

Abstract
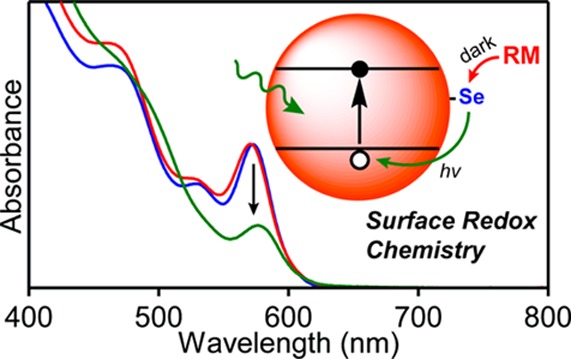
Understanding the structural and compositional origins of midgap states in semiconductor nanocrystals is a longstanding challenge in nanoscience. Here, we report a broad variety of reagents useful for photochemical reduction of colloidal CdSe quantum dots, and we establish that these reactions proceed via a dark surface prereduction step prior to photoexcitation. Mechanistic studies relying on the specific properties of various reductants lead to the proposal that this surface prereduction occurs at oxidized surface selenium sites. These results demonstrate the use of small-molecule inorganic chemistries to control the physical properties of colloidal QDs and provide microscopic insights into the identities and reactivities of their localized surface species.
Improving redox control over semiconductor nanocrystal quantum dots (QDs) is essential for the application of such QDs in electronic and optical technologies and in photocatalysis. Delocalized carriers in metal-oxide and metal-chalcogenide QDs have been introduced using chemical,1 electrochemical,2−6 and photochemical methods.7,8 Studies of these chemistries are often complicated by contributions from redox-active midgap states. Depending on their redox state, such surface species can trap carriers, introduce fast nonradiative decay pathways for core QD photoexcited states, or participate in irreversible nanocrystal degradation. Localized surface redox chemistry has been shown to affect QD photoluminescence (PL) and blinking9,10 and can sustain picosecond trap-assisted Auger recombination dynamics.11 A greater understanding of this surface redox chemistry is necessary for advancing QD applications.
Recently, we communicated that CdSe QDs can be photodoped upon treatment with Li[Et3BH] (Figure 1A) to yield high-optical-quality n-type colloidal QDs.7 Photodoping allows the redox properties of free-standing colloidal QDs to be studied under well-controlled conditions without depositing them onto electrodes or altering their native surface ligation. The Cd2+/Sex– ratios at CdSe QDs surfaces impact the band-edge potentials dramatically and alter the slow (milliseconds to hours) spontaneous surface trapping of the delocalized conduction-band (CB) electrons,12 a phenomenon potentially related to PL blinking. More detailed investigations into the redox chemistries of colloidal QDs that enable photodoping and govern electron trapping will provide new insights into this important class of materials and will advance our ability to control their functional properties relevant to various QD applications. Here, we report a much broader array of reducing agents that can be used for photodoping of CdSe QDs than examined previously. We further show that most of these photodoping reactions are initiated by dark thermal prereduction of surface sites and that photodoping then proceeds via photooxidation of these reduced surface species.
Figure 1.
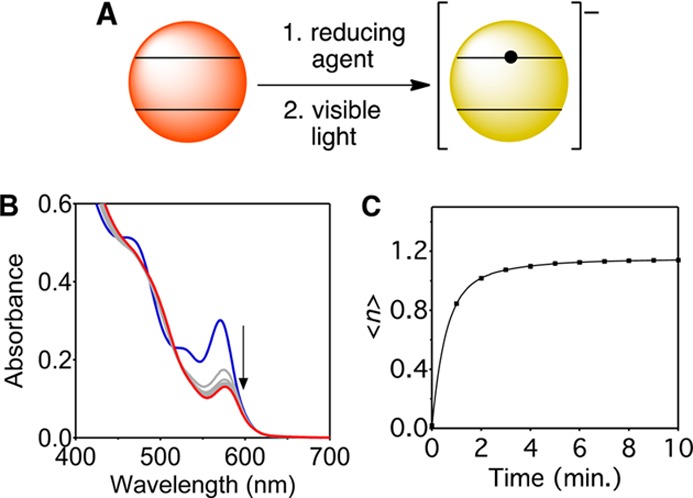
(A) Schematic illustration of QD photodoping. (B) Absorption spectra of colloidal d ≈ 3.6 nm CdSe QDs during photodoping (50 equiv/QD Li[Et3BH], 5 mW of 405 nm excitation). (C) Average number of CB electrons ⟨n⟩ vs time, reflecting photodoping up to a steady-state value of ⟨nmax⟩. Line is a guide to the eye.
Figure 1B illustrates the characteristic absorption bleach at the first exciton observed upon photodoping. Occupation of the CB by excess delocalized electrons bleaches the visible band-edge absorption and introduces new infrared intraband absorption. The magnitude of the exciton absorption bleach is proportional to the average number of electrons per QD, given by ⟨n⟩ = 2(A0 – A)/A0 = −2ΔA/A0.6,13Figure 1C illustrates that continuous photoexcitation adds CB electrons to the ensemble of CdSe QDs up to a maximum value of ⟨nmax⟩. ⟨nmax⟩ is reached at steady state when the photodoping rate equals the rate of subsequent surface electron trapping.12
Li[Et3BH] is a “complex” reductant and does not display reversible one-electron chemistry.14 The photodoping process described in Figure 1 must therefore be more complicated than a simple electron-transfer reaction. The mechanism of hole quenching was explored by monitoring the impact of triethylborohydride on the CdSe QD PL. Figure 2A summarizes the PL intensity and decay dynamics at various concentrations of Li[Et3BH] in the dark before photodoping. Although the integrated PL intensity decreases with increasing [Et3BH–], consistent with hole quenching, the PL decay dynamics are found to remain approximately independent of [Et3BH–]. This result indicates that the visible PL quenching by Li[Et3BH] does not result from a diffusion-limited bimolecular reaction but instead involves a static, or preassociation, mechanism. NMR spectroscopy rules out the possibility that the Et3BH– is simply adsorbed to the QD surfaces. Figure 2B compares the 11B NMR spectrum of a solution of Li[Et3BH] in C6D6 (δ = −12 ppm) with the spectrum of a C6D6 solution of CdSe QDs treated with 100 equiv/QD of Li[Et3BH] in the dark. In the QD sample, no Et3BH– is detected; instead, only free triethylborane is observed (δ = 84 ppm).15 These experiments implicate a dark reaction between the CdSe QDs and Li[Et3BH], allowing the conclusion that the product of this dark reaction is responsible for hole quenching during photodoping, rather than electron transfer from Et3BH– itself.
Figure 2.
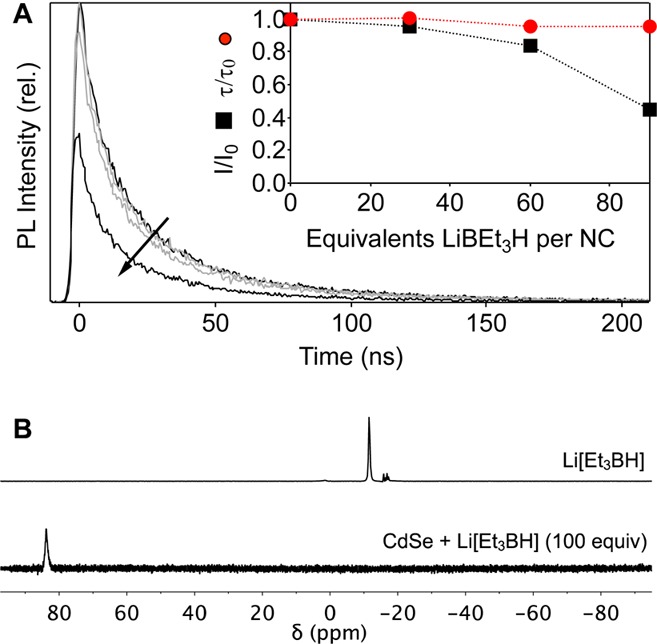
(A) Time-resolved PL from a stirred anaerobic solution of unphotodoped CdSe QDs (0.5 μM in toluene, d ≈ 4.1 nm) with added Li[Et3BH] (0, 30, 60, 90 equiv/QD, arrow). Inset: Normalized integrated PL intensities (I/I0) and PL single-exponential decay time constants (τ/τ0) vs Li[Et3BH], neglecting the first few nanoseconds of decay. (B) 11B NMR spectra of Li[Et3BH] (top)16 and of CdSe QDs treated with Li[Et3BH] (100 equiv) in C6D6 (bottom).
The dark reaction of CdSe QDs with Li[Et3BH] could occur at three possible reducible sites: (i) coordinated carboxylate ligands could be reduced to the corresponding alkoxides.17 Alcohols such as ethanol have been shown to photodope metal-oxide NCs (e.g., ZnO)8,18−23 and have been used as sacrificial reductants for semiconductor–metal heterostructure photocatalysis.24−26 (ii) Cd2+ centers on the QD surfaces could be reduced to Cd0. This reduction has been proposed to be a key step for CdSe photocatalysis.27 (iii) Partially oxidized surface selenium moieties could be reduced to Se2–. Se2– oxidation on CdSe QD surfaces has been observed, for example, forming surface oxides or interparticle diselenide bonds.28,29
Oleate reduction to the corresponding alkoxide upon treatment of CdSe QDs with Li[Et3BH] (50 equiv/QD) is ruled out by 1H NMR spectroscopy, although H2 formation was detected (see SI). When the CdSe QD mixtures were treated with isotopically labeled Li[Et3BD], D2 rather than HD was found to be the major product, although both species were detected by NMR spectroscopy. HD could be a side product formed by protonation of Li[Et3BD] by a protic species in solution such as oleic acid. D2 could be formed in several ways: by reaction of free D• radicals generated via outer-sphere electron transfer to the CdSe surface, by reductive elimination from a cadmium-deuteride species, or by deprotonation of a surface selenol-type Se–D bond by an additional equivalent of Li[Et3BD]. Regardless of mechanism, D2 would not be an expected byproduct of carboxylate reduction to alkoxides. Moreover, CdSe QDs treated with thousands of equivalents of sodium ethoxide display much less photodoping (⟨n⟩ ≈ 0.25) than the same QDs treated with only tens of equivalents of Li[Et3BH] (⟨n⟩ ≈ 1). These results demonstrate that reduced ligands cannot be the primary origin of this photodoping.
To test the possibility that redox-active surface species participate in photodoping, a sample of CdSe QDs was treated with the strong reductant sodium naphthalenide, (EO = −3.10 V vs Fc+/Fc in THF).14 Sodium naphthalenide is capable of directly injecting electrons into the CdSe CB.1 For one sample of CdSe QDs, an excess of sodium naphthalenide (∼60 equiv/QD) was required before stable CB electrons were observed optically. When the same QDs were treated with fewer equivalents of sodium naphthalenide (∼40 equiv/QD), no band-edge absorption bleach was observed until the sample was photoexcited, at which point CB electrons appeared. We conclude that these photodoping reactions proceed by a two-step mechanism in which surface species are first reduced in a dark reaction and are then reoxidized by photogenerated holes. Reducible surface states in CdSe QDs have been detected spectroscopically13 and by direct chemical titration of photodoped CdSe QDs.7 Because they constitute a minority of the QD atoms and likely possess inhomogeneous speciation, it has been challenging to deduce their chemical identity using typical spectroscopic or electrochemical methods.
To broaden the scope of this photodoping chemistry, which helps to elucidate the mechanism and may also potentially allow a broader range of QDs to be photodoped, we tested a number of different reductants (Table 1). The results of these experiments suggest surface selenium reduction rather than cadmium reduction as the critical dark reaction. We find that nucleophilic organometallic reagents such as tert-butyllithium, methylmagnesium bromide, and mesitylmagnesium bromide can be used to photodope CdSe QDs (Table 1, entries 4–6). The QDs can be treated with tens of equivalents of these reagents in the dark without any change in the QD absorption spectra, although excess reductant results in QD precipitation or etching. Other organometallic reagents like diethylzinc also can be used to photodope the CdSe QDs (Table 1, entry 7). Even though only tens of equivalents are sufficient for photodoping, thousands of equivalents of Et2Zn can be added with no spectroscopic degradation. In this extreme case, prolonged photoexcitation generates a gray precipitate that is likely Zn0 metal. A similar metallic precipitate was observed during photodoping of ZnO QDs in the presence of a large excess of Li[Et3BH].8
Table 1. Results from Photodoping CdSe QDs Using Various Reducing Agentsa.
| entry | reducing agent | equiv/QD | ⟨nmax⟩b |
|---|---|---|---|
| 1 | Li[Et3BH] | 50 | 1.1 |
| 2 | Na[Et3BH] | 50 | 0.9 |
| 3 | Na[C10H8] | 40 | 0.8 |
| 4 | tert-BuLi | 50 | 1.3 |
| 5 | MeMgBr | 25 | 1.2 |
| 6 | MesMgBr | 25 | 1.9 |
| 7 | Et2Zn | 50 | 1.4 |
| 8c | PEt3/NaOH | 5000 | 0.3 |
| 9d | Me3SnSnMe3 | 9000 | 1.2 |
| 10e | MeN(H)NH2 | 30,000 | 0.7 |
Reaction conditions: CdSe QDs (d ≈ 3.6 nm, 1.8 μM in toluene), excitation with a 5 mW, 405 nm diode over 15 min while stirring. All data here are for the same synthetic batch of QDs, but specific results vary from batch to batch.
Estimated using ⟨n⟩ = 2(A0 – A)/A0.
Irradiation for 2 min using 100 W photolysis lamp with 480 nm longpass filter.
Excitation over 60 min.
First-exciton feature blue-shifted by 0.05 eV.
We also examined other, nonorganometallic, reagents known to selectively reduce selenium compounds. For example, organodiselenide compounds can be treated with tributylphosphine and sodium hydroxide to form the corresponding trialkylphosphine oxide and the reduced sodium organoselenide.30 Under the same conditions, cadmium oleate is not reduced to cadmium metal (see SI). We find that CdSe QDs suspended in 2:1 THF/toluene, when treated with a large excess of PBu3 or PEt3 followed by a degassed solution of aqueous NaOH, can indeed be photodoped (eq 1), while the same QDs in the absence of either reagent do not photodope.31 This result suggests that photodoping proceeds specifically via reduced surface selenium moieties and not through cadmium reduction.
| 1 |
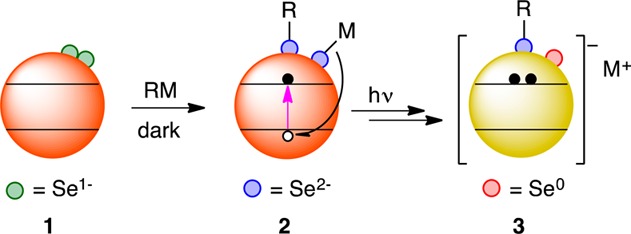
Scheme 1 diagrams the proposed surface selenium chemistry involved in photodoping. We hypothesize that as-prepared CdSe QDs (1) have a population of oxidized surface selenium species. These may include diselenides, as proposed computationally.32 The oxidized surface selenium species can be attacked by Grignards or other reagents to form reduced selenides terminated either by organic groups or metal cations (2).33 Under photoexcitation, these reduced selenide moieties can quench photogenerated valence-band (VB) holes, yielding CdSe QDs with delocalized CB electrons (3). Importantly, localization of these CB electrons back into these specific surface traps is very slow.12 We note that EPR signals are not detected during photodoping, which may suggest that a secondary electron-transfer step follows VB hole quenching, as proposed for “current doubling” in ZnO photodoping.34 This second electron-transfer step would likely be coupled to a structural rearrangement, which would be consistent with the observation that the CB electron is not immediately recaptured by the now-empty surface trap.
Scheme 1. Proposed Reduction of Surface Selenium Moieties upon Treatment with Organometallic Reagents, Followed by Photodoping.
Selenium reduction promoted by organometallic reagents could conceivably proceed by one- or two-electron processes. To test the possibility of a one-electron selenium reduction route, other reagents that are known to follow one-electron radical pathways were tested for QD photodoping. We find that organotin reagents such as hexamethylditin and tributyltin hydride are indeed effective at photodoping CdSe QDs (Table 1, entry 9), but their reactions are much slower than those of the other reagents in Table 1 and require many more equivalents of reductant. We propose that surface radicals are formed under photoexcitation that then react with these organotin compounds to form reduced surface selenide sites. Excitation of CdSe QDs in the presence of the spin trap phenyl N-tert-butylnitrone confirms that radicals are indeed formed under irradiation, possibly by homolytic dissociation of surface diselenides or by disproportionation of Se0 and Se2– centers. Because the reaction between surface radicals and the organotin compounds would be diffusion-limited, photodoping of CdSe QDs with these reagents is slow and requires a high concentration of the organotin compound. Additionally, we find that CdSe QD surfaces can be prereduced via dark thermal treatment with tributyltin hydride and azobis(isobutyronitrile) (AIBN), included as a radical initiator, and that these prereduced QDs then exhibit photodoping rates similar to those treated with the organometallic reductants (see SI). Surface prereduction can thus proceed via radical intermediates when using organotin reductants. This finding does not rule out the possibility of two-electron surface reduction when using other reductants in Table 1.
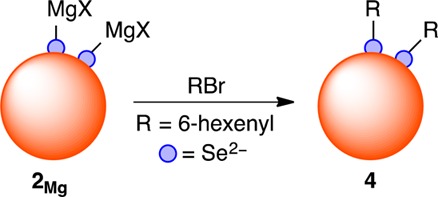
The motif of reduced surface selenide moieties counterbalanced by metal cations proposed here (2 in Scheme 1) resembles molecular nucleophilic selenide species.33 To explore this analogy, CdSe QDs with prereduced surfaces (2) were investigated for alkylation reactivity with electrophiles. Scheme 2 diagrams one such reaction. After surface reduction with MesMgBr (50 equiv/QD) to form the proposed reduced selenides with magnesium halide counterions (2Mg), these QDs were then treated with 6-bromo-1-hexene as the alkyl halide electrophile, and the mixture was refluxed in THF for 24 h. After acid digestion and extraction of the organics, the corresponding di(5-hexenyl) diselenide compound was detected by GC–MS, consistent with QD surface alkylation (4). The same reaction of 6-bromo-1-hexene with unreduced CdSe QDs (1) followed by acid digestion still forms some of the corresponding diselenide compound, but in much lower yield (<40% conversion). These results support the proposal that treatment of CdSe QDs with the organometallic reagents of Table 1 forms reduced surface selenide moieties as illustrated in Scheme 1. This chemistry also suggests an intriguing new method of QD surface functionalization.
Scheme 2. Alkylation of CdSe QDs after Surface Selenium Reduction.
CdSe photodoping is proposed to proceed through similar reduced surface selenide moieties for all reductants examined here. Table 1 shows that ⟨nmax⟩ depends on the identity of the reducing agent, however. Reductant-dependent variations in ⟨nmax⟩ were also observed for ZnO QD photodoping.8,35 In CdSe QDs, ⟨nmax⟩ is determined kinetically by competing photodoping and electron-trapping processes.12 Differences in ⟨nmax⟩ could result from variations in the surface density of reduced selenide moieties, the metal cation (Li+, Na+, [MgX]+, etc.), or steric access of the reducing agents to the surface. Unexpectedly, MesMgBr, a sterically hindered Grignard reagent, generated nearly twice as many CB electrons compared to MeMgBr. Bimesityl was identified as a reaction byproduct (GC–MS), suggesting the possibility that MesMgBr can reduce surface Sex– via outer-sphere electron transfer to form the reduced selenide and an aryl radical. In contrast, no ethane was detected during the reaction of MeMgBr with CdSe QDs.
To further clarify the dependence of ⟨nmax⟩ on the sterics of the reducing agent, CdSe QD photodoping by a series of substituted hydrazine reductants was examined. Rather than displaying a dependence on N–H bond strength, ⟨nmax⟩ correlates most strongly with the bulkiness of the hydrazine reductant, decreasing from MeN(H)NH2 > Me2NNH2 > PhN(H)NH2. The observation that hydrazine reagents can be used to photodope CdSe QDs is important in part because hydrazine is commonly used during the preparation of QD films for conductivity or photoconductivity studies.36−40 Prior studies have focused on the roles of hydrazine for reducing interparticle spacing40 and “charge-transfer” n-doping.36,41 Computational studies have claimed that hydrazine is not able to directly access the PbSe QD CB.42 The results here raise the possibility of unintentional photodoping of QD films after treatment with hydrazine and exposure to ambient photoexcitation. Indeed, given the number and variety of chemical reagents (Table 1) active for photoreduction of CdSe QDs, unintentional photodoping is likely a relatively common occurrence.
In summary, CdSe QD photodoping is demonstrated with a broad variety of reductants. The photodoping reaction is shown to proceed through dark prereduction of the QD surfaces prior to photoexcitation. The experimental results suggest that this dark reduction step occurs at oxidized selenium moieties. Surface selenium reduction can proceed through one- or two-electron processes and using several different reducing agents, including borohydrides, organometallic reagents, organotin compounds, and hydrazine compounds. The rich chemistries of these surface moieties suggest intriguing new possibilities for tuning functionally relevant QD physical properties via targeted surface modification involving redox transformations. These results have important implications for future fundamental studies and applications of colloidal semiconductor QDs.
Acknowledgments
This research was supported by the NSF (CHE-1506014 to D.R.G. and Graduate Research Fellowship DGE-1256082 to K.H.H.) and the NIH (Postdoctoral Fellowship F32GM110876 to E.Y.T.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06548.
Additional experimental details and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shim M.; Guyot-Sionnest P. Nature 2000, 407, 981–983. 10.1038/35039577. [DOI] [PubMed] [Google Scholar]
- Wehrenberg B. L.; Guyot-Sionnest P. J. Am. Chem. Soc. 2003, 125, 7806–7807. 10.1021/ja035369d. [DOI] [PubMed] [Google Scholar]
- Haram S. K.; Quinn B. M.; Bard A. J. J. Am. Chem. Soc. 2001, 123, 8860–8861. 10.1021/ja0158206. [DOI] [PubMed] [Google Scholar]
- Myung N.; Ding Z.; Bard A. J. Nano Lett. 2002, 2, 1315–1319. 10.1021/nl0257824. [DOI] [Google Scholar]
- Boehme S. C.; Wang H.; Siebbeles L. D. A.; Vanmaekelbergh D.; Houtepen A. J. ACS Nano 2013, 7, 2500–2508. 10.1021/nn3058455. [DOI] [PubMed] [Google Scholar]
- Wang C.; Shim M.; Guyot-Sionnest P. Science 2001, 291, 2390–2392. 10.1126/science.291.5512.2390. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Schimpf A. M.; Weaver A. L.; Cohn A. W.; Gamelin D. R. J. Am. Chem. Soc. 2013, 135, 18782–18785. 10.1021/ja410825c. [DOI] [PubMed] [Google Scholar]
- Schimpf A. M.; Gunthardt C. E.; Rinehart J. D.; Mayer J. M.; Gamelin D. R. J. Am. Chem. Soc. 2013, 135, 16569–16577. 10.1021/ja408030u. [DOI] [PubMed] [Google Scholar]
- Weaver A. L.; Gamelin D. R. J. Am. Chem. Soc. 2012, 134, 6819–6825. 10.1021/ja301102h. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Weaver A. L.; Gamelin D. R. J. Am. Chem. Soc. 2012, 134, 16175–16177. 10.1021/ja307996b. [DOI] [PubMed] [Google Scholar]
- Cohn A. W.; Janßen N.; Mayer J. M.; Gamelin D. R. J. Phys. Chem. C 2012, 116, 20633–20642. 10.1021/jp3075942. [DOI] [Google Scholar]
- Tsui E. Y.; Carroll G. M.; Miller B.; Marchioro A.; Gamelin D. R. Manuscript in preparation, 2016.
- Shim M.; Wang C.; Guyot-Sionnest P. J. Phys. Chem. B 2001, 105, 2369–2373. 10.1021/jp0035683. [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- The triethylborane formed was collected by vacuum transfer and quantified using an internal NMR standard to account for ∼60% of the triethylborohydride added. This number is a lower limit, as some triethylborane was lost during the transfer.
- A small amount of [Et2BH2]− anion is present as an impurity but likely does not affect reactivity, as multiple sources of Li[Et3BH] reagent result in similar photodoping behavior.
- Jang E.; Jun S.; Chung Y.; Pu L. J. Phys. Chem. B 2004, 108, 4597–4600. 10.1021/jp049475t. [DOI] [Google Scholar]
- Haase M.; Weller H.; Henglein A. J. Phys. Chem. 1988, 92, 482–487. 10.1021/j100313a047. [DOI] [Google Scholar]
- Wood A.; Giersig M.; Mulvaney P. J. Phys. Chem. B 2001, 105, 8810–8815. 10.1021/jp011576t. [DOI] [Google Scholar]
- Liu W. K.; Whitaker K. M.; Kittilstved K. R.; Gamelin D. R. J. Am. Chem. Soc. 2006, 128, 3910–3911. 10.1021/ja060488p. [DOI] [PubMed] [Google Scholar]
- Liu W. K.; Whitaker K. M.; Smith A. L.; Kittilstved K. R.; Robinson B. H.; Gamelin D. R. Phys. Rev. Lett. 2007, 98, 186804. 10.1103/PhysRevLett.98.186804. [DOI] [PubMed] [Google Scholar]
- Schimpf A. M.; Ochsenbein S. T.; Buonsanti R.; Milliron D. J.; Gamelin D. R. Chem. Commun. 2012, 48, 9352–9354. 10.1039/c2cc34635d. [DOI] [PubMed] [Google Scholar]
- Schrauben J. N.; Hayoun R.; Valdez C. N.; Braten M.; Fridley L.; Mayer J. M. Science 2012, 336, 1298–1301. 10.1126/science.1220234. [DOI] [PubMed] [Google Scholar]
- Costi R.; Saunders A. E.; Elmalem E.; Salant A.; Banin U. Nano Lett. 2008, 8, 637–641. 10.1021/nl0730514. [DOI] [PubMed] [Google Scholar]
- Amirav L.; Alivisatos A. P. J. Phys. Chem. Lett. 2010, 1, 1051–1054. 10.1021/jz100075c. [DOI] [Google Scholar]
- Ruberu T. P. A.; Nelson N. C.; Slowing I. I.; Vela J. J. Phys. Chem. Lett. 2012, 3, 2798–2802. 10.1021/jz301309d. [DOI] [Google Scholar]
- Zhao J.; Holmes M. A.; Osterloh F. E. ACS Nano 2013, 7, 4316–4325. 10.1021/nn400826h. [DOI] [PubMed] [Google Scholar]
- Katari J. E. B.; Colvin V. L.; Alivisatos A. P. J. Phys. Chem. 1994, 98, 4109–4117. 10.1021/j100066a034. [DOI] [Google Scholar]
- Pala I. R.; Arachchige I. U.; Georgiev D. G.; Brock S. L. Angew. Chem., Int. Ed. 2010, 49, 3661–3665. 10.1002/anie.201000034. [DOI] [PubMed] [Google Scholar]
- Sakakibara M.; Katsumata K.; Watanabe Y.; Toru T.; Ueno Y. Synthesis 1992, 1992, 377–379. 10.1055/s-1992-26116. [DOI] [Google Scholar]
- Treatment of CdSe QDs with NaOH in the absence of trialkylphosphine results in a red shift (∼30 meV) of the excitonic absorbance, likely due to unknown surface effects, but no bleach is observed under photoexcitation.
- Voznyy O.; Thon S. M.; Ip A. H.; Sargent E. H. J. Phys. Chem. Lett. 2013, 4, 987–992. 10.1021/jz400125r. [DOI] [PubMed] [Google Scholar]
- Iwaoka M.; Tomoda S. Top. Curr. Chem. 2000, 208, 55–80. 10.1007/3-540-48171-0_3. [DOI] [Google Scholar]
- Morrison S. R.; Freund T. J. Chem. Phys. 1967, 47, 1543–1551. 10.1063/1.1712115. [DOI] [Google Scholar]
- Carroll G. M.; Schimpf A. M.; Tsui E. Y.; Gamelin D. R. J. Am. Chem. Soc. 2015, 137, 11163–11169. 10.1021/jacs.5b06715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talapin D. V.; Murray C. B. Science 2005, 310, 86–89. 10.1126/science.1116703. [DOI] [PubMed] [Google Scholar]
- Steiner D.; Azulay D.; Aharoni A.; Salant A.; Banin U.; Millo O. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 80, 195308. 10.1103/PhysRevB.80.195308. [DOI] [Google Scholar]
- Luther J. M.; Beard M. C.; Song Q.; Law M.; Ellingson R. J.; Nozik A. J. Nano Lett. 2007, 7, 1779–1784. 10.1021/nl0708617. [DOI] [PubMed] [Google Scholar]
- Law M.; Luther J. M.; Song Q.; Hughes B. K.; Perkins C. L.; Nozik A. J. J. Am. Chem. Soc. 2008, 130, 5974–5985. 10.1021/ja800040c. [DOI] [PubMed] [Google Scholar]
- Vercelli B.; Zotti G.; Berlin A. J. Phys. Chem. C 2011, 115, 4476–4482. 10.1021/jp110223f. [DOI] [Google Scholar]
- Talapin D. V.; Black C. T.; Kagan C. R.; Shevchenko E. V.; Afzali A.; Murray C. B. J. Phys. Chem. C 2007, 111, 13244–13249. 10.1021/jp074156y. [DOI] [Google Scholar]
- Kutana A.; Erwin S. C. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 83, 235419. 10.1103/PhysRevB.83.235419. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.