

Summary
Objective
To assess the efficacy and safety of eslicarbazepine acetate (ESL) as monotherapy in North American patients with partial‐onset seizures (POS).
Methods
This multicenter, randomized, double‐blind “withdrawal to monotherapy” study used historical control data as the comparator. Adults with POS medically uncontrolled by one to two antiepileptic drugs gradually converted to ESL monotherapy. Following an 8‐week baseline period, patients were randomized 2:1 to receive ESL 1,600 mg (n = 128) or 1,200 mg QD (n = 65) for 18 weeks. The primary end point was the proportion of patients meeting predefined exit criteria (signifying worsening seizure control). Treatment was considered effective if the 95% upper confidence limit (UCL) for the Kaplan‐Meier estimated exit rate was lower than the exit rate threshold calculated from the historical control (65.3%).
Results
Kaplan‐Meier estimated exit rates were: ESL 1,600 mg, 28.7% (95% CI 21.2–38.1%) and 1,200 mg, 44.4% (32.5–58.3%). The difference between doses was not significant (p = 0.07). For both doses, the 95% UCLs for the exit rate were ˂65.3%; ESL monotherapy was considered superior to the historical control. There was no statistically significant increase in the risk of study exit related to carbamazepine use. Nine (7.6%) and five patients (8.3%) remained seizure‐free during the 10‐week monotherapy period, while taking ESL 1,600 and 1,200 mg, respectively. The reductions in median standardized seizure frequency (seizures per 28 days) between baseline and the 18‐week treatment period were: ESL 1,600 mg, 42% and 1,200 mg, 31%. Treatment‐emergent adverse events (TEAEs) occurring in ≥10% of patients were dizziness, headache, fatigue, somnolence, nausea, and nasopharyngitis. The TEAE most frequently leading to discontinuation was hyponatremia (2.1%).
Significance
ESL was efficacious and well tolerated as monotherapy in North American patients, and led to a reduction in seizure frequency. Exit rates for ESL 1,600 and 1,200 mg QD were superior to the historical control; the difference in exit rates between doses was not statistically significant.
Keywords: Monotherapy, Antiepileptic drugs, Eslicarbazepine acetate, Refractory epilepsy, Partial‐onset seizures, Anticonvulsants
Antiepileptic drug (AED) monotherapy is generally preferred to polytherapy, due to the potential for a greater incidence of adverse events and adverse drug interactions that occur with combination therapy.1 Moreover, some patients, including the elderly and those with comorbid conditions, particularly benefit from AED monotherapy, as AED toxicity and drug interactions can have specific consequences.2, 3 There is an unmet need for effective and well‐tolerated AEDs2 suitable for use as monotherapy in patients with epilepsy.
Eslicarbazepine acetate (ESL) is a member of the dibenzazepine family of AEDs, with structural and metabolic differences from the other dibenzazepine AEDs, carbamazepine (CBZ), and oxcarbazepine (OXC).4, 5, 6, 7 Following oral administration, ESL undergoes first‐pass hydrolysis in the liver and is rapidly metabolized to its active metabolite, eslicarbazepine.6, 8 Ninety‐one percent of oral ESL exposure is accounted for by eslicarbazepine, 5% by (R)‐licarbazepine and 1% by OXC.9 In contrast, only 78% of oral OXC exposure is accounted for by eslicarbazepine, 18% by (R)‐licarbazepine and 3% by OXC.10 Thus, use of ESL limits exposure to (R)‐licarbazepine and OXC. It is not known whether these differences in exposure have an influence on the clinical profiles of these agents.
Eslicarbazepine inhibits sodium currents by stabilizing the inactivated state of voltage‐gated sodium channels (VGSCs), with a higher affinity for the inactivated state than the resting state compared with CBZ and OXC.11 Eslicarbazepine has a 20‐ to 24‐h apparent half‐life in cerebrospinal fluid, supporting once‐daily (QD) dosing.10
ESL is a QD oral AED, approved in the United States and the European Union as adjunctive treatment for partial‐onset seizures (POS). ESL is not approved for monotherapy use. Conversion to ESL monotherapy was recently investigated in a historical control trial conducted in North America (25% of patients) and other locations (Study 093‐046, NCT01091662),12 which demonstrated that ESL (1,600 and 1,200 mg QD) was superior to the historical control. No previous AED trials of this type have been conducted in an exclusively North American (NA) population. In a historical control trial of conversion to lamotrigine extended release (XR) monotherapy, the exit rate was 42% for U.S. patients compared with 30% for non‐U.S. patients.13 In sensitivity and worst‐case analyses of the data conducted by the U.S. Food and Drug Administration (FDA), lamotrigine XR did not demonstrate superiority versus the historical control in the U.S. subgroup.13 Consequently, because of the potential difference in response to AED therapy between U.S. and non‐U.S. populations, the current study was conducted exclusively with North American patients. This trial (Study 093‐045; http://ClinicalTrials.gov identifier: NCT00866775), which was identical in design to study 093‐046, evaluated the efficacy of ESL monotherapy in NA patients with POS medically uncontrolled by current AEDs. The safety and tolerability of ESL monotherapy were also evaluated.
Methods
Trial design
Study 093‐045 adopted a “withdrawal to monotherapy” design; a historical control group14 was used for evaluation of efficacy. The use of a historical control (based on the exit rates in eight previous monotherapy studies of similar design and duration to the current study, including randomization as done in the original studies) eliminates the need for a placebo control (an ethical challenge in monotherapy trials of patients with epilepsy), allowing all trial participants to receive active treatment. In this trial, the efficacy and safety of ESL were evaluated in adults with POS not controlled by one to two AEDs. The trial was performed between April 2009 and May 2013; patients were screened at 89 investigational sites (86 in the U.S., and 3 in Canada).
The inclusion and exclusion criteria for this study were identical to Study 093‐046,12 and are shown in Table S1.
Patients who successfully completed screening entered into an 8‐week baseline period for assessment of seizure frequency (Fig. S1). Those eligible for randomization (Table S1) were randomized in a 2:1 ratio to receive ESL 1,600 or 1,200 mg QD and began the 18‐week double‐blind treatment period. Randomization to treatment group was performed using an interactive voice‐response system to associate each patient with double‐blind clinical trial material (“kits”) and a randomization number. The randomization list was prepared by a third party using a random number generator, and followed a permutated‐block design (block size = 6). Placebo capsules to match overencapsulated ESL 400 and 600 mg were supplied to maintain the blind. Patients were randomized at 67 sites (64 in the U.S., and 3 in Canada).
ESL was titrated to the target dose during the first 2 weeks of double‐blind treatment (Fig. S1), after which, doses of baseline AEDs were reduced (by 50% in the first 3 weeks, and to zero during the next 3 weeks). At the end of this conversion period, patients continued to receive ESL as monotherapy for 10 weeks. Patients who completed the first 3 weeks of double‐blind treatment, and subsequently completed, discontinued, or exited the study were eligible to participate in a long‐term open‐label extension trial; alternatively, patients entered a 1‐week taper period, during which original background AEDs were reintroduced.
This trial was designed and conducted in accordance with all relevant regulations and guidelines. Prior to obtaining written consent from all patients, the protocol was approved by the institutional review board and an independent ethics committee.
Assessments
Primary end point
Seizure data were collected using daily seizure diaries completed by patients. The primary efficacy end point was the exit rate for patients meeting at least one of five prospective exit criteria (signifying worsening seizure control; Table 1) during the 16‐week study period (from start of AED conversion period to end of double‐blind monotherapy period). Discontinuations for nonexit reasons, if >10%, were randomly reassigned as exits.
Table 1.
Reasons for exit (EFF population)
| ESL 1,200 mg (n = 60) | ESL 1,600 mg (n = 118) | Total (n = 178) | |
|---|---|---|---|
| Met one of the 5 exit criteria, n (%) | 23 (38.3) | 17 (14.4)a | 40 (22.5) |
| 1. One episode of status epilepticus | 0 | 1 (0.8)b | 1 (0.6)b |
| 2. One secondary generalized partial seizure (for patients without generalized seizures during 6 months prior to screening) | 4 (6.7) | 1 (0.8) | 5 (2.8) |
| 3. Twofold increase from baseline in consecutive 28‐day seizure rate | 6 (10.0) | 5 (4.2) | 11 (6.2) |
| 4. Twofold increase from baseline in consecutive 2‐day seizure rate | 5 (8.3) | 6 (5.1) | 11 (6.2) |
| 5. Worsening of seizures or increase in seizure frequency (as judged by investigator) | 8 (13.3) | 4 (3.4) | 12 (6.7) |
EFF, efficacy; ESL, eslicarbazepine acetate.
Two additional patients (ESL 1,600 mg group) met exit criteria, based on poststudy programmatic assessment; patients whose seizure diaries indicated that maximum average 2‐ or 28‐day seizure rate exceeded the threshold of doubling during the 16‐week study period compared to the baseline period, were reassigned as exits, even if the investigator had allowed them to remain in the study.
One patient with status epilepticus took 19% of the assigned daily dose as determined by count of returned tablets.
The primary comparison was between the cumulative exit rate after 112 days of double‐blind ESL monotherapy in the ESL 1,600 mg treatment arm and the combined exit rate calculated from eight historical control trials.14 Treatment was considered effective (and the null hypothesis rejected) if the upper confidence limit (UCL) for the exit rate (estimated using Kaplan‐Meier [KM] methodology) was below the lower limit of the prespecified prediction interval calculated from historical controls (65.3%).14
Secondary end points
The key secondary efficacy end point was the percentage of seizure‐free patients during the 10‐week monotherapy period. Other prospectively specified secondary efficacy end points included proportion of patients meeting each exit criterion; proportion of seizure‐free patients during the last 4 weeks of monotherapy; change in standardized seizure frequency (SSF) from baseline (for the 18‐week treatment and 10‐week monotherapy periods); responder rate (proportion of patients with a reduction in SSF ≥ 50% from baseline); change in seizure frequency from baseline according to seizure severity; change in 31‐item Quality of Life in Epilepsy (QOLIE‐31) total scores; change in Montgomery‐Asberg Depression Rating Scale (MADRS) scores (for all patients, and for patients with a score ≥14 at randomization); and completion rate (proportion of patients completing the 18‐week treatment and 10‐week monotherapy periods).
Safety and tolerability
Adverse events (AEs) were noted at each clinic visit. AEs were coded according to their respective system organ class and preferred term, using the Medical Dictionary for Regulatory Activities version 13.1. Treatment‐emergent AEs (TEAEs) were defined as AEs that occurred on or after the first dose of study drug. All AEs were recorded by the investigator and classified with regard to intensity (mild, moderate, or severe).
Statistical analyses
For the primary efficacy end point (exits based on seizure criteria), a sequential testing procedure was used to control type I errors. The first comparison in the sequence was for ESL 1,600 mg versus historical control, then ESL 1,200 mg versus historical control, followed by a comparison between the two treatment groups using a log‐rank test. French et al.14 calculated a 95% prediction interval (PI) based on the exit rates reported in eight historical trials. At a type I error rate of ≤5%, the lower bound of the PI for a single study is an exit rate of 65.3% at 112 days.14 Therefore, if the 95% UCL for a treatment group was <65.3%, the null hypothesis (that the exit rate for the test group equals the combined exit rate derived from the historical controls) could be rejected. The exit rate was estimated using KM analysis, using the time to exit observed for each patient.
Patients were censored if they withdrew from the study for reasons other than meeting the exit criteria, prior to reaching 112 days. The rate of early withdrawal reported in historical control trials was approximately 10%. Consequently, the protocol specified that if the withdrawal rate exceeded 10%, any further withdrawals would be reassigned as having met the exit criteria, using random sampling. A secondary analysis of the primary end point was performed in which censored patients were not reassigned as exits.
For subgroups of patients taking different AEDs at baseline, descriptive statistics for the exit rate (%s and 95% confidence intervals [CIs]) were calculated for each group (for AEDs used in ≥20% of patients).
The statistical analyses of the secondary efficacy end points and safety end points are described in the Appendix S1.
All statistical procedures were performed using SAS v.9.2 (SAS Institute, Cary, NC, U.S.A.) or higher. All statistical tests were two sided.
Determination of sample size
Patients were randomized in a 2:1 ratio to either 1,600 or 1,200 mg ESL. The exit rate is assumed to be 54% for the 1,600 mg QD arm, corresponding to a weekly hazard rate of 4.85%. This exit rate represents a 39% reduction in the exit hazard rate based on the lower end of the 80% PI established for replication in two clinical studies (72.2%; weekly hazard rate of 8%) and a 20% reduction in the exit hazard rate based on the lower end of the 95% PI established for a single clinical study (65.3%; weekly hazard rate of 6.62%). For 116 patients randomized to ESL 1,600 mg, and assuming a 10% discontinuation rate without observed exit events, there was ≥95% chance that the UCL for the observed exit rate would fall below 72.2% and a 70% chance that the UCL would fall below 65.3%. Assuming an early dropout rate of 20%, approximately 202 patients were required to enter the baseline period to achieve a minimum of 174 randomized patients.
Study populations
The intent‐to‐treat (ITT) population consisted of all randomized patients who received at least one dose of the study drug; this population was used to evaluate patient disposition, baseline demographics and characteristics, and safety outcomes. Primary and secondary efficacy analyses were conducted for the efficacy (EFF) population (all ITT patients who entered the AED conversion period). An additional analysis of the primary efficacy end point was conducted for the per‐protocol (PP) population (patients in the EFF population with no important protocol deviations).
Results
Patient demographics, baseline characteristics, and disposition
The ITT, EFF, and PP population treatment groups were generally well balanced in terms of demographics and baseline characteristics (Table 2). Overall, the ITT population (n = 193 [ESL 1,600 mg: n = 128; 1,200 mg: n = 65]) had a median age of 39 years; approximately 50% of patients were male; most were white (76%) and were treated at a U.S. center (97%); 17% were Hispanic/Latino. Most patients (74%) were taking one AED when they entered the trial; levetiracetam (LEV) and CBZ were the most commonly used AEDs. Benzodiazepines were used intermittently (emergency use) by 10.8% of patients taking ESL 1,200 mg and by 12.5% of patients taking ESL 1,600 mg. The mean maximum seizure rate during the baseline period was 3.8 per 2 days, or 13.3 per 28 days.
Table 2.
Demographic and clinical characteristics at baseline (ITT population)
| Characteristic | ESL 1,200 mg (n = 65) | ESL 1,600 mg (n = 128) | Total (n = 193) |
|---|---|---|---|
| Age, years; median (range) | 39 (16–67) | 40 (16–68) | 39 (16–68) |
| Gender, male; n (%) | 31 (47.7) | 61 (47.7) | 92 (47.7) |
| Race, n (%) | |||
| White | 53 (81.5) | 94 (73.4) | 147 (76.2) |
| Black or African American | 4 (6.2) | 18 (14.1) | 22 (11.4) |
| Other (including multiple) | 8 (12.3) | 16 (12.5) | 24 (12.4) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 14 (21.5) | 18 (14.1) | 32 (16.6) |
| Region, n (%) | |||
| U.S. | 64 (98.5) | 123 (96.1) | 187 (96.9) |
| Non‐U.S. | 1 (1.5) | 5 (3.9) | 6 (3.1) |
| BMI, kg/m2; median (range) | 27.6 (17.6–53.7) | 28.8 (17.9–109.2) | 28.7 (17.6–109.2) |
| Maximum consecutive 2‐day baseline seizure rate, | |||
| mean ± SD | 3.7 ± 2.84a | 3.8 ± 2.36b | 3.8 ± 2.53c |
| Maximum consecutive 28‐day baseline seizure rate, | |||
| mean ± SD | 12.1 ± 8.31a | 13.8 ± 9.10b | 13.3 ± 8.86c |
| Baseline AEDs used by ≥20% patientsd, n (%) | |||
| Carbamazepine | 17 (26.2) | 34 (26.6) | 51 (26.4) |
| Levetiracetam | 19 (29.2) | 38 (29.7) | 57 (29.5) |
| Number of AEDs at baselined, n (%) | |||
| 1 | 50 (76.9) | 92 (71.9) | 142 (73.6) |
| 2 | 15 (23.1) | 36 (28.1) | 51 (26.4) |
ITT, intent‐to‐treat; ESL, eslicarbazepine acetate; BMI, body mass index; SD, standard deviation; AED, antiepileptic drug; U.S., United States.
Percentages are calculated based on the number of patients with nonmissing data in the ITT population in each column.
n = 63.
n = 126.
n = 189.
An AED was considered to be used at baseline if it was started at any time before the first dose of study drug and continued into the titration period.
Of the 397 patients screened for eligibility, 193 received double‐blind ESL treatment (ITT population), 178 patients entered the AED conversion period (EFF population), and 138 patients entered the ESL monotherapy period, with 105 patients completing (Fig. S2). Reasons for study withdrawal are listed in Table S2.
Efficacy
Primary end point
Overall, based on the investigators' determinations, 40 patients (22.5%) met one of the five seizure‐related exit criteria and exited the trial (Table 1). Half of these (n = 20) exited during the AED conversion period and the other half during the monotherapy period.
Two additional patients in the ESL 1,600 mg treatment group were reassigned as study exits based on information from seizure diaries. In addition, as determined by the study protocol, once the withdrawal rate had reached 10%, subsequent withdrawals were reassigned as seizure‐related exits using random sampling (13 patients in the ESL 1,600 mg group; two patients in the 1,200 mg group).
Primary analysis
The KM estimated exit rate for the EFF population (n = 178 [ESL 1,600 mg: n = 118; ESL 1,200 mg: n = 60]) was 28.7% (95% CI 21.2–38.1%) for ESL 1,600 mg and 44.4% (32.5–58.3%) for ESL 1,200 mg (Fig. 1). For both the 1,600 and 1,200 mg groups, the 95% UCLs for the exit rates were below the prespecified threshold of 65.3% (Fig. 1). Therefore the exit rates for ESL 1,600 and 1,200 mg were lower than the exit rate for the historical controls; the difference between dose groups was not significant (log‐rank test: p = 0.07).
Figure 1.
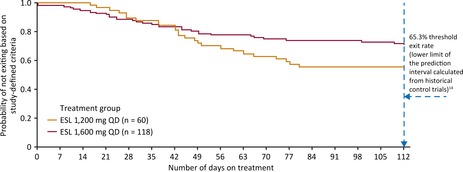
Kaplan‐Meier plot of time to exit (EFF population). EFF, efficacy; ESL, eslicarbazepine acetate; QD, once daily.
Secondary analyses
The KM estimated exit rate for the PP population (n = 139) was similar to that for the EFF population: 29.6% (95% CI 21.2–40.4%) for ESL 1,600 mg and 43.1% (29.8–59.5%) for ESL 1,200 mg (difference between groups, p = 0.18). Without reassignment of withdrawals, KM exit rates were 18.9% for ESL 1,600 mg and 42.0% for ESL 1,200 mg (difference between groups, p = 0.0019); for both doses, the 95% UCLs for the exit rate were below the prespecified threshold of 65.3% (ESL 1,600 mg, 28.1%; ESL 1,200 mg, 56.3%). This analysis, without reassignment of withdrawals, is comparable to that conducted in previous historical control trials of conversion to monotherapy.
A worse‐case analysis was also conducted, in which patients who withdrew without meeting exit criteria were reassigned as seizure‐related exits unless seizure‐related causes could be excluded. In this analysis, KM exit rates were 29.0% for ESL 1,600 mg and 44.3% for ESL 1,200 mg (difference between groups, p = 0.068). Again, for both doses, the 95% UCLs for the exit rate were below the prespecified threshold of 65.3% (ESL 1,600 mg, 38.4%; ESL 1,200 mg, 58.2%).
Influence of AEDs used during the baseline period: KM‐estimated exit rates
KM exit rates were 34.7% (ESL 1,600 mg; 95% CI 20.3–55.0%) and 46.4% (ESL 1,200 mg; 25.4–73.5%) for patients taking CBZ during the baseline period, versus 26.9% (18.6–38.0%) and 45.7% (32.1–62.0%), respectively, for those not taking CBZ (Fig. 2). For patients taking LEV during the baseline period, KM exit rates were 42.2% (ESL 1,600 mg; 27.5–60.6%) and 43.8% (ESL 1,200 mg; 23.8–70.5%) versus 23.6% (15.5–34.8%) and 46.8% (32.9–63.2%), respectively, for those not taking LEV (Fig. 2). Only a small proportion of patients were taking OXC during baseline (6.7%). Consequently, exit rates for these patients could not be accurately determined.
Figure 2.
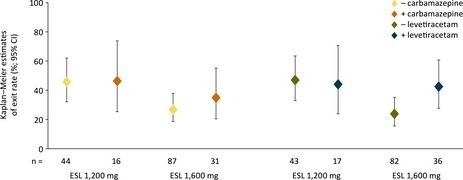
Kaplan‐Meier estimates of exit rate with/without carbamazepine and levetiracetam (EFF population). EFF, efficacy; CI, confidence interval; ESL, eslicarbazepine acetate.
Secondary end points
During the 10‐week monotherapy period, nine patients (7.6%; 95% CI 3.5–14.0%) in the ESL 1,600 mg group, and five (8.3%; 2.8–18.4%) in the ESL 1,200 mg group, remained seizure‐free; seizure‐free rates during the last 4 weeks of monotherapy were 14.4% (95% CI 8.6–22.1%) for ESL 1,600 mg and 13.3% (5.9–24.6%) for ESL 1,200 mg.
Marked reductions in SSF (seizures per 28 days), and substantial responder rates (percentage of patients with ≥50% reduction in SSF from baseline) occurred in the ESL 1,600 and 1,200 mg treatment arms, for both the 18‐week treatment period and the 10‐week monotherapy period (Table 3). Figure S3A shows the reductions in SSF between the baseline and monotherapy periods according to seizure severity. The most marked reduction in seizure frequency occurred in patients with partial seizures evolving to secondarily generalized seizures (ESL 1,600 mg, 80%; ESL 1,200 mg, 49%). A large proportion of patients achieved a ≥50% reduction in SSF, irrespective of seizure severity and ESL dose (Fig. S3B).
Table 3.
Percentage change from baseline in standardized seizure frequency during the monotherapy and double‐blind periods, and responder rate by study period (EFF population)
| Study period | ESL 1,200 mg QD (n = 60) | ESL 1,600 mg QD (n = 118) |
|---|---|---|
| Change in SSF from baseline; median (%) | ||
| 18‐week double‐blind period | −30.9 | −41.5 |
| 10‐week monotherapy period | −48.7 | −38.6 |
| Responders, n (% [95% CIa]) | ||
| 18‐week double‐blind period | 22 (36.7 [24.6–50.1]) | 47 (39.8 [30.9–49.3]) |
| Titration period | 28 (46.7 [33.7–60.0]) | 61 (51.7 [42.3–61.0]) |
| AED conversion period | 25 (41.7 [29.1–55.1]) | 51 (43.2 [34.1–52.7]) |
| 10‐week monotherapy period | 21 (35.0 [23.1–48.4]) | 38 (32.2 [23.9–41.4]) |
EFF, efficacy; ESL, eslicarbazepine acetate; QD, once daily; SSF, standardized seizure frequency (seizure frequency is standardized to a 28‐day frequency); AED, antiepileptic drug; CI, confidence interval.
A response was defined as ≥50% reduction in standardized seizure frequency from baseline. Percentages of responders and exact 95% CIs are based on the number of patients with post‐baseline seizure data.
The 95% CI values are for the percent of responders.
Increases in QOLIE‐31 scores were apparent between baseline and the end of the monotherapy period (ESL 1,600 mg, 6.4 [SD 12.6]; ESL 1,200 mg, 7.8 [SD 13.8]). A reduction in total MADRS scores occurred between baseline and the end of the monotherapy period (ESL 1,600 mg, −2.3 [95% CI −3.3, −1.4]; ESL 1,200 mg, −1.2 [−2.6, 0.2]). Reductions were also apparent in patients with MADRS scores ≥14 at baseline (ESL 1,600 mg, −9.6 ± 8.7; ESL 1,200 mg, −6.8 ± 6.7).
For the 18‐week treatment period, the completion rates were 64% (ESL 1,600 mg) and 48% (ESL 1,200 mg); for the 10‐week monotherapy period, completion rates were 82% and 64%, respectively.
Safety
Overall, 171 patients (89%) had ≥1 TEAE (ESL 1,600 mg, 91%; ESL 1,200 mg, 85%). Of these, 78% had a TEAE that was considered potentially related to the trial drug (ESL 1,600 mg, 82%; ESL 1,200 mg, 69%). The most commonly reported TEAEs are presented in Table 4. TEAEs were reported more often during the titration period (72%) than during the AED conversion (61%) and monotherapy periods (49%).
Table 4.
TEAEs,a severe TEAEs, and SAEs (ITT population)
| ESL 1,200 mg QD (n = 65) | ESL 1,600 mg QD (n = 128) | Total (n = 193) | |
|---|---|---|---|
| TEAE, n (%)b | |||
| Dizziness | 18 (27.7) | 31 (24.2) | 49 (25.4) |
| Headache | 14 (21.5) | 26 (20.3) | 40 (20.7) |
| Fatigue | 13 (20.0) | 20 (15.6) | 33 (17.1) |
| Somnolence | 7 (10.8) | 23 (18.0) | 30 (15.5) |
| Nausea | 7 (10.8) | 19 (14.8) | 26 (13.5) |
| Nasopharyngitis | 7 (10.8) | 14 (10.9) | 21 (10.9) |
| Vomiting | 6 (9.2) | 12 (9.4) | 18 (9.3) |
| Vision blurred | 6 (9.2) | 12 (9.4) | 18 (9.3) |
| Back pain | 10 (15.4) | 6 (4.7) | 16 (8.3) |
| Diarrhea | 5 (7.7) | 9 (7.0) | 14 (7.3) |
| Hyponatremia | 4 (6.2) | 9 (7.0) | 13 (6.7) |
| Diplopia | 5 (7.7) | 6 (4.7) | 11 (5.7) |
| Insomnia | 3 (4.6) | 8 (6.3) | 11 (5.7) |
| Contusion | 4 (6.2) | 7 (5.5) | 11 (5.7) |
| Influenza | 4 (6.2) | 5 (3.9) | 9 (4.7) |
| Anxiety | 2 (3.1) | 7 (5.5) | 9 (4.7) |
| Abdominal discomfort | 4 (6.2) | 4 (3.1) | 8 (4.1) |
| Gastroenteritis viral | 4 (6.2) | 4 (3.1) | 8 (4.1) |
| Partial seizures with secondary generalization | 7 (10.8) | 1 (0.8) | 8 (4.1) |
| Fall | 4 (6.2) | 3 (2.3) | 7 (3.6) |
| Musculoskeletal pain | 4 (6.2) | 3 (2.3) | 7 (3.6) |
| Pain in extremity | 5 (7.7) | 2 (1.6) | 7 (3.6) |
| Memory impairment | 5 (7.7) | 2 (1.6) | 7 (3.6) |
| Hypertension | 0 | 7 (5.5) | 7 (3.6) |
| Toothache | 4 (6.2) | 1 (0.8) | 5 (2.6) |
| Any severe TEAE | 5 (7.7) | 12 (9.4) | 17 (8.8) |
| Any treatment‐emergent SAE | 4 (6.2) | 8 (6.3) | 12 (6.2) |
ITT, intent‐to‐treat; ESL, eslicarbazepine acetate; QD, once daily; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
The following severe TEAEs occurred in one patient each in the ESL 1,600 mg group: diarrhea; dyspepsia; nausea; vomiting; sinusitis; accidental overdose; hyponatremia; arthralgia; back pain; amnesia; dizziness; headache; simple partial seizures; status epilepticus; agitation; anxiety; confused state; and changes in mental status.
The following severe TEAEs occurred in one patient each in the ESL 1,200 mg group: cardiogenic shock; myocardial infarction; multiple injuries; back pain; pancreatic neoplasm; dizziness; migraine; agitation; pulmonary edema; and rash.
Hyponatremia was reported as an SAE in two patients in the ESL 1,600 mg QD group.
The following SAEs were reported for one patient each, in the ESL 1,600 mg QD group: nausea; vomiting; accidental overdose; cerebral cyst; complex partial seizures; hypoesthesia; status epilepticus; anxiety; depression; dyspnea; and hypertension.
The following SAEs were reported for one patient each in the ESL 1,200 mg QD group: cardiogenic shock; edema peripheral (downgraded to a nonserious AE by the investigator after database lock); multiple injuries (death; patient died in a car accident); hypokalemia; pancreatic neoplasm; partial seizures with secondary generalization; and pulmonary edema.
Affecting ≥5% of patients in any ESL dose group.
Some patients had more than one TEAE. Patients with multiple occurrences of a single TEAE were counted only once for that TEAE.
Most TEAEs were mild or moderate in severity, but 17 patients (8.8%) had ≥1 severe TEAEs (12 in the ESL 1,600 mg group; 5 in the ESL 1,200 mg group); see Table 4 footnote for the list of severe TEAEs. Treatment‐emergent serious AEs (SAEs) occurred in 12 patients (6.2%; Table 4); hyponatremia was reported in two patients (1.0%), all other SAEs occurred in one patient each (see footnote to Table 4 for a list of SAEs). One patient died during the trial (SAE: multiple injuries), in a car accident (considered not treatment‐related by the physician). Overall, SAEs and TEAEs of severe intensity were reported infrequently during the trial.
A total of 31 patients (16%) discontinued the trial due to a TEAE (ESL 1,600 mg, 18%; ESL 1,200 mg, 12%). Hyponatremia was the TEAE that most frequently led to discontinuation (four patients [2.1%]). Discontinuations due to TEAEs were less frequent during the monotherapy period (3.6%) than the titration period (6.7%) or the AED conversion period (5.2%). Dose reductions due to TEAEs occurred more frequently with ESL 1,600 mg (9.4%) than with ESL 1,200 mg (4.6%). Altogether, 2.6% of patients had dose reductions during the monotherapy period, compared with 4.1% during the AED conversion period.
Overall, 11.7% of patients taking ESL 1,600 mg and 20.3% of those taking ESL 1,200 mg had a reduction in plasma sodium concentration ≥10 mEq/L from baseline during the 18‐week treatment period; 3.9% and 4.7% respectively had plasma sodium values ≤125 mEq/L. There were no clinically relevant changes in vital signs, no orthostatic effects, no significant abnormalities in physical and neurologic examinations in either treatment group, and no clinically significant electrocardiogram findings.
During the 18‐week treatment period, suicidality (assessed using the Columbia Suicide Severity Rating Scale [C‐SSRS]) occurred in four patients in each dose group (ESL 1,600 mg, 3.1%; ESL 1,200 mg, 6.2%). Suicidality was related mainly to suicidal ideation (3.6%) rather than suicidal behavior (0.5%, one subject who prepared for an attempt); no patients carried out an actual suicide attempt.
Discussion
The present trial demonstrated efficacy of ESL 1,600 and 1,200 mg taken once daily as monotherapy (the doses currently recommended for use as adjunctive therapy are 800 and 1,200 mg QD).9, 15 The full 18‐week treatment period was completed by 59% of patients, whereas the completion rate for patients who entered the monotherapy period was 76%. These rates are similar to those reported in other conversion to monotherapy trials.16, 17, 18 These data demonstrate that ESL monotherapy was both effective and well tolerated by epilepsy patients after conversion from adjunctive AED therapy.
The KM estimated exit rates for patients taking ESL 1,200 and 1,600 mg QD were significantly lower than the exit rates for historical controls. The exit rate was lower with ESL 1,600 mg than ESL 1,200 mg, although the difference between treatment groups was not statistically significant. For the primary end point, the comparison between doses was corrected for the effect of multiplicity, but the study was not powered to detect a difference between doses. In addition, the differences in SSF and responder rates between the two dose groups do not suggest that the 1,600 mg dose is substantially more effective. However, this does not exclude the possibility that some patients may benefit from the higher ESL dose. Furthermore, the decrease in SSF during the 18‐week treatment period was similar for different severities of POS, suggesting that monotherapy conversion to ESL can be performed irrespective of seizure severity.
This trial employed a “historical control withdrawal to monotherapy” design.14 This study design is appropriate for AED monotherapy trials, provided the design, patient population, evaluation criteria, and analysis methods are comparable to those of the historical trials.16 In these respects, the current trial was comparable to the eight historical trials. The study region (North America), mean ages, median duration of epilepsy, and median baseline seizure frequencies were similar in all nine trials; a smaller proportion of patients were taking CBZ during baseline in the current trial.14 A possible limitation of this study is the lack of a concurrent control.
In an identical study (093‐046) carried out in international cente rs (75% of which were located outside of North America), ESL monotherapy (1,600 and 1,200 mg QD) was also shown to be superior to historical controls.12 The KM‐estimated exit rates (95% CI) for study 093‐046 were 12.8% (7.5–21.5%) for ESL 1,600 mg and 15.6% (8.1–28.7%) for ESL 1,200 mg. Together, these two trials demonstrate successful conversion to ESL monotherapy, in both an international and an NA population. The exit rates were higher in this NA study (1,600 mg, 28.7%; 1,200 mg, 44.4%) than in the international study 093‐046.12 The two studies were conducted concurrently, with identical protocols, therefore, the only identifiable difference between them is the geographic region of recruitment. The reason for the difference in exit rates remains unknown, but could relate to differences in clinical characteristics between the NA and non‐NA populations, or may reflect differences in the management of epilepsy. In the 093‐046 (international) trial, baseline seizure rates were lower than in the current trial, and the mean duration of epilepsy was shorter, suggesting a less severely affected population. Use of LEV was less frequent in non‐NA patients, reflecting differences in patient management.
In the ESL 1,600 mg dose group, use of CBZ as a baseline AED was associated with an increased KM estimated exit rate (34.7% compared with 26.9%). This modest increase may be due to overlap in mechanisms of action of the two compounds, leading to an additive effect with respect to side effects; an increased incidence of side effects may lead to reduced adherence, which in turn could exacerbate seizures and increase study exit rate. In the ESL 1,200 mg group, the rates were comparable (using CBZ, 46.4%; not using CBZ, 45.7%). In their analysis of the eight historical trials, French et al.14 found that the use of CBZ at baseline did not significantly increase the likelihood of exit, but increased the hazard rate for trial exit by 8.0% (95% CI −19.4 to 35.4). ESL patient exit rates for those who had or had not been taking LEV during the baseline period of this study were not markedly different.
The improvements in QOLIE‐31 scores observed in both ESL treatment groups exceeded five points. According to Borghs et al.,19 this suggests that the improvements in quality of life may have been clinically meaningful. An improvement in depressive symptoms, as indicated by a reduction in MADRS score, was observed for both ESL dose groups. However, only the reduction in MADRS score with ESL 1,600 mg (2.4‐point reduction) appeared to be clinically relevant, as it exceeded the minimal clinically important difference (−1.6 to −1.9) described by Duru and Fantino.20 This effect has been observed in other AED trials,21, 22 and may result from seizure reduction during conversion to monotherapy, or other causes.
No new safety issues or trends with ESL were detected in this trial; the safety data resembled those generated with adjunctive ESL use.23 The most common TEAEs of dizziness and headache were mainly mild or moderate in severity. The overall incidence of TEAEs appeared to be dose related, as observed in the adjunctive ESL trials.23 Although reductions in plasma sodium levels ≥10 mEq/L were detected in 15% of patients, these reductions necessitated withdrawal from the study only infrequently. Patients with plasma sodium values ˂125 mEq/L were required by the protocol to withdraw from the study, regardless of whether they were symptomatic; 1.6% of patients taking ESL 1,600 mg, and 3.1% of patients taking ESL 1,200 mg QD discontinued due to hyponatremia (reported as a TEAE). In line with the 2008 FDA report on AEDs and suicide risk,24 the association of suicidality with ESL treatment was analyzed in this study. Suicidality was reported infrequently, and no clear association with ESL was observed, similar to the conclusions regarding AED use and suicidality drawn by Mula et al.25
Other AEDs have also been investigated using a “conversion to monotherapy” trial design with a historical control: levetiracetam XR,26 lamotrigine XR,16 lacosamide,17 pregabalin,18 and brivaracetam (NCT00698581 and NCT00699283); the two brivaracetam trials were terminated after an interim analysis demonstrated trial futility. There are differences between the published monotherapy studies with regard to the maximum dose of a second AED allowed during screening (for example, the levetiracetam and lacosamide studies allowed up to half of the defined daily dose, whereas the lamotrigine XR study excluded use of a second AED). Furthermore, the studies of other AEDs did not include reassignment of withdrawals as seizure exits in their primary end point. All previous historical control studies of AED monotherapy (including study 093‐046) enrolled patients from a mix of U.S. and non‐U.S. populations, whereas the current study and those comprising the historical control14 were conducted exclusively in North America. It is unclear whether these factors had a major influence on the current findings. Therefore caution must be used when comparing exit rates in the different monotherapy studies.
Conclusion
The results of the current trial suggest that ESL monotherapy improves seizure control in NA patients with partial (focal) epilepsy not previously controlled by one or two AEDs, and is well tolerated.
Conflict of Interest
Michael Sperling has received honoraria for consultancy from UCB Pharma, ElectroCore, and Accorda Therapeutics; an honorarium from Wiley Blackwell for serving as an associate editor for Epilepsia; and grants from National Institute of Neurological Diseases and Stroke (NINDS)), UCB Pharma, Sunovion Pharmaceuticals Inc., Eisai, SK Life Science, Upsher‐Smith, Medtronics, Lundbeck, Visualase, and Brain Sentinel. Jay Harvey has received financial support for research from Sunovion Pharmaceuticals Inc., UCB Pharma, Upsher‐Smith, Marinus, SK Life Science, Cyberonics, Brain Sentinel, and Eisai; speaker fees from Sunovion Pharmaceuticals Inc. and UCB Pharma, and honoraria for consultancy from Sunovion Pharmaceuticals Inc. Todd Grinnell, Hailong Cheng, and David Blum are paid employees of Sunovion Pharmaceuticals Inc. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1. Study design.
Figure S2. Patient disposition.
Figure S3. Change from baseline in standardized seizure frequency and responder rate according to seizure severity (monotherapy period; efficacy [EFF] population).
Table S1. Patients: inclusion and exclusion criteria.
Table S2. Patient disposition: Reasons for early study withdrawal.
Appendix S1. Statistical analyses.
Appendix S2. The Study 045 Team.
Acknowledgments
This study was supported by Sunovion Pharmaceuticals Inc., Marlborough, Massachusetts, U.S.A. The authors would like to acknowledge the writing assistance of Mallory Gough of FireKite, part of KnowledgePoint360, an Ashfield Company, for support in drafting this manuscript. Medical writing support was funded by Sunovion Pharmaceuticals Inc.
Biography
Dr. Michael R. Sperling is Baldwin Keyes Professor of Neurology at Thomas Jefferson University.

045 Study Team members are in Appendix S2.
References
- 1. Perucca E. Designing clinical trials to assess antiepileptic drugs as monotherapy. CNS Drugs 2008;22:917–938. [DOI] [PubMed] [Google Scholar]
- 2. Perucca E, Tomson T. The pharmacological treatment of epilepsy in adults. Lancet Neurol 2011;10:446–456. [DOI] [PubMed] [Google Scholar]
- 3. St. Louis EK, Rosenfeld WE, Bramley T. Antiepileptic drug monotherapy: the initial approach in epilepsy management. Curr Neuropharmacol 2009;7:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bialer M, Soares‐da‐Silva P. Pharmacokinetics and drug interactions of eslicarbazepine acetate. Epilepsia 2012;53:935–946. [DOI] [PubMed] [Google Scholar]
- 5. Almeida L, Bialer M, Soares‐da‐Silva P. Eslicarbazepine acetate In Shorvon S, Peruca E, Engel J. (Eds) The treatment of epilepsy. Oxford: Blackwell Publishing, 2009:485–498. [Google Scholar]
- 6. Brown ME, El‐Mallakh RS. Role of eslicarbazepine in the treatment of epilepsy in adults with partial‐onset seizures. Ther Clin Risk Manag 2010;6:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perucca E, French J, Bialer M. Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol 2007;6:793–804. [DOI] [PubMed] [Google Scholar]
- 8. Almeida L, Potgieter JH, Maia J, et al. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur J Clin Pharmacol 2008;64:267–273. [DOI] [PubMed] [Google Scholar]
- 9. Aptiom® (eslicarbazepine acetate) prescribing information. 2013. Sunovion Pharmaceuticals Inc., Marlborough, Massachusetts, USA.
- 10. Bonifacio MJ, Sheridan RD, Parada A, et al. Interaction of the novel anticonvulsant, BIA 2‐093, with voltage‐gated sodium channels: comparison with carbamazepine. Epilepsia 2001;42:600–608. [DOI] [PubMed] [Google Scholar]
- 11. Nunes T, Rocha JF, Falcão A, et al. Steady‐state plasma and cerebrospinal fluid pharmacokinetics and tolerability of eslicarbazepine acetate and oxcarbazepine in healthy volunteers. Epilepsia 2013;54:108–116. [DOI] [PubMed] [Google Scholar]
- 12. Pazdera L, Jacobson MP, Bhatia P, et al. Conversion to monotherapy with eslicarbazepine acetate in adults with partial‐onset seizures. Epilepsy Curr 2014;14(Suppl. 1):108 (Abstract 1.228). [Google Scholar]
- 13. Ling X, U.S. Food and Drug Administration . Lamictal® XR™ (lamotrigine) historical‐controlled trial. Peripheral and Central Nervous System Drugs Advisory Committee Meeting March 10, 2011. Available at: http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/peripheralandcentralnervoussystemdrugsadvisorycommittee/ucm247490.pdf. Accessed March 31, 2014.
- 14. French JA, Wang S, Warnock B, et al. Historical control monotherapy design in the treatment of epilepsy. Epilepsia 2010;51:1936–1943. [DOI] [PubMed] [Google Scholar]
- 15. Zebinix® (eslicarbazepine acetate) summary of product characteristics. 2014. BIAL‐Portela & Ca, S.A., Mamede do Coronado, Portugal.
- 16. French JA, Temkin NR, Shneker BF, et al. Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapeutics 2012;9:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wechsler RT, Li G, French J, et al. Conversion to lacosamide monotherapy in the treatment of focal epilepsy: results from a historical‐controlled, multicenter, double‐blind study. Epilepsia 2014;55:1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. French JA, Kwan P, Fakhoury T, et al. Pregabalin monotherapy in patients with partial‐onset seizures. A historical‐controlled trial. Neurology 2014;82:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borghs S, de la Loge C, Cramer A. Defining minimally important change in QOLIE‐31 scores: estimates from three placebo‐controlled lacosamide trials in patients with partial‐onset seizures. Epilepsy Behav 2012;23:230–234. [DOI] [PubMed] [Google Scholar]
- 20. Duru G, Fantino B. The clinical relevance of changes in the Montgomery‐Asberg depression rating scale using the minimum clinically important difference approach. Curr Med Res Opin 2008;24:1329–1335. [DOI] [PubMed] [Google Scholar]
- 21. Mazza M, Martini A, Scoppetta M, et al. Effect of levetiracetam on depression and anxiety in adult epileptic patients. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:539–543. [DOI] [PubMed] [Google Scholar]
- 22. Kalogjera‐Sackellares D, Sackellares JC. Improvement in depression associated with partial epilepsy in patients treated with lamotrigine. Epilepsy Behav 2002;3:510–516. [DOI] [PubMed] [Google Scholar]
- 23. Rogin J, Abou‐Khalil B, Blum D, et al. Eslicarbazepine acetate as adjunctive treatment for refractory partial‐onset seizures: pooled analysis of safety data from three phase III controlled trials. Epilepsy Curr 2014;14(Suppl. 1):209 (Abstract 2.126). [Google Scholar]
- 24. U.S. Food and Drug Administration . Information for healthcare professionals: suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100192.htm. Last updated August 15, 2013. Accessed February 13, 2014.
- 25. Mula M, Kanner AM, Schmitz B, et al. Antiepileptic drugs and suicidality: an expert consensus statement from the task force on therapeutic strategies of the ILAE commission on neuropsychobiology. Epilepsia 2013;54:199–203. [DOI] [PubMed] [Google Scholar]
- 26. Chung S, Ceja H, Gawłowicz J, et al. Levetiracetam extended release conversion to monotherapy for the treatment of patients with partial‐onset seizures: a double‐blind, randomised, multicentre, historical control study. Epilepsy Res 2012;101:92–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Study design.
Figure S2. Patient disposition.
Figure S3. Change from baseline in standardized seizure frequency and responder rate according to seizure severity (monotherapy period; efficacy [EFF] population).
Table S1. Patients: inclusion and exclusion criteria.
Table S2. Patient disposition: Reasons for early study withdrawal.
Appendix S1. Statistical analyses.
Appendix S2. The Study 045 Team.