

Abstract
The A-kinase anchoring protein (AKAP) GSK3β interaction protein (GSKIP) is a cytosolic scaffolding protein binding protein kinase A (PKA) and glycogen synthase kinase 3β (GSK3β). Here we show that both the AKAP function of GSKIP, i.e. its direct interaction with PKA, and its direct interaction with GSK3β are required for the regulation of β-catenin and thus Wnt signaling. A cytoplasmic destruction complex targets β-catenin for degradation and thus prevents Wnt signaling. Wnt signals cause β-catenin accumulation and translocation into the nucleus, where it induces Wnt target gene expression. GSKIP facilitates control of the β-catenin stabilizing phosphorylation at Ser-675 by PKA. Its interaction with GSK3β facilitates control of the destabilizing phosphorylation of β-catenin at Ser-33/Ser-37/Thr-41. The influence of GSKIP on β-catenin is explained by its scavenger function; it recruits the kinases away from the destruction complex without forming a complex with β-catenin. The regulation of β-catenin by GSKIP is specific for this AKAP as AKAP220, which also binds PKA and GSK3β, did not affect Wnt signaling. We find that the binding domain of AKAP220 for GSK3β is a conserved GSK3β interaction domain (GID), which is also present in GSKIP. Our findings highlight an essential compartmentalization of both PKA and GSK3β by GSKIP, and ascribe a function to a cytosolic AKAP-PKA interaction as a regulatory factor in the control of canonical Wnt signaling. Wnt signaling controls different biological processes, including embryonic development, cell cycle progression, glycogen metabolism, and immune regulation; deregulation is associated with diseases such as cancer, type 2 diabetes, inflammatory, and Alzheimer's and Parkinson's diseases.
Keywords: A-kinase anchoring protein (AKAP), beta-catenin (β-catenin), cell compartmentalization, glycogen synthase kinase 3 (GSK-3), Wnt signaling, GSKIP, glycogen synthase kinase 3β interaction protein, protein kinase
Introduction
A-kinase anchoring proteins (AKAPs) 3 are a family of about 50 scaffolding proteins. Their conserved function is the compartmentalization of protein kinase A (PKA). PKA holoenzyme consists of a dimer of regulatory (RIα, RIβ, RIIα, or RIIβ) and two catalytic subunits each bound to one R subunit. AKAPs directly interact with R subunits and tether the kinase to defined cellular compartments such as vesicles, the sarcoplasmic reticulum, or the cytoskeleton. This compartmentalization confers a tight spatiotemporal control to PKA signaling, and enables PKA to elicit a specific cellular response to each of the many stimuli that cause cAMP elevation and thereby lead to activation of this ubiquitous kinase. AKAPs directly interact with further signaling proteins, thus mediating crosstalk between signaling systems: phosphatases, dephosphorylating PKA-phosphorylated substrates, adenylyl cyclases, synthesizing cAMP, and phosphodiesterases (PDEs), hydrolyzing cAMP. Several AKAPs bind further kinases such as protein kinase C (PKC), which are activated by signals other than cAMP, e.g. Ca2+. AKAPs and their interactions play key roles in a variety of physiological processes such as vasopressin-mediated water reabsorption in renal principal cells and cardiac myocyte contractility (1–5, 7, 8).
A new example of an AKAP that mediates crosstalk is the cytoplasmic GSK3β interaction protein (GSKIP). It binds PKA and GSK3β (9). The ubiquitously expressed serine/threonine protein kinase GSK3β is a component of multiple signaling systems such as canonical Wnt, insulin, Hedgehog, Notch, and TGFβ signaling. GSK3β is constitutively active. It can be inactivated by phosphorylation of Ser-9 by multiple kinases including protein PKA (10), PKB (11), and p38 MAPK (12). GSK3β is involved in the regulation of different biological processes, e.g. embryonic development, cell cycle progression, glycogen metabolism, and immune regulation. Deregulation of GSK3β is associated with pathologies such as cancer, type 2 diabetes, bipolar disorder, cardiac hypertrophy inflammatory, and Alzheimer's and Parkinson's diseases (13–16). The diversity of GSK3β functions is also reflected by its presence in different cellular compartments; GSK3β is located in the cytosol, at the plasma membrane, in the nucleus, and in mitochondria (17, 18).
In canonical Wnt signaling, GSK3β assembles with Axin, β-catenin, adenomatous polyposis coli (APC), and casein kinase 1 (CK1) in the destruction complex in the cytosol. In the absence of a Wnt signal, GSK3β phosphorylates Axin-bound β-catenin at Thr-41, Ser-33 and Ser-37 (19–23). This targets β-catenin for ubiquitination and proteasomal degradation and inhibits Wnt signaling (24). Wnt signaling is activated by binding of Wnt ligands to receptor complexes at the plasma membrane, consisting of LRP5/6 single-pass transmembrane protein and G protein-like receptors of the Frizzled (Fz) family. The β-catenin destruction complex is recruited to the LRP5/6 receptor, accompanied by an inhibition of degradation of β-catenin. Then β-catenin accumulates and translocates into the nucleus where it induces expression of Wnt target genes (25, 26).
GSKIP seems able to induce β-catenin accumulation in the cytoplasm and the nucleus and to activate transcription through direct interaction with GSK3β when overexpressed in HeLa and neuroblastoma SH-SY5Y cells (27, 28). In SH-SY5Y cells, the GSKIP overexpression blocked neurite outgrowth during retinoic acid-mediated differentiation. Although the data were obtained in cell systems overexpressing GSKIP, they suggested that GSKIP acts as a regulator in canonical Wnt signaling.
We observed that GSKIP facilitates the phosphorylation of GSK3β by PKA at Ser-9 and thereby its inhibition (9), and that GSKIP deficiency modulates this phosphorylation and thus GSK3β activity during development (29). The loss of GSKIP in mice causes a cleft palate, resembling the one in Gsk3β−/− mice (30), and perinatal lethality through respiratory distress (29). Knock-out of any of the subunits of PKA does not cause a cleft palate. Thus GSKIP and GSK3β are crucial for normal craniofacial development. This most likely does not involve Wnt signaling, as Wnt signals apparently do not influence phosphorylation of Ser-9 of GSK3β (31).
Here we show that GSKIP affects specific phosphorylations of β-catenin and thus Wnt signaling through its direct interactions with PKA and GSK3β. To our knowledge, this is the first time that a physiological function can be ascribed to a cytosolic AKAP-PKA interaction.
Results
Endogenous GSKIP Regulates Wnt Signaling through Phosphorylation of β-Catenin by PKA
The scaffolding protein assembling the destruction complex is Axin. Axin exists in two isoforms, Axin1 and Axin2: Axin1 is constitutively expressed, and Axin2 expression is regulated through Wnt signaling (32, 33). GSK3β phosphorylates Axin-bound β-catenin within the destruction complex at Ser-33/Ser-37/Thr-41, targeting it for proteasomal degradation. We initially examined whether endogenous GSKIP influences the composition of the destruction complex. We knocked down GSKIP in HEK293 cells using siRNA and immunoprecipitated endogenous GSK3β (Fig. 1). As expected, GSK3β co-immunoprecipitated with Axin1 in the presence of a control non-targeting siRNA (siNT). The siRNA-mediated reduction of GSKIP increased the co-immunoprecipitation of GSK3β and Axin1, indicating that GSKIP affects the composition of the complex by recruiting GSK3β.
FIGURE 1.
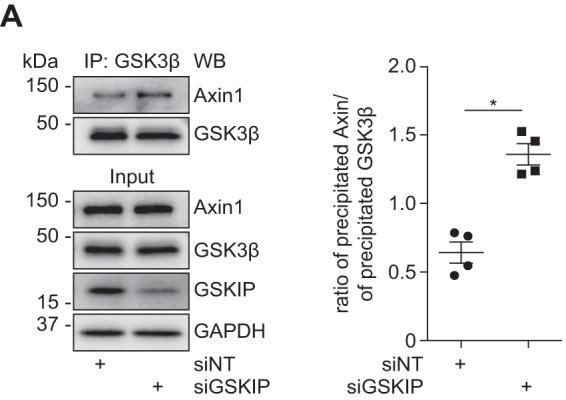
GSKIP recruits GSK3β from Axin. HEK293 cells were treated with siRNA to knock down the expression of GSKIP (siGSKIP) or with control non-targeting siRNA (siNT). GSK3β was immunoprecipitated, and GSK3β and co-immunoprecipitated Axin1 were detected by Western blotting (WB). Signals were semi-quantitatively analyzed by densitometry, and the ratio of precipitated Axin1 to GSK3β was calculated; n = 4, mean ± S.E., ANOVA, *, p < 0.05.
Next, we performed siRNA experiments to assess the role of endogenous GSKIP in coordinating PKA in Wnt signaling. PKA phosphorylates β-catenin at Ser-675 and thereby stabilizes the protein (34, 35). Cytosols from HEK293, A549, and HeLa cells were analyzed by Western blotting. In the absence of a Wnt signal, the level of β-catenin, unphosphorylated or phosphorylated at Ser-675, was low (Fig. 2, A–C). Wnt3a stimulation increased β-catenin expression, which was enhanced by the knockdown of GSKIP. Thus the reduction of the scaffold GSKIP apparently sets free the kinase, which then phosphorylates its substrate. Inhibition of PKA with the stearate-coupled and thus membrane-permeant specific heat-stable PKA inhibitor peptide (PKI) or H89, either alone or in combination with the GSKIP siRNA, inhibited the increase in Ser-675 phosphorylation.
FIGURE 2.

Endogenous GSKIP enhances Wnt signaling through controlling PKA phosphorylation of β-catenin at Ser-675. HEK293, A549, and HeLa cells were left untreated or treated as indicated with siRNA to knock down GSKIP (siGSKIP), control siRNA (siNT), Wnt3a-enriched medium, or control medium in the absence or presence of the PKA inhibitors, PKA inhibitor peptide (PKI) or H89, or the proteasome inhibitor MG-132. A–C, cytosol fractions were purified from the three cell types and analyzed by Western blotting (WB) for expression of the indicated proteins. The signals were densitometrically analyzed, and ratios of β-catenin/GAPDH and Ser(p)675-β-catenin/GAPDH were calculated; n ≥ 6, mean ± S.E., ANOVA, **, p < 0.01, ***, p < 0.001. D, TOPflash/FOPflash (TOP/FOP) luciferase assay. HEK293 cells were stimulated with Wnt-enriched or control medium; n = 4, mean ± S.E., ANOVA, **, p < 0.01. E, cell lysates were purified from HEK293 cells, and the indicated proteins were detected by Western blotting. Semi-quantitative densitometric analysis of Western blots is depicted as ratios of Ser(P)1490 LRP6/GAPDH and Ser(P)9 GSK3β/GAPDH; n = 6, mean ± S.E., ANOVA, *, p < 0.05, ***, p < 0.001. F, cytosolic fractions were purified, and the indicated proteins were detected by Western blotting. Semi-quantitative densitometric analysis of Western blots shows the ratios of β-catenin/GAPDH and (Ser(P)33/Ser-37/Thr-41)-β-catenin/GAPDH; n = 6, mean ± S.E., ANOVA, ***, p < 0.001. pS33/S37/T41, Ser(P)33/Ser-37/Thr-41. G, the enrichment of the indicated proteins in the cytosolic fractions of HEK293 cells that were used in A was confirmed by Western blotting. The cells were treated with Wnt3a-enriched or control medium. Shown are representative blots from n ≥ 4 experiments.
To measure Wnt signaling activity, we performed Wnt reporter assays using the TOPflash/FOPflash reporter system (36). HEK293 cells were transfected with siRNAs together with the reporter vectors and a vector encoding Renilla luciferase, used as an internal standard. Luciferase activity was measured 48 h later. The measurements showed that the knockdown of GSKIP and the associated increase in Ser-675 phosphorylation caused an increase in luciferase activity, indicative of an increase in β-catenin-mediated transcription (Fig. 2D).
Binding of Wnt to its receptor causes phosphorylation of LRP6 at Ser-1490 by GSK3β, and other kinases. This phosphorylation was not affected by the knockdown of GSKIP (Fig. 2E). The knockdown also did not result in a significant change of the level of Ser(P)-9 GSK3β, indicating that Ser-9 is not involved in control of GSK3β over Wnt signaling if GSKIP is reduced (Fig. 2E). In addition, GSKIP knockdown did not alter the phosphorylation of β-catenin at Ser-33/Ser-37/Thr-41 in the presence of the proteasome inhibitor, MG-132 (Fig. 2F). MG-132 was used to obtain signals in the Western blot as the phosphorylation marks β-catenin for proteasomal degradation. The actual enrichment of cytosolic proteins in the cytosolic fraction used for the experiments such as those depicted in Fig. 2, A–C, was confirmed by Western blotting (Fig. 2G).
Direct Interactions of GSKIP with Both PKA and GSK3β Are Required for Control of Wnt Signaling under Resting Conditions
To confirm the requirement of the direct interaction between GSKIP and PKA for the control of β-catenin, we generated a PKA-binding-deficient (GSKIP-N42I) variant of GSKIP, and as a control, the previously described GSK3β-binding-deficient GSKIP variant, GSKIP-L130P (27, 37). We initially analyzed the interaction of the GSKIP variants with PKA by cAMP-agarose precipitation. cAMP-agarose precipitates regulatory subunits of PKA and the associated AKAPs. When compared with the wild type and GSKIP-L130P, little GSKIP-N42I was detectable in the precipitates, confirming its inability to interact with PKA (Fig. 3A). To analyze binding of the GSKIP variants to endogenous GSK3β, tagged proteins were expressed in HEK293 cells. Endogenous GSK3β was immunoprecipitated, and Western blotting was used to detect GSKIP or the variants (Fig. 3B). Wild type GSKIP and GSKIP-N42I co-precipitated with GSK3β, whereas GSKIP-L130P did not. Similar experiments where GSKIP was immunoprecipitated via its FLAG tag were also performed (Fig. 3C).
FIGURE 3.

Elevated levels of GSKIP cause up-regulation of Wnt signaling under resting conditions through interactions with both PKA and GSK3β. A, HEK293 cells transiently expressing an empty control vector or the indicated GSKIP variants were subjected to cAMP-agarose precipitations. GSKIP, regulatory RIIα subunits of PKA, and HSP90 (as a control) were detected by Western blotting (WB). Semi-quantitative densitometric analysis of results is shown as the ratio of the GSKIP pulldown (PD) to GSKIP in the input. The interaction of regulatory RIIα subunits of PKA with GSKIP-N42I is strongly reduced, but not that with the GSK3β-binding-deficient mutant CFP-GSKIP-L130P; n = 5, mean ± S.E., ANOVA, **, p < 0.01. B and C, endogenous GSK3β (B) or FLAG-tagged GSKIP versions (C) were immunoprecipitated (IP; IgG, negative control immunoprecipitation). Precipitated proteins were detected by Western blotting, and signals were analyzed by semi-quantitative densitometry. Depicted are the ratios of the GSKIP IP to the GSK3β IP × GSKIP input (B) and of the GSK3β IP to the GSKIP IP (C); n = 3, mean ± S.E., ANOVA, *, p < 0.05, **, p < 0.01. D, TOPflash/FOPflash (TOP/FOP) luciferase assays using HEK293 cells expressing the indicated proteins in the absence of a Wnt signal (Wnt; n = 4, mean ± S.E., ANOVA, *, p < 0.05, **, p < 0.01.
Only the wild type GSKIP triggered a significant increase in basal β-catenin-induced transcription, whereas no changes were observed in the presence of the PKA-binding-deficient or GSK3β-binding-deficient variants of GSKIP (Fig. 3D). These findings show that the interactions of GSKIP with both PKA and GSK3β are required for β-catenin-dependent transcription under resting conditions.
Interactions of GSKIP with Both PKA and GSK3β Are Required for Wnt-stimulated Transcriptional Activation through β-Catenin
HEK293 cells were transfected with GSKIP, GSKIP-L130P, or GSKIP-N42I together with TOPflash/FOPflash. Stimulation for 24 h with Wnt3a-conditioned medium increased Wnt signaling in the presence of wild type GSKIP (Fig. 4A). Ablation of binding to either PKA or GSK3β prevented the Wnt-induced transcription. We further examined whether the PKA-binding-deficient GSKIP-N42I would affect the PKA phosphorylation of β-catenin (Fig. 4, B and C). Surprisingly, and in contrast to the increase in Ser-675 phosphorylation if GSKIP was down-regulated, the loss of PKA binding to GSKIP did not alter the phosphorylation of Ser-675 in the presence of Wnt. We also investigated the influence of GSKIP on Wnt signaling following stimulation with recombinant Wnt3a in a concentration-dependent manner for 24 h. Wnt3a and GSKIP expression activated Wnt signaling additively (Fig. 4D).
FIGURE 4.

The interaction of GSKIP with both GSK3β and PKA is required for enhancing Wnt signaling in the presence of a Wnt stimulus. A, HEK293 cells expressing the indicated proteins were subjected to TOPflash/FOPflash (TOP/FOP) luciferase assays to detect Wnt signaling activity in the presence of Wnt-conditioned medium. n = 4, mean ± S.E., ANOVA, *, p < 0.05. B, cytosolic fractions were purified, and the indicated proteins were detected by Western blotting. C, the signals were semi-quantitatively analyzed by densitometry; n = 5, mean ± S.E., ANOVA, *, p < 0.05, **, p < 0.01, ***, p < 0.001. D, HEK293 cells expressing an empty vector control or wild type CFP-GSKIP were stimulated with increasing amounts of recombinant Wnt3a, and TOPflash/FOPflash luciferase assays were performed to determine Wnt signaling activity; n = 4, mean ± S.E., ANOVA, ***, p < 0.001.
We inhibited the proteasomal degradation of proteins with MG-132 using HEK293 cells that expressed the GSKIP variants. MG-132 increased the levels of β-catenin phosphorylated at Ser-33/Ser-37/Thr-41 (19, 38) and of β-catenin (Fig. 5A). However, only in the presence of wild type GSKIP, Ser(P)-33/Ser-37/Thr-41 β-catenin was reduced significantly and cytosolic β-catenin increased (Fig. 5, A and B).
FIGURE 5.

Elevated levels of GSKIP enhance Wnt signaling through controlling the GSK3β-dependent phosphorylation of β-catenin at Ser-33/Ser-37/Thr-41. A, HEK293 cells expressing an empty vector control or wild type FLAG-GSKIP were left untreated or treated with the proteasome inhibitor, MG-132. Cytosolic fractions were purified, and the indicated proteins were detected by Western blotting. Semi-quantitative densitometric analyses of the signals depict the ratios of β-catenin to GAPDH and of (Ser(P)-33/Ser-37/Thr-41)-β-catenin to GAPDH; n = 5, mean ± S.E., ANOVA, **, p < 0.01, ***, p < 0.001. pS33/S37/T41, Ser(P)-33/Ser-37/Thr-41. B, HEK293 cells expressing an empty vector control or the indicated FLAG-GSKIP variants were left untreated or treated with MG-132, and cytosolic fractions were purified and subjected to Western blotting. Semi-quantitative densitometric analysis of the signals is depicted as the ratio of (Ser(P)-33/Ser-37/Thr-41)-β-catenin to GAPDH; n = 8, mean ± S.E., ANOVA, **, p < 0.01, ***, p < 0.001. C, GSKIP and β-catenin do not form a complex in cells. HEK293 cells expressing FLAG-GSKIP or an empty control vector were subjected to an immunoprecipitation using anti-FLAG antibody. Precipitated FLAG-GSKIP and endogenous β-catenin were detected by Western blotting (WB).
Thus collectively, it appears that GSKIP must interact with both kinases to regulate Wnt-dependent transcription through β-catenin. It involves phosphorylation of Ser-33/Ser-37/Thr-41 and Ser-675 of β-catenin by GSK3β and PKA, respectively. When GSKIP expression is knocked down, i.e. probing the endogenous role of GSKIP, the mode of action is independent from the previously observed Ser-9 phosphorylation of GSK3β by PKA, which GSKIP facilitates (9). GSKIP and β-catenin did not co-immunoprecipitate, indicating that stable complex formation of the two proteins is not required for the observed GSKIP-mediated control of β-catenin phosphorylation (Fig. 5C).
GSKIP-dependent Regulation of β-Catenin Is Specific for this AKAP
Besides GSKIP, two other AKAPs, MAP2 and AKAP220 (AKAP11), bind both PKA and GSK3β (9, 39–41). AKAP220 and GSKIP both facilitate phosphorylation of GSK3β at Ser-9 by PKA and thus inhibition of GSK3β (9, 39). The binding sites for PKA are conserved within the AKAP family. The site in AKAP220 was previously mapped (41). We examined whether AKAP220 binds GSK3β in a similar way as other GSK3β-interacting proteins, namely by a GSK3β interaction domain (GID). AKAP220 binds GSK3β directly in a region that spans amino acid residues 1017–1386 (39). We performed protein sequence alignment of mammalian Axin1/2, GSKIP, and AKAP220, which suggested a putative GID in this region, because they show strong conservation among the homologs and contain central FLL motifs that are identical at the critical positions (Fig. 6A). In addition, the motif was predicted to form an α-helix, as is true of other GIDs (42). We spot-synthesized this region of AKAP220 as 25-mer overlapping peptides and investigated interactions of the peptides with recombinant GST-GSK3β that had been pre-exposed to peptides representing the GSK3β-binding domain of GSKIP (GSKIPtide) or with the GSK3β-binding-deficient variant GSKIPtide-L130P (9, 27) (Fig. 6B). Remarkably, binding of GSK3β to spots A8, A9, and A10 of human AKAP220 was observed in the presence of GSKIPtide-L130P, but binding was strongly reduced in the presence of GSKIPtide. We performed alanine scans to identify single amino acid residues of the AKAP220 GID that contribute to GSK3β binding (Fig. 6C). Substitutions revealed that Phe-1162, Leu-1166, and Leu-1170 were each essential for binding. This shows that as in Axin1/2 and GSKIP, an FLL motif within the GID of AKAP220 is required for the interaction with GSK3β.
FIGURE 6.

GSK3β binds AKAP220 at a conserved GID. A, alignment of the GIDs from the indicated human (Hs), rat (Rn), and mouse (Mm) proteins, and prediction of AKAP220 three-dimensional structure in the 1162–1176 region through secondary structure analysis using PSIPRED at the University College London (UCL) Department Of Computer Science. aa, amino acid. B, amino acid residues 1017–1386 of AKAP220 were spot-synthesized as 25-mer overlapping peptides and overlaid with GST alone or with GST-GSK3β that was pre-incubated with either the inactive control peptide GSKIPtide-L130P or the peptide GSKIPtide, which blocks the interaction of GSK3β with spot-synthesized peptides. Interaction of GSK3β with peptides was detected using anti-GSK3β antibodies. The sequences of the GSK3β-binding peptides in positions A8–A10 of the peptide array are shown. The sequence common in all three peptides is highlighted. C, the GID of AKAP220, amino acids 1155–1179, was spot-synthesized as 25-mer peptides (wild type (wt)) or as peptides where each amino acid was substituted by an alanine. The peptides were overlaid with recombinant GSK3β, and interaction was detected using anti-GSK3β antibodies.
GSKIP, Axin, and AKAP220 Compete for GSK3β Binding
Differently tagged GSK3β, AKAP220, Axin1, GSKIP, and combinations thereof were produced in HEK293 cells, followed by co-immunoprecipitation and Western blotting. GSK3β precipitated with AKAP220, Axin, and GSKIP (Fig. 7A). The level of AKAP220 was reduced upon co-expression of Axin1, and GSKIP co-expression strongly diminished the AKAP220 level. Co-immunoprecipitated AKAP220 was then calculated as the ratio to the input of the competitors, which was reduced when cells co-expressed Axin1 and even more so with GSKIP (Fig. 7A). We then examined whether GSKIP competes with Axin1/2 for GSK3β binding by co-immunoprecipitation and Western blotting, as GSKIP and Axin1/2 share a common motif for binding GSK3β (Fig. 7B) (27). In cells expressing Axin1 or Axin2, the amount of GSKIP bound to GSK3β was significantly reduced (Fig. 7B). These results indicate that AKAP220, GSKIP, and Axin1 compete for the binding of GSK3β. As these proteins may assemble with GSK3β in different cellular compartments, Axin, AKAP220, and GSKIP might establish functionally different pools of GSK3β.
FIGURE 7.

GSKIP, Axin1, Axin2, and AKAP220 compete for GSK3β binding. A, immunoprecipitation of YFP-GSK3β from HEK293 cells and detection of the indicated proteins by Western blotting (WB). The semi-quantitative analysis of co-immunoprecipitated HA-AKAP220 is depicted as the ratio of precipitated HA-AKAP220 to HA-AKAP220 in the input; data are normalized to cells expressing YFP-GSK3β and HA-AKAP220; n = 6, mean ± S.E., ANOVA, *, p < 0.05. B, co-immunoprecipitation of YFP-GSK3β and the indicated proteins from HEK293 cells. The proteins were detected by Western blotting. Semi-quantitative densitometric analysis of the signals is depicted as the ratio of precipitated GSKIP to precipitated GSK3β; n = 5, mean ± S.E., ANOVA, **, p < 0.01.
GSKIP and AKAP220 Play Different Roles in Wnt Signaling
We compared the influence of GSKIP and AKAP220 on β-catenin-dependent transcription. Fig. 3D shows that only GSKIP activated the luciferase reporter system in HEK293 cells under resting conditions. In HEK293 cells stimulated for 4 h with Wnt, only GSKIP enhanced the reporter system but not AKAP220 (Fig. 4A). The cytosolic fractions of untreated HEK293 cells and cells treated with Wnt3a were enriched, and β-catenin levels were monitored by Western blotting (Fig. 8). These levels were strongly increased in the Wnt-stimulated samples. The effect of Wnt on β-catenin levels significantly increased in the presence of GSKIP, but not in the presence of AKAP220. As in the presence of elevated levels of GSKIP (Fig. 4), the elevation of AKAP220 levels did not affect the phosphorylation of Ser-675 of β-catenin (Fig. 8). Axin1 up-regulation was observed upon expression of wild type GSKIP in unstimulated cells but not in the presence of AKAP220. This was not seen upon Wnt stimulation. Thus GSKIP and AKAP220 exert different effects on β-catenin, although both bind GSK3β and PKA. This suggests that they control different pools of GSK3β and PKA that influence the Wnt signaling destruction complex in different ways.
FIGURE 8.
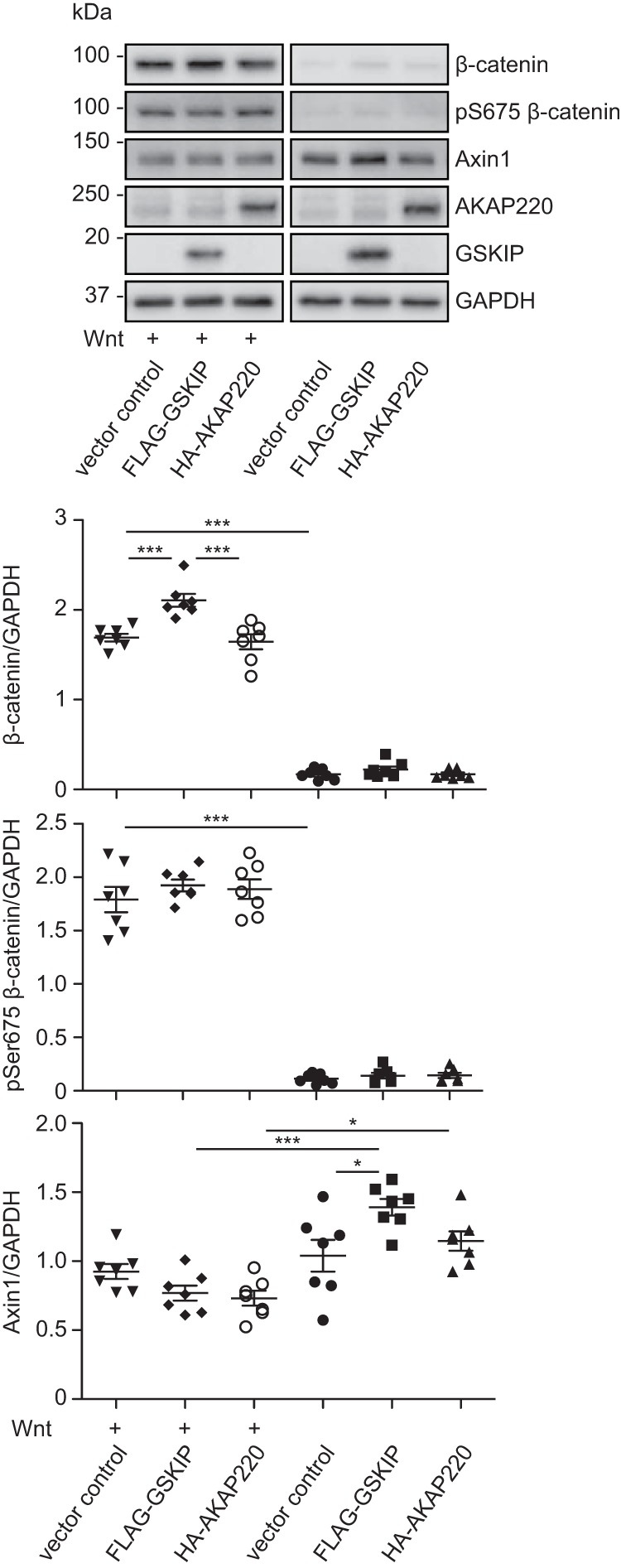
GSKIP but not AKAP220 elevates the level of β-catenin in the cytosol in response to a Wnt signal. HEK293 cells expressing the wild type GSKIP or AKAP220 or an empty vector control were stimulated with control or Wnt3a-enriched medium, cytosolic fractions were prepared, and the indicated proteins were detected by Western blotting. The signals were semi-quantitatively analyzed by densitometry and depicted as ratios; n = 7, mean ± S.E., ANOVA, *, p < 0.05, ***, p < 0.001. pS675, Ser(P)-675.
Discussion
Our findings demonstrate that GSKIP regulates Wnt signaling through its direct protein-protein interactions with both GSK3β and PKA. The two GSKIP-bound kinases differentially modulate the phosphorylation and thereby the stability of β-catenin (Fig. 9). GSKIP apparently has a “scavenger” activity for GSK3β and PKA that is independent of a direct interaction of GSKIP with the destruction complex. Instead, GSKIP establishes an independent GSK3β/PKA pool. We observed a role of GSKIP in Wnt signaling in three different cell lines. The ubiquitous expression of all three, GSKIP, GSK3β, and PKA, suggests that the complex regulates canonical Wnt signaling in a broad range of physiological and disease contexts in which the pathway has been implicated.
FIGURE 9.
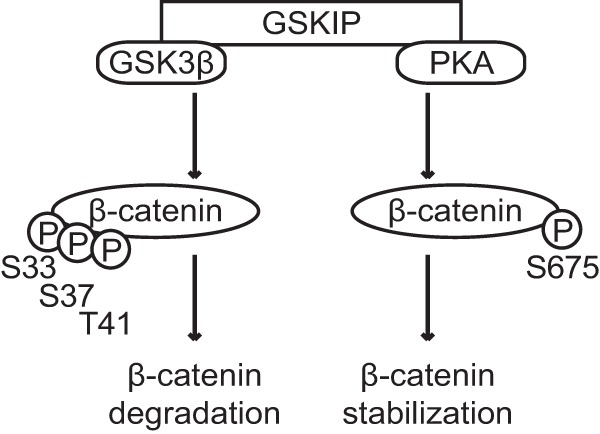
Model of GSKIP-directed regulation of β-catenin and thus of Wnt signaling. GSKIP directly binds GSK3β and PKA. It controls the stability of β-catenin and thus its transcriptional activity through facilitating its phosphorylation (indicated by circled P) by the two kinases. GSK3β phosphorylates Ser-33/Ser-37/Thr-41 in the cytoplasm and targets it for proteasomal degradation. The phosphorylation of Ser-675 by PKA stabilizes β-catenin and enhances Wnt-induced signaling.
Most AKAPs possess unique targeting domains, which direct the AKAP-PKA complex to defined cellular compartments including membranes, cytoskeletal components, or other structures (2, 3, 44, 45). Some AKAPs, however, lack canonical targeting domains. They can both reside in the cytosol and constitutively associate with other compartments, or they can reside in the cytosol and reversibly associate with other compartments through dynamic regulatory mechanisms. For example, AKAP18δ is a cytosolic protein, but positively charged amino acids distributed across its surface permit it to associate with membrane lipids in renal collecting duct principal cells (46, 47). The AKAP Gravin is released from the plasma membrane into the cytosol in response to an elevation of intracellular calcium in HEC-1A endometrial cancer cells (48). GSKIP is cytosolic (Fig. 2G) (9), and there is no evidence of a stimulus-dependent redistribution to another compartment. Its location is not altered, for example, by retinoic acid (28), an elevation of cAMP (9), or Wnt stimulation. Thus for the first time, our data ascribe a physiological function to a strictly cytosolic AKAP-PKA interaction. Based on the conservation and ubiquitous expression of GSKIP (9), the interaction presumably plays a role in Wnt signaling in many types of cells. Because GSKIP is an RII-specific AKAP (9), Wnt signaling must be controlled by PKA type II, the PKA that contains RII subunits. PKA is involved in the control of Wnt signaling in various processes. For example, parathyroid hormone stimulation promotes osteoblast differentiation, a process that involves phosphorylation of LRP5/6 by PKA (49), and PKA-dependent phosphorylation and thus inhibition of GSK3β (50). Whether such processes require an interaction between PKA and AKAPs, and specifically with GSKIP, remains to be determined.
We asked whether the effects observed for GSKIP are specific for this one AKAP, or whether an effect on Wnt signaling can also be achieved through a simple scaffolding of PKA and GSK3β by another AKAP such as AKAP220. GSKIP and AKAP220 differ in their cellular location: GSKIP is cytoplasmic (9, 27), whereas AKAP220 is predominantly found at the plasma membrane and on peroxisomes (41, 51–53). GSKIP does not interact with β-catenin, whereas AKAP220 forms a complex with β-catenin and cadherin at the plasma membrane, which competes for β-catenin in Wnt signaling (51, 54). However, a direct competition would affect Wnt signaling activity and cytosolic β-catenin levels. This effect does not seem to explain our observations and emphasizes the neutrality of AKAP220 in Wnt signaling. This is also in line with observations that the role of the AKAP220-cadherin complex in endothelial barrier function is independent of Wnt signaling (51). These effects can best be explained by the fact that the two AKAPs are localized to different cellular compartments, which most likely leads to distinct pools of GSK3β that are regulated in different ways. This would explain why AKAP220, in contrast to GSKIP, does not influence the availability of GSK3β for the cytoplasmic destruction complex.
Here we also show that a common GID mediates the interaction of AKAP220, Axin1/2, and GSKIP with GSK3β. A crystal structure of the complex of GSK3β with the GID of Axin has revealed the molecular determinants of this interaction (PDB: 1O9U) (42). The GID of Axin forms an amphipathic helix whose hydrophobic face docks into a hydrophobic groove on the surface of GSK3β. Like Axin 1/2 and GSKIP, the GID of human AKAP220 contains an FLL motif and thus is likely to interact with GSK3β in a similar way. The presence of the GID domain explains the competition between Axin1/2, AKAP220, and GSKIP for binding to GSK3β and provides further evidence that the scaffolding proteins define distinct pools of this kinase. An additional motif was recently discovered in a nearby region in AKAP220, identifying Thr-1132, which can be phosphorylated by GSK3β itself to permit binding (41). However, a previous study demonstrated binding between kinase-dead GSK3β and AKAP220 (39), which suggests the existence of further points of contact between AKAP220 and GSK3β (41), likely the FLL motif we identified.
By adding a new level to our understanding of Wnt signaling control, our findings may have important clinical implications. Increased Wnt signaling activity plays a role in various cancers, neurological diseases, fibrosis, and other diseases (55, 56). A duplication of the chromosomal region encoding GSKIP predisposes to myeloid lymphoma, presumably due to increased GSKIP protein, inhibition of GSK3β, and increased Wnt signaling (57). A general challenge in targeting the Wnt signaling pathway for therapeutic purposes is its complex crosstalk with MAPK, bone morphogenetic protein (BMP), Hedgehog, Notch, cAMP/PKA, and other pathways (50, 56, 58, 59). Targeting GSK3β itself is equally challenging as it would likely cause side effects due its broad involvement in several of these pathways. An option might be to displace a specific pool of GSK3β from a cellular compartment where it has specific, undesirable effects on Wnt signaling. GSKIP, AKAP220, and Axin establish different pools of GSK3β, but GSK3β interacts with further proteins including MAP2, FRAT1, -2, and -3, and DISC1. Our work opens a door on understanding the interplay of these GSK3β interactions with regard to Wnt signaling. The interaction of GSKIP with GSK3β in the cytosol increases Wnt signaling. It would be worthwhile to evaluate the therapeutic potential of pharmacological interference with this interaction, because GSKIP is up-regulated in breast cancer, melanoma, and other forms of cancer that also exhibit a deregulation of Wnt signaling. Targeting AKAP-dependent protein-protein interactions is feasible and may be a novel approach in the treatment of various diseases (7, 45, 60).
Experimental Procedures
GSKIP Variants
Mutagenesis was carried out using a pECFP-GSKIP plasmid (9) with the QuikChange II Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer's recommendation. To prevent the interaction of GSKIP with PKA, asparagine 42 was replaced by isoleucine. To generate a GSKIP variant deficient for binding of GSK3β, leucine 130 was replaced by proline (37). The following primers were used: N42IFwd, CTC GAA GCT GAA GCA GTT GTA AAT GAT GTT CTC TTT GCT; N42IRev, TCG TTT CTC TTG TAG TAA ATG TTG ACG AAG TCG AAG CTC; L130PFwd, CGC ACT GCT TCA AAG ACC GGA AGC TTT GAA AAG AG; and L130PRev, CTC TTT TCA AAG CTT CCG GTC TTT GAA GCA GTG CG, thus generating the plasmids pECFP-GSKIP-N42I and pECFP-GSKIP-L130P. The cDNAs were cloned into pCMV6ev (OriGene) to generate the vectors pCMV6-GSKIP-FLAG, pCMV6-GSKIP-L130P-FLAG, and pCMV6-GSKIP-N42I-FLAG.
Cell Culture, Transfection, and Fractionation
HEK293, A549, and HeLa cells were grown in DMEM (GlutaMAX, 10% FCS, Thermo Fisher Scientific) and transfected with the above mentioned plasmids, the vectors pECFP, pEYFP, pEYFP-GSK3β, pΔECFP-Myc-GSKIP (CFP deleted), pcDNA3.1-FLAG-Axin1, pcDNA3.1-FLAG-Axin2, TOPflash, FOPflash, pRL-SV40, and pCGN-HA-AKAP220 (kindly provided by K. Taskén, The Biotechnology Centre of Oslo, University of Oslo, Norway), SMARTpool siGENOME GSKIP siRNA, and siGENOME Non-Targeting siRNA #2 (GE Healthcare, Chalfont St Giles, UK) using Lipofectamine2000 (Thermo Fisher Scientific) (9).
For cell fractionation and cytosol purification, a digitonin semi-permeabilization protocol was adapted (61). Briefly, HEK293 cells were treated with Wnt-enriched or control medium for 4 h, washed twice with PBS, and semi-permeabilized with Digitonin (61). The lysis buffer containing the cytosol was cleared by centrifugation (supernatants; 5,000 × g, 5 min). The adherent, permeabilized cells were washed twice with PBS and lysed in radioimmunoprecipitation assay buffer. Purity of the cytosolic fractions was confirmed by Western blotting with antibodies directed against pan-Cadherin, LaminA/C and GAPDH.
Immuno- and cAMP-Agarose Precipitations, and Western Blotting
Immunoprecipitations were carried out as described (9, 46). Cells were lysed in standard lysis buffer (10 mm K2HPO4, 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 1% Triton X-100, 0,2% sodium deoxycholate, pH 7.4) containing protease inhibitors (cOmplete; Roche Applied Science, Rotkreuz, Switzerland) and phosphatase inhibitors (PhosSTOP, Roche Applied Science). The lysates were cleared by centrifugation (20,000 × g, 4 °C, 10 min). Proteins were immunoprecipitated with anti-GFP (62, 63) or anti-GSK3β (L-17, Goat, Santa Cruz Biotechnology, Dallas, TX) antibodies, and protein A-conjugated agarose (Sigma-Aldrich) or anti-FLAG-coupled magnetic beads (Sigma-Aldrich).
As described previously (9), for cAMP-agarose precipitations, the lysates were incubated with 8-AHA-cAMP-agarose (4 °C, 3 h; BioLog, Bremen, Germany). Agarose-bound proteins were washed four times with lysis buffer and eluted with Laemmli sample buffer.
Western blotting was carried out as described (9, 46). The following antibodies were used: HSP90 mouse (AC88; Enzo Life Sciences, Lörrach, Germany); GFP mouse (JL-8; Takara Bio Inc., Shiga, Japan); GFP-01, rabbit (custom-made (62)); GSKIP, rabbit, (custom-made (9)); LaminA/C, goat (N-18; Santa Cruz Biotechnology); HA High Affinity, rat (3F10; Roche Applied Science); FLAG (M2), mouse and pan-Cadherin, mouse (Sigma-Aldrich); Axin1 (C7B12), rabbit; Myc tag (9B11), rabbit; GAPDH (H-12), rabbit; GSK3β (27C10), rabbit; phospho-GSK3β (S9), rabbit; phospho-β-catenin (S675), rabbit; β-catenin (#9562), rabbit; and phospho-β-catenin (Ser-33/Ser-37/Thr-41), rabbit (all from Cell Signaling, Cambridge, UK); and RIIα antibody (mouse, BD Biosciences). Signals were detected using EMD Millipore Immobilon Western Chemiluminescent HRP Substrate (Thermo Fisher Scientific) and an Odyssey Fc Imaging System (LI-COR Biotechnology, Lincoln, NE).
Peptide Spot Synthesis and Overlay with Recombinant GSK3β
25-mer peptides were spot-synthesized as described using an Intavis ResPep SL spot synthesizer (Intavis, Cologne, Germany) and incubated with a recombinant fusion protein of GSK3β and glutathione S-transferase (GST; Cell Signaling; 1 μg/ml) or GST alone (9, 43, 64, 65).
Luciferase Assays
HEK293 cells were transfected with plasmids encoding the GSKIP variants together with pRL-SV40 and TOPflash or FOPflash vectors and stimulated with recombinant human Wnt3a (R&D Systems, Minneapolis, MN) or Wnt3a-conditioned medium 24 h after transfection (6). The cells were lysed 24 h later, and luciferase activity was measured using the Dual-Luciferase Assay Kit (Promega, Fitchburg, WI) and a Centro XS3 LB 960 luminometer (Berthold Technologies, Bad Wildbad, Germany). Luciferase activity was induced by binding of β-catenin to the reporter system. Activity of the TOPflash and FOPflash vectors was normalized to activity of the pRL-SV40 vector, and normalized TOPflash activity was divided by the normalized FOPflash activity to obtain a TOPflash/FOPflash ratio. For siRNA experiments, 24 h after knockdown of GSKIP, the cells were transfected a second time with the reporter vectors. After an additional 24 h, the cells were stimulated for a further 24 h with Wnt3a-conditioned medium.
Statistical Analysis
GraphPad Prism 5.0 (GraphPad Software) was used to perform statistical analyses.
Author Contributions
A. D. and M. F. S. carried out cloning, immunoprecipitations, and Wnt signaling activity measurements. E. P., M. C. M., and V. A. D. carried out immunoprecipitations. P. S. performed peptide spot and overlay experiments and immunoprecipitations. W. B. provided plasmids for Wnt signaling analysis and contributed to writing the manuscript. M. F. S., A. D., and E. K. designed experiments and wrote the manuscript.
Acknowledgments
We thank Russ Hodge for carefully and critically reading the manuscript. We are grateful for technical help from Andrea Geelhaar and Beate Eisermann.
This work was supported by grants from the Else Kröner-Fresenius-Stiftung (2013_A145), the German-Israeli Foundation (G.I.F. I-1210-286.13/2012), and the German Centre for Cardiovascular Research (DZHK 81X210012) (to E. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- AKAP
- A-kinase anchoring protein
- GSKIP
- GSK3β interaction protein
- GSK3β
- glycogen synthase kinase 3β
- siNT
- control non-targeting siRNA
- GID
- GSK3β interaction domain
- ANOVA
- analysis of variance
- 8-AHA-cAMP
- 8-(6-aminohexyl)aminoadenosine-3′,5′-cyclic monophosphate
- IP
- immunoprecipitation.
References
- 1. Scott J. D., Dessauer C. W., and Taskén K. (2013) Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53, 187–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Langeberg L. K., and Scott J. D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skroblin P., Grossmann S., Schäfer G., Rosenthal W., and Klussmann E. (2010) Mechanisms of protein kinase a anchoring. Int. Rev. Cell Mol. Biol. 283, 235–330 [DOI] [PubMed] [Google Scholar]
- 4. Taylor S. S., Ilouz R., Zhang P., and Kornev A. P. (2012) Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McSorley T., Stefan E., Henn V., Wiesner B., Baillie G. S., Houslay M. D., Rosenthal W., and Klussmann E. (2006) Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur. J. Cell Biol. 85, 673–678 [DOI] [PubMed] [Google Scholar]
- 6. Willert K., Brown J. D., Danenberg E., Duncan A. W., Weissman I. L., Reya T., Yates J. R. 3rd, and Nusse R. (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452 [DOI] [PubMed] [Google Scholar]
- 7. Deák V. A., and Klussmann E. (2016) Pharmacological interference with protein-protein interactions of A-kinase anchoring proteins as a strategy for the treatment of disease. Curr. Drug Targets 17, 1147–1171 [DOI] [PubMed] [Google Scholar]
- 8. Dema A., Perets E., Schulz M. S., Deák V. A., and Klussmann E. (2015) Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal. 27, 2474–2487 [DOI] [PubMed] [Google Scholar]
- 9. Hundsrucker C., Skroblin P., Christian F., Zenn H. M., Popara V., Joshi M., Eichhorst J., Wiesner B., Herberg F. W., Reif B., Rosenthal W., and Klussmann E. (2010) Glycogen synthase kinase 3β interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 285, 5507–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fang X., Yu S. X., Lu Y., Bast R. C. Jr, Woodgett J. R., and Mills G. B. (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 97, 11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haq S., Michael A., Andreucci M., Bhattacharya K., Dotto P., Walters B., Woodgett J., Kilter H., and Force T. (2003) Stabilization of β-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc. Natl. Acad. Sci. U.S.A. 100, 4610–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thornton T. M., Pedraza-Alva G., Deng B., Wood C. D., Aronshtam A., Clements J. L., Sabio G., Davis R. J., Matthews D. E., Doble B., and Rincon M. (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science 320, 667–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rayasam G. V., Tulasi V. K., Sodhi R., Davis J. A., and Ray A. (2009) Glycogen synthase kinase 3: more than a namesake. Br. J. Pharmacol. 156, 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Antos C. L., McKinsey T. A., Frey N., Kutschke W., McAnally J., Shelton J. M., Richardson J. A., Hill J. A., and Olson E. N. (2002) Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forde J. E., and Dale T. C. (2007) Glycogen synthase kinase 3: a key regulator of cellular fate. Cell. Mol. Life Sci. 64, 1930–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Force T., and Woodgett J. R. (2009) Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 284, 9643–9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Diehl J. A., Cheng M., Roussel M. F., and Sherr C. J. (1998) Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12, 3499–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoshi M., Takashima A., Noguchi K., Murayama M., Sato M., Kondo S., Saitoh Y., Ishiguro K., Hoshino T., and Imahori K. (1996) Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3β in brain. Proc. Natl. Acad. Sci. U.S.A. 93, 2719–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sadot E., Conacci-Sorrell M., Zhurinsky J., Shnizer D., Lando Z., Zharhary D., Kam Z., Ben-Ze'ev A., and Geiger B. (2002) Regulation of S33/S37 phosphorylated β-catenin in normal and transformed cells. J. Cell Sci. 115, 2771–2780 [DOI] [PubMed] [Google Scholar]
- 20. MacDonald B. T., Tamai K., and He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amit S., Hatzubai A., Birman Y., Andersen J. S., Ben-Shushan E., Mann M., Ben-Neriah Y., and Alkalay I. (2002) Axin-mediated CKI phosphorylation of β-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16, 1066–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz-Romond T., Asbrand C., Bakkers J., Kühl M., Schaeffer H. J., Huelsken J., Behrens J., Hammerschmidt M., and Birchmeier W. (2002) The ankyrin repeat protein Diversin recruits Casein kinase Iϵ to the β-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 16, 2073–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davidson G., Wu W., Shen J., Bilic J., Fenger U., Stannek P., Glinka A., and Niehrs C. (2005) Casein kinase 1 γ couples Wnt receptor activation to cytoplasmic signal transduction. Nature 438, 867–872 [DOI] [PubMed] [Google Scholar]
- 24. Aberle H., Bauer A., Stappert J., Kispert A., and Kemler R. (1997) β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 26. Klaus A., and Birchmeier W. (2008) Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 8, 387–398 [DOI] [PubMed] [Google Scholar]
- 27. Chou H. Y., Howng S. L., Cheng T. S., Hsiao Y. L., Lieu A. S., Loh J. K., Hwang S. L., Lin C. C., Hsu C. M., Wang C., Lee C. I., Lu P. J., Chou C. K., Huang C. Y., and Hong Y. R. (2006) GSKIP is homologous to the Axin GSK3β interaction domain and functions as a negative regulator of GSK3β. Biochemistry 45, 11379–11389 [DOI] [PubMed] [Google Scholar]
- 28. Lin C. C., Chou C. H., Howng S. L., Hsu C. Y., Hwang C. C., Wang C., Hsu C. M., and Hong Y. R. (2009) GSKIP, an inhibitor of GSK3β, mediates the N-cadherin/β-catenin pool in the differentiation of SH-SY5Y cells. J. Cell. Biochem. 108, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 29. Deák V. A., Skroblin P., Dittmayer C., Knobeloch K. P., Bachmann S., and Klussmann E. (2016) The A-kinase anchoring protein GSKIP regulates GSK3β activity and controls palatal shelf fusion in mice. J. Biol. Chem. 291, 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu K. J., Arron J. R., Stankunas K., Crabtree G. R., and Longaker M. T. (2007) Chemical rescue of cleft palate and midline defects in conditional GSK-3β mice. Nature 446, 79–82 [DOI] [PubMed] [Google Scholar]
- 31. Wu D., and Pan W. (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lustig B., Jerchow B., Sachs M., Weiler S., Pietsch T., Karsten U., van de Wetering M., Clevers H., Schlag P. M., Birchmeier W., and Behrens J. (2002) Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 22, 1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jho E. H., Zhang T., Domon C., Joo C. K., Freund J. N., and Costantini F. (2002) Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hino S., Tanji C., Nakayama K. I., and Kikuchi A. (2005) Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase stabilizes β-catenin through inhibition of its ubiquitination. Mol. Cell. Biol. 25, 9063–9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taurin S., Sandbo N., Qin Y., Browning D., and Dulin N. O. (2006) Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 281, 9971–9976 [DOI] [PubMed] [Google Scholar]
- 36. Korinek V., Barker N., Morin P. J., van Wichen D., de Weger R., Kinzler K. W., Vogelstein B., and Clevers H. (1997) Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 [DOI] [PubMed] [Google Scholar]
- 37. Howng S. L., Hwang C. C., Hsu C. Y., Hsu M. Y., Teng C. Y., Chou C. H., Lee M. F., Wu C. H., Chiou S. J., Lieu A. S., Loh J. K., Yang C. N., Lin C. S., and Hong Y. R. (2010) Involvement of the residues of GSKIP, AxinGID, and FRATtide in their binding with GSK3β to unravel a novel C-terminal scaffold-binding region. Mol. Cell. Biochem. 339, 23–33 [DOI] [PubMed] [Google Scholar]
- 38. Li V. S., Ng S. S., Boersema P. J., Low T. Y., Karthaus W. R., Gerlach J. P., Mohammed S., Heck A. J., Maurice M. M., Mahmoudi T., and Clevers H. (2012) Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 39. Tanji C., Yamamoto H., Yorioka N., Kohno N., Kikuchi K., and Kikuchi A. (2002) A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3β (GSK-3β) and mediates protein kinase A-dependent inhibition of GSK-3β. J. Biol. Chem. 277, 36955–36961 [DOI] [PubMed] [Google Scholar]
- 40. Flynn M. P., Maizels E. T., Karlsson A. B., McAvoy T., Ahn J. H., Nairn A. C., and Hunzicker-Dunn M. (2008) Luteinizing hormone receptor activation in ovarian granulosa cells promotes protein kinase A-dependent dephosphorylation of microtubule-associated protein 2D. Mol. Endocrinol. 22, 1695–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whiting J. L., Nygren P. J., Tunquist B. J., Langeberg L. K., Seternes O. M., and Scott J. D. (2015) Protein kinase A opposes the phosphorylation-dependent recruitment of glycogen synthase kinase 3β to A-kinase anchoring protein 220. J. Biol. Chem. 290, 19445–19457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dajani R., Fraser E., Roe S. M., Yeo M., Good V. M., Thompson V., Dale T. C., and Pearl L. H. (2003) Structural basis for recruitment of glycogen synthase kinase 3β to the axin-APC scaffold complex. EMBO J. 22, 494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meng D., Lynch M. J., Huston E., Beyermann M., Eichhorst J., Adams D. R., Klussmann E., Houslay M. D., and Baillie G. S. (2009) MEK1 binds directly to βarrestin1, influencing both its phosphorylation by ERK and the timing of its isoprenaline-stimulated internalization. J. Biol. Chem. 284, 11425–11435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szaszák M., Christian F., Rosenthal W., and Klussmann E. (2008) Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell. Signal. 20, 590–601 [DOI] [PubMed] [Google Scholar]
- 45. Tröger J., Moutty M. C., Skroblin P., and Klussmann E. (2012) A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 166, 420–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Henn V., Edemir B., Stefan E., Wiesner B., Lorenz D., Theilig F., Schmitt R., Vossebein L., Tamma G., Beyermann M., Krause E., Herberg F. W., Valenti G., Bachmann S., Rosenthal W., and Klussmann E. (2004) Identification of a novel A-kinase anchoring protein 18 isoform and evidence for its role in the vasopressin-induced aquaporin-2 shuttle in renal principal cells. J. Biol. Chem. 279, 26654–26665 [DOI] [PubMed] [Google Scholar]
- 47. Horner A., Goetz F., Tampé R., Klussmann E., and Pohl P. (2012) Mechanism for targeting the A-kinase anchoring protein AKAP18δ to the membrane. J. Biol. Chem. 287, 42495–42501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schott M. B., and Grove B. (2013) Receptor-mediated Ca2+ and PKC signaling triggers the loss of cortical PKA compartmentalization through the redistribution of gravin. Cell. Signal. 25, 2125–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wan M., Yang C., Li J., Wu X., Yuan H., Ma H., He X., Nie S., Chang C., and Cao X. (2008) Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 22, 2968–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzuki A., Ozono K., Kubota T., Kondou H., Tachikawa K., and Michigami T. (2008) PTH/cAMP/PKA signaling facilitates canonical Wnt signaling via inactivation of glycogen synthase kinase-3β in osteoblastic Saos-2 cells. J. Cell. Biochem. 104, 304–317 [DOI] [PubMed] [Google Scholar]
- 51. Radeva M. Y., Kugelmann D., Spindler V., and Waschke J. (2014) PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS One 9, e106733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Logue J. S., Whiting J. L., Tunquist B., Sacks D. B., Langeberg L. K., Wordeman L., and Scott J. D. (2011) AKAP220 protein organizes signaling elements that impact cell migration. J. Biol. Chem. 286, 39269–39281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lester L. B., Coghlan V. M., Nauert B., and Scott J. D. (1996) Cloning and characterization of a novel A-kinase anchoring protein. AKAP 220, association with testicular peroxisomes. J. Biol. Chem. 271, 9460–9465 [DOI] [PubMed] [Google Scholar]
- 54. Heuberger J., and Birchmeier W. (2010) Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2, a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anastas J. N., and Moon R. T. (2013) WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 [DOI] [PubMed] [Google Scholar]
- 56. Kahn M. (2014) Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saliba J., Saint-Martin C., Di Stefano A., Lenglet G., Marty C., Keren B., Pasquier F., Valle V. D., Secardin L., Leroy G., Mahfoudhi E., Grosjean S., Droin N., Diop M., Dessen P., et al. (2015) Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 47, 1131–1140 [DOI] [PubMed] [Google Scholar]
- 58. Bertrand F. E., Angus C. W., Partis W. J., and Sigounas G. (2012) Developmental pathways in colon cancer: crosstalk between WNT, BMP, Hedgehog and Notch. Cell cycle 11, 4344–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y., Pizzute T., and Pei M. (2014) A review of crosstalk between MAPK and Wnt signals and its impact on cartilage regeneration. Cell Tissue Res. 358, 633–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Esseltine J. L., and Scott J. D. (2013) AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 34, 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu X., and Fagotto F. (2011) A method to separate nuclear, cytosolic, and membrane-associated signaling molecules in cultured cells. Sci. Signal 4, pl2. [DOI] [PubMed] [Google Scholar]
- 62. Alken M., Rutz C., Köchl R., Donalies U., Oueslati M., Furkert J., Wietfeld D., Hermosilla R., Scholz A., Beyermann M., Rosenthal W., and Schülein R. (2005) The signal peptide of the rat corticotropin-releasing factor receptor 1 promotes receptor expression but is not essential for establishing a functional receptor. Biochem. J. 390, 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nedvetsky P. I., Stefan E., Frische S., Santamaria K., Wiesner B., Valenti G., Hammer J. A. 3rd, Nielsen S., Goldenring J. R., Rosenthal W., and Klussmann E. (2007) A role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic 8, 110–123 [DOI] [PubMed] [Google Scholar]
- 64. Maass P. G., Aydin A., Luft F. C., Schächterle C., Weise A., Stricker S., Lindschau C., Vaegler M., Qadri F., Toka H. R., Schulz H., Krawitz P. M., Parkhomchuk D., Hecht J., Hollfinger I., et al. (2015) PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 47, 647–653 [DOI] [PubMed] [Google Scholar]
- 65. Klussmann E. (2016) Protein-protein interactions of PDE4 family members: functions, interactions and therapeutic value. Cell. Signal. 28, 713–718 [DOI] [PubMed] [Google Scholar]