and Yu J. (2008) PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2, 576–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galluzzi L., Vitale I., Vacchelli E., and Kroemer G. (2011) Cell death signaling and anticancer therapy. Front. Oncol. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gudkov A. V., and Komarova E. A. (2010) Radioprotection: smart games with death. J. Clin. Invest. 120, 2270–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mathew R., Degenhardt K., Haramaty L., Karp C. M., and White E. (2008) Immortalized mouse epithelial cell models to study the role of apoptosis in cancer. Methods Enzymol. 446, 77–106 [DOI] [PubMed] [Google Scholar]
- 46. Degenhardt K., and White E. (2006) A mouse model system to genetically dissect the molecular mechanisms regulating tumorigenesis. Clin. Cancer Res. 12, 5298–5304 [DOI] [PubMed] [Google Scholar]