

Abstract
Within the ovarian follicle, granulosa cells (GCs) surround and support immature oocytes. FSH promotes the differentiation and proliferation of GCs and is essential for fertility. We recently reported that ERK activation is necessary for FSH to induce key genes that define the preovulatory GC. This research focused on the phosphoregulation by FSH of ERK within GCs. FSH-stimulated ERK phosphorylation on Thr202/Tyr204 was PKA-dependent, but MEK(Ser217/Ser221) phosphorylation was not regulated; rather, MEK was already active. However, treatment of GCs with the EGF receptor inhibitor AG1478, a dominant-negative RAS, an Src homology 2 domain-containing Tyr phosphatase inhibitor (NSC 87877), or the MEK inhibitor PD98059 blocked FSH-dependent ERK(Thr202/Tyr204) phosphorylation, demonstrating the requirement for upstream pathway components. We hypothesized that FSH via PKA enhances ERK phosphorylation by inhibiting the activity of a protein phosphatase that constitutively dephosphorylates ERK in the absence of FSH, allowing MEK-phosphorylated ERK to accumulate in the presence of FSH because of inactivation of the phosphatase. GCs treated with different phosphatase inhibitors permitted elimination of both Ser/Thr and Tyr phosphatases and implicated dual specificity phosphatases (DUSPs) in the dephosphorylation of ERK. Treatment with MAP kinase phosphatase (MKP3, DUSP6) inhibitors increased ERK(Thr202/Tyr204) phosphorylation in the absence of FSH to levels comparable with ERK phosphorylated in the presence of FSH. ERK co-immunoprecipitated with Myc-FLAG-tagged MKP3(DUSP6). GCs treated with MKP3(DUSP6) inhibitors blocked and PKA inhibitors enhanced dephosphorylation of recombinant ERK2-GST in an in vitro phosphatase assay. Together, these results suggest that FSH-stimulated ERK activation in GCs requires the PKA-dependent inactivation of MKP3(DUSP6).
Keywords: dual specificity phosphoprotein phosphatase, ERK, FSH, gene expression, ovary, PKA, MAP kinase phosphatase 3 (MKP3), dual specificity phosphatase 6 (DUSP6), granulosa cells
Introduction
FSH acts selectively on granulosa cells (GCs) 2 contained within ovarian follicles to promote the proliferation and differentiation of GCs to a preovulatory phenotype (reviewed in Ref. 1) as well as meiotic competency of the enclosed oocyte (2, 3). Luteinizing hormone then initiates meiosis and promotes ovulation and differentiation of remaining follicular cells into luteal cells. Although it is well recognized that the ERK signaling pathway plays a predominant role in the ovulatory response to luteinizing hormone in preovulatory GCs (4), the functional significance of the ERK signaling pathway in immature GCs is less understood. We recently showed that the ERK signaling pathway in immature GCs is required for the induction of at least a subset of FSH gene targets that define the mature preovulatory GC, including Inha (which encodes the α subunit of the hormone inhibin), Lhcgr (which encodes the luteinizing hormone receptor), Egfr (which encodes the EGF receptor (EGFR) required for ovulation), and Cyp19a1 (which encodes the rate-limiting enzyme in estrogen biosynthesis) (5). ERK-dependent gene expression in immature GCs is mediated in part by phosphorylation of the transcriptional activator Y-box binding protein 1 (YB-1) on Ser102 (5). Based on the relevance of the ERK signaling pathway to immature GC maturation, we sought to better understand the mechanism by which FSH activates ERK.
ERK is canonically activated by receptor tyrosine kinases (RTKs). Ligand-dependent activation of RTKs recruits the RAS guanine exchange factor SOS to the plasma membrane, resulting in RAS and, subsequently, RAF activation (reviewed in Ref. 6). The Ser/Thr kinase RAF then phosphorylates/activates the dual specificity kinase MEK that phosphorylates ERK. ERK then phosphorylates downstream kinases like ribosomal S6 kinase (RSK-2) (7). Phosphorylation of ERK by MEK on both Thr202 and Tyr204 is required for ERK activation; hence, dephosphorylation of either site by protein-tyrosine phosphatases (PTPs), Ser/Thr phosphatases, or dual specificity phosphatases (DUSPs) inactivates ERK (reviewed in Ref. 8).
In GCs, FSH activates ERK in a PKA-dependent manner (9–12). We reported previously that the canonical pathway upstream of ERK is constitutively active in immature GCs but that ERK phosphorylation/activity is restrained by a PTP (10). We hypothesized that FSH inactivated this phosphatase, thereby permitting the accumulation of MEK-phosphorylated/active ERK. We identified the PTP as a 100-kDa putative member of the PTP-straital-enriched protein tyrosine phosphatase (STEP)-like (SL) family (13, 14) based on the following criteria: Western blotting of GC extracts using rabbit polyclonal anti-PTP-SL antibody (15) revealed a signal at 100 kDa; an in-gel PTP assay detected a signal at 100 kDa in concentrated ovarian extracts; an anti-PTP-SL-reactive signal at 100 kDa was selectively immunoprecipitated with ERK-conjugated agarose from GC extracts; in GCs loaded with 32Pi, FSH stimulated the phosphorylation of a protein at 100 kDa immunoprecipitated with anti-PTP-SL antibody that was inhibited by the PKA, PKC, PKG (ACG) kinase family inhibitor H89 (16); and in an ERK-agarose immunoprecipitation, the anti-PTL-SL-reactive band at 100 kDa was reduced by ∼50% in GCs treated with FSH. Previous results by Pulido and co-workers (17, 18) showed that PTP-SL was phosphorylated on Ser231 in a PKA-dependent manner, relieving inhibition of ERK and its restriction to the cytoplasm and allowing ERK to translocate to the nucleus.
However, our recent results question our interpretation of our previous results. As we show below, the pan-PTP inhibitor Na2VO3 (19, 20), which should inhibit PTP-SL (15), does not block FSH-stimulated ERK phosphorylation. PTP-SL has a molecular weight of ∼55 kDa, not 100 kDa (21). A second antibody directed against PTPBR7, another phosphatase sharing protein sequence homology with PTP-SL except for a 127-amino acid insertion in the N terminus (14), does not detect a signal on Western blotting analyses of GC extracts at 100 kDa (10). Although the Ptprr gene that encodes PTP-SL is expressed in GCs (based on RNA sequencing results (22)), we thus question whether the PTP activity at 100 kDa that is detected by an anti-PTL-SL antibody is the phosphatase that constitutively dephosphorylates MEK-phosphorylated ERK in the absence of FSH.
The results below confirm that the ERK signaling pathway upstream of MEK is constitutively active in GCs, that ERK is constitutively phosphorylated by MEK but actively dephosphorylated in the absence of FSH, that MEK-phosphorylated ERK accumulates in the presence of FSH because of the inactivation of a phosphatase, and that FSH-stimulated ERK but not MEK phosphorylation is PKA-dependent. FSH-stimulated ERK activation is not inhibited either by inhibitors of the Ser/Thr protein phosphatase PP1 or PP2 or by the pan-PTP inhibitor Na2VO3. Rather, using a panel of DUSP inhibitors, our results show that inhibitors of MKP3(DUSP6) selectively enhance the phosphorylation of ERK in the absence of FSH to levels equivalent to those of ERK phosphorylated in the presence of FSH. ERK co-immunoprecipitates with Myc-FLAG-tagged MKP3(DUSP6), MKP3(DUSP6) inhibitors block ERK phosphatase activity in GC extracts in the absence of FSH in an in vitro phosphatase assay, and the selective PKA inhibitor PKI blocks the inactivation of the ERK phosphatase in the presence of FSH in an in vitro phosphatase assay. Together, these results suggest that ERK in GCs is maintained in a dephosphorylated state in the absence of FSH by MKP3(DUSP6) and that PKA inhibits the activity of this DUSP.
Results
Both Canonical Signaling to MEK and PKA Activity Are Required for FSH-stimulated Phosphorylation of ERK(Thr202/Tyr204)
We initially sought to confirm that the canonical ERK signaling pathway in GCs upstream of MEK is tonically active and independent of FSH. Our previous results demonstrated an apparent requirement for the tyrosine kinase activity of the EGFR based on results showing that the EGFR inhibitor AG1478 (23) blocked FSH-stimulated ERK(Thr202/Tyr204) phosphorylation. However, we did not evaluate the effect of AG1478 on MEK(Ser217/Ser221) phosphorylation (10). To this end, GCs were pretreated with AG1478, followed by treatment without or with FSH. The results (Fig. 1A) show that MEK(Ser217/Ser221) phosphorylation is readily detected in vehicle-treated GCs and independent of FSH, whereas ERK(Thr202/Tyr204) phosphorylation depends on FSH, and that AG1478 not only abolishes ERK(Thr202/Tyr204) phosphorylation (95.1% ± 0.8% inhibition, n = 5) but also abolishes MEK(Ser217/Ser221) phosphorylation (89.0% ± 3.8% inhibition, n = 2). AKT(Ser308) phosphorylation served as a negative control, as FSH-dependent activation of the PI3K/AKT pathway is independent of the EGFR in GCs. 3 These results indicate that the signaling pathway that promotes MEK phosphorylation is active in the absence of FSH and that this pathway requires the tyrosine kinase activity of the EGFR.
FIGURE 1.

The Canonical MAPK signaling pathway is active in GCs independent of FSH stimulation and is necessary for phosphorylation of ERK1/2(Thr202/Tyr204). A, GCs were treated for 15 min without (DMSO) or with 250 nm AG1478, an EGFR kinase inhibitor, followed by treatment without (veh) or with 50 ng/ml FSH for 15 min. Samples were heat-denatured after collection in SDS sample buffer, and proteins were separated by SDS/PAGE. A blot of whole cell extracts was probed with the indicated antibodies. The results are representative of five independent experiments. B, GCs were transduced with Ad-GFP or Ad-(S17N)RAS overnight, followed by treatment without (veh) or with FSH 15 min. Samples were collected as described in A. The results are representative of four independent experiments. The dotted lines between lanes represent cropped images. C, GCs were treated without (DMSO) or with 20 mm NSC 87877, a SHP2 inhibitor, for 3 h, followed by treatment without (veh) or with FSH for 15 min. Samples were collected as described in A. The results are representative of five independent experiments.
Although AG1478 primarily inhibits the kinase activity of the EGFR, it is also reported to inhibit non-kinase targets (24). As the EGFR canonically activates RAS, we utilized two additional approaches to confirm that the signaling pathway downstream of the EGFR was required for FSH to activate ERK and was tonically active in the absence of FSH. We transduced GCs with a dominant negative adenoviral (Ad)-(S17N)-RAS or control Ad-GFP and then treated GCs with vehicle or FSH. The results (Fig. 1B) show that the dominant negative RAS blocked MEK(Ser217/Ser221) phosphorylation (75.2% ± 4.7% inhibition, n = 3) both in the absence and presence of FSH as well as FSH-stimulated ERK(Thr202/Tyr204) phosphorylation (78.3% ± 10.0% inhibition, n = 4). cAMP-response element-binding protein(Ser133) phosphorylation served as a negative control that is independent of EGFR/RAS signaling (25). The tyrosine phosphatase SRC homology 2 domain-containing tyrosine phosphatase (SHP2) is also required for RTK signaling into MEK/ERK and is generally believed to contribute to the activation of RAS (26–28). We determined whether SHP2 activity was required for the phosphorylation of MEK(Ser217/Ser221) by pretreating GCs without or with NSC 87877, a compound that selectively inhibits SHP1 (which is not expressed in rat GCs based on RNA sequencing results (22)) and SHP2 (29). NSC 87877 markedly attenuated MEK(Ser217/Ser221) phosphorylation (82.8% ± 2.5% inhibition, n = 2) in vehicle- and FSH-treated GCs and, hence, similarly reduced FSH-stimulated ERK(Thr202/Tyr204) phosphorylation (Fig. 1C) (92.9% ± 5.5% inhibition, n = 5). GAB2(Ser159) phosphorylation served as a negative control that is independent of SHP2 signaling (30); FSH-stimulated GAB2(Tyr452) dephosphorylation shows the selectivity of the SHP2 PTP activity. Taken together, these results show that the signaling pathway that promotes MEK(Ser217/Ser221) phosphorylation in GCs is tonically active in the absence of FSH and requires the EGFR, RAS, and SHP2.
FSH-stimulated ERK(Thr202/Tyr204) phosphorylation is also recognized to be dependent on PKA (9, 10, 12). In the following experiment, we sought to confirm that, although the PKA inhibitor PKI, which functions as a PKA catalytic subunit pseudosubstrate (31), blocks FSH-stimulated ERK(Thr202/Tyr204) phosphorylation, it does not affect MEK(Ser217/Ser221) phosphorylation. As shown in Fig. 2, although transduction of GCs with Ad-PKI blocked FSH-stimulated ERK(Thr202/Tyr204) phosphorylation (89.3% ± 10.3% inhibition, n = 4), MEK(Ser217/Ser221) phosphorylation was unaffected. Taken together, these results demonstrate that the effect of PKA must be downstream of MEK at the level of ERK phosphorylation. Active MEK should constitutively phosphorylate ERK; however, phosphorylated ERK is not detected in the absence of FSH. This result suggests that a phosphatase is actively dephosphorylating ERK in the absence of FSH. It then follows that FSH via PKA must inactivate the ERK phosphatase.
FIGURE 2.
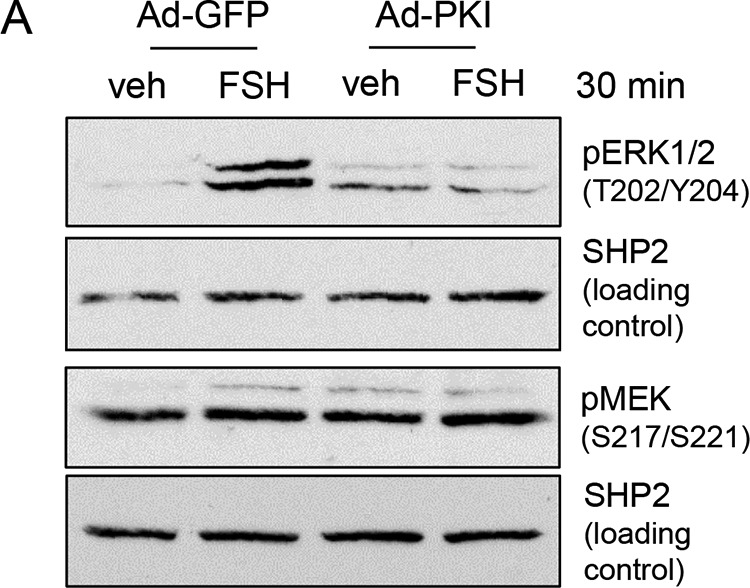
Phosphorylation of ERK1/2 on Thr202/Tyr204, but not of MEK on Ser217/Ser221, is dependent on PKA activity. GCs were transduced overnight with Ad-GFP or Ad-PKI, a PKA inhibitor. Cells were then treated without (veh) or with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of four independent experiments.
ERK Phosphorylation Is Not Directly Regulated by a Ser/Thr Phosphatase or PTP
In the following experiments, we determined whether the phosphatase that dephosphorylates ERK on Thr202 and/or Tyr204 in GCs is a Ser/Thr protein phosphatase (PP1 or PP2) or a Tyr phosphatase. Pretreatment of GCs with the PP2 inhibitor okadaic acid (at 200 nm (32–35)) resulted in no change in phosphorylation of ERK(Thr202/Tyr204) (Fig. 3A). Phosphorylation of GSK3β(Ser9) increased with okadaic acid alone treatment (Fig. 3A, lane 3) (5-fold increase over veh, n = 3]. GSK3β(Ser9) is a recognized PP2 target (36) and is used as a positive control.
FIGURE 3.

FSH-stimulated phosphorylation of ERK1/2(Thr202/Tyr204) is not regulated by Ser/Thr and Tyr phosphatases. A, GCs were pretreated without (EtOH) or with 0.2 μm okadaic acid, a PP2 inhibitor, for 1 h, followed by treatment without (veh) and with FSH for 15 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments. The results for pGSK3β and its S6 loading control were published previously (58). B, GCs were pretreated without (EtOH) or with 1 μm tautomycin, a PP1 inhibitor, for 5.5 h, followed by treatment without (veh) and with FSH for 15 min. Samples were collected as described in Fig. 1A. The results are representative of six independent experiments. C, GCs were pretreated without (H2O) or with 50 μm Na2VO3, a pan tyrosine phosphatase inhibitor, for 12 h, followed by treatment without (veh) and with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments. The results for the SHP2 loading control were published previously (58).
Pretreatment of GCs with the PP1 inhibitor tautomycin (at 1 μm (32–35)) promoted a decrease in both FSH-stimulated ERK(Thr202/Tyr204) phosphorylation (70.9% ± 3.5% inhibition, n = 6) and basal MEK(Ser217/Ser221) phosphorylation (70.3% ± 9.9% inhibition, n = 2) (Fig. 3B). Blockade of FSH-stimulated myosin light chain(Ser19) dephosphorylation by tautomycin, a recognized PP1 target (37, 38), served as a positive control for tautomycin. These results indicate that PP1 is necessary for signaling into MEK and acts upstream of MEK, not on ERK, based on the inability of the PP1 inhibitor tautomycin to raise ERK(Thr202/Tyr204) phosphorylation in vehicle-treated cells (Fig. 3B, lane 3).
Pretreatment of GCs with the pan-PTP inhibitor Na2VO3 (19, 20) resulted in an increase in both basal and FSH-stimulated ERK(Thr202/Tyr204) phosphorylation (5-fold increase, n = 3) (Fig. 3C). Concomitantly, MEK(Ser217/Ser221) phosphorylation also increased (2.5-fold increase over veh, n = 2) with Na2VO3 treatment. Phosphorylation of insulin receptor substrate 1 (IRS1(Tyr989)), an RTK target (39), served as a positive control. The target(s) of the PTP is thus upstream of MEK, likely at the level of the EGFR, and not at the level of ERK itself. Taken together, these results suggest that the phosphatase that dephosphorylates MEK-phosphorylated ERK in the absence of FSH is neither a Ser/Thr phosphatase nor a PTP.
ERK Phosphorylation Is Regulated by a DUSP
The inability of selective inhibitors of PP1, PP2, or PTPs to preferentially enhance ERK phosphorylation in the absence of FSH suggested that the relevant ERK phosphatase must be a member of the family of DUSPs. Indeed, in a recent report, DUSP27 blocked ERK but not MEK phosphorylation in a rat luteal cell line in response to prolactin treatment (40). However, DUSP27 is not expressed in rat GCs based on both microarray (41) and RNA sequencing (22) results. Although 61 DUSPs have been identified (as reviewed in Ref. 42), preantral GCs only express DUSPs 1, 3, 6, 7, 10, 11, 12, 16, 18, 19, and 22 as well as cell division cycle (CDC) dual specificity phosphatase CDC25A, 25B, and 25C mRNAs based on RNA sequencing results (22). Only a subgroup of the DUSPs are classified as MKPs based on the presence of a CDC25-like domain that confers specificity toward the TXY motif in MAPKs (as reviewed in Ref. 42). These include MKP1(DUSP1), MKP3(DUPS6), MKPX/PYST2(DUSP7), MKP5(DUSP10), MKP7(DUSP16), and CDC25A, CDC25B, and CDC25C. Of those, MKP5(DUSP10) and MKP7(SP16) are selective for c-Jun N-terminal kinase and p38 MAPK over ERK (as reviewed in Ref. 42) and were not investigated. Based on the availability of commercially available selective inhibitors and protein detection by Western blotting in GCs (data not shown), in the following experiments we tested the contributions of MKP1(DUSP1), MKP3(DUSP6) and CDC25A, CDC25B and CDC25C to the level of ERK phosphorylation in GCs. (Commercially available inhibitors for the DUSPs were determined according to Jeffrey et. al. (80), and at time of publication, there are no commercially available inhibitors for MKPX/PYST2(DUSP7)).
GCs were pretreated without or with NSC 663284, a selective CDC25A, CDC25B, and CDC25C inhibitor that does inhibit MKP1(DUSP1) or MKP3(DUSP6) (43, 44). The results (Fig. 4A) show that ERK(Thr202/Tyr204) phosphorylation in the absence or presence of FSH is not affected (lanes 3 and 4 versus lanes 1 and 2) by this competitive CDC25 inhibitor. Although we were unable to identify a positive control for this inhibitor, as rat GCs do not divide in primary culture (45–47) and, hence, do not express the cyclins necessary for cyclin-dependent kinase 2 phosphorylation (48), this result suggests that CDC25s are not functioning as ERK phosphatases in GCs. Additional indirect evidence that the CDC25 DUSPs are not functioning as ERK phosphatases in GCs is a report that Na2VO3 inhibits CDC25 DUSPs (49), whereas this phosphatase inhibitor does not inhibit the ERK phosphatase in GCs (Fig. 3C).
FIGURE 4.

DUSP inhibitor studies suggest that MKP3(DUSP6) actively dephosphorylates MEK-phosphorylated ERK1/2(Thr202/Tyr204) in the absence of FSH. A, GCs were pretreated without (DMSO) or with 5 μm NSC 663284, a selective CDC25A, CDC25B, and CDC25C inhibitor (which does not inhibit MKP1(DUSP1) or MKP3(DUSP6)) for 30 min, followed by treatment without (veh) and with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments. B, GCs were pretreated without (DMSO) or with 5 μm BCI, a selective MKP1(DUSP1) and MKP3(DUSP6) inhibitor, for 30 min, followed by treatment without (veh) and with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments. C, GCs were treated without (veh) and with FSH for the indicated times. Samples were collected as described in Fig. 1A. D, GCs were pretreated without (DMSO) or with 10 μm triptolide, a selective MKP1(DUSP1) inhibitor, for 1 h, followed by treatment without (veh) and with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments, except for the phospho-p38 blot, which is representative of two independent experiments. E, GCs were pretreated without (DMSO) or with 5 μm NSC 295642, a selective MKP3(DUSP6) and CDC25A and CDC25B inhibitor, for 30 min, followed by treatment without (veh) and with FSH for 30 min. Samples were collected as described in Fig. 1A. The results are representative of three independent experiments. The dotted lines between lanes represent cropped images.
GCs were next pretreated without or with BCI (NSC 150117), a selective allosteric inhibitor of MKP1(DUSP1) and MKP3(DUSP6) which does not inhibit CDC25 (50). The results (Fig. 4B) show that BCI increased ERK(Thr202/Tyr204) phosphorylation in vehicle-treated GCs (lane 3) to levels equivalent to those of FSH-treated cells (lane 2) (10-fold increase over veh, n = 3), whereas the FSH response was lost (lane 4). MEK(Ser217/Ser221) phosphorylation in the presence of BCI (Fig. 4B, lanes 3 and 4) was equivalent to that of vehicle- and FSH-treated GCs in the absence of BCI (Fig. 4B, lane 2). These results suggest that either MKP1(DUSP1) or MKP3(DUPS6) may be the relevant ERK DUSP in GCs.
We next sought to verify the expression of both MKP1(DUSP1) and MKP3(DUSP6) proteins within GCs, especially because MKP1(DUSP1) is most commonly induced as an immediate early gene upon RTK activation of ERK (as reviewed in Ref. 42). GCs were treated with vehicle or FSH for the indicated times. The results show that both MKP1(DUSP1) and MKP3(DUSP6) are readily detected in GCs, and total protein levels do not change within 60 min of FSH treatment (Fig. 4C).
To distinguish between MKP1(DUSP1) and MKP3(DUSP6) as regulators of ERK(Thr202/Tyr204) phosphorylation, GCs were pretreated with the selective MKP1(DUSP1) inhibitor triptolide (NSC 163062) (51, 52). Treatment with triptolide had no effect on ERK(Thr202/Tyr204) phosphorylation in the absence or presence of FSH (Fig. 4D, compare lanes 3 and 4 with lanes 1 and 2). However, triptolide enhanced the phosphorylation of p38 MAPK(Thr180/Tyr182) (Fig. 4D, lane 3; 6.1-fold over veh, n = 2) to levels equivalent to those of FSH-treated cells (Fig. 4D, lanes 2 and 4). This result suggests, but does not prove, that, like ERK, FSH promotes the phosphorylation of p38 MAPK by inhibiting an MKP, in this case MKP1. Treatment of GCs with the MKP1(DUSP1)-selective inhibitor chelerythrine chloride (25 μm, 30 min) (53) also did not affect ERK(Thr202/Tyr204) phosphorylation (data not shown). These results suggest that the relevant ERK DUSP in GCs is likely MKP3(DUSP6) and not MKP1(DUSP1).
As we were unable to identify a selective MKP3(DUPS6) inhibitor, we pretreated GCs with NSC 295642, a partial MKP3(DUSP6), CDC25A, and CDC25B inhibitor (54). The results (Fig. 4E) show that NSC 295642 promoted a 5.9-fold increase in ERK(Thr202/Tyr204) phosphorylation in vehicle-treated GCs (lane 3), nearly equivalent to that of FSH-treated cells (lane 2) (6.2-fold increase over veh, n = 3). FSH treatment did not further enhance ERK phosphorylation in the presence of NSC 295642 (Fig. 4E, lanes 3 and 4). Because the results from NSC 663284 treatments (Fig. 4A) allowed us to rule out contributions from CDC25A and CDC25B, we can conclude from these inhibitor studies that MKP3(DUSP6) is likely the DUSP that dephosphorylates ERK(Thr202/Tyr204) in the absence of FSH. Collectively, these results indicate that MKP3(DUSP6) is dephosphorylating ERK in untreated GCs and that FSH promotes inactivation of MKP3(DUSP6) to allow MEK-phosphorylated ERK(Thr202/Tyr204) to accumulate.
ERK and MKP3(DUSP6) Interact within GCs
If MKP3(DUSP6) is dephosphorylating MEK-phosphorylated ERK in the absence of FSH, then MKP3(DUSP6) should bind to ERK in GCs. As the MKP3(DUSP6) antibody does not readily immunoprecipitate MKP3(DUSP6) (data not shown), GCs were transiently transfected with an MKP3-Myc-FLAG plasmid to determine whether MKP3(DUPS6) and ERK interact. Immunoprecipitation of ERK2 from vehicle- and FSH-treated GCs showed the presence of the FLAG-tagged MKP3(DUPS6) in ERK2-immunoprecipitated samples but not in IgG controls (Fig. 5A). Conversely, immunoprecipitation of Myc-tagged MKP3 with anti-Myc antibody from vehicle- and FSH-treated cells showed that the anti-Myc antibody selectively pulls down ERK (Fig. 5B). These results suggest that MKP3(DUSP6) and ERK1/2 interact within GCs independent of FSH treatment.
FIGURE 5.
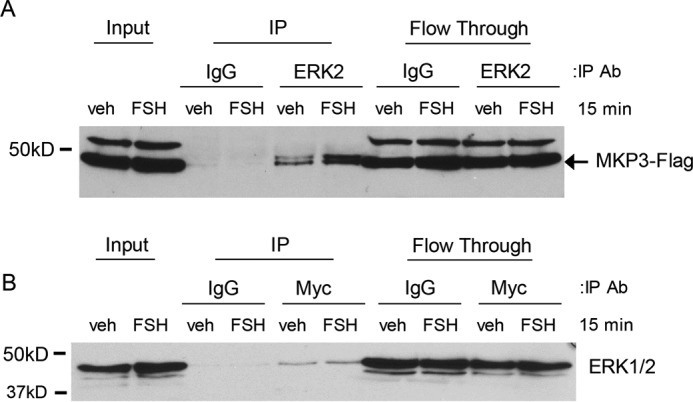
A, GCs were transfected with 500 ng of MKP3-Myc-FLAG plasmid for 6 h as described under “Experimental Procedures.” Following an overnight recovery, GCs were treated without (veh) or with FSH for 1 h and then collected in immunoprecipitation (IP) lysis buffer and sonicated. After removal of the insoluble particles, the soluble fraction was rotated overnight with 20 μg of agarose-conjugated anti-ERK2 (SC 1647 AC) or agarose-conjugated IgG (5). Inputs, bound (IP), and unbound (Flow Through) samples were separated by SDS/PAGE, and the Western blot was probed with the indicated antibody (Ab). The arrow indicates the protein band of interest. The results are representative of two independent experiments. B, soluble lysates were incubated with 20 μg of agarose-conjugated anti-Myc (SC 40-AC) to immunoprecipitate Myc-FLAG-tagged MKP3. For more details, see A. The results are representative of two independent experiments.
ERK(Thr202/Tyr204) Dephosphorylation Is Dependent on MKP3(DUSP6) Activity
In the following experiments, we sought to demonstrate that MKP3(DUSP6), but not MKP1(DUSP1), is able to dephosphorylate ERK in vitro. Using the MKP1 and MKP3 inhibitors discussed above, whole GC lysate phosphatase assays were performed using GST-tagged phospho-ERK2. GCs were pretreated without or with the indicated inhibitors, followed by treatment with vehicle or FSH. Lysates were then assayed for phosphatase activity, evidenced by their ability to dephosphorylate GST-tagged phospho-ERK2, as detected by Western blotting with phospho- ERK(Thr202/Tyr204) antibody. Pretreatment of GCs with BCI, the dual MKP1(DUSP1) and MKP3(DUSP6) inhibitor, blocked the DUSP that dephosphorylated ERK(Thr202/Tyr204) in the absence of FSH (Fig. 6A, compare lanes 3 and 1). Equivalent results were obtained upon pretreatment of GCs with NSC 295642, the MKP3(DUSP6) inhibitor (Fig. 6B). Taken together, these results support the conclusion that MKP3(DUSP6) is the DUSP that dephosphorylates ERK in the absence of FSH.
FIGURE 6.
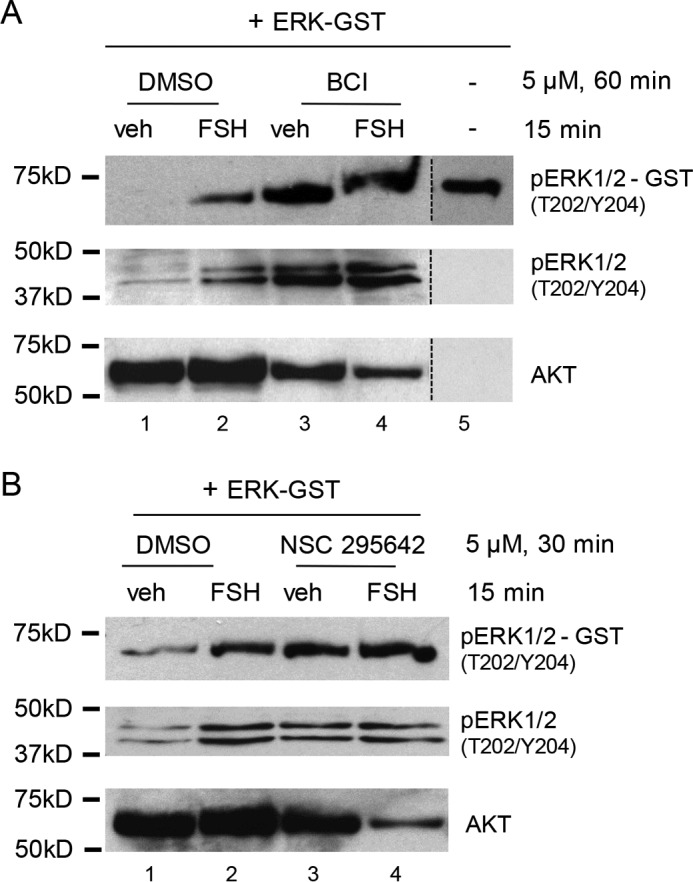
Active ERK2-GST dephosphorylation is blocked by MKP3(DUSP6) inhibitors. A, GCs were pretreated without (DMSO) or with 5 μm BCI for 60 min, followed by treatment without (veh) and with FSH for 15 min. Samples were collected in phosphatase assay buffer as described under “Experimental Procedures.” Active ERK2-GST (EMD Millipore, 14-173) was added to the samples and incubated for 60 min. The samples were separated by SDS/PAGE, and the Western blot was probed with the indicated antibodies. The results are representative of two independent experiments. The migration of phosphorylated ERK2-GST is shown in lane 5. The dotted lines between lanes represent cropped images. B, GCs were pretreated without (DMSO) or with 5 μm NSC 295642 for 30 min, followed by treatment without (veh) and with FSH for 15 min. Samples were collected as described in A. The results are representative of two independent experiments.
Inactivation of the ERK Phosphatase by FSH Is Dependent upon PKA Activity
We sought to determine whether the ability of MKP3(DUSP6) to dephosphorylate ERK(Thr202/Tyr204) in vitro was dependent on PKA using the selective PKA inhibitor PKI (31). GCs were transduced with Ad-E or Ad-PKI overnight, treated with vehicle or FSH, and whole GC lysate phosphatase assays were performed using GST-tagged phospho-ERK2 as described above. Transduction of GCs with Ad-PKI promoted the dephosphorylation of ERK2(Thr202/Tyr204)-GST in both vehicle- and FSH-treated cells (Fig. 7, compare lanes 3 and 4 with lanes 1 and 2). These data indicate that FSH-dependent PKA activation is necessary for inactivation of the ERK DUSP.
FIGURE 7.
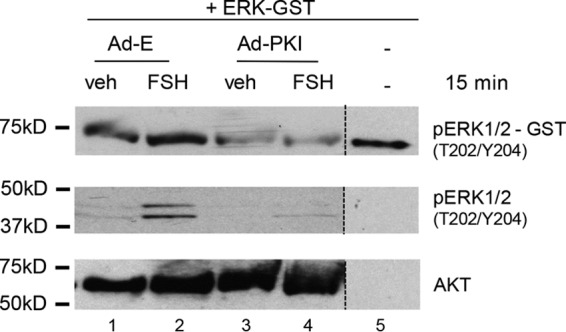
Active ERK2-GST dephosphorylation is dependent on PKA activity. GCs were transduced with Ad-Empty (E) or Ad-PKI overnight. Following the medium replacement outlined under “Experimental Procedures,” cells were treated without (veh) and with FSH for 15 min. Samples were collected as described in Fig. 6A. The results are representative of two independent experiments. The dotted lines between lanes represent cropped images.
Discussion
Our results confirm that the canonical signaling pathway upstream of ERK, which includes the EGFR, RAS, SHP2, and MEK, is constitutively active in GCs and that the activity of these upstream components is necessary but not sufficient for FSH-dependent ERK(Thr202/Tyr204) phosphorylation, as modeled in Fig. 8. FSH-dependent ERK(Thr202/Tyr204) phosphorylation also requires the inhibition of a DUSP, most likely MKP3(DUPS6), based on the ability of MKP3(DUPS6) inhibitors to raise ERK(Thr202/Tyr204) phosphorylation in the absence of FSH to levels comparable with those of FSH-treated cells and on the loss of an FSH response in the presence of the inhibitors. Additionally, ERK selectively co-immunoprecipitates with tagged-MKP3(DUPS6), and tagged-MKP3(DUSP6) selectively co-immunoprecipitates with ERK. The in vitro phosphatase assay results show that MKP3(DUPS6) inhibitors block dephosphorylation of ERK2-GST in the absence of FSH and that the selective PKA inhibitor PKI abrogates the ability of FSH to inhibit the ERK DUSP. Together, these results strongly suggest that ERK is dephosphorylated in the absence of FSH by MKP3(DUPS6) and that FSH via PKA endorses the MEK-dependent phosphorylation of ERK by inhibiting this DUSP.
FIGURE 8.
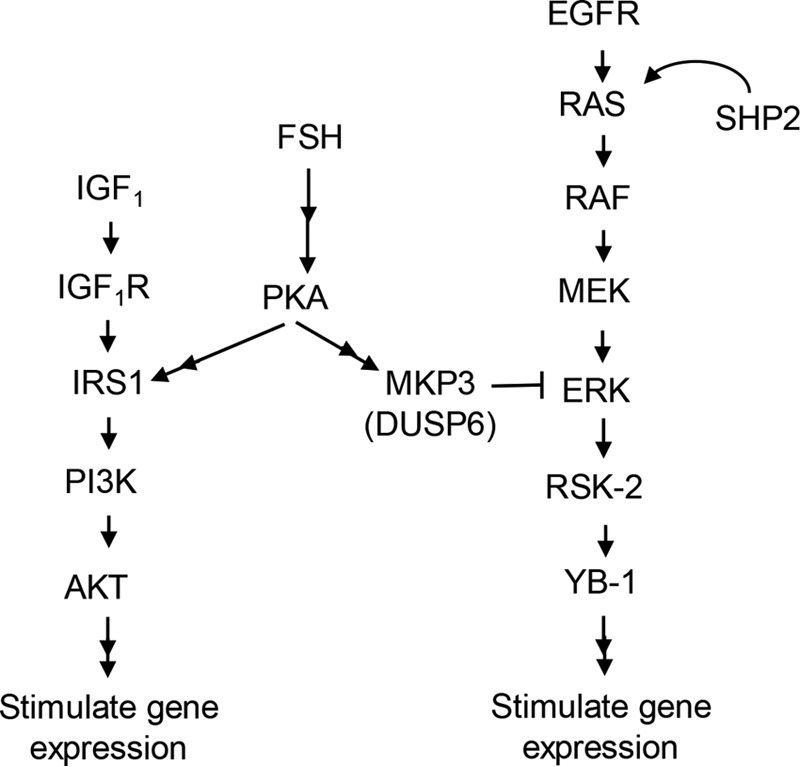
Model of the signaling pathway by which FSH, via PKA, regulates the PI3K and ERK pathways in GCs. The results presented here support the contributions of constitutively active pathway components upstream of ERK, the ability of MKP3(DUSP6) to dephosphorylate MEK-phosphorylated ERK in the absence of FSH, and the ability of FSH, in a PKA-dependent manner, to inhibit the phosphatase activity of the likely ERK DUSP, MKP3. The ability of ERK to promote phosphorylation/activation of the transcriptional factor YB1 is based on a prior publication (5). The ability of PKA to sensitize IRS1 to the tyrosine kinase activity of the insulin-like growth factor 1 receptor (IGF1R) is also based on prior publications (30, 58).
Recent studies suggest that the majority of the responses in GCs elicited by FSH are mediated by PKA, based not only on the ability of PKA inhibitors to block FSH-activated signaling pathways and gene expression (reviewed in Ref. 1) but also on the ability of a constitutively active PKA catalytic subunit mutant to mimic both acute FSH signaling and delayed gene expression (12, 41). Acute signaling responses include not only direct phosphorylation of target proteins by PKA, including cAMP-response element-binding protein (55), histone H3 (56), and β-catenin (57), but also activation by PKA of classical RTK-activated pathways such as the PI3K/AKT and MEK/ERK pathways (modeled in Fig. 8) (5, 10, 30). However, until recently, the mechanisms by which PKA accomplished the regulation of these canonical signaling pathways remained elusive. Our results indicate that, through regulation of MKP3(DUPS6), FSH is able promote the accumulation of MEK-phosphorylated ERK(Thr202/Tyr204), whereas the upstream components of the MEK/ERK signaling cascade remain active and unregulated by FSH treatment. Interestingly, our laboratory recently published results demonstrating that FSH activates the PI3K/AKT signaling pathway by promoting the PKA-dependent activation of PP1, which sensitizes IRS1 to the RTK activity of the insulin-like growth factor 1 receptor to activate PI3K in GCs (58). We also recently reported that FSH, in a PKA-, ERK-, and RSK-2-dependent manner, promotes phosphorylation of the transcriptional factor YB-1 on Ser102, which appears to be mediated primarily by the ability of RSK-2 to inhibit the phosphatase activity of PP1 (5). Collectively, these results indicate that FSH, via PKA, regulates at least two established RTK signaling cascades through regulation of phosphatases, seemingly hijacking these cascades. FSH, via PKA, also seems to enhance the transcriptional events by promoting the phosphorylation of at least one transcriptional factor via the inhibition of a phosphatase (5). Together, these results indicate that, at least in GCs, PKA either directly phosphorylates target proteins or enhances the phosphorylation of key proteins in RTK pathways by regulating the activity of protein phosphatases.
MKP3(DUPS6) is a dual specificity phosphatase and member of the MKP subfamily whose substrates are the MAPKs (ERK, p38 MAPK, and JNK) (reviewed in Ref. 59). MKP3(DUPS6) has been shown to have a substrate preference for ERK over p38 MAPK and JNK (60–62). Regulation of MKP3(DUPS6) expression is most often placed downstream of ERK signaling (63, 64) and is thought to constitute a negative feedback loop to down-regulate mitogenic signaling. Consistent with this conclusion, Mkp3/Dusp6 is often induced following ERK activation (65) and requires regulation by the transcription factor Ets1 (64, 65). Upon expression, MKP3 is catalytically activated via its physical association with ERK (62, 66, 67). We showed that MKP3(DUSP6) protein is readily expressed in GCs and that levels remain relatively constant following FSH treatment, giving no indication of a large change in total protein expression (Fig. 4C). These results indicate that, in GCs at the time points checked, initial regulation of MKP3(DUSP6) is not at the transcriptional level, as protein levels remain stable up to 1 h following treatment with FSH.
MKP3(DUSP6) is also reported to undergo posttranslational modifications that promote reduced activity or degradation. For example, MKP3(DUPS6) can be phosphorylated by ERK on Ser159 (SSSS159PP[rat]) and Ser197 (SDGS197SL[rat]), resulting in proteasomal degradation (68). Ser159 on MKP3(DUSP6) is also reported to be phosphorylated downstream of PI3K to promote MKP3(DUSP6) degradation (69). Casein kinase 2α has also been reported to bind MKP3, displacing ERK, and to phosphorylate MKP3(DUSP6) on at least two sites, resulting in an inhibition of its phosphatase activity (70).
It is our hypothesis that MKP3(DUSP6) becomes phosphorylated in GCs in a FSH- and PKA-dependent manner, resulting in its inactivation. Although it is possible that MKP3(DUSP6) becomes phosphorylated on Ser159 and Ser197 in response to the FSH/PKA-dependent activation of ERK in GCs, possibly sustaining the inactivation of MKP3(DUSP6), the initial inactivation of MKP3(DUPS6) must be mediated by another PKA-dependent event. As casein kinase 2α is a constitutively active kinase (71), it does not appear to be a PKA-regulated candidate to inhibit MKP3(DUPS6) in GCs. Although FSH activates PI3K in a PKA-dependent manner (30, 58), phosphorylation of YB-1(Ser102) downstream of ERK and RSK-2 is not inhibited by the PI3K inhibitor wortmannin (5). These results thus indicate that PKA activation of the PI3K pathway in GCs is not the mechanism by which PKA inactivates MKP3(DUPS6). Moreover, if MKP3(DUSP6) is degraded in response to FSH, reduced protein levels of MKP3(DUSP6) are not detected within the first hour following FSH treatment of GCs (Fig. 4C).
There are no reports, to our knowledge, that MKP3(DUPS6) is directly phosphorylated by PKA or that PKA can regulate its activity. Rat MKP3(DUSP6) contains one potential, albeit weak, PKA phosphorylation motif (RR/XX/S/T): RSVThr302V. Consistent with our results, we thus hypothesize that MKP3(DUSP6) is phosphorylated by PKA on Thr302, resulting in an inhibition of its phosphatase activity. However, our inability to directly immunoprecipitate MKP3(DUSP6) and a lack of phosphospecific antibodies to MKP3(Thr302) have prevented us from directly testing this hypothesis.
Although our results support MKP3(DUSP6) as the relevant DUSP that regulates the activity of ERK in response to FSH in GCs, the MKP3(DUSP6) global knockout mouse is fertile (72). We predict that another ERK-selective DUSP, such as MKPX/PYST2(DUSP7) (73), is able to compensate for the absence of MKP3(DUSP6) in GCs. The MKPX/PYST2(DUSP7) (The Jackson Laboratory, Bar Harbor, ME) and MKP1(DUSP1) (74) global knockout mice are also fertile. Formal proof that MKP3(DUSP6) is the DUSP that dephosphorylates constitutively phosphorylated ERK in GCs requires a genetic approach in which at least MKP3(DUSP6) and MKPX/PYST2(DUSP7) are deleted.
In summary, we have shown that FSH-dependent ERK(Thr202/Tyr204) phosphorylation is PKA-dependent as well as dependent on the constitutively active canonical upstream MEK/ERK signaling components that include the EGFR, RAS, SHP2, and MEK. We conclude that, in the absence of FSH, constitutively phosphorylated ERK is maintained in a dephosphorylated state by a DUSP, most likely by MKP3(DUPS6). FSH in a PKA-dependent manner inactivates the DUSP, allowing for the accumulation of MEK-phosphorylated ERK.
Experimental Procedures
Materials
The following were purchased: ovine FSH (oFSH-19) from the National Hormone and Pituitary Agency of the NIDDK, National Institutes of Health (Torrance, CA); human fibronectin from BD Biosciences; anti-phospho-AKT (Ser308, CST 9275), anti-AKT (CST 9272), anti-phospho-ERK (Thr202/Tyr204, CST 9107), anti-ERK (CST 9107), anti-phospho-MEK (Ser217/Ser221, CST 9121), anti-S6 (CST 2217), anti-phospho-cAMP-response element-binding protein (Ser133, CST 9191), anti-phospho-GRB2-associated binding protein 2 (GAB2, Ser159, CST 3844; Tyr452, CST 3881), anti-phospho-glycogen synthase kinase (GSK) 3β (Ser9, CST 9336), anti-phospho-myosin light chain (Ser19, CST 3671), anti-GAPDH (CST 5144), and anti-phospho-p38 MAPK (Thr180/Tyr182, CST 4511) from Cell Signaling Technology; SHP2 (SC 7384), anti-cMyc AC (SC 40 AC), anti-ERK2 AC (SC 1647 AC), anti-MKP1 (SC 10769), anti-MKP3 (SC 28902), anti-phospho-IRS1 (Tyr989, SC 17200), protein A/G Plus-agarose (SC 2003), NSC 295642 (SC 253204), and triptolide (NSC 163062, SC 200122) from Santa Cruz Biotechnology; anti-RAS (catalog no. R02120) from BD Transduction Laboratories; tautomycin, okadaic acid, BCI (NSC150117), NSC 663284, and active phospho-ERK2-GST (14–173) from Calbiochem/EMD Millipore; NSC 87877 from ACROS Organics; Na2VO3 from Sigma-Aldrich; and chelerythrine chloride (BML-EI225) from Enzo Life Sciences. Inhibitor concentrations were based on previous publications indicated in the text.
Animals
Sprague-Dawley, CD-outbred rats (breeders from Charles River Laboratories) were from a breeder colony maintained by our laboratory in a pathogen-free facility at Washington State University. The facility is maintained in accordance with the Guidelines for the Care and Use of Laboratory Animals using protocols approved by the Washington State University Institutional Animal Care and Use Committee.
GC Cultures
Immature female rats were primed with subcutaneous injections of 1.5 mg of estradiol-17β (E2) in propylene glycol on postnatal days 21–23. Ovaries were collected following 3 days of injections. GCs were collected by puncturing individual follicles using 27-gauge needles (75). Cells were plated on fibronectin-coated plates at a density of ∼1 × 106 cells/ml of serum-free medium supplemented with 1 nm E2, 100 units/ml penicillin (P), and 100 μg/ml streptomycin (S). The indicated treatments were added to cells ∼20 h following plating and terminated by aspirating the medium and washing once with PBS, followed by sample collection.
Western Blotting
Total cell extracts were collected by scraping cells in a SDS sample buffer (76) at 50 μl/1 × 106 cells, followed by heat denaturation. Equal protein loading was accomplished by plating equal numbers of cells and collecting in a standardized SDS collection volume. Equal volumes of protein extract were loaded per gel lane, and equal loading was verified by probing for total SHP2 or AKT, as indicated. Proteins were separated by SDS/PAGE and transferred onto either a Hybond C-extra or Protran (Amersham Biosciences) nitrocellulose membrane (56). Western blotting analyses were scanned using an Epson Perfection V500 scanner and Adobe Photoshop CS2 9.0 software with minimal processing and quantified using Quantity One software (Bio-Rad). Experimental densitometric values were divided by load control protein values and expressed relative to vehicle values.
Adenoviral Transductions of GCs
Transduction with adenoviruses was done as described previously (47). Briefly, GCs were plated in 35-mm plates at 1.5 × 106 cells/2 ml in DMEM/F12 + E/PS. Four hours after plating, the indicated adenoviruses were added to the cells. The next morning, the adenovirus was removed, the cells were washed with PBS, and fresh DMEM/F12 + E/PS was added. The cells were treated as indicated. Adenoviral optical particle unit concentration per milliliter of viral stock was calculated from the A260 as described previously (30). Results are expressed as optical particle unit per cell and based on the number of GCs plated and the volume of virus added. Ad-PKI was kindly provided by Marco Conti (University of California, San Francisco, CA) (77). Ad-(S17N)-RAS was kindly provided by Valina Dawson (Johns Hopkins University School of Medicine, Baltimore, MD) (78).
Transfection of GCs
Full-length MKP3-Myc-FLAG was purchased from OriGene (Rockville, MD). Cells were plated on fibronectin-coated plates at a density of 1 × 106 cells/ml in OptiMEM + P/S + E2 with expression constructs (500 ng/1 × 106 cells) and transfected using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. After 6 h of incubation, the medium was removed, and fresh DMEM + P/S + E2 was added. Cells were treated as indicated following an ∼16-h recovery period.
Immunoprecipitation
Immunoprecipitations were performed as described previously (5). Inputs represent ∼2% and bound sample ∼98% of total sample.
Whole Cell Lysate Phosphatase Assay
The protocol was adapted from a previous report (79) as detailed below. Cells were treated as indicated and then collected in 0.2 ml of phosphatase assay buffer (10 mm EDTA, 10 mm EGTA, 50 mm HEPES (pH 7.6), 1 mm phenylmethylsulfonyl fluoride, 0.1 mg/ml leupeptin, and 1 mg/ml aprotinin). Lysates were subjected to five rounds of freezing/thawing, followed aspiration through a 25-guage needle or sonication. Lysates were clarified by centrifugation at 13,000 × g for 30 min at 4 °C. To assess phosphatase activity, 50 ng of active ERK2-GST (EMD Millipore, 14-173) was added to each sample, and samples were incubated by rotating for 30 min at 30 °C. The reaction was halted by adding 100 μl of 3× SDS sample buffer (76), and samples were subjected to SDS/PAGE and Western blotting.
Statistics
Results were analyzed using GraphPad Prism, and significance was determined using either a one-way analysis of variance with Tukey's multiple comparison test or one-tailed Student's t test.
Author Contributions
E. M. D. and M. E. H. D. designed the experiments. E. M. D. and N. C. L. performed the experiments. E. M. D. and M. E. H. D. analyzed the data and wrote the manuscript.
Acknowledgments
We thank Drs. John H. Nilson and Maria Herndon for many helpful discussions. We thank Timothy Woodiwiss for performing the preliminary DUSP inhibitor experiments.
This work was supported by National Institutes of Health Grants R01HD065859 and R01HD62503 (to M. E. H. D.) and NIGMS, National Institutes of Health Training Grant T32GM083864 (to E. M. D). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M. E. Hunzicker-Dunn, unpublished data.
- GC
- granulosa cell
- EGFR
- EGF receptor
- RTK
- receptor tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- DUSP
- dual specificity phosphatase
- SL
- striatal-enriched protein tyrosine phosphatase [STEP]-like
- Ad
- adenoviral
- veh
- vehicle
- CDC
- cell division cycle
- GSK
- glycogen synthase kinase
- P
- penicillin
- S
- streptomycin
- BCI
- (E)-2-Benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one
- DUSP
- dual specificity phosphatase
- PKI
- PKA inhibitor
- RKS-2
- ribosomal S6 kinase 2
- YB-1
- Y-box binding protein 1.
References
- 1. Hunzicker-Dunn M., and Mayo K. E. (2015) in Physiology of Reproduction (Plant T. M., and Zeleznik A. J., eds.) pp. 895–992, Elsevier Academic Press, New York. [Google Scholar]
- 2. Erickson G. F., and Sorensen R. A. (1974) In vitro maturation of mouse oocytes isolated from late, middle, and pre-antral graafian follicles. J. Exp. Zool. 190, 123–127 [DOI] [PubMed] [Google Scholar]
- 3. Chesnel F., and Eppig J. J. (1995) Synthesis and accumulation of p34cdc2 and cyclin B in mouse oocytes during acquisition of competence to resume meiosis. Mol. Reprod. Dev. 40, 503–508 [DOI] [PubMed] [Google Scholar]
- 4. Fan H. Y., Liu Z., Shimada M., Sterneck E., Johnson P. F., Hedrick S. M., and Richards J. S. (2009) MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science 324, 938–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Donaubauer E. M., and Hunzicker-Dunn M. E. (2016) Extracellular signal-regulated kinase (ERK)-dependent phosphorylation of Y-box-binding protein 1 (YB-1) enhances gene expression in granulosa cells in response to follicle-stimulating hormone (FSH). J. Biol. Chem. 291, 12145–12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roskoski R., Jr.(2012) MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 417, 5–10 [DOI] [PubMed] [Google Scholar]
- 7. Dalby K. N., Morrice N., Caudwell F. B., Avruch J., and Cohen P. (1998) Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 273, 1496–1505 [DOI] [PubMed] [Google Scholar]
- 8. Roskoski R., Jr.(2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143 [DOI] [PubMed] [Google Scholar]
- 9. Andric N., and Ascoli M. (2006) A delayed gonadotropin-dependent and growth factor-mediated activation of the extracellular signal-regulated kinase 1/2 cascade negatively regulates aromatase expression in granulosa cells. Mol. Endocrinol. 20, 3308–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cottom J., Salvador L. M., Maizels E. T., Reierstad S., Park Y., Carr D. W., Davare M. A., Hell J. W., Palmer S. S., Dent P., Kawakatsu H., Ogata M., and Hunzicker-Dunn M. (2003) Follicle-stimulating hormone activates extracellular signal-regulated kinase but not extracellular signal-regulated kinase kinase through a 100-kDa phosphotyrosine phosphatase. J. Biol. Chem. 278, 7167–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das S., Maizels E. T., DeManno D., St Clair E., Adam S. A., and Hunzicker-Dunn M. (1996) A stimulatory role of cyclic adenosine 3′,5′-monophosphate in follicle-stimulating hormone-activated mitogen-activated protein kinase signaling pathway in rat ovarian granulosa cells. Endocrinology 137, 967–974 [DOI] [PubMed] [Google Scholar]
- 12. Puri P., Little-Ihrig L., Chandran U., Law N. C., Hunzicker-Dunn M., and Zeleznik A. J. (2016) Protein kinase A: a master kinase of granulosa cell differentiation. Sci. Rep. 6, 28132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ogata M., Oh-hora M., Kosugi A., and Hamaoka T. (1999) Inactivation of mitogen-activated protein kinases by a mammalian tyrosine-specific phosphatase, PTPBR7. Biochem. Biophys. Res. Commun. 256, 52–56 [DOI] [PubMed] [Google Scholar]
- 14. Van Den Maagdenberg A. M., Bächner D., Schepens J. T., Peters W., Fransen J. A., Wieringa B., and Hendriks W. J. (1999) The mouse Ptprr gene encodes two protein tyrosine phosphatases, PTP-SL and PTPBR7, that display distinct patterns of expression during neural development. Eur. J. Neurosci. 11, 3832–3844 [DOI] [PubMed] [Google Scholar]
- 15. Pulido R., Zúñiga A., and Ullrich A. (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 17, 7337–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hidaka H., Watanabe M., and Kobayashi R. (1991) Properties and use of H-series compounds as protein kinase inhibitors. Methods Enzymol. 201, 328–339 [DOI] [PubMed] [Google Scholar]
- 17. Blanco-Aparicio C., Torres J., and Pulido R. (1999) A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J. Cell Biol. 147, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zúñiga A., Torres J., Ubeda J., and Pulido R. (1999) Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J. Biol. Chem. 274, 21900–21907 [DOI] [PubMed] [Google Scholar]
- 19. Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsaprailis G., Gresser M. J., and Ramachandran C. (1997) Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272, 843–851 [DOI] [PubMed] [Google Scholar]
- 20. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 21. Noordman Y. E., Augustus E. D., Schepens J. T., Chirivi R. G., Ríos P., Pulido R., and Hendriks W. J. (2008) Multimerisation of receptor-type protein tyrosine phosphatases PTPBR7 and PTP-SL attenuates enzymatic activity. Biochim. Biophys. Acta 1783, 275–286 [DOI] [PubMed] [Google Scholar]
- 22. Herndon M. K., Law N. C., Donaubauer E. M., Kyriss B., and Hunzicker-Dunn M. (2016) Forkhead box O member FOXO1 regulates the majority of follicle-stimulating hormone responsive genes in ovarian granulosa cells. Mol. Cell. Endocrinol. 434, 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levitzki A., and Gazit A. (1995) Tyrosine kinase inhibition: an approach to drug development. Science 267, 1782–1788 [DOI] [PubMed] [Google Scholar]
- 24. Choi B. H., Choi J. S., Rhie D. J., Yoon S. H., Min D. S., Jo Y. H., Kim M. S., and Hahn S. J. (2002) Direct inhibition of the cloned Kv1.5 channel by AG-1478, a tyrosine kinase inhibitor. Am. J. Physiol. Cell Physiol. 282, C1461–1468 [DOI] [PubMed] [Google Scholar]
- 25. Montminy M. (1997) Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 66, 807–822 [DOI] [PubMed] [Google Scholar]
- 26. Dance M., Montagner A., Salles J. P., Yart A., and Raynal P. (2008) The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 20, 453–459 [DOI] [PubMed] [Google Scholar]
- 27. De Rocca Serra-Nédélec A., Edouard T., Tréguer K., Tajan M., Araki T., Dance M., Mus M., Montagner A., Tauber M., Salles J. P., Valet P., Neel B. G., Raynal P., and Yart A. (2012) Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. U.S.A. 109, 4257–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mashima R., Hishida Y., Tezuka T., and Yamanashi Y. (2009) The roles of Dok family adapters in immunoreceptor signaling. Immunol. Rev. 232, 273–285 [DOI] [PubMed] [Google Scholar]
- 29. Chen L., Sung S. S., Yip M. L., Lawrence H. R., Ren Y., Guida W. C., Sebti S. M., Lawrence N. J., and Wu J. (2006) Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 70, 562–570 [DOI] [PubMed] [Google Scholar]
- 30. Hunzicker-Dunn M. E., Lopez-Biladeau B., Law N. C., Fiedler S. E., Carr D. W., and Maizels E. T. (2012) PKA and GAB2 play central roles in the FSH signaling pathway to PI3K and AKT in ovarian granulosa cells. Proc. Natl. Acad. Sci. U.S.A. 109, E2979–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kemp B. E., Cheng H. C., and Walsh D. A. (1988) Peptide inhibitors of cAMP-dependent protein kinase. Methods Enzymol. 159, 173–183 [DOI] [PubMed] [Google Scholar]
- 32. Favre B., Turowski P., and Hemmings B. A. (1997) Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 272, 13856–13863 [DOI] [PubMed] [Google Scholar]
- 33. Resjö S., Oknianska A., Zolnierowicz S., Manganiello V., and Degerman E. (1999) Phosphorylation and activation of phosphodiesterase type 3B (PDE3B) in adipocytes in response to serine/threonine phosphatase inhibitors: deactivation of PDE3B in vitro by protein phosphatase type 2A. Biochem. J. 341, 839–845 [PMC free article] [PubMed] [Google Scholar]
- 34. Yan Y., and Mumby M. C. (1999) Distinct roles for PP1 and PP2A in phosphorylation of the retinoblastoma protein: PP2a regulates the activities of G1 cyclin-dependent kinases. J. Biol. Chem. 274, 31917–31924 [DOI] [PubMed] [Google Scholar]
- 35. Chatfield K., and Eastman A. (2004) Inhibitors of protein phosphatases 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem. Biophys. Res. Commun. 323, 1313–1320 [DOI] [PubMed] [Google Scholar]
- 36. Arif M., Wei J., Zhang Q., Liu F., Basurto-Islas G., Grundke-Iqbal I., and Iqbal K. (2014) Cytoplasmic retention of protein phosphatase 2A inhibitor 2 (I2PP2A) induces Alzheimer-like abnormal hyperphosphorylation of Tau. J. Biol. Chem. 289, 27677–27691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bollen M., Peti W., Ragusa M. J., and Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flynn M. P., Maizels E. T., Karlsson A. B., McAvoy T., Ahn J. H., Nairn A. C., and Hunzicker-Dunn M. (2008) Luteinizing hormone receptor activation in ovarian granulosa cells promotes protein kinase A-dependent dephosphorylation of microtubule-associated protein 2D. Mol. Endocrinol. 22, 1695–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shoelson S. E., Chatterjee S., Chaudhuri M., and White M. F. (1992) YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. U.S.A. 89, 2027–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Devi Y. S., Seibold A. M., Shehu A., Maizels E., Halperin J., Le J., Binart N., Bao L., and Gibori G. (2011) Inhibition of MAPK by prolactin signaling through the short form of its receptor in the ovary and decidua: involvement of a novel phosphatase. J. Biol. Chem. 286, 7609–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Escamilla-Hernandez R., Little-Ihrig L., Orwig K. E., Yue J., Chandran U., and Zeleznik A. J. (2008) Constitutively active protein kinase A qualitatively mimics the effects of follicle-stimulating hormone on granulosa cell differentiation. Mol. Endocrinol. 22, 1842–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patterson K. I., Brummer T., O'Brien P. M., and Daly R. J. (2009) Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J. 418, 475–489 [DOI] [PubMed] [Google Scholar]
- 43. Vogt A., McDonald P. R., Tamewitz A., Sikorski R. P., Wipf P., Skoko J. J. 3rd, and Lazo J. S. (2008) A cell-active inhibitor of mitogen-activated protein kinase phosphatases restores paclitaxel-induced apoptosis in dexamethasone-protected cancer cells. Mol. Cancer Ther. 7, 330–340 [DOI] [PubMed] [Google Scholar]
- 44. Lazo J. S., Aslan D. C., Southwick E. C., Cooley K. A., Ducruet A. P., Joo B., Vogt A., and Wipf P. (2001) Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J. Med. Chem. 44, 4042–4049 [DOI] [PubMed] [Google Scholar]
- 45. Miró F., and Hillier S. G. (1996) Modulation of granulosa cell deoxyribonucleic acid synthesis and differentiation by activin. Endocrinology 137, 464–468 [DOI] [PubMed] [Google Scholar]
- 46. El-Hefnawy T., and Zeleznik A. J. (2001) Synergism between FSH and activin in the regulation of proliferating cell nuclear antigen (PCNA) and cyclin D2 expression in rat granulosa cells. Endocrinology 142, 4357–4362 [DOI] [PubMed] [Google Scholar]
- 47. Park Y., Maizels E. T., Feiger Z. J., Alam H., Peters C. A., Woodruff T. K., Unterman T. G., Lee E. J., Jameson J. L., and Hunzicker-Dunn M. (2005) Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J. Biol. Chem. 280, 9135–9148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Solomon M. J., Lee T., and Kirschner M. W. (1992) Role of phosphorylation in p34cdc2 activation: identification of an activating kinase. Mol. Biol. Cell 3, 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pestell K. E., Ducruet A. P., Wipf P., and Lazo J. S. (2000) Small molecule inhibitors of dual specificity protein phosphatases. Oncogene 19, 6607–6612 [DOI] [PubMed] [Google Scholar]
- 50. Molina G., Vogt A., Bakan A., Dai W., Queiroz de Oliveira P., Znosko W., Smithgall T. E., Bahar I., Lazo J. S., Day B. W., and Tsang M. (2009) Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 5, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao Q., Shepherd E. G., Manson M. E., Nelin L. D., Sorokin A., and Liu Y. (2005) The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 280, 8101–8108 [DOI] [PubMed] [Google Scholar]
- 52. Srivastava N., Sudan R., and Saha B. (2011) CD40-modulated dual-specificity phosphatases MAPK phosphatase (MKP)-1 and MKP-3 reciprocally regulate Leishmania major infection. J. Immunol. 186, 5863–5872 [DOI] [PubMed] [Google Scholar]
- 53. Vogt A., Tamewitz A., Skoko J., Sikorski R. P., Giuliano K. A., and Lazo J. S. (2005) The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J. Biol. Chem. 280, 19078–19086 [DOI] [PubMed] [Google Scholar]
- 54. Vogt A., Cooley K. A., Brisson M., Tarpley M. G., Wipf P., and Lazo J. S. (2003) Cell-active dual specificity phosphatase inhibitors identified by high-content screening. Chem. Biol. 10, 733–742 [DOI] [PubMed] [Google Scholar]
- 55. Mukherjee A., Park-Sarge O. K., and Mayo K. E. (1996) Gonadotropins induce rapid phosphorylation of the 3′,5′-cyclic adenosine monophosphate response element binding protein in ovarian granulosa cells. Endocrinology 137, 3234–3245 [DOI] [PubMed] [Google Scholar]
- 56. Salvador L. M., Park Y., Cottom J., Maizels E. T., Jones J. C., Schillace R. V., Carr D. W., Cheung P., Allis C. D., Jameson J. L., and Hunzicker-Dunn M. (2001) Follicle-stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading to select gene activation in ovarian granulosa cells. J. Biol. Chem. 276, 40146–40155 [DOI] [PubMed] [Google Scholar]
- 57. Law N. C., Weck J., Kyriss B., Nilson J. H., and Hunzicker-Dunn M. (2013) Lhcgr expression in granulosa cells: roles for PKA-phosphorylated β-catenin, TCF3, and FOXO1. Mol. Endocrinol. 27, 1295–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Law N. C., and Hunzicker-Dunn M. E. (2016) Insulin receptor substrate 1, the hub linking follicle-stimulating hormone to phosphatidylinositol 3-kinase activation. J. Biol. Chem. 291, 4547–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keyse S. M. (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol. 12, 186–192 [DOI] [PubMed] [Google Scholar]
- 60. Muda M., Boschert U., Dickinson R., Martinou J. C., Martinou I., Camps M., Schlegel W., and Arkinstall S. (1996) MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem. 271, 4319–4326 [DOI] [PubMed] [Google Scholar]
- 61. Zhou B., Wang Z. X., Zhao Y., Brautigan D. L., and Zhang Z. Y. (2002) The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J. Biol. Chem. 277, 31818–31825 [DOI] [PubMed] [Google Scholar]
- 62. Zhao Y., and Zhang Z. Y. (2001) The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 276, 32382–32391 [DOI] [PubMed] [Google Scholar]
- 63. Eblaghie M. C., Lunn J. S., Dickinson R. J., Münsterberg A. E., Sanz-Ezquerro J. J., Farrell E. R., Mathers J., Keyse S. M., Storey K., and Tickle C. (2003) Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr. Biol. 13, 1009–1018 [DOI] [PubMed] [Google Scholar]
- 64. Nunes-Xavier C. E., Tárrega C., Cejudo-Marín R., Frijhoff J., Sandin A., Ostman A., and Pulido R. (2010) Differential up-regulation of MAP kinase phosphatases MKP3/DUSP6 and DUSP5 by Ets2 and c-Jun converge in the control of the growth arrest versus proliferation response of MCF-7 breast cancer cells to phorbol ester. J. Biol. Chem. 285, 26417–26430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ekerot M., Stavridis M. P., Delavaine L., Mitchell M. P., Staples C., Owens D. M., Keenan I. D., Dickinson R. J., Storey K. G., and Keyse S. M. (2008) Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 412, 287–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Camps M., Nichols A., Gillieron C., Antonsson B., Muda M., Chabert C., Boschert U., and Arkinstall S. (1998) Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 280, 1262–1265 [DOI] [PubMed] [Google Scholar]
- 67. Farooq A., Chaturvedi G., Mujtaba S., Plotnikova O., Zeng L., Dhalluin C., Ashton R., and Zhou M. M. (2001) Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol. Cell 7, 387–399 [DOI] [PubMed] [Google Scholar]
- 68. Marchetti S., Gimond C., Chambard J. C., Touboul T., Roux D., Pouysségur J., and Pagès G. (2005) Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell Biol. 25, 854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bermudez O., Marchetti S., Pagès G., and Gimond C. (2008) Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene 27, 3685–3691 [DOI] [PubMed] [Google Scholar]
- 70. Castelli M., Camps M., Gillieron C., Leroy D., Arkinstall S., Rommel C., and Nichols A. (2004) MAP kinase phosphatase 3 (MKP3) interacts with and is phosphorylated by protein kinase CK2α. J. Biol. Chem. 279, 44731–44739 [DOI] [PubMed] [Google Scholar]
- 71. Blanquet P. R. (2000) Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 60, 211–246 [DOI] [PubMed] [Google Scholar]
- 72. Maillet M., Purcell N. H., Sargent M. A., York A. J., Bueno O. F., and Molkentin J. D. (2008) DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 283, 31246–31255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kidger A. M., and Keyse S. M. (2016) The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 50, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dorfman K., Carrasco D., Gruda M., Ryan C., Lira S. A., and Bravo R. (1996) Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene 13, 925–931 [PubMed] [Google Scholar]
- 75. Carr D. W., DeManno D. A., Atwood A., Hunzicker-Dunn M., and Scott J. D. (1993) Follicle-stimulating hormone regulation of A-kinase anchoring proteins in granulosa cells. J. Biol. Chem. 268, 20729–20732 [PubMed] [Google Scholar]
- 76. Hunzicker-Dunn M. (1981) Selective activation of rabbit ovarian protein kinase isozymes in rabbit ovarian follicles and corpora lutea. J. Biol. Chem. 256, 12185–12193 [PubMed] [Google Scholar]
- 77. Bruss M. D., Richter W., Horner K., Jin S. L., and Conti M. (2008) Critical role of PDE4D in β2-adrenoceptor-dependent cAMP signaling in mouse embryonic fibroblasts. J. Biol. Chem. 283, 22430–22442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gonzalez-Zulueta M., Feldman A. B., Klesse L. J., Kalb R. G., Dillman J. F., Parada L. F., Dawson T. M., and Dawson V. L. (2000) Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc. Natl. Acad. Sci. U.S.A. 97, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Woods D. C., and Johnson A. L. (2006) Phosphatase activation by epidermal growth factor family ligands regulates extracellular regulated kinase signaling in undifferentiated hen granulosa cells. Endocrinology 147, 4931–4940 [DOI] [PubMed] [Google Scholar]
- 80. Jeffrey K. L., Camps M., Rommel C., and Mackay C. R. (2007) Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug. Discov. 6, 391–403 [DOI] [PubMed] [Google Scholar]