

Abstract
Non-proliferating cells oxidize respiratory substrates in mitochondria to generate a protonmotive force (Δp) that drives ATP synthesis. The mitochondrial membrane potential (ΔΨ), a component of Δp, drives release of mitochondrial ATP4− in exchange for cytosolic ADP3− via the electrogenic adenine nucleotide translocator (ANT) located in the mitochondrial inner membrane, which leads to a high cytosolic ATP/ADP ratio up to >100-fold greater than matrix ATP/ADP. In rat hepatocytes, ANT inhibitors, bongkrekic acid (BA), and carboxyatractyloside (CAT), and the F1FO-ATP synthase inhibitor, oligomycin (OLIG), inhibited ureagenesis-induced respiration. However, in several cancer cell lines, OLIG but not BA and CAT inhibited respiration. In hepatocytes, respiratory inhibition did not collapse ΔΨ until OLIG, BA, or CAT was added. Similarly, in cancer cells OLIG and 2-deoxyglucose, a glycolytic inhibitor, depolarized mitochondria after respiratory inhibition, which showed that mitochondrial hydrolysis of glycolytic ATP maintained ΔΨ in the absence of respiration in all cell types studied. However in cancer cells, BA, CAT, and knockdown of the major ANT isoforms, ANT2 and ANT3, did not collapse ΔΨ after respiratory inhibition. These findings indicated that ANT did mediate mitochondrial ATP/ADP exchange in cancer cells. We propose that suppression of ANT contributes to low cytosolic ATP/ADP, activation of glycolysis, and a Warburg metabolic phenotype in proliferating cells.
Keywords: ATP, glycolysis, mitochondria, voltage-dependent anion channel (VDAC), Warburg effect, ANT, ATP/ADP turnover, bongkrekic acid, carboxyatractyloside
Introduction
Cellular bioenergetics in cancer and other proliferating cells is characterized by enhanced aerobic glycolysis and suppression of mitochondrial metabolism despite adequate oxygenation (1, 2). As described by Otto Warburg in the early 20th century, tumors produce excess lactic acid, the end-product of glycolysis, in comparison to normal post-mitotic tissues (3). By contrast in non-proliferating cells, pyruvate generated by glycolysis becomes fully oxidized to CO2 and H2O inside mitochondria. Thus, lactic acid production by oxygenated tumors signifies aerobic glycolysis and suppressed oxidation of pyruvate by mitochondria. It is now widely accepted that the Warburg metabolic phenotype is pro-proliferative by providing carbon atoms required for synthesis of new proteins, lipids, and nucleic acids (4–6). Despite the initial postulate of Warburg that damage to metabolism now known to be catalyzed in mitochondria might be the origin of cancer, mitochondria from cancer cells are functional as assessed by measurements of respiration, mitochondrial membrane potential (ΔΨ), and activity of the respiratory chain (7–11).
ATP synthesized by mitochondria must move across the inner membrane into the cytosol in exchange for cytosolic ADP and Pi. ATP/ADP exchange is catalyzed by the adenine nucleotide translocator (ANT), 2 a member of the solute carrier family (SLC), also known as the ATP/ADP carrier (AAC), whereas Pi exchanges for OH− via the phosphate transporter (12). The one-to-one exchange of mitochondrial matrix ATP4− for cytosolic ADP3− is highly selective (12, 13). Importantly, ATP4−/ADP3− exchange is electrogenic and thus driven by the mitochondrial membrane potential (ΔΨ). Consequently, ANT coupled to ΔΨ can drive an ATP/ADP ratio in the cytosol that is up to 100× greater than matrix ATP/ADP.
ANT in humans comprises four isoforms, ANT1 through ANT4, encoded by different nuclear genes: SLC25A4 (ANT1), SLC25A5 (ANT2), SLC25A6 (ANT3), and SLC25A31 (ANT4) (14). ANT1 is expressed in heart, skeletal muscle, and brain; ANT2 is expressed mostly in proliferating cells and liver; weakly expressed ANT3 is ubiquitous (15, 16). The recently discovered isoform ANT4 is found mainly in liver, testis, and brain (17).
ANT2, specifically expressed in proliferating cells and overexpressed in various types of cancer cells, is considered a marker of cell proliferation. By contrast, most non-proliferating tissues with the exception of the liver have low or very low expression of ANT2 (18, 19). In cancer cell lines originating from colon (HT29), breast (MCF7), and liver (HepG2), ANT2 mRNA is more abundant than ANT1 (20). Human tumors from bladder, thyroid gland, lung, ovary, breast, and testis also overexpress ANT2 in comparison to corresponding normal tissues (21).
Yeast express three isoforms of ANT: AAC1, AAC2, and AAC3. Only AAC1 and AAC2 are expressed under aerobic conditions, whereas AAC3, the yeast equivalent of ANT2 in mammalian cells, is exclusively expressed in anaerobiosis and is essential to maintain cell proliferation on a fermentable substrate (22–24). This finding led to the hypothesis that AAC3 in yeasts, and possibly ANT2 in mammals, imports glycolytic ATP into mitochondria to support the anabolic functions of mitochondria.
The biological relevance of ANT2 in proliferating cells is supported also by the fact that ANT2−/− mice are embryonically lethal, whereas ANT1 disruption causes mitochondrial myopathy (25). Nonetheless, here using chemical inhibitors of ANT and knockdown of the major isoforms ANT2/3 in cancer cells, we show that ADP/ATP exchange and the import of glycolytic ATP into mitochondria of cancer cells is ANT-independent.
Results
Bongkrekic Acid and Carboxyatractyloside Decrease Respiration in Rat Hepatocytes but Not in Cancer Cells
Respiration by cultured rat hepatocytes was measured in a Seahorse XF24 extracellular flux analyzer under basal conditions and after the addition of ureagenic substrates (3 mm NH4Cl, 5 mm l-ornithine, and 5 mm lactate) (26). Because bongkrekic acid (BA), carboxyatractyloside (CAT) and oligomycin (OLIG) do not alter maximal uncoupler-stimulated respiration, we did not measure respiration in the presence of BA, CAT, or OLIG after protonophoric uncoupling. Ureagenic substrates stimulated respiration by 110–150%. BA (10 μm) and CAT (20 μm), specific inhibitors of ANT, and OLIG (10 μg/ml), an ATP synthase inhibitor, were added after the ureagenic stimulus. BA, CAT, and OLIG each decreased respiration to at or below basal non-induced levels, indicating that ureagenic respiration depends on ATP synthesis driven by the ATP synthase and that ATP turnover depends on ANT function (Fig. 1A).
FIGURE 1.

Bongkrekic acid and carboxyatractyloside decrease ATP turnover-linked respiration in rat hepatocytes but not in cancer cells. A, rat hepatocytes were maintained in Krebs-Ringer-HEPES buffer as described under “Experimental Procedures.” Respiratory rates were measured before and after the addition of ureagenic substrates (3 mm NH4Cl, 5 mm ornithine, and 5 mm lactate). BA (10 μm), CAT (10 μm), OLIG (10 μg/ml), or vehicle (VEH) was added after ureagenic substrates. B and C, cancer cells were incubated in MHBSS. After measurement of baseline respiration, BA, CAT, OLIG, or VEH was added to A549 cells (B) and HepG2 cells (C).
In cancer cells, OLIG (10 μg/ml) decreased basal respiration by ∼50% in A549 human lung adenocarcinoma cells and HepG2 human hepatoma cells (Fig. 1, B and C) and by 26 and 34% in INS-1 rat insulinoma cells and UMSCC22 human head and neck squamous carcinoma cells, respectively (not shown). By contrast, BA and CAT did not decrease respiration in any of the cancer cell lines studied (Fig. 1, B and C, and not shown). These findings imply that although ATP synthase drives respiration in cancer cells, such ATP synthase-dependent respiration is not dependent on mitochondrial ATP/ADP exchange via ANT.
Bongkrekic Acid and Carboxyatractyloside Inhibit Respiration in Permeabilized Cancer Cells
HepG2 cells in intracellular buffer were permeabilized with digitonin (10 μm) in the presence of ADP-containing intracellular buffer supplemented with succinate. Respiration increased by ∼90% after 20 min. Subsequent addition of either OLIG (10 μg/ml), BA (10 μm), or CAT (20 μm) decreased respiration to at or slightly below basal levels before digitonin (Fig. 2). Sensitivity to BA and CAT of ADP-stimulated respiration after permeabilization indicated that ANT2 and ANT3, the two major isoforms of ANT in HepG2 cells (see below), became functional and participated in ATP/ADP exchange. This finding implicated the presence of a soluble factor inhibiting ANT that was released after plasma membrane permeabilization by digitonin.
FIGURE 2.

Bongkrekic acid and carboxyatractyloside inhibit respiration in permeabilized cancer cells. HepG2 cells were permeabilized with digitonin (DIG, 10 μm) in intracellular buffer as described under “Experimental Procedures.” OLIG, BA, CAT, or vehicle (VEH) was added as indicated. Digitonin (DIG) was not added in VEH-treated group.
Bongkrekic Acid and Carboxyatractyloside Collapse Mitochondrial Membrane Potential in Rat Hepatocytes after Respiratory Inhibition
Rat hepatocytes were loaded with tetramethylrhodamine methyl ester (TMRM) and imaged by confocal microscopy. Myxothiazol (MYX, 10 μm), an inhibitor of the Complex III of the respiratory chain, decreased TMRM fluorescence by 11%, indicating a small drop in ΔΨ. Because MYX inhibits respiration supported by all known physiological substrates, maintenance of ΔΨ after MYX was presumably due to ATP hydrolysis by the ATP synthase working in reverse. Thus, as expected, OLIG (10 μg/ml) after MYX decreased TMRM fluorescence by 60–70% (Fig. 3, A and D). Similar to OLIG, inhibition of ANT by BA (10 μm) or CAT (20 μm) after MYX also collapsed ΔΨ (Fig. 3, B–D). By contrast, in the absence of respiratory inhibition, ΔΨ was maintained after OLIG and only slightly increased after BA or CAT (Fig. 4). As expected, subsequent addition of MYX after OLIG, BA, or CAT collapsed ΔΨ of hepatocytes. These results showed that hydrolysis of cytosolic ATP entering mitochondria via ANT by the ATP synthase, working in reverse, maintained mitochondrial ΔΨ in hepatocytes after respiratory inhibition. Conversely, respiration alone also maintained mitochondrial ΔΨ when the ATP synthase or ANT was inhibited.
FIGURE 3.
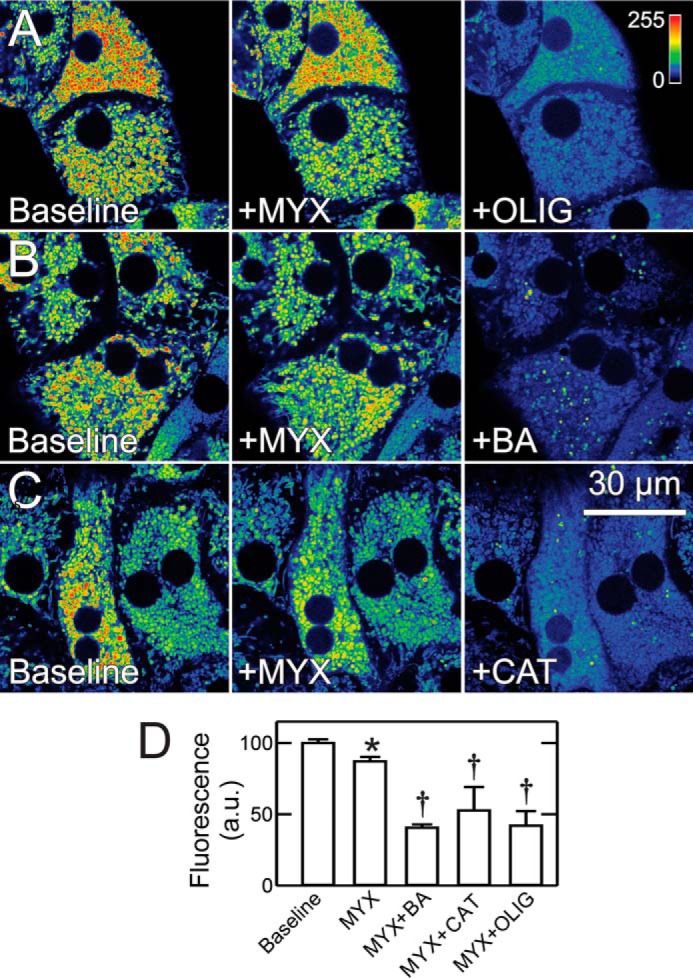
Bongkrekic acid, carboxyatractyloside, and oligomycin collapse mitochondrial membrane potential in hepatocytes after respiratory inhibition. A–C, rat hepatocytes in Krebs-Ringer-HEPES buffer were loaded with TMRM, as described under “Experimental Procedures.” After collection of baseline images, MYX (10 μm) was added, and another image was collected after 30 min. OLIG, BA, or CAT was then added, and images were collected after additional 30 min. Image intensity is pseudocolored according to the reference bar. D, average changes of fluorescence quantified as described under “Experimental Procedures” after various treatments are plotted. *, p < 0.05 versus baseline; †, p < 0.05 versus MYX. a.u., arbitrary units.
FIGURE 4.
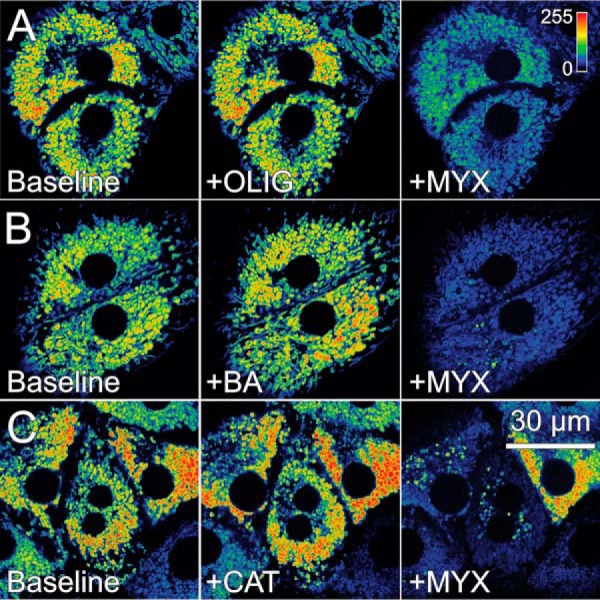
Mitochondrial membrane potential in rat hepatocytes is maintained after bongkrekic acid, carboxyatractyloside and oligomycin but collapsed by subsequent respiratory inhibition. Rat hepatocytes were loaded as described in Fig. 3. OLIG (A), BA (B), or CAT (C) was added for 30 min before MYX for 20 min.
Bongkrekic Acid and Carboxyatractyloside Fail to Decrease Mitochondrial Membrane Potential after Respiratory Inhibition in Cancer Cells
MYX decreased ΔΨ measured by TMRM fluorescence by 12% in A549 cells. Subsequent addition of OLIG then decreased TMRM fluorescence by 84% (Fig. 5, A and D). By contrast, neither BA nor CAT added after MYX caused ΔΨ to decrease. Thus, cytosolic ATP entering mitochondria by a BA- and CAT-insensitive pathway supported maintenance of ΔΨ in A549 cells after respiratory inhibition. To show that mitochondrial ATP hydrolysis was still ongoing after ANT inhibitors, OLIG was added after MYX plus BA and MYX plus CAT, which decreased TMRM uptake by 89 and 85%, respectively (Fig. 5, B–D). Similar results after treatments with OLIG, BA, and CAT were obtained in HepG2 cells (Fig. 6) and in UMSCC22 and INS-1 cells (not shown). In separate experiments in HepG2 cells, OLIG alone increased TMRM uptake by ∼64%, whereas BA and CATR each decreased TMRM uptake by 13 and 27%, respectively (not shown). Increased ΔΨ after OLIG signified that mitochondria were actively producing ATP by oxidative phosphorylation, whereas the inability of BA and CAT to increase TMRM uptake like OLIG indicated that ANT was not involved in the ATP turnover associated with oxidative phosphorylation.
FIGURE 5.
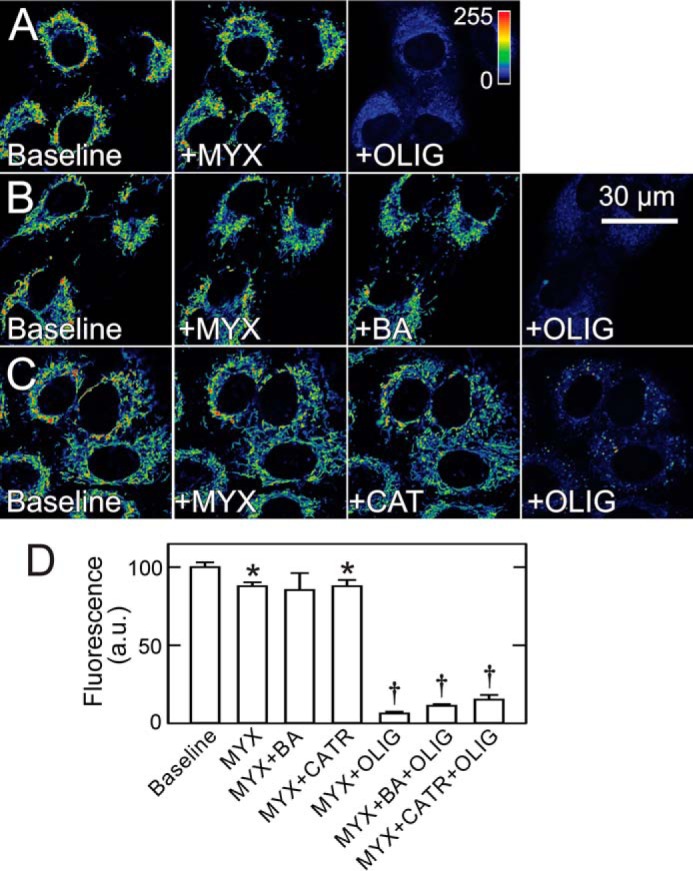
Bongkrekic acid and carboxyatractyloside do not decrease mitochondrial membrane potential in cancer cells after respiratory inhibition. A–C, A549 cells in MHBSS were loaded with TMRM, as described under “Experimental Procedures.” After collection of baseline images, MYX was added, and another image was collected after 30 min. In A, OLIG was added for 30 min. In B and C, BA or CAT was added for 30 min followed by OLIG for another 30 min. D shows changes of fluorescence after different treatments. *, p < 0.05 versus baseline; †, p < 0.05 versus MYX + BA, CAT, or OLIG. a.u., arbitrary units.
FIGURE 6.
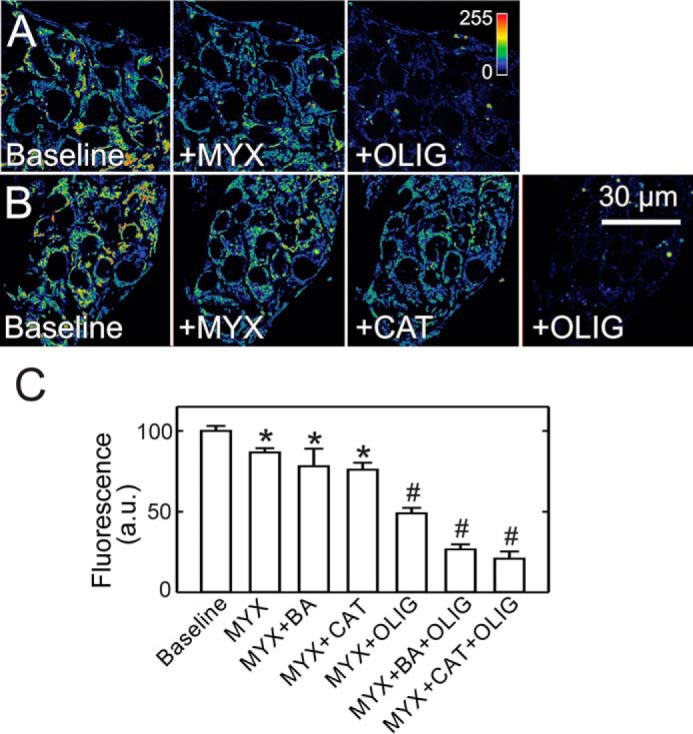
Bongkrekic acid and carboxyatractyloside do not decrease mitochondrial membrane potential in HepG2 cells after respiratory inhibition. Conditions are as in Fig. 5, except HepG2 cells were used. After collection of baseline images, MYX was added followed by OLIG (A). In B, MYX was followed by CAT and subsequent OLIG. C, average fluorescence as percent of baseline is plotted for various treatments. *, p < 0.05 versus baseline. †, p < 0.05 versus MYX + BA, CAT, or OLIG. a.u., arbitrary units.
Glycolytic ATP Contributes to Maintenance of Mitochondrial Membrane Potential in Cancer Cells
After respiratory inhibition, mitochondrial ΔΨ depended on ATP hydrolysis by the reversible F1F0-ATP synthase, as OLIG added after respiratory inhibition promptly collapsed ΔΨ (Fig. 5A). To show that the source of ATP hydrolyzed after respiratory inhibition originated from glycolysis, we treated A549 cells with the glycolytic inhibitor 2-deoxyglucose (2DG, 50 mm) alone and after respiratory inhibition with MYX. 2DG alone decreased TMRM fluorescence by 21% (Fig. 7, A and C). Subsequent addition of BA or CAT did not cause further changes of TMRM uptake (not shown). By contrast, 2DG added after MYX decreased TMRM fluorescence by 85%, indicating that generation of ΔΨ after respiratory inhibition required ATP synthesized by glycolysis in the cytosol (Fig. 7, B and C). Similar results were obtained in HepG2 cells (not shown).
FIGURE 7.
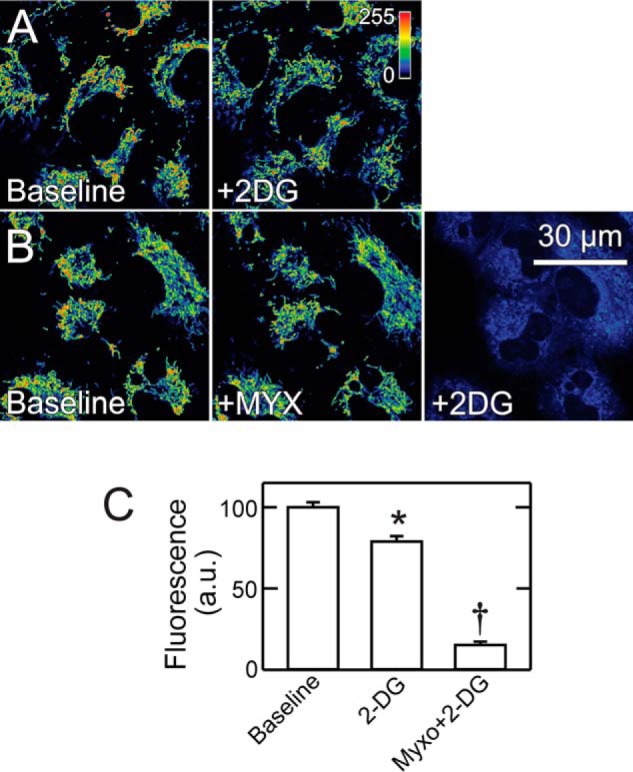
2-Deoxyglucose collapses mitochondrial membrane potential after respiratory inhibition in A549 cells. A549 cells were loaded as in Fig. 5. In A, a baseline image was collected, and 2DG (50 mm) was added followed by another image after 30 min. B, images were collected at baseline, 30 min after MYX, and 30 min after subsequent 2DG. C, average fluorescence as percent of baseline is plotted for various treatments. *, p < 0.05 versus Baseline; †, p < 0.05 versus 2DG. a.u., arbitrary units.
Tubulin Is Not the Soluble Factor Inhibiting ANT in Cancer Cells
To determine if free tubulin was the soluble factor inhibiting ANT, we treated HepG2 cells with the microtubule stabilizer paclitaxel (10 μm). Paclitaxel decreases cytosolic free tubulin, which leads to opening of VDAC and an increase of mitochondrial ΔΨ in cancer cells (27). After paclitaxel, subsequent inhibition of respiration with MYX (10 μm) only slightly decreased ΔΨ, indicating that ΔΨ was still maintained by hydrolysis of ATP. Further addition of both BA (10 μm) and CAT (20 μm) after MYX failed to collapse ΔΨ, which indicated that mitochondrial ATP/ADP exchange continued even after exposure to ANT inhibitors. Subsequently, OLIG (10 μm) collapsed ΔΨ, which confirmed that ATP hydrolysis by the ATP synthase working in reverse had been maintaining ΔΨ (Fig. 8).
FIGURE 8.
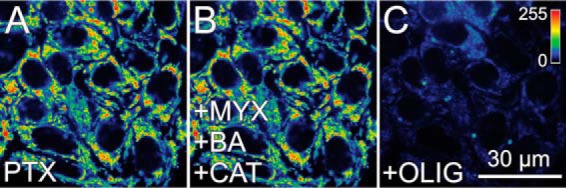
Depletion of cytosolic free tubulin by paclitaxel does not lead to sensitivity to bongkrekic acid and carboxyatractyloside in cancer cells. A549 cells were loaded as described in Fig. 5, and paclitaxel (PTX; 10 μm) was added for 30 min (A). Subsequently, MYX (20 min) and then BA plus CAT (30 min) were added (B) followed by OLIG (20 min) (C).
Knockdown of ANT2 and ANT3 in Cancer Cells Does Not Prevent Import of Glycolytic ATP into Mitochondria
In cancer cells, ANT2 and ANT3 are the major ANT isoforms with virtually no expression of ANT1 (20). Consistent with these previous observations, relative mRNA expression of ANT1, -2, and -3 assessed by qPCR in HepG2 cells was <1%, 70%, and 30%. Similarly, in A549 cells, ANT1, -2, and -3 mRNA expression was 1%, 56% and 44%, respectively. Thus, ANT2 was the major isoform in both cell lines with somewhat lesser expression of ANT3. A549 cells were transfected with isoform-specific siRNAs (5 nm) targeting ANT2 and ANT3, leading to an mRNA knockdown of 98 and 97% for ANT2 and ANT3, respectively (Fig. 9A). Knockdown of ANT2 did not alter expression of ANT3, whereas knockdown of ANT3 caused a 24% decrease in ANT2 expression (Fig. 9A). Expression of the barely detectable isoform ANT1 did not increase after ANT2 or ANT3 knockdown (not shown). Double knockdown of ANT2 and ANT3 also substantially decreased mRNA levels by 95 and 93% for ANT2 and ANT3, respectively, without increasing ANT1 mRNA expression (Fig. 9A and not shown). Using an antibody cross-reacting with both ANT2 and ANT3, immunoblots revealed a 60–70% decrease of ANT protein expression after siRNA knockdown of either ANT2 or ANT3 in A549 cells, whereas ANT2/3 double knockdown led to virtually complete loss of ANT protein expression (Fig. 9B).
FIGURE 9.

mRNA and protein expression of ANT1 and ANT2 after single and double knockdown. A549 cells were transfected with non-target siRNA or siRNA against ANT2, ANT3, or ANT2 plus ANT3. After 48 h, mRNA and protein expression of the two isoforms were assessed by qPCR (A) and immunoblotting (B). *, p < 0.05 versus non-target.
Single knockdown of ANT2 and ANT3 decreased TMRM fluorescence by 18 and 19%, respectively, compared with non-target siRNA, whereas ANT2/3 double knockdown decreased TMRM fluorescence by 7%, indicating modest but statistically significant contributions of both ANT isoforms to the maintenance of ΔΨ (not shown). However, differences between individual single ANT1 and ANT2 knockdowns and the ANT2/3 double knockdown were not statistically significant. To determine if import of glycolytic ATP into mitochondria depended on expression of ANT2 and/or ANT3, we treated ANT2 and ANT3 single knockdown cells and ANT2/3 double knockdown cells with MYX followed by 2DG. After respiratory inhibition with MYX, the decrease of TMRM fluorescence in ANT2 and ANT3 single knockdown cells and in ANT2/3 double knockdown cells was ∼25%, virtually identical to that after MYX treatment of wild type cells (Fig. 10, A–D, compare with Fig. 7). Moreover, the addition of 2DG after MYX caused a similar large release of mitochondrial TMRM in all single and double knockdown cells compared with wild type cells (Fig. 10, A–D). These results signified that entry of glycolytic ATP into mitochondria for ΔΨ maintenance by reverse ATP synthase activity during respiratory inhibition was not via ANT2 or ANT3. Namely, import of glycolytic ATP into mitochondria did not depend on the function of ANT2 or ANT3.
FIGURE 10.
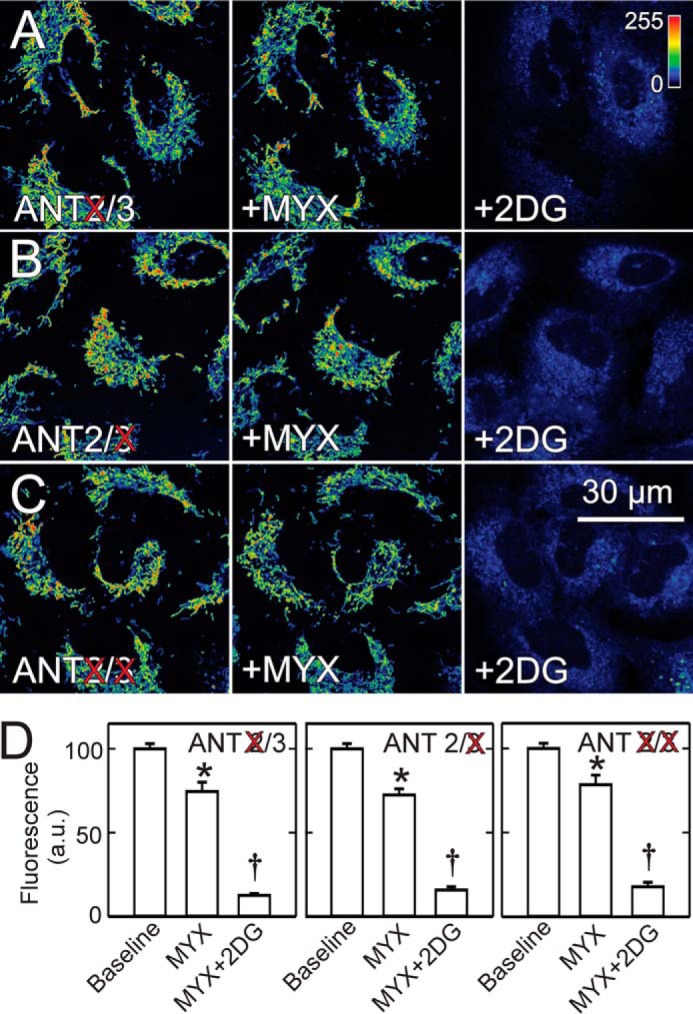
2-Deoxyglucose collapses mitochondrial membrane potential after respiratory inhibition in ANT2/3 single and double knockdown cells. MYX and 2DG were added sequentially to A549 cells after 48 h of siRNA knockdown of ANT2 (A), ANT3 (B), and both ANT2 and ANT3 (C). Average intensity of TMRM fluorescence after knockdowns is plotted in comparison with non-target (D). *, p < 0.05 versus Baseline; †, p < 0.05 versus MYX.
Discussion
In mitochondrial oxidative phosphorylation, respiration drives the formation of ΔΨ (and to a lesser extent ΔpH) across the mitochondrial inner membrane, which in turns drives ATP formation by the F1FO-ATP synthase (12). ATP so formed is released into the matrix space and then exported to the cytosol in exchange for ADP by ANT. This process is reversible, and ANT-dependent import of extramitochondrial ATP and subsequent hydrolysis by the ATP synthase working in reverse can power ΔΨ formation and even drive reverse electron transfer across Site 1 (Complexes I and II) and Sites 1 + 2 (Complexes I and III). Here, we confirm bidirectional exchange of ATP for ADP through ANT in intact cultured hepatocytes. By contrast, in four lines of cancer cells derived from different tissues (liver, lung, pancreatic islets, oral epithelium), we make the unexpected observation that ANT does not participate in mitochondrial transport of ATP.
Using Seahorse technology to measure cellular oxygen consumption, the ATP synthase inhibitor, OLIG, and the ANT inhibitors, BA and CAT, potently blocked ureagenic respiration in hepatocytes (Fig. 1A). Ureagenesis requires mitochondrial ATP generation and release to the cytosol, and the nearly equal respiratory inhibition by OLIG, BA, and CAT implied that ATP release from mitochondria was via ANT. By contrast, in cancer cell lines, OLIG inhibited respiration by 25–50% depending on the cell line, but BA and CAT had no effect, suggesting that ANT does not participate in ATP turnover (Fig. 1, B and C).
We also examined ATP-driven ΔΨ formation. In hepatocytes, respiratory inhibition with MYX caused a small decrease of ΔΨ, but subsequent OLIG, BA, and CAT, each, caused virtually complete mitochondrial depolarization (Fig. 3, A–D). These findings signified that cytosolic ATP entered mitochondria via ANT after respiratory inhibition to drive ΔΨ formation catalyzed by the mitochondrial ATP synthase working in reverse, as expected. In cancer cells, ΔΨ was likewise preserved after MYX, and subsequent OLIG then produced nearly complete depolarization (Fig. 5A). However, BA and CAT added after MYX did not depolarize the mitochondria of cancer cells (Fig. 5, B and C). Nonetheless, glycolytic ATP supported ΔΨ formation after MYX, as the glycolytic inhibitor, 2DG, collapsed mitochondrial ΔΨ when added subsequent to MYX (Fig. 7B). Thus, ATP entered the mitochondria of cancer cells after respiratory inhibition by a pathway different from ANT.
The evidence also showed that mitochondria of cancer cells generate ATP under basal conditions in the absence of respiratory inhibition. First, OLIG inhibited basal oxygen uptake, signifying that the respiration inhibited by OLIG was linked to mitochondrial ATP generation (Fig. 1, A and B). Second, OLIG added to cancer cells increased ΔΨ, which signified that active ATP formation by oxidative phosphorylation consumed ΔΨ (not shown). Interestingly, BA and CAT each added alone caused a small decrease of ΔΨ. The basis for this decrease is unexplained but suggests that ANT may have a different metabolic role in cancer cells.
The clear-cut effects of BA and CAT on both oxygen consumption and ΔΨ in hepatocytes demonstrated that these inhibitors can cross the plasma membrane and reach their mitochondrial target. Previous work also shows that BA and atractyloside (a CAT derivative) inhibit and enhance, respectively, apoptosis of intact cells, which further documents cellular penetrance of these agents (28, 29). Indeed, BA must be membrane-permeant, as BA must cross the mitochondrial inner membrane to bind to ANT from the matrix side of the membrane (30).
To characterize the possible involvement of specific ANT isoforms in ATP import into the mitochondria of cancer cells, we used siRNA for single and double knockdowns of ANT2 and ANT3, the predominant ANT isoforms in cancer cells (Fig. 9). Although knockdown efficiency was >93%, mitochondrial import of glycolytic ATP after respiratory inhibition was not diminished for either single ANT2/3 knockdown cells or double knockdown cells, as the decrease of ΔΨ after MYX was not different from wild type cells (Fig. 10). Although ANT2 has been suggested to mediate ATP import into mitochondria of cancer cells (16, 20), our data indicate that this is not the case, at least in the cancer cell lines studied.
Nonetheless, cancer cells robustly expressed ANT2 and ANT3 (56 and 44% relative expression, respectively, as assessed by mRNA expression), and the mechanism preventing their ATP/ADP exchange activity remains unknown. However, a soluble cytoplasmic factor may be involved, as plasma membrane permeabilization with digitonin restored ADP-stimulated, BA/CAT-inhibited respiration (Fig. 2). These findings also indicated that ANT2 and/or ANT3 were not post translationally modified to become insensitive to BA and ANT. Thus, after permeabilization, the affinity of the major isoforms ANT2 and ANT3 for BA and CAT in cancer cells was similar to that observed in rat hepatocytes.
Previously, we showed that high free tubulin in the cytoplasm of proliferating cancer cells closes voltage-dependent anion channels (VDAC) in the mitochondrial outer membrane, confirming tubulin inhibition shown for VDAC reconstituted into lipid bilayers (27, 31–33). Paclitaxel, a microtubule stabilizer, decreases free tubulin in the cytoplasm, leading to VDAC opening. However, paclitaxel did not restore sensitivity to BA and CAT in causing mitochondrial depolarization in HepG2 cells after respiratory inhibition (Fig. 8). Thus, free tubulin seems not to be the soluble factor mediating inhibition of ATP/ADP exchange by ANT in our cancer cell lines.
The pathway for ANT-independent ATP/ADP exchange in our cancer cell lines remains to be determined. A strong candidate is the ATP/Mg-Pi carrier, which was first described as a pathway for net uptake of adenine nucleotides by mitochondria (34, 35). The ATP-Mg/Pi carrier that catalyzes an electroneutral exchange of Mg2+-ATP4− for HPO42− and Mg2+-ADP3− for H2PO4− is encoded by three paralogues in mammals, SLC25A23, SLC25A24, and SLC25A25 (36). A more recent report shows that the dominant isoform SLC25A24 is overexpressed in several cancer cell lines and in >60 tumor tissue types compared with their normal tissue counterparts (37). The ATP/Mg-PI carrier is proposed to promote cancer survival by preventing mitochondrial permeability transition-dependent cell death induced by calcium accumulation during oxidative stress (37). Because inhibitors of the ATP-Mg/Pi carrier are not presently available, future studies will need to use genetic knockdown/knock-out strategies to determine whether any of the three paralogues of the ATP-Mg/Pi carrier are responsible for ATP turnover in cancer cells.
In differentiated post-mitotic cells including heart, brain, liver, and kidney, electrogenic ATP/ADP exchange by ANT acts to generate cytosolic ATP/ADP ratios that are as much as 100× greater than ATP/ADP in the mitochondrial matrix (12, 38). Such high ATP/ADP ratios inhibit glycolysis, as decreased ATP/ADP after inhibition of oxidative phosphorylation by anoxia or other stress rapidly and profoundly activates glycolytic flux. Here, we found that ANT does not mediate ATP/ADP exchange between mitochondria and cytosol in cancer cells. Rather, an alternative pathway, such as the ATP/Mg-Pi carrier, is employed. If, like the ATP/Mg-Pi carrier, this alternative pathway is non-electrogenic, then ATP/ADP in proliferating cancer cells will be much lower than in typical post-mitotic cells. We propose that lower ATP/ADP promotes glycolysis by releasing the brake on glycolysis provided by high ATP/ADP. Thus, the Warburg metabolic phenotype of aerobic glycolysis, which is characteristic of cancer cells both in vivo and in vitro (39, 40), is a consequence at least in part of a switch from electrogenic ANT-mediated mitochondrial ATP/ADP exchange to non-electrogenic exchange.
Many other processes are regulated or in near equilibrium with ATP/ADP. Thus, a switch from electrogenic to non-electrogenic mitochondrial ATP/ADP exchange would have far-reaching effects on other aspects of cancer cell metabolism. The switch between electrogenic versus non-electrogenic ATP turnover may also affect the bioenergetics of cancer cells exposed to different levels of oxygen, as occurs during tumor growth in vivo. Future studies will be needed to determine the contribution of ANT and the ATP-Mg/Pi carrier to the bioenergetics of cells grown under normoxic versus hypoxic conditions.
In conclusion, our findings indicate that ATP turnover and import of glycolytic ATP into mitochondria are independent of ANT in cancer cells. We propose that an alternative exchanger, possibly the non-electrogenic ATP/Mg-Pi carrier, takes over exchange of ATP and ADP. Such non-electrogenic ATP/ADP exchange substantially decreases cytosolic ATP/ADP ratios of proliferating cancer cells in comparison to post-mitotic differentiated cells. Low ATP/ADP removes the brake on glycolysis applied by high ATP/ADP and thus contributes to the Warburg metabolic phenotype of aerobic glycolysis.
Experimental Procedures
Materials
siRNAs were purchased from Ambion (Austin, TX). A549 cells, HepG2 cells, INS-1 cells, and UMSCC22 cells, Eagle's minimum essential medium, Kaighn's modification of Ham's F12K medium, and fetal bovine serum were from American Tissue Culture Collection (Manassas, VA). Bongkrekic acid and Protein Inhibitor Mixture Set III were from Calbiochem. Nonfat dry milk, ANT2 rabbit mAb #14671S, and β-tubulin rabbit mAb were from Cell Signaling (Danvers, MA). Novex NuPAGE35 gels, Lipofectamine RNAiMAX Transfection Reagent, Multispeck Multispectral Fluorescence Microscopy Standards kits, and TMRM were from Invitrogen. Dexamethasone was from LyphoMed (Rosemont, IL). Glass-bottomed Petri dishes were from MatTek Corp. (Ashland, MA). Goat anti-rabbit IgG-horseradish peroxidase #SC2004 was from Santa Cruz Biotechnology (Dallas, TX). l-Ornithine, lactate, CAT, MYX, 2DG, digitonin, PhosSTOP, succinate, rotenone, OLIG, and other analytical grade reagents were purchased from Sigma. Insulin was from Squibb-Novo, (Princeton, NJ). Immobilon PVDF membranes, the Pierce BCA Protein Assay kit, and SuperSignal West Dura Extended Duration Substrate #34075 were from Thermo Fisher (Waltham, MA).
Hepatocyte Isolation and Culture
Hepatocytes were isolated by collagenase perfusion from 200–250 g of overnight-fasted male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) as described previously (41). Hepatocytes were cultured overnight on 24-well plates or MatTek dishes coated with 20 μl/well 0.1% Type 1 rat tail collagen dissolved in 0.1% acetic acid (Sigma) and maintained in Waymouth's MB-752/1 growth medium supplemented with 10% fetal bovine serum, 100 nm insulin, 100 nm dexamethasone, 100 units/ml penicillin, and 100 μg/ml of streptomycin. Viability of cells was typically >90% as determined using a Cellometer Vision cell counter (Nexcelcom Biosciences, Lawrence, MA).
Cancer Cell Culture
HepG2, INS-1, and UMSCC22 cells were grown in Eagle's minimum essential medium, and A549 cells were grown in modified F12K medium. All media were supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in 5% CO2/air at 37 °C. For confocal microscopy, cells were plated 48 h on MatTek dishes. Live cell imaging was performed in 5% CO2/air at 37 °C in modified Hank's balanced salt solution (MHBSS) containing 137 mm NaCl, 0.35 mm Na2HPO4, 5.4 mm KCl, 1 mm KH2PO4, 0.81 mm MgSO4, 0.95 mm Ca2Cl, 5.5 mm glucose, 25 mm NaHCO3, and 20 mm HEPES, pH 7.4, as described (27).
Measurement of Hepatocyte Respiration
Hepatocytes (3 × 104 cells in 0.1 ml/well) were plated on 24-well Seahorse V28 plates (Seahorse Biosciences, Billerica, MA) coated with rat tail collagen, as described above, and maintained for 2 h in humidified 5% CO2/95% air at 37 °C. Subsequently, 0.4 ml of warm complete growth medium was added, and the hepatocytes were incubated for 16–18 h in humidified 5% CO2/95% air at 37 °C. Respiration (oxygen consumption rate) was measured in a Seahorse Bioscience XF-24 extracellular flux analyzer, and respiratory rates were calculated using Seahorse XF-24 software and the Direct ACOS fast algorithm with continuous averaging, as described (42). Hepatocytes were rinsed and maintained throughout the duration of the experiments in modified Krebs-Ringer-HEPES buffer containing 115 mm NaCl, 5 mm KCl, 1 mm CaCl2,1 mm KH2PO4, 1.2 mm MgSO4, 25 mm HEPES buffer (pH 7.4 with KOH), and 27 mm NaHCO3. Respiratory rates were expressed as the percentage of baseline.
Measurement of Cancer Cell Respiration
A549, HepG2, INS-1, and UMSCC22 cells (6 × 104 in 0.1 ml/well) were plated in Seahorse V7 plates in humidified 5% CO2/95% air at 37 °C. After 2 h, 0.4 ml of growth medium was added. Seahorse respirometry measurements were performed in MHBSS as described above at 48 h after plating. Permeabilization experiments in HepG2 cells were performed in intracellular buffer containing 120 mm KCl, 10 mm NaCl, 1 mm KH2PO4, 2 mm Mg-ATP, and 20 mm HEPES/NaOH supplemented with 5 mm succinate, 0.5 mm ADP, 1 mg/ml cytochrome c, and 10 μm rotenone.
Relative Abundance of ANT Isoforms, siRNA Treatment, and Real-time qPCR
A549 cells grown to 70–80% of confluency were transfected with siRNA (5 nm) targeting ANT2 and ANT3 or control non-target siRNA. Non-target siRNA was Silencer Select Negative Control #1 siRNA (catalogue #4390844). siRNA for ANT2 and ANT3 were SLC25A5 Silencer Select Pre-designed siRNA (catalogue #4392420 ID s1376 and SLC25A6 Silencer Select Pre-designed siRNA catalogue #4392420 ID s1377, respectively). siRNAs were transfected using Lipofectamine RNAiMAX, as described previously (31). mRNA levels of ANT1, ANT2, and ANT3 were assessed by two-step qPCR. Total RNA was isolated using a Qiagen RNeasy Mini kit and quantified using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, Inc.). cDNA was synthesized from total RNA using a Bio-Rad iScript cDNA Synthesis kit. Primers for ANT isoforms were designed using the Mfold webserver (UNAFfold) for nucleic acid folding prediction and Primer 3 for primer design. Forward and reverse primers were: 18S ribosomal RNA (r18S), GAG GGA GCC TGA GAA ACG G and GTC GGG AGT GGG TAA TTT GC; ANT1, ACA GAT TGT GTG GTT T and TTT TGT GCA TTA AGT GGT CTT T; ANT2, CTC GGT GAC TGC CTG GTT and CCA GCT GAT GAC GAT GTG AG; ANT3, TCG AGA AAT TCC AGT TGT CTT T and AGA ACA CGA CTT GGC TCC TAC. qPCR was performed as previously described (31).
Western Blotting
Proteins were extracted in 0.5 m NaCl, 50 mm Hepes, pH 7.4, 1 mm DTT, 1% Triton X-100, and 0.5% Tween 20 with phosphatase and protease inhibitors. Proteins (25 μg/lane) were separated on 4–12% Bis-Tris discontinuous gels, transferred onto PVDF membranes, and blocked for 60 min in 5% nonfat milk. Blots were probed with antibodies against ANT and β-tubulin (1:1000) and developed with goat anti-rabbit IgG conjugated to horseradish peroxidase (1:10:000) using a Supersignal chemiluminescence kit for detection. Intensity of bands was quantified using a Kodak Carestream 4000MM Pro Image Station (Stoneham, MA) and normalized to β-tubulin. Three independent experiments were used for calculations.
Loading of Tetramethylrhodamine Methyl Ester and Confocal Microscopy
Cells in MHBSS were loaded 30 min at 37 °C with 200 nm TMRM. After loading and washing, subsequent incubations were performed with 50 nm TMRM to maintain equilibrium distribution of the fluorophore (43–46). Cells incubated in MHBSS in humidified 5% CO2/air at 37 °C were imaged with a Zeiss LSM 510 NLO inverted laser scanning confocal/multiphoton microscope (Thornwood, NY) using a 63× 1.4 N.A. planapochromat oil immersion lens (27). Fluorescence of TMRM was excited at 543 nm and detected through a 560-nm long-pass filter and a 1-Airy-unit-diameter pinhole. Fluorescent polystyrene microspheres (4-μm) were used as fiducial markers. Microspheres were prediluted in MHBSS and added to TMRM-loaded cells at a final concentration of 30,000/ml. Images were taken 20 min after the addition to allow sedimentation of the microspheres.
TMRM fluorescence was quantified using Zeiss LSM and Photoshop CS4 (Adobe Systems, San Jose, CA) software. Mitochondrial TMRM uptake in a region of interest was estimated after subtracting average nuclear TMRM fluorescence from total average fluorescence and multiplying by the number of pixels. For each experimental condition, a minimum of 4 random fields containing ∼4–8 cells was analyzed. A minimum of three independent experiments were used to make final calculations.
Statistics
Differences between groups were analyzed by Student's t test using p < 0.05 as the criterion of significance. Data points are the means ± S.E. of 3–6 independent experiments. Images in figures are representative of three or more independent experiments.
Author Contributions
E. N. M. designed and coordinated the study, wrote the paper, and prepared the figures. D. N. D., J. P., S. K., and M. B. G. performed and analyzed the experiments. J. J. L. supervised the conception and design of the experiments and revised the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Craig C. Beeson for advice on Seahorse experiments and to Gregory L. Lovelace for technical assistance. Imaging facilities were supported, in part, by NIH Grant P30 CA138313.
This work was supported, in whole or in part, by National Institutes of Health Grants CA184456 and P20 GM103542 (to E. N. M.) and DK37034, DK073336, and AA021191 (to J. J. L.). This work was also supported by American Cancer Society Grant 13-043-01-IRG (to E. N. M.) and by Grant 14.Z50.31.0028 from the Russian Federation (to J. J. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ANT
- adenine nucleotide translocator
- SLC
- solute carrier family
- AAC
- ATP/ADP carrier
- ΔΨ
- mitochondrial membrane potential
- BA
- bongkrekic acid
- CAT
- carboxyatractyloside
- OLIG
- oligomycin
- TMRM
- tetramethylrhodamine methyl ester
- MYX
- myxothiazol
- 2DG
- 2-deoxyglucose
- VDAC
- voltage-dependent anion channel
- MHBSS
- modified Hank's balanced salt solution
- qPCR
- quantitative PCR
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Harvey A. J., Kind K. L., and Thompson J. G. (2002) REDOX regulation of early embryo development. Reproduction 123, 479–486 [DOI] [PubMed] [Google Scholar]
- 2. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 3. Warburg O., Wind F., and Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deberardinis R. J., Sayed N., Ditsworth D., and Thompson C. B. (2008) Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zong W. X., Rabinowitz J. D., and White E. (2016) Mitochondria and cancer. Mol. Cell 61, 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mathupala S. P., Ko Y. H., and Pedersen P. L. (2010) The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 1797, 1225–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakashima R. A., Paggi M. G., and Pedersen P. L. (1984) Contributions of glycolysis and oxidative phosphorylation to adenosine 5′-triphosphate production in AS-30D hepatoma cells. Cancer Res. 44, 5702–5706 [PubMed] [Google Scholar]
- 9. Singleterry J., Sreedhar A., and Zhao Y. (2014) Components of cancer metabolism and therapeutic interventions. Mitochondrion 17, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pedersen P. L. (1978) Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 22, 190–274 [DOI] [PubMed] [Google Scholar]
- 11. Moreno-Sánchez R., Marín-Hernández A., Saavedra E., Pardo J. P., Ralph S. J., and Rodríguez-Enríquez S. (2014) Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 50, 10–23 [DOI] [PubMed] [Google Scholar]
- 12. Klingenberg M. (2008) The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 1778, 1978–2021 [DOI] [PubMed] [Google Scholar]
- 13. Pfaff E., Heldt H. W., and Klingenberg M. (1969) Adenine nucleotide translocation of mitochondria: kinetics of the adenine nucleotide exchange. Eur. J. Biochem. 10, 484–493 [DOI] [PubMed] [Google Scholar]
- 14. Clémençon B., Babot M., and Trézéguet V. (2013) The mitochondrial ADP/ATP carrier (SLC25 family): pathological implications of its dysfunction. Mol. Aspects Med. 34, 485–493 [DOI] [PubMed] [Google Scholar]
- 15. Stepien G., Torroni A., Chung A. B., Hodge J. A., and Wallace D. C. (1992) Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 267, 14592–14597 [PubMed] [Google Scholar]
- 16. Chevrollier A., Loiseau D., Chabi B., Renier G., Douay O., Malthièry Y., and Stepien G. (2005) ANT2 isoform required for cancer cell glycolysis. J. Bioenerg. Biomembr. 37, 307–316 [DOI] [PubMed] [Google Scholar]
- 17. Dolce V., Scarcia P., Iacopetta D., and Palmieri F. (2005) A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization, and tissue distribution. FEBS Lett. 579, 633–637 [DOI] [PubMed] [Google Scholar]
- 18. Barath P., Luciakova K., Hodny Z., Li R., and Nelson B. D. (1999) The growth-dependent expression of the adenine nucleotide translocase-2 (ANT2) gene is regulated at the level of transcription and is a marker of cell proliferation. Exp. Cell Res. 248, 583–588 [DOI] [PubMed] [Google Scholar]
- 19. Battini R., Ferrari S., Kaczmarek L., Calabretta B., Chen S. T., and Baserga R. (1987) Molecular cloning of a cDNA for a human ADP/ATP carrier which is growth-regulated. J. Biol. Chem. 262, 4355–4359 [PubMed] [Google Scholar]
- 20. Giraud S., Bonod-Bidaud C., Wesolowski-Louvel M., and Stepien G. (1998) Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J. Mol. Biol. 281, 409–418 [DOI] [PubMed] [Google Scholar]
- 21. Le Bras M., Borgne-Sanchez A., Touat Z., El Dein O. S., Deniaud A., Maillier E., Lecellier G., Rebouillat D., Lemaire C., Kroemer G., Jacotot E., and Brenner C.(2006) Chemosensitization by knockdown of adenine nucleotide translocase-2. Cancer Res. 66, 9143–9152 [DOI] [PubMed] [Google Scholar]
- 22. Kolarov J., Kolarova N., and Nelson N. (1990) A third ADP/ATP translocator gene in yeast. J. Biol. Chem. 265, 12711–12716 [PubMed] [Google Scholar]
- 23. Lawson J. E., and Douglas M. G. (1988) Separate genes encode functionally equivalent ADP/ATP carrier proteins in Saccharomyces cerevisiae: isolation and analysis of AAC2. J. Biol. Chem. 263, 14812–14818 [PubMed] [Google Scholar]
- 24. Drgon T., Sabová L., Nelson N., and Kolarov J. (1991) ADP/ATP translocator is essential only for anaerobic growt h f yeast Saccharomyces cerevisiae. FEBS Lett. 289, 159–162 [DOI] [PubMed] [Google Scholar]
- 25. Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., MacGregor G. R., and Wallace D. C. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holmuhamedov E. L., Czerny C., Beeson C. C., Lemasters J. J. (2012) Ethanol suppresses ureagenesis in rat hepatocytes: role of acetaldehyde. J. Biol. Chem. 287, 7692–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maldonado E. N., Patnaik J., Mullins M. R., and Lemasters J. J. (2010) Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 70, 10192–10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown J., Higo H., McKalip A., and Herman B. (1997) Human papillomavirus (HPV) 16 E6 sensitizes cells to atractyloside-induced apoptosis: role of p53, ICE-like proteases and the mitochondrial permeability transition. J. Cell. Biochem. 66, 245–255 [DOI] [PubMed] [Google Scholar]
- 29. Zamzami N., Marchetti P., Castedo M., Hirsch T., Susin S. A., Masse B., and Kroemer G. (1996) Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 384, 53–57 [DOI] [PubMed] [Google Scholar]
- 30. Klingenberg M., Appel M., Babel W., and Aquila H. (1983) The binding of bongkrekate to mitochondria. Eur. J. Biochem. 131, 647–654 [DOI] [PubMed] [Google Scholar]
- 31. Maldonado E. N., Sheldon K. L., DeHart D. N., Patnaik J., Manevich Y., Townsend D. M., Bezrukov S. M., Rostovtseva T. K., and Lemasters J. J. (2013) Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J. Biol. Chem. 288, 11920–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maldonado E. N., and Lemasters J. J. (2012) Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 342, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rostovtseva T. K., Sheldon K. L., Hassanzadeh E., Monge C., Saks V., Bezrukov S. M., and Sackett D. L. (2008) Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. U.S.A. 105, 18746–18751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joyal J. L., and Aprille J. R. (1992) The ATP-Mg/Pi carrier of rat liver mitochondria catalyzes a divalent electroneutral exchange. J. Biol. Chem. 267, 19198–19203 [PubMed] [Google Scholar]
- 35. Fiermonte G., De Leonardis F., Todisco S., Palmieri L., Lasorsa F. M., and Palmieri F. (2004) Identification of the mitochondrial ATP-Mg/Pi transporter: bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 279, 30722–30730 [DOI] [PubMed] [Google Scholar]
- 36. del Arco A., and Satrústegui J. (2004) Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J. Biol. Chem. 279, 24701–24713 [DOI] [PubMed] [Google Scholar]
- 37. Traba J., Del Arco A., Duchen M. R., Szabadkai G., and Satrústegui J. (2012) SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca2+ buffering. Cell Death Differ 19, 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heldt H. W., Klingenberg M., Milovancev M. (1972) Differences between the ATP-ADP ratios in the mitochondrial matrix and in the extramitochondrial space. Eur. J. Biochem. 30, 434–440 [DOI] [PubMed] [Google Scholar]
- 39. Gambhir S. S. (2002) Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693 [DOI] [PubMed] [Google Scholar]
- 40. Gatenby R. A., and Gillies R. J. (2004) Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899 [DOI] [PubMed] [Google Scholar]
- 41. Qian T., Nieminen A. L., Herman B., and Lemasters J. J. (1997) Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am. J. Physiol. 273, C1783–C1792 [DOI] [PubMed] [Google Scholar]
- 42. Gerencser A. A., Neilson A., Choi S. W., Edman U., Yadava N., Oh R. J., Ferrick D. A., Nicholls D. G., and Brand M. D. (2009) Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 81, 6868–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lemasters J. J., Ramshesh V. K. (2007) Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Methods Cell Biol. 80, 283–295 [DOI] [PubMed] [Google Scholar]
- 44. Farkas D. L., Wei M. D., Febbroriello P., Carson J. H., and Loew L. M. (1989) Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys. J. 56, 1053–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scaduto R. C. Jr, and Grotyohann L. W. (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 76, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zahrebelski G., Nieminen A. L., al-Ghoul K., Qian T., Herman B., and Lemasters J. J. (1995) Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: a laser scanning confocal microscopic study. Hepatology 21, 1361–1372 [PubMed] [Google Scholar]