

Abstract
Desulfurivibrio alkaliphilus strain AHT2T is a strictly anaerobic sulfidogenic haloalkaliphile isolated from a composite sediment sample of eight hypersaline alkaline lakes in the Wadi al Natrun valley in the Egyptian Libyan Desert. D. alkaliphilus AHT2T is Gram-negative and belongs to the family Desulfobulbaceae within the Deltaproteobacteria. Here we report its genome sequence, which contains a 3.10 Mbp chromosome. D. alkaliphilus AHT2T is adapted to survive under highly alkaline and moderately saline conditions and therefore, is relevant to the biotechnology industry and life under extreme conditions. For these reasons, D. alkaliphilus AHT2T was sequenced by the DOE Joint Genome Institute as part of the Community Science Program.
Keywords: Deltaproteobacteria, Soda lake, Sediment, Sulfur cycle, Sulfur disproportionation
Introduction
Soda lakes are extreme environments with high salinity and highly alkaline pH values. They are formed in arid regions where high rates of evaporation lead to the accumulation of sodium carbonate salts, which are dominant in these distinctive lakes. Soda lakes support an active microbial sulfur cycle, enhanced by the stability of intermediate sulfur species such as thiosulfate and polysulfides and much lower toxicity of sulfide at these elevated pH conditions. Correspondingly, a wide variety of anaerobic haloalkaliphiles active in the reductive sulfur cycle have been isolated from these lakes [1]. Insights into sulfur redox processes will contribute to understanding how haloalkaliphilic organisms survive and thrive under dual extreme conditions. Some metabolic processes within the reductive sulfur cycle are more favorable under alkaline pH conditions than under circumneutral conditions, such as the disproportionation of elemental sulfur [2]. These sulfur redox processes are not only relevant in natural haloalkaline environments, some wastewater and gas desulfurization treatment plants are often operated at high salt concentrations and pH values where haloalkaliphiles play a role in the remediation of the affected areas. Thus, the haloalkaliphile Desulfurivibrio alkaliphilus strain AHT2T was sequenced for its relevance to sulfur cycling and the environmental biotechnology sector by the DOE-JGI Community Science Program.
Organism information
Classification and features
D. alkaliphilus AHT2T is the type strain of the Desulfurivibrio alkaliphilus species and was isolated from a mixed sediment sample from eight hypersaline alkaline lakes in the Wadi al Natrun valley in the Libyan Desert (Egypt) [3]. The cells are Gram-negative, non-motile, curved rods that do not form spores (Fig. 1). D. alkaliphilus AHT2T tolerates sodium carbonate concentrations ranging from 0.2 - 2.5 M total Na+ and grows within a pH range of 8.5 - 10.3 (optimum at pH 9.5) [3]. Phylogenetic analysis showed that the strain belongs to the family Desulfobulbaceae within the Deltaproteobacteria and is most closely related to a, so far undescribed, haloalkaliphilic chemoautotrophic sulfur-disproportionator within the same genus: Desulfurivibrio sp. strain AMeS2 [2]. Strains AMeS2 and AHT2T are, so far, the only known representatives of the Desulfurivibrio genus (Fig. 2). The closest sequenced relative to this novel genus, is another soda lake isolate delta proteobacterium sp. MLMS-1, which has been enriched as an arsenate-dependent sulfide oxidizer [4]. D. alkaliphilus AHT2T is able to reduce thiosulfate and elemental sulfur [3] and plays a role in the reductive sulfur cycle in soda lake environments [1]. D. alkaliphilus AHT2T is also capable of chemolithoautotrophic growth through the disproportionation of elemental sulfur under alkaline pH conditions without iron(III) oxides [2], which are normally required by neutrophilic sulfur disproportionators. More classifications and features are listed in Table 1.
Fig. 1.
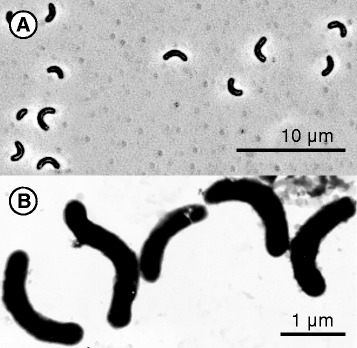
Morphology of D. alkaliphilus AHT2T. a A phase contrast micrograph of the D. alkaliphilus AHT2T cells. b A scanning electron microscope image of the D. alkaliphilus AHT2T cells
Fig. 2.
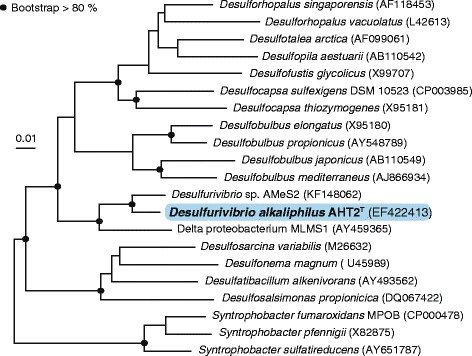
Neighbour joining tree based on 16S rRNA gene sequences showing the phylogenetic position of D. alkaliphilus AHT2T to other species within the Deltaproteobacteria class. The Firmicutes were used as an outgroup and subsequently pruned from the tree. The black dots indicate a bootstrap value between 80 and 100 %. The scale bar indicates a 1 % sequence difference. The tree was constructed with the ARB software package [37] and the SILVA database [19]. The bootstrap values were calculated using MEGA-6 [38]
Table 1.
Classification and general features of D. alkaliphilus AHT2T
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [39] | |
| Phylum: Proteobacteria | TAS [40, 41] | ||
| Class: Deltaproteobacteria | TAS [42, 43] | ||
| Order: Desulfobacterales | TAS [43, 44] | ||
| Family: Desulfobulbaceae | TAS [43, 45] | ||
| Genus: Desulfurivibrio | TAS [3, 46] | ||
| Species: Desulfurivibrio alkaliphilus | TAS [3, 46] | ||
| Type strain: AHT2T | TAS [3] | ||
| Gram stain | negative | ||
| Cell shape | rod-shaped | ||
| Motility | non-motile | ||
| Sporulation | nonsporulating | ||
| Temperature range | mesophile | ||
| Optimum temperature | 35 | ||
| pH range; Optimum | 8.5–10.3; 9.5 | TAS [3] | |
| Carbon source | acetate, HCO3 − | TAS [3] | |
| GS-6 | Habitat | hypersaline alkaline lake sediments | |
| MIGS-6.3 | Salinity | moderately salt-tolerant | |
| MIGS-22 | Oxygen requirement | anaerobe | |
| MIGS-15 | Biotic relationship | free living | |
| MIGS-14 | Pathogenicity | none | |
| MIGS-4 | Geographic location | Wadi al Natrun, Libyan Desert (Egypt) | |
| MIGS-5 | Sample collection | September 2000 | |
| MIGS-4.1 | Latitude – Longitude | 30° 24′ N | |
| MIGS-4.2 | 30° 18′ E | ||
| MIGS-4.3 | Depth | 0–10 cm | TAS [3] |
| MIGS-4.4 | Altitude | −20 m |
Genome sequencing information
Genome project history
D. alkaliphilus AHT2T was sequenced by the DOE Joint Genome Institute [5] based on its relevance to the biotechnology industry. It is part of the Community Science Program (CSP_788492) entitled ‘Haloalkaliphilic sulfate-, thiosulfate- and sulfur-reducing bacteria’. The project is registered in the Genomes Online Database (Ga0028523) [6] and the complete genome sequence is deposited in GenBank (GCA_000092205). Sequencing and assembly were performed at the DOE Joint Genome Institute using state of the art sequencing technology [7]. A summary of the project information is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Solexa, 454 |
| MIGS-29 | Sequencing platforms | 454, Illumina |
| MIGS-31.2 | Fold coverage | 39.9 × 454, 98 × Illumina |
| MIGS-30 | Assemblers | Newbler,Velvet, phrap |
| MIGS-32 | Gene calling method | Prodigal [17] |
| Locus Tag | DaAHT2 | |
| Genbank ID | CP001940 | |
| Genbank Date of Release | 01.28.2014 | |
| GOLD ID | Gp0003395 | |
| BIOPROJECT | PRJNA33629 | |
| MIGS-13 | Project relevance | biotechnological |
Growth conditions and genomic DNA preparation
D. alkaliphilus AHT2T was grown anaerobically at 30 °C in Na-carbonate buffered mineral medium containing 0.6 M total Na+ with a pH of 10. 4 mM NH4Cl, 1 mM MgCl2 x 6H2O, 1 ml L−1 trace element solution [8], 2 mM Na-acetate as C-source and ~5 g/L powdered sulfur (electron acceptor) were added after sterilization. 2 L culture was grown in a 10 L bottle mounted on a magnetic stirrer with an 0.5 bar H2 (electron donor) overpressure head-space. The cells from 1 L culture were harvested by centrifugation at 13,000 g for 30 min, washed with 1 M NaCl and stored at −80 °C. The DNA was extracted and purified from frozen pellets by the phenol-chloroform method after pre-treatment with SDS-proteinase K according to Murmur [9]. The purity and molecular weight of the DNA was checked by UV spectroscopy and gel electrophoresis, respectively.
Genome sequencing and assembly
The total size of the D. alkaliphilus AHT2T genome sequence assembly was 3.1 Mbp. The draft genome of D. alkaliphilus AHT2T was generated at the DOE Joint Genome Institute using a combination of Illumina [10] and 454 DNA sequencing technologies [11]. An Illumina GAii shotgun library was constructed, which generated 3,998,684 reads and a 454 Titanium standard library, which generated 517,041 reads totalling 123.6 Mb of 454 data. The initial draft assembly contained 57 contigs in 1 scaffold. The 454 Titanium data were assembled with Newbler, 2.0.00.20-PostRelease-11-05-2008-gcc-3.4.6. The Newbler consensus sequences were computationally shredded into 2 kb overlapping fake reads (shreds). Illumina sequencing data was assembled with VELVET, version 1.0.13 [12], and the consensus sequences were computationally shredded into 1.5 kb overlapping fake reads (shreds). We integrated the 454 Newbler consensus shreds and the Illumina VELVET consensus shreds using parallel Phrap, version SPS - 4.24 (High Performance Software, LLC). The software Consed [13] was used in the finishing process as described previously [14]. The final assembly is based on 123.6 Mb of 454 draft data which provides an average 39.9x coverage of the genome and 303.9 Mb of Illumina draft data providing an average 98x coverage of the genome.
Genome annotation
The complete genome sequence was annotated using the JGI Prokaryotic Automatic Annotation Pipeline [15] with additional manual review using the Integrated Microbial Genomes - Expert Review platform [16]. Genes were predicted using Prodigal [17], followed by a round of manual curation using the JGI GenePRIMP pipeline [18]. Ribosomal RNAs were detected using models built from SILVA [19] and tRNAs were predicted with tRNAScanSE [20]. The predicted coding sequences were translated and used to search the National Center for Biotechnology Information non-redundant database, UniProt, TIGRFam, Pfam, KEGG, COG and InterPro databases. Further annotation was performed using the Integrated Microbial Genomes platform. The final annotated genome is available from the Integrated Microbial Genome system [21].
Genome properties
The genome is 3,097,763 bp long with GC content of 60.29 % (Table 3). 2732 genes were found, of which 2676 are annotated as protein-coding genes and 56 are RNA genes (47 tRNA genes). A total of 75 % of the protein-coding genes have been assigned a function prediction and 62.26 % have been assigned to a COG (Table 3). The number of genes assigned to each functional COG category is listed in Table 4.
Table 3.
Nucleotide content and gene count levels of the genome
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 3,097,763 | 100.00 |
| DNA coding (bp) | 2,806,423 | 90.60 |
| DNA G + C (bp) | 1,867,527 | 60.29 |
| DNA scaffolds | 1 | 100.00 |
| Total genes | 2,732 | 100.00 |
| Protein coding genes | 2,676 | 97.95 |
| RNA genes | 56 | 2.05 |
| Pseudo genes | 56 | 2.05 |
| Genes in internal clusters | 103 | 3.77 |
| Genes with function prediction | 2,049 | 75 |
| Genes assigned to COGs | 1,701 | 62.26 |
| Genes with Pfam domains | 2,280 | 83.46 |
| Genes with signal peptides | 175 | 6.41 |
| Genes with transmembrane helices | 672 | 24.60 |
| CRISPR repeats | 2 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % of total | Description |
|---|---|---|---|
| J | 180 | 9.50 | Translation, ribosomal structure and biogenesis |
| A | NA | RNA processing and modification | |
| K | 72 | 3.80 | Transcription |
| L | 84 | 4.43 | Replication, recombination and repair |
| B | 2 | 0.11 | Chromatin structure and dynamics |
| D | 26 | 1.37 | Cell cycle control, cell division, chromosome partitioning |
| V | 44 | 2.32 | Defense mechanisms |
| T | 134 | 7.07 | Signal transduction mechanisms |
| M | 149 | 7.86 | Cell wall/membrane biogenesis |
| N | 82 | 4.33 | Cell motility |
| U | 50 | 2.64 | Intracellular trafficking and secretion |
| O | 93 | 4.91 | Posttranslational modification, protein turnover, chaperones |
| C | 139 | 7.34 | Energy production and conversion |
| G | 67 | 3.54 | Carbohydrate transport and metabolism |
| E | 129 | 6.81 | Amino acid transport and metabolism |
| F | 53 | 2.80 | Nucleotide transport and metabolism |
| H | 132 | 6.97 | Coenzyme transport and metabolism |
| I | 52 | 2.74 | Lipid transport and metabolism |
| P | 130 | 6.86 | Inorganic ion transport and metabolism |
| Q | 20 | 1.06 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 134 | 7.07 | General function prediction only |
| S | 70 | 3.69 | Function unknown |
| - | 1031 | 37.74 | Not in COGs |
Extended insights from the genome sequence
Carbon fixation
In order to grow chemolithoautotrophically, D. alkaliphilus AHT2T assimilates inorganic carbon from the environment. The genome of D. alkaliphilus AHT2T contains the key genes necessary for the WL pathway, a mode of carbon fixation from CO2, which can run in the reductive and oxidative direction [22]. In the reductive direction, carbon is fixed from inorganic CO2 to cell material. The WL pathway functions in this direction in many representatives of sulfate-reducing bacteria within the Deltaproteobacteria. Some organisms may couple the reverse, or oxidative, direction to sulfate reduction. The WL gene clusters have previously been defined for delta proteobacterium sp. MLMS-1 from Mono Lake [23], the closest sequenced relative of D. alkaliphilus AHT2T (Fig. 2). Here we identified the WL genes necessary for carbon fixation by comparing the corresponding delta proteobacterium sp. MLMS-1 gene clusters to those present in D. alkaliphilus AHT2T using the JGI IMG database (Fig. 3). The first step in the reductive pathway is the reduction of CO2 to formate, by formate dehydrogenase (DaAHT2_0823 and an accessory protein DaAHT2_0820). This is followed by formyl-THF synthetase (DaAHT2_0837) and a methylene-THF dehydrogenase/cyclohydrolase (DaAHT2_0828) and a methylene-THF reductase (DaAHT2_0827). The acs gene cluster is necessary for the carbonyl branch of the reaction [22], which starts with the reduction of CO2 to carbon monoxide by a carbon monoxide dehydrogenase (DaAHT2_0826). In the last step, the products of the carbonyl and methyl branch are combined to form the product acetyl-CoA, by a CO dehydrogenase/acetyl-CoA synthase complex (DaAHT2_0825 and DaAHT2_0824). The end product of the WL cycle is typically acetate, however, the genes needed to convert acetyl-CoA to the end product acetate are absent in the D. alkaliphilus AHT2T genome, resulting in acetyl CoA being the carbon end product which can be incorporated into biomass.
Fig. 3.
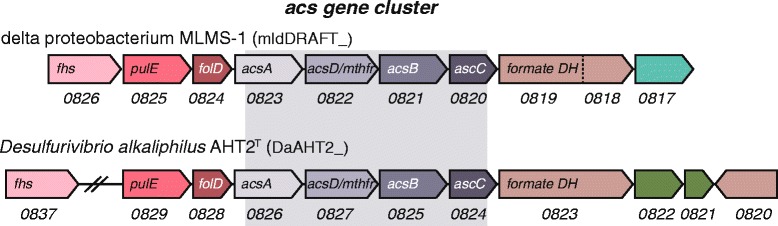
D. alkaliphilus AHT2T Wood-Ljungdahl pathway genes, including the acs gene cluster, based on delta proteobacterium sp. MLMS-1 [23]. The gene locus tags are depicted beneath the illustrated gene representations
Sulfur cycle
Culture studies have provided evidence that D. alkaliphilus AHT2T is able to reduce a number of different sulfur redox species to conserve energy [4]. The dsr cluster catalyzes sulfite reduction to sulfide [24, 25], which is also present in the D. alkaliphilus AHT2T genome consisting of dsrABC (DaAHT2_0296, DaAHT2_0297, DaAHT2_2041) and dsrMK(JOP) (DaAHT2_2298-DaAHT2_2302). D. alkaliphilus AHT2T also has genes which may be involved in the oxidative branch of sulfite disproportionation: a sulfate adenylyltransferase sat (DaAHT2_0293) and two adenylylsulfate reductase subunits aprAB (alpha: DaAHT2_1471 and beta: DaAHT2_1472). In the haloalkaline environment from which D. alkaliphilus AHT2T was isolated, intermediate redox species of sulfur such as polysulfides and thiosulfate are abundantly present. The genes for the reduction of elemental sulfur (polysulfides) and thiosulfate (psr/phs) are annotated together as a single KEGG ortholog, namely K08352 [26]. However, the psr and phs genes have been identified individually in different organisms and are responsible for different reactions.
The molybdenum-containing polysulfide reductase gene psrA (WS0116 / Ga0076602_11110) was first identified in the sulfur/polysulfide-reducing epsilonproteobacterium Wolinella succinogenes [27, 28]. The thiosulfate reductase operon phs (STY2271-STY2269) was first identified in the enteric bacterium Salmonella typhimurium [29, 30]. The genome of D. alkaliphilus AHT2T contains two molybdopterin oxidoreductases (DaAHT2_0547 and DaAHT2_0420) (Fig. 4a). In order to determine whether the D. alkaliphilus AHT2T gene cluster is a psr or a phs operon, we used eggNOG 4.5 [31] to find 446 orthologs of psrA (WS0116 / Ga0076602_11110) in 233 species, from which a phylogenetic neighbor-joining tree was constructed and trimmed (Fig. 4b). The molybdopterin oxidoreductase sequences of D. alkaliphilus AHT2T (DaAHT2_0420 and DaAHT2_0547) did not cluster within the psr or phs branch (Fig. 4b). Nevertheless, they are part of the same orthologous group as the W. succinogenespsrA (ENOG4107QY8) with which they share 24,80 % (DaAHT2_0547) and 31,75 % (DaAHT2_0420) identity. The S. typhimuriumphsA is clustered in the same orthologous group and is 27,34 identical to DaAHT2_0547 and 29,79 % identical to DaAHT2_0420 (Fig. 4a). Only one of the D. alkaliphilus AHT2TphsA/psrA genes is located within an operon of three subunits (Fig. 4a). This means that the D. alkaliphilus AHT2T gene with the locus tag DaAHT2_0420 is most probably the active psrA/phsA. Laboratory culture evidence points towards the D. alkaliphilus AHT2T DaAHT2_4020 – DaAHT2_0418 operon being functional as a sulfur reductase, as it is unable to grow on thiosulfate in absence of H2 as electron donor [3]. In addition, the operon is directly adjacent to a sulfur transferase rhodanese domain (DaAHT2_0417), which has been suggested to be essential for the binding, stabilizing and transferring sulfur to the psrA subunit [32]. However, more research is needed to define this gene operon as either a psr or a phs gene cluster.
Fig. 4.
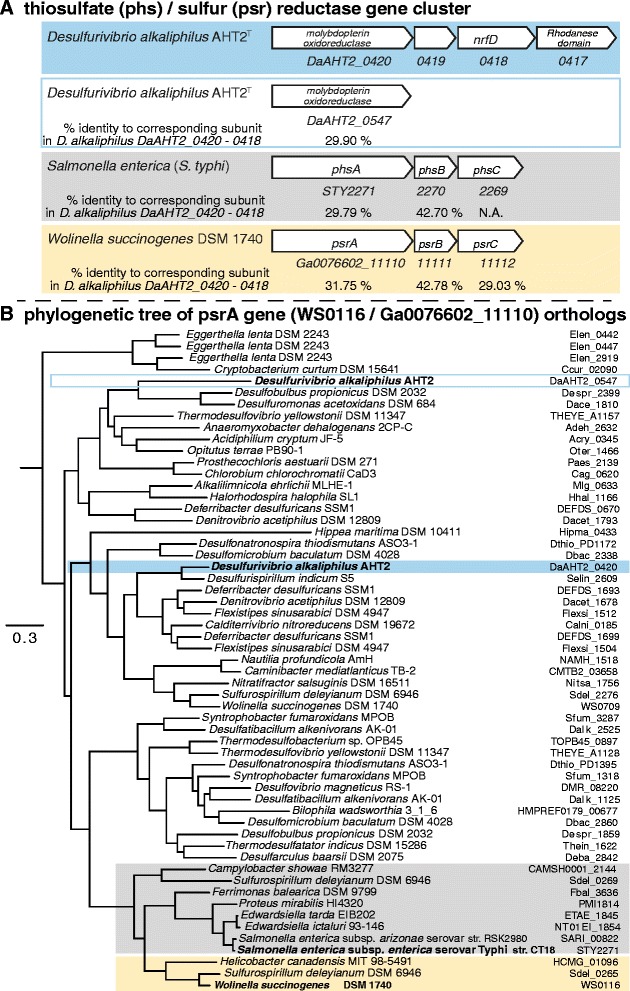
A comparison of the thiosulfate reductase (phs) and sulfur reductase (psr) gene annotation. a The phs (grey) and psr (yellow) gene clusters and how similar they are to a phs/psr gene cluster in D. alkaliphilus AHT2T (blue) based on BLAST analysis [47]. b A phylogenetic tree of an orthologous group of the psrA gene derived from EggNOG (ENOG4107QY8) [31]. Sequences annotated as phs are indicated in grey and sequences annotated as psr are coloured in yellow. The orthologous genes in D. alkaliphilus AHT2T are coloured in blue, and white with a blue outline
Adaptations to the haloalkaline environment
There are several adaptations that haloalkaliphiles can use to survive in the haloalkaline environment: bioenergetic adaptations, structural membrane adaptations and the use of osmoprotectants to retain osmotic balance [1]. The genome of D. alkaliphilus AHT2T contains a voltage gated sodium channel gene ncbA (DaAHT2_0077) and the electrogenic sodium/proton antiporter mrpBCDEFG operon (DaAHT2_2362 to DaAHT2_2357). The nqr operon encodes a sodium pumping NADH: quinone oxidoreductase (alternative to H+-pumping conventional NADH-quionone oxidoreductases) that shuttles electrons from NADH to ubiquinone [33, 34]. The D. alkaliphilus AHT2T genome contains the first account of the nqr operon in anaerobic haloalkaliphiles [35, 36]. The locus tags of the nqr gene cluster nqrA-nqrF in D. alkaliphilus AHT2T are DaAHT2_0042 – DaAHT2_0047, and we also found this cluster in D. alkaliphilus AHT2T’s closest sequenced relative delta proteobacterium sp. MLMS-1 (mldDRAFT_0493-0498) (Fig. 5). The D. alkaliphilus AHT2T genome does not contain genes for the synthesis of ectoine or betaine, which function as common osmoprotectants in haloalkaliphiles, but it does have a choline/betaine transporter (DaAHT2_1056).
Fig. 5.

The sodium dependent NADH ubiquinone oxidoreductase (nqr) gene cluster. Vibrio alginolyticus ATCC 17749T [33, 48] was used as a reference for the delta proteobacterium sp. MLMS-1 and D. alkaliphilus AHT2T gene clusters
Conclusions
In this manuscript we give a short description of the D. alkaliphilus AHT2T genome, which was isolated from hypersaline soda lake sediments in the Libyan Desert in Egypt. Its ability to perform inorganic sulfur disproportionation reactions in laboratory cultures indicates that the necessary gene pathways are present in the genome of this organism. The metabolic pathways of disproportionation are so far poorly understood; therefore, further investigation of the D. alkaliphilus AHT2T genome may lead to insights which genes are essential to this metabolism. In addition, a more in depth genome sequence analysis might provide more insights into autotrophic carbon metabolism in haloalkaline environments.
Acknowledgements
Emily Denise Melton, Lex Overmars and Gerard Muyzer are supported by ERC Advanced Grant PARASOL (No. 322551); Dimitry Y. Sorokin was supported by the Gravitation SIAM grant 24002002 and the RFBR grant 16-04-00035. Alla L. Lapidus is supported by the RSF grant 14-50-00069. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, was supported under Contract No. DE-AC02-05CH11231.
Authors’ contributions
EDM drafted and wrote the manuscript. DYS, LO, GM, NCK and ALL contributed to the written manuscript. DYS, LO and GM stimulated critical discussions. DS cultured D. alkaliphilus and extracted the DNA. The sequencing and annotation of the genome were performed at the JGI by OC, AC, MP, NI, NS, NCK, TW and all. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- acsA
Carbon monoxide dehydrogenase
- acsB
Acetyl-CoA synthase
- acsC
Corrinoid iron-sulfur protein large subunit
- Formate DH
Formate dehydrogenase
- fhs
Formyl-H4-folate synthase
- folD
Formyl-H4folate cyclohydrolase/methylene-H4folate dehydrogenase
- mthfr/acsD
Methylene-H4folate reductase/corrinoid iron-sulfur protein small subunit fusion
- pulE
Type II secretory pathway ATPase PulE
- THF
Tetrahydrofolate
- WL
Wood Ljungdahl
References
- 1.Sorokin DY, Berben T, Melton ED, Overmars L, Vavourakis CD, Muyzer G. Microbial diversity and biogeochemical cycling in soda lakes. Extremophiles. 2014;18:791–809. doi: 10.1007/s00792-014-0670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poser A, Lohmayer R, Vogt C, Knoeller K, Planer-Friedrich B, Sorokin D, et al. Disproportionation of elemental sulfur by haloalkaliphilic bacteria from soda lakes. Extremophiles. 2013;17:1003–1012. doi: 10.1007/s00792-013-0582-0. [DOI] [PubMed] [Google Scholar]
- 3.Sorokin DY, Tourova TP, Mussmann M, Muyzer G. Dethiobacter alkaliphilus gen. nov. sp. nov., and Desulfurivibrio alkaliphilus gen. nov. sp. nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles. 2008;12:431–439. doi: 10.1007/s00792-008-0148-8. [DOI] [PubMed] [Google Scholar]
- 4.Hoeft SE, Kulp TR, Stolz JF, Hollibaugh JT, Oremland RS. Dissimilatory arsenate reduction with sulfide as electron donor: experiments with Mono Lake water and isolation of strain MLMS-1, a chemoautotrophic arsenate respirer. Appl Environ Microbiol. 2004;70:2741–2747. doi: 10.1128/AEM.70.5.2741-2747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joint Genome Institute [jgi.doe.gov].
- 6.Reddy TBK, Thomas AD, Stamatis D, Bertsch J, Isbandi M, Jansson J, Mallajosyula J, Pagani I, Lobos EA, Kyrpides NC. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucl Acids Res. 2014;43:1099–106. doi: 10.1093/nar/gku950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, Cottingham RW, Kyrpides NC. The fast changing landscape of sequencing technologies and their impact on microbial assemblies and annotations. PLoS One. 2012;7:e48837. doi: 10.1371/journal.pone.0048837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfennig N, Lippert KD. Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch Mikrobiol. 1966;55:245–256. doi: 10.1007/BF00410246. [DOI] [Google Scholar]
- 9.Marmur J. A procedure for isolation of DNA from microorganisms. J Mol Biol. 1961;3:208–214. doi: 10.1016/S0022-2836(61)80047-8. [DOI] [Google Scholar]
- 10.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 11.Margulies M, Egholm M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:326–327. doi: 10.1038/437326a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 14.Sims D, Brettin T, Detter JC, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome sequence of Kytococcus sedentarius type strain (541T) Stand Genomic Sci. 2009;1:12–20. doi: 10.4056/sigs.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K, Szeto E, Pillay M, Chen IM-A, Pati A, Nielsen T, Markowitz VM, Kyrpides NC. The standard operating procedure of the DOE-JGI microbial genome annotation pipeline (MGAP v.4) Stand Genomic Sci. 2015;10:86. doi: 10.1186/s40793-015-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG-ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 17.Hyatt D, Chen G, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for microbial genomes. Nat Methods. 2010;7:455–457. doi: 10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- 19.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 2013;41(D1):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe TM, Eddy SR. TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markowitz VM, Chen I-M A, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–22. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC, et al. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum) Environ Microbiol. 2008;10:2550–2573. doi: 10.1111/j.1462-2920.2008.01679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pires RH, Venceslau SS, Morais F, Teixeira M, Xavier AV, Pereira IAC. Characterization of the Desulfovibrio desulfuricans ATCC 27774 dsrMKJOP complex – a membrane bound redox complex involved in the sulfate respiration pathway. Biogeosciences. 2006;45:249–262. doi: 10.1021/bi0515265. [DOI] [PubMed] [Google Scholar]
- 25.Venceslau SS, Stockdreher Y, Dahl C, Pereira IAC. The bacterial heterodisulfide dsrC is a key protein in dissimilatory sulfur metabolism. Biochim Biophys Acta. 1837;2014:1148–1164. doi: 10.1016/j.bbabio.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucl Acids Res. 2016;44:D457–462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krafft T, Bokranz M, Klimmek O, Schroeder I, Fahrenholz F, Kojro E, Kroeger A. Cloning and nucleotide sequence of the psrA gene of Wolinella succinogenes polysulphide reductase. Eur J Biochem. 1992;206:503–510. doi: 10.1111/j.1432-1033.1992.tb16953.x. [DOI] [PubMed] [Google Scholar]
- 28.Baar C, Eppinger M, Raddatz G, Simon J, Lanz C, Klimmek O, et al. Complete genome sequence and analysis of Wolinella succinogenes. Proc Natl Acad Sci U S A. 2003;100:11690–11695. doi: 10.1073/pnas.1932838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinzinger NK, Fujimoto SY, Clark MA, Moreno MS, Barrett EL. Sequence analysis of the phs operon in Salmonella typhimurium and the contribution of thiosulfate reduction to anaerobic energy metabolism. J Bacteriol. 1995;177:2813–2820. doi: 10.1128/jb.177.10.2813-2820.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 31.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. EggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucl Acids Res. 2016;44:D286–93. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorokin DY, Kublanov IV, Gavrilov SN, Rojo D, Roman P, Golyshin PN, et al. Elemental sulfur and acetate can support life of a novel strictly anaerobic haloarchaeon. ISME J. 2016;10:240–252. doi: 10.1038/ismej.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi M, Nakayama Y, Unemoto T. Recent progress in the Na+-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biochim Biophys Acta. 2001;1505:37–44. doi: 10.1016/S0005-2728(00)00275-9. [DOI] [PubMed] [Google Scholar]
- 34.Reyes-Prieto A, Barquera B, Juárez O. Origin and evolution of the sodium-pumping NADH: ubiquinone oxidoreductase. PlosOne. 2014;9:e96696. doi: 10.1371/journal.pone.0096696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol. 2011;2:69. doi: 10.3389/fmicb.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol. 2015;66:55–321. doi: 10.1016/bs.ampbs.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucl Acids Res. 2004;32(4):1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2005. p. 1. [Google Scholar]
- 41.Validation of publication of new names and new combinations previously effectively published outside the IJSEM. Int J Syst Evol Microbiol. 2005;55:2235–2238. [DOI] [PubMed]
- 42.Kuever J, Rainey FA, Widdel F. Class IV. Deltaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2005. p. 922. [Google Scholar]
- 43.Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006;56:1–6. [DOI] [PubMed]
- 44.Kuever J, Rainey FA, Widdel F. Order III. Desulfobacterales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2005. p. 959. [Google Scholar]
- 45.Kuever J, Rainey FA, Widdel F. Family II. Desulfobulbaceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2005. p. 988. [Google Scholar]
- 46.Validation List no. 123 List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2008;58:1993–1994. [DOI] [PubMed]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Cao Y, Zhang H, Chen Y, Hu C. Complete genome sequence of Vibrio alginolyticus ATCC 17749T. Genomea. 2015;3:e01500–14. doi: 10.1128/genomeA.01500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]