

Abstract
Despite a clinical, economic, and regulatory imperative to develop companion diagnostics, precious few new biomarkers have been successfully translated into clinical use, due in part to inadequate protein assay technologies to support large-scale testing of hundreds of candidate biomarkers in formalin-fixed paraffin embedded (FFPE) tissues. While the feasibility of using targeted, multiple reaction monitoring-mass spectrometry (MRM-MS) for quantitative analyses of FFPE tissues has been demonstrated, protocols have not been systematically optimized for robust quantification across a large number of analytes, nor has the performance of peptide immuno-MRM been evaluated. To address this gap, we used a test battery approach coupled to MRM-MS with the addition of stable isotope labeled standard peptides (targeting 512 analytes) to quantitatively evaluate the performance of three extraction protocols in combination with three trypsin digestion protocols (i.e. 9 processes). A process based on RapiGest buffer extraction and urea-based digestion was identified to enable similar quantitation results from FFPE and frozen tissues. Using the optimized protocols for MRM-based analysis of FFPE tissues, median precision was 11.4% (across 249 analytes). There was excellent correlation between measurements made on matched FFPE and frozen tissues, both for direct MRM analysis (R2 = 0.94) and immuno-MRM (R2 = 0.89). The optimized process enables highly reproducible, multiplex, standardizable, quantitative MRM in archival tissue specimens.
Keywords: targeted proteomics, peptide assays, FFPE, archived tissue, immunoaffinity enrichment, multiple reaction monitoring, mass spectrometry
INTRODUCTION
Despite a clinical, economic, and regulatory imperative1 to develop companion diagnostics, precious few new tissue biomarkers have been translated into clinical use.2 Clinical validation studies must be performed on large numbers of candidate biomarkers for a single novel biomarker of clinical utility to be identified.3–5 The handful of biomarkers that have successfully reached the clinic were identified mostly through retrospective analysis of archival formalin-fixed paraffin-embedded (FFPE) biospecimens.2 The most widely used technique for detecting proteins in FFPE tissues is immunohistochemistry (IHC). Although IHC is the mainstay of biomarker determinations in clinical pathology, this technology is inadequate to support large-scale testing of hundreds of candidate biomarkers in retrospective validation studies, due to the high costs and long lead times for the development and analytical validation of new IHC assays. Additionally, even with multi-parameter fluorescence detection, the multiplex capabilities of IHC remain limited and would allow testing of only small numbers of candidate biomarkers in each assay.6 Furthermore, as currently widely deployed, IHC assay results are semi-quantitative at best, leading to difficulties interpreting intermediate results, and hampering the ability to assemble multivariate panels as diagnostics. Finally, in the clinical setting, multiple sources of variation have resulted in poor inter-laboratory concordance of tissue markers determined by IHC.7–13 We desperately need the development of a multiplexed quantitative platform to analyze FFPE archival tissues.14
To date, several studies have demonstrated the feasibility of using targeted, multiple reaction monitoring-mass spectrometry (MRM-MS) for quantitative proteomic analyses of FFPE tissues.15–30 Although protein localization is not preserved, MRM has many desirable characteristics for quantification, as it is already established in clinical laboratories,31–33 incorporates internal isotopic standards,34 and enables highly multiplex, precise, specific, and standardizable proteomic quantification that can be harmonized across laboratories.35–38 Furthermore, MRM can be coupled to immuno-enrichment of proteins or peptides (immuno-MRM) to provide excellent sensitivity in plasma or solid tissues.5,39–45
Although there are several widely used protocols for the extraction and trypsin proteolysis of proteins from FFPE tissues,15–30 these various methods have never been tested in head-to-head comparisons with the endpoint of achieving analytically robust MRM-based quantification. Indeed, the majority of proteomic methods development has been done in the setting of shotgun MS/MS,46–55 and none has examined the use of peptide immuno-MRM assays, nor optimized protocols based on the quantitative performance of MRM. To address these gaps, we used a test battery approach to evaluate three methods for protein extraction and antigen retrieval in combination with three methods for trypsin digestion, to identify the combination of protocols (i.e. “process”) providing the most sensitive and reproducible recovery of peptide analytes. The comparison is not a comprehensive evaluation of all protocols for extraction of FFPE, but is a detailed evaluation of common mass spectrometry-compatible approaches for analyzing the soluble proteome. Furthermore, we compare the performances of immuno-MRM assays in FFPE and in frozen tissue, and use panels of targeted LC-MRM and immuno-MRM assays to show equivalent quantitative measurements of endogenous peptides in FFPE samples with those from matched fresh frozen samples. The optimized process provides standardized protocols for quantitative analysis of proteins, enabling verification studies of tissue biomarkers in archival biospecimens. Furthermore, the protocol provides a benchmark for comparison of other methods developed for analysis of proteins in FFPE tissue.
EXPERIMENTAL METHODS
Reagents
Xylene (catalog #422685000) and 2,2,2 Trifluoroethanol (TFE, #1397510000) were purchased from Acros Organics, part of ThermoFisher Scientific (Pittsburgh, PA). RapiGest SF surfactant (#186001861) was purchased from Waters (Milford, MA). The Liquid Tissue MS Protein Prep Kit (#10001-023) was purchased from Expression Pathology (Rockville, MD). Urea (#U0631), iodoacetamide (IAM, #A3221), tris(2-carboxyethyl)phosphine (TCEP, #77720), EGTA (#E0396), EDTA (#E7889), Trizma buffer solution (Tris, # T2694), and phosphatase inhibitor cocktails #1 (#P2850) and #2 (#P5726) were purchased from Sigma Aldrich (St. Louis, MO). Sequencing grade trypsin was purchased from Promega (Madison, WI). MS grade acetonitrile (MeCN, #A955), MS grade water (#W6), ethanol (EtOH, #04-355-223), 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS) detergent (#28300) and 10X PBS (#BP-399) were purchased from Thermo Fisher Scientific (Waltham, MA). Formic acid (FA, #11670-1) was purchased from Millipore (Billerica, MA). Custom monoclonal anti-peptide antibodies for immuno-MRM assays were generated by Epitomics, an Abcam company (Burlingame, CA), using recombinant B-cell technology56 or hybridoma screening.57
Synthetic heavy and light peptides were purchased from Thermo Fisher Scientific, New England Peptide (Gardner, MA), and 21st Century Biochemicals (Malborough, MA). Heavy stable isotope-labeled standards (SIS) and matched light versions were handled according to published recommendations.34 Peptide purity was >95% by HPLC. Heavy peptides incorporated a fully atom labeled 13C and 15N isotope at the C-terminal lysine (K) or arginine (R) position of each (tryptic) peptide, resulting in a mass shift of +8 or +10 Da, respectively. Quantification by amino acid analysis was performed and aliquots were stored in 5-30% acetonitrile/0.1% formic acid at −80 °C until use.
Breast cancer tissues
Archived FFPE-treated ER+/Her2+ breast cancer tissue samples were obtained from Stanford University Medical Center (SUMC) under IRB approval (#348), and FFPE and fresh frozen ER+/Her2+ breast cancer tissue samples were obtained from NWBioTrust (NWBT) under IRB approval (#7077). FFPE samples were prepared according to ASCO/CAP guidelines.58,59
Breast cancer sample preparation
FFPE and fresh frozen tissue samples were prepared from three individual breast cancer tissues that were collected, quartered, and then sectioned into pieces > 5mg (wet tumor weight). Alternating tissue pieces were frozen in liquid nitrogen or fixed at room temperature in 10% (v/v) neutral buffered formaldehyde for 18 – 27 hours, processed by a Leica Peloris Processor (Buffalo Grove, IL) over eight hours (rehydrated with 100% EtOH (Richard Allen Scientific #8101), 90% EtOH (Richard Allen Scientific #8201), permeated in xylene (Richard Allen Scientific #8101) and paraffin embedded), and stored at room temperature. 10 μm sections of the FFPE samples were cut using a microtome and mounted on glass slides. Every 10th FFPE slide was a 4 μm section stained with hematoxylin and eosin for pathology review. The frozen samples were anchored in OCT compound, and 10 μm sections were cut on a cryostat at −20 °C, such that no OCT was included in the slice.
Deparaffination and rehydration of FFPE samples
Slide-mounted FFPE tissue sections were distributed evenly among the days and replicates (to control for the microheterogeneity of the sample across the slides) and then placed in a 24 slide holder. The slides were incubated three times in xylene for 3 min followed by 100% (v/v) ethanol twice for 3 min. The tissue was then hydrated twice in 85% (v/v) ethanol for 3 min, 70% (v/v) ethanol for 3 min, and distilled water for 3 min. The tissue was then blotted and scraped off of the slide into a screw cap microfuge tube.
Protein extraction of FFPE samples with RapiGest
To each sample containing three FFPE 10 μm tissue sections, extraction buffer (0.2% RapiGest in 50 mM NH4HCO3) was added and incubated at 95 °C for 30 minutes with mixing at 1000 rpm (Thermomixer, Eppendorf). The samples were then cooled on ice for 5 minutes and sonicated twice in a cup horn probe (filled with ice water) at 50% power for 30 s. The samples were then incubated at 80 °C for 120 minutes with mixing at 1000 rpm and then cooled on ice for 5 min. 100 μL of 50 mM NH4HCO3, pH 8.0 was added, and the samples were sonicated twice in the cup horn probe (filled with ice water) at 50% power for 30 s. Following processing, all samples were stored at −80 °C until the day of digestion.
Protein extraction of FFPE samples with TFE
To each sample containing three FFPE 10 μm tissue sections, extraction buffer (100 μL of 100 mM NH4HCO3) was added and incubated at 80 °C for 120 minutes with mixing at 1000 rpm (Thermomixer, Eppendorf). Samples were then cooled on ice for 5 min. 100 μL of TFE was added and the samples were sonicated twice in a cup horn probe (filled with ice water) at 50% power for 30 s. The samples were then incubated at 60 °C for 60 minutes with mixing at 1000 rpm and then cooled on ice for 5 min. The samples were sonicated twice in cup horn probe (filled with ice water) at 50% power for 30 s. Following processing, all samples were stored at −80 °C until the day of digestion.
Protein extraction of FFPE samples with Liquid Tissue
To each sample containing three FFPE 10 μm tissue sections, extraction buffer (62.5 μL of Liquid Tissue) was added and the sample was incubated at 95 °C for 90 minutes with mixing at 1000 rpm (Thermomixer, Eppendorf). Samples were then cooled on ice for 5 min. The samples were spun at 10000 rcf for 1 min at 20 °C and 137.5 μL of 50 mM NH4HCO3, pH 8.0 was added and vortexed. Following processing, all samples were stored at −80 °C until the day of digestion.
Protein extraction of frozen tissue samples
To each sample containing the equivalent of three intact tissue sections, 200 μL of lysis buffer (25 mM Tris, 6 M Urea, 1 mM EDTA, 1 mM EGTA, 1 mM TCEP, 1% Sigma phosphatase inhibitor cocktail 1, 1% Sigma phosphatase inhibitor cocktail 2) was added. The sample was vortexed for 10-15 s and sonicated three times in a cup horn probe (filled with ice water) at 50% power for 30 s. The samples were stored in liquid nitrogen until the day of digestion.
Trypsin digestion of FFPE samples with Rapigest
At least 30 μg (by μBCA) of cell lysate was diluted to 200 μL with 0.1% RapiGest in 50 mM NH4HCO3. Lysates were reduced in 15 mM TCEP for 30 minutes at 37 °C with shaking, followed by alkylation with 40 mM IAM in the dark for 30 minutes at room temperature. Lysates were then diluted to 1.1 mL with 50 mM NH4HCO3 before trypsin was added at a 1:50 trypsin:protein ratio by mass and incubated for 2 hours at 37 °C with mixing at 600 rpm (Thermomixer, Eppendorf). After 2 hours, a second aliquot was added at 1:100 enzyme:substrate. Digestion was carried out overnight at 37 °C with mixing at 600 rpm. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume).
Trypsin digestion of FFPE samples with TFE
At least 30 μg (by μBCA) of cell lysate was diluted to 200 μL with 50% TFE in 50 mM NH4HCO3. Lysates were reduced in 15 mM TCEP for 30 minutes at 37 °C with shaking, followed by alkylation with 40 mM IAM in the dark at room temperature. Lysates were then diluted to 1.1 mL with 50 mM NH4HCO3 before trypsin was added at a 1:50 trypsin:protein ratio by mass and incubated for 2 hours at 37 °C with mixing at 600 rpm (Thermomixer, Eppendorf). After 2 hours, a second aliquot was added at 1:100 enzyme:substrate. Digestion was carried out overnight at 37 °C with mixing at 600 rpm. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume).
Trypsin digestion of FFPE samples with Urea
At least 30 μg (by μBCA) of cell lysate was diluted to 200 μL with 6M urea in 50 mM NH4HCO3. Lysates were reduced in 15 mM TCEP for 30 minutes at 37 °C with shaking, followed by alkylation with 40 mM IAM in the dark at room temperature. Lysates were then diluted to 1.1 mL with 200 mM TRIS before trypsin was added at a 1:50 trypsin:protein ratio by mass and incubated for 2 hours at 37 °C with mixing at 600 rpm (Thermomixer, Eppendorf). After 2 hours, a second aliquot was added at 1:100 enzyme:substrate. Digestion was carried out overnight at 37 °C with mixing at 600 rpm. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume).
Trypsin digestion of frozen tissue samples
At least 30 μg (by μBCA) of cell lysate was diluted to 200 μL with lysis buffer. Lysates were reduced in 15 mM TCEP for 30 minutes at 37 °C with shaking, followed by alkylation with 40 mM IAM in the dark at room temperature. Lysates were then diluted to 1.1 mL with 200 mM TRIS, pH 8, before trypsin was added at a 1:50 trypsin:protein ratio (by mass), and the lysate was incubated for 2 hours at 37 °C with mixing at 600 rpm (Thermomixer, Eppendorf). After 2 hours, a second aliquot of trypsin was added at 1:100 enzyme:substrate. Digestion was carried out overnight at 37 °C with mixing at 600 rpm. After 16 hours, the reaction was quenched with formic acid (final concentration 1% by volume).
Nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS)
The system was composed of a nanoAcquity HPLC (Waters) coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific). The LC system consisted of a 75 μm × 250 mm C18, 130 Å, 1.7 μm, column (Waters Cat.#186003545) with mobile phases of 0.1% FA in water (A) and 0.1% FA in MeCN (B). The peptide sample was diluted to 1 μg protein/μL in 0.1% FA, 3% MeCN and 1 μL was loaded onto the column and separated at a flow rate of 300 nL/min, with the following gradient (time, %B): 0, 3%; 120, 40%; 122, 90%; 132, 90%; re-equilibration for 23 min at 3%B. The mass spectrometer used an Advance CaptiveSpray source (Michrom Bioresources, Auburn, CA) operated in positive ion mode. A spray voltage of 1700 V was applied to the nanospray tip. MS/MS analysis consisted of 1 full scan MS from 300-2000 m/z at resolution 30,000 followed by data dependent MS/MS scans using 35% normalized collision energy of the 15 most abundant ions. Selected ions were dynamically excluded for 15 seconds after a repeat count of 1.
Data were searched against version 3.69 of the Human International Protein Index (IPI) sequence database with decoy sequences using the X!Tandem database search engine with a previously described score plugin,3 performed with tryptic enzyme constraint set for up to two missed cleavages, oxidized methionine set as a variable modification and carbamidomethylated cysteine set as a static modification. Peptide MH+ mass tolerances were set at ±2.0 Da with post search filtering of precursor mass to 50 ppm and fragment MH+ mass tolerances were set at ±0.5 Da. Identifications were made with an FDR < 0.01 based on a decoy database search.
LC-MRM-MS with the addition of stable isotope labeled standard peptides
A mix of SIS peptides was prepared at 100nM in 0.1% FA, 3% ACN, divided into aliquots and stored at −80 °C. Separate individual aliquots were used for each day of the experiment. After digestion, this mix was spiked into the individual samples at 10 fmol/μg of protein lysate for analytes targeted by LC-MRM and 1 fmol/μg for those targeted by immuno-MRM, and the samples were desalted as described in the Supplemental Methods. Spike levels were high enough above the LLOQ so as not to contribute unnecessarily to the assay CV and were designed to be close to expected endogenous levels so that the peak area ratio was not outside of the range of 100:1 and 1:100. Quantitative LC-MRM-MS and immuno-MRM-MS data were collected using the method described below. Sample run order was staggered so the types of samples were not grouped together.
Response curves
Quantitative assays were characterized in background matrices consisting of an equal mix (by protein mass) of three individual FFPE or frozen samples. Digestion was performed as described above. Reverse curves were prepared in triplicate by varying SIS peptide concentration over 8 concentration points (LC-MRM: 200, 22.2, 2.47, 0.82, 0.27, 0.09, 0.03, 0.01 fmol/μg; immuno-MRM: 10.0, 1.11, 0.124, 0.041, 0.014, 0.0046, 0.0015, 0.0005 fmol/μg). Light peptide was added at a constant concentration of 5 fmol/μg for LC-MRM analytes and 1 fmol/μg for immuno-MRM analytes. Blanks contained no SIS peptide. Linear regression was performed using a 1/x2 weighting on all points within the linear range, defined as those points such that the standard error of the slope of the linear regression was less than 10%. Limit of detection (LOD) was obtained by using the average of the three blank measurements plus three times the standard deviation of the noise. Lower limit of quantification (LLOQ) was reported as the lowest point in the response curve measured with CV≤20%. The upper limit of quantification (ULOQ) was determined by the highest concentration point of the response curve that was maintained in the linear range of the response.
Nano-liquid chromatography-tandem mass spectrometry (LC-MRM)
Targeted LC-MRM-MS analysis of peptide analytes was performed by a trap-elute configuration on a nanoLC- 2D system with AS1 autosampler (Eksigent Technologies, Dublin, CA) coupled to a 5500 QTRAP mass spectrometer (ABSciex, Foster City, CA) by an Advance CaptiveSpray source (Michrom Bioresources, Auburn, CA). Mobile phases consisted of 0.1% FA in water (A) and 90% MeCN with 0.1% FA (B).
LC-MRM used a cHiPLC-Nanoflex system (Eksigent) with the following method: 1 μL of sample was loaded onto a 200 μm × 0.5 mm ChromXP C18-CL 3 μm 120 Å column (Eksigent) at 10 μL/min and 3%B for 3 minutes. At 3 minutes, the sample was injected onto the analytical column and separated by a 75 μm × 15 cm ChromXP C18-CL 3 μm 120 Å column (Eksigent Technologies, Dublin, CA) using the following gradient method: hold at 3% B for 8 minutes, gradient from 3 to 10% B for 3 minutes, gradient from 10 to 30% B for 27 minutes, gradient from 30 to 40% B for 7 minutes, gradient from 40 to 60% B for 1 minutes, hold 60% B for 2 minutes, gradient from 60 to 90% B for 1 minute, hold 90% B for 5 minutes, gradient from 90 to 3% B for 1 minute, re-equilibrate at 3% B for 25 minutes. The flow rate was 300 nL/min. The column temperature was 40 °C.
Immuno-MRM used the following method: Samples were loaded onto a 300 μm × 5 mm trap column (Acclaim PepMap 100 C18, 5 μm, 100 Å, Dionex Cat #160454, Sunnyvale, CA) at 6.0 μL/min (2% B) for 4 minutes. Peptides were eluted at 300 nL/min from a 75 μm × 10 cm IntegraFrit column packed with ReproSil-Pur C18-AQ, 3 μm particles, using the following gradient (time, %B): 4, 2%; 22, 50%; 23, 90%; 24, 90%; re-equilibrate for 14 minutes at 2%B. The analytical column was held at 45 °C.
The mass spectrometer was used in positive ion mode with ion spray voltage 1200-1300 V, curtain gas 0-10, nebulizer gas 0-10, interface heater temperature 110 °C, and collision gas Medium. Q1 and Q3 resolutions were Unit/Unit, the settling time was 0 milliseconds, and the pause between mass ranges was 5.007 milliseconds. CE was set by Skyline as described previously,4,5 DP was set to 90, EP was set to 10, and CXP was set to 10. Scheduled MRMs used a 150 second detection window and a target cycle time of 1.5 seconds.
MRM mass spectrometry data analysis
Full details of the data analysis are provided in the Supplemental Methods. Briefly, MRM peak integration was performed by Skyline,60 and the integrations were manually inspected to ensure correct peak detection, absence of interferences, and accurate integration. Specificity was confirmed by equivalent retention time and relative areas of light and heavy transitions. Peptide concentrations are calculated as the peak area ratio of the sum of all transitions with no interferences multiplied by the concentration of isotope-labeled SIS peptide analog. Precision was determined by measuring the coefficient of variation (CV, standard deviation divided by the mean) and expressed as a percentage.
Public access to mass spectrometry data
The LC-MS/MS data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD003770 (username: reviewer00908@ebi.ac.uk; password: JzwRamIg). All MRM data (LC-MRM method optimization, LC-MRM and immuno-MRM FFPE-frozen comparison and response curves) are available at https://panoramaweb.org/labkey/FFPE_MRM_optimization.url (sign in with username:panorama+paulovich@proteinms.net; password: awH7@"U>).
RESULTS AND DISCUSSION
Overview of Study
The goal of this study was to identify an optimized process supporting analytically robust (e.g. sensitive, precise, reproducible) MRM-based proteomic quantification from archival, FFPE tissues, using flash frozen tissue as the gold standard comparator. We used a test battery approach61 (Figure 1A) to evaluate 9 processes representing all possible combinations of 3 previously described protocols for protein extraction (Table 1), coupled with 3 previously described protocols for trypsin digestion (Table 2) of lysates from FFPE tissues.29,41,51,62 To standardize the approach and comparison, each protocol was adapted to extracting and analyzing tissue sections mounted on slides. To assess the generalizability of the processes, we used shotgun mass spectrometry in addition to a large panel of 512 MRM assays that we previously characterized in breast cancer cell lines.35 Additionally, quantitative results obtained in FFPE samples were compared to identical measurements made in matched flash frozen tissues (i.e. gold-standard) (Figure 1B). To assess peptide antigen retrieval, the optimized process was also tested for performance with a 42-plex immuno-MRM assay, with results being directly compared to MRM-based measurements for the identical analytes.
Figure 1. Test battery approach for finding the optimal process for protein extraction and digestion from FFPE.

A. A test battery approach for evaluation of FFPE processing protocols using an ER+/HER2+ breast cancer specimen cut in 10 μm sections. Each combination of protein extraction and protein digestion protocols was performed in triplicate (using a single process replicate from 3 separate days, yielding 27 total samples). Performance of the protocols was evaluated by LC-MS/MS ‘shotgun’ analysis and LC-MRM targeted analysis. B. Performance of the method was compared in analysis of FFPE and frozen tissue prepared from a single specimen. Alternating sections were used for fixation or freshly frozen. Direct LC-MRM and immuno-MRM assays were applied for quantitative analysis of targeted peptides.
Table 1.
Comparison of protocols evaluated for protein extraction and extraction from FFPE tissue.
| Procedural Step | Methoda |
||
|---|---|---|---|
| TFE / Ammonium bicarbonate |
RapiGest / Ammonium bicarbonate |
Liquid Tissue | |
| extraction buffer | 100 mM NH4HCO3 |
0.2% RapiGest in 50 mM NH4HCO3 |
Liquid Tissue |
| incubation temperature |
80°C | 95°C | 95°C |
| incubation time | 120 min | 30 min | 90 min |
| additional buffer | TFE | ||
| sonication | 30 sec | 30 sec | |
| incubation temperature |
60°C | 80°C | |
| incubation time | 60 min | 120 min | |
| additional buffer | 50 mM NH4HCO3 | ||
| sonication | 30 sec | 30 sec | |
| centrifuge | 10000 rcf 1min | ||
| additional buffer | 50 mM NH4HCO3 | ||
| storage | −80°C | −80°C | −80°C |
Table 2.
Comparison of protocols evaluated for protein digestion from FFPE tissue.
| Methoda |
|||
|---|---|---|---|
| Procedural Step | Urea | TFE | RapiGest |
| denaturing buffer |
6M Urea in 50 mM NH4HCO3 |
50% TFE in 50 mM NH4HCO3 |
0.1% RaipGest in 50 mM NH4HCO3 |
| reducing agent | 15 mM TCEP | 15 mM TCEP | 15 mM TCEP |
| alkylation agent | 40 mM IAM | 40 mM IAM | 40 mM IAM |
| dilution buffer | 200 mM TRIS | 50 mM NH4HCO3 | 50 mM NH4HCO3 |
| trypsin ratio | 1:50, followed by 1:100 |
1:50, followed by 1:100 |
1:50, followed by 1:100 |
| temperature | 37°C | 37°C | 37°C |
| duration | overnight | overnight | overnight |
Optimization of FFPE sample processing
We first assessed the performances of all 9 processes representing combinations of protein extraction and trypsin digestion using a test battery approach61 (Figure 1A). Of note, extraction and proteolysis protocols were tested together in pairwise combinations (i.e. “processes”) because protocols for extraction may have an effect on digestion efficiency,62 and we sought to identify the combination that achieves the most reproducible recovery of peptides. Each process was evaluated in triplicate (i.e. one complete process replicate on each of three separate days) on a single FFPE-treated, ER+/Her2+ breast cancer biospecimen. The input for each replicate consisted of three 10 micron tissue sections, mounted on slides. Individual sections were systematically distributed across the replicates to minimize the variability associated with tissue microheterogeneity (see Supplemental Figure S1).
Performances of all nine processes were evaluated with respect to the following metrics: i) protein recovery, ii) digestion efficiency/fidelity, and iii) sensitivity, precision, and repeatability of MRM-based measurements. Protein recovery was measured by the micro bicinchoninic acid (BCA) assay (Figure 2A). The RapiGest/ammonium bicarbonate method extracted the most protein, averaging 268 μg per sample, while methods using TFE and Liquid Tissue extracted averages of 50 and 43 μg per sample, respectively. Note the low yields achieved for the TFE and Liquid Tissue protocols were lower than previously reported.46,51 It is possible that extraction from slide-mounted tissue differs from extraction performed in tubes.
Figure 2. Evaluation of processes for quantitative proteomics on FFPE.

Results are from the analysis of an FFPE, ER+/HER2+ breast cancer. Each sample consists of three 10 μm sections. All error bars reflect the standard deviation of three analytical process replicates (i.e. de-paraffinization, protein extraction, digestion, enrichment, analysis performed on three separate days). A. Protein yield measured by micro-BCA assay. B. Unique peptide identifications obtained from shotgun LC-MS/MS results. Equal amount of protein (2 μg) was loaded on-column for each process. C. Trypsin digestion efficiency and fidelity measured by the percentage of identifications containing missed cleavages (internal K or R) or mis-cleavages (non-tryptic cut site). D. LC-MRM results showing the number of observable peptides (>LOD in 2 out of 3 replicates). Heavy peptide standards were added to1 μg of the digested lysates at 10 fmol/μg protein and analyzed in process triplicate. E. LC-MRM results showing the distribution of CVs for observable peptides. F. Distribution of peptide amounts measured by LC-MRM. Box plots show the median (bar), inner quartiles (box), 5-95th percentiles (line), and outliers (points).
The different amounts of extracted protein were normalized to 1.0 μg/μL prior to MS analysis to allow comparison of the analytical results from the different processes. To test trypsin digestion efficiency and fidelity, two microgram aliquots of each individual digest were analyzed by ‘shotgun’ LC-MS/MS. Figure 2B shows the number of unique peptides identified. Overall, there was a large range (643 - 2721 unique peptides) in identifications amongst the protocols. RapiGest protein extraction produced a significantly higher number of identifications (2397 on average; p-value < 0.001). Within each extraction protocol, TFE digestion produced the highest number of identifications. The percentages of missed cleavages (i.e. peptides containing an internal Arg or Lys residue) and mis-cleavages (i.e. non-tryptic cuts) in the LC-MS/MS results were used as indicators of trypsin digestion efficiency and fidelity (Figure 2C). Overall, RapiGest extraction methods performed significantly better than the TFE and Liquid Tissue extraction methods, with an average of 3.3% missed cleavages (p-value = 0.022 and p-value < 0.001 for TFE, Liquid Tissue comparisons) and 10.5% mis-cleavages (p values < 0.001 for both comparisons) for the digestion protocols studied (Figure 2C).
Quantitative, MRM mass spectrometry with the addition of stable isotope labeled standard peptides was used to evaluate the sensitivity, precision, and repeatability of the nine processes, targeting 512 peptide analytes using multiplexed assays that we previously characterized in breast cancer-related cell lines.35 Each process was run in triplicate (i.e. one complete process replicate on each of three separate days). The number of peptides with observable endogenous signal (i.e., detected above LOD in at least 2 out of 3 replicates) are shown in Figure 2D, and the CV of those measurements are shown in Figure 2E. Differences in the number of peptides detected in cell lines and tissue are likely the result of differences in expression levels and dilution of the epithelial component in tissue due to tissue heterogeneity. RapiGest followed by urea digest provided the highest number of detectable peptides (117 peptides measured at endogenous levels). The precision of the RapiGest/Urea and RapiGest/TFE processes were very similar, with median CVs for urea of 4.1% (ranging <1% to 50%) and median CVs for TFE of 4.0% (ranging <1% to 51%).
We also examined the recovery of endogenous peptides in the quantitative MRM experiment. Because the heavy peptides were added following digestion, differences in the amount of endogenous peptides measured by the nine processes are indicative of the relative recovery of the peptides. There were 27 peptides with measurable endogenous signal detected in all nine processes, and the amount of endogenous peptide measured for these analytes is plotted in Figure 2F. Overall, the same pattern as seen in the total number of observed peptides is evident, namely the RapiGest/Urea process had the highest recovery and lowest CV.
Based on these data, the RapiGest protein extraction buffer outperformed the others in overall protein yield, shotgun MS-based metrics (total peptide identifications and trypsin performance), and targeted MS-based metrics (peptide yield, recovery, and quantitative precision). While the combination of RapiGest buffer extraction and TFE as a denaturant for trypsin digestion provided the most identifications by shotgun MS/MS, the combined quantitative performance (i.e. number of peptides and precision of the measurements) for the RapiGest/Urea process was superior. For these reasons, we selected the RapiGest/Urea process as the optimum FFPE sample preparation method. (We note that since the majority of the targeted peptides are from soluble proteins, further work would need to be done to optimize a protocol for membrane proteins).
Analytical performances of MRM and immuno-MRM assays in FFPE tissue (using the RapiGest/Urea process) compared with matched frozen tissue
We next compared the analytical performances of MRM-based assays run on optimally prepared flash frozen tissues to the performances of the same assays run on the corresponding FFPE samples using the RapiGest/Urea process. We used two multiplexed assay panels, one direct LC-MRM (i.e., run on neat cell lysate, without enrichment) and one immuno-MRM assay. The direct LC-MRM assay targets 512 peptides over four multiplex assay groups.35 The immuno-MRM assay utilizes 42 monoclonal antibody-based assays targeting cancer-related targets.41,56 Response curves were generated in background matrices of FFPE and matching fresh frozen tissue using a pooled sample from three individual ER+/Her2+ breast cancers. (Matching frozen and FFPE tissues were used to characterize the performance of the assays, using the best possible matrix to characterize potential interferences or suppression effects.) Care was taken to minimize microheterogeneity of the input samples by using adjacent sections of tissue for frozen and fixed specimens. Response curves were used to characterize the linear range, limit of detection (LOD), upper and lower limits of quantification (ULOQ & LLOQ), and precision (CV) for comparison across the two sample types (frozen vs FFPE).
The performances of the assays are very similar in the matrices tested. Example response curves in FFPE and frozen tissue background matrix are shown in Figure 3A and 3B for the peptide GLQSLPTHDPSPLQR from the Her2 protein (response curves for all analytes with detectable endogenous signal are plotted individually in Supplemental Figure S2, and performance figures of merit are reported in Supplemental Table S1). As shown in Figure 3A and 3B, curves measured in frozen and fixed tissue are nearly overlaid. Each of the assays shows good linearity (>3 orders of magnitude) and equivalent response in the two matrices. (Differences in the FFPE and frozen curves are likely the result of small variations in the amount of endogenous light peptide detected in the samples. Best efforts were taken to provide equal input for the frozen and fixed specimens, but because of microheterogeneity in the tumor tissue, it is not possible to obtain identical specimens for comparison.)
Figure 3. Characterization of assays in frozen and FFPE matrices.
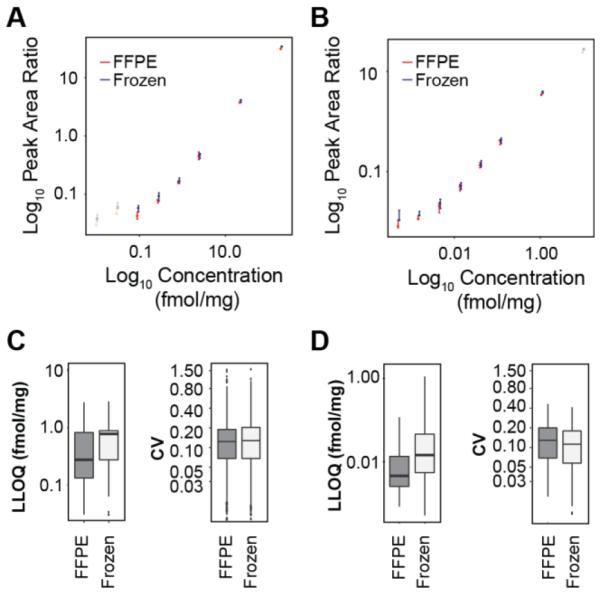
LC-MRM and immuno-MRM assays were characterized by reverse response curves in frozen or FFPE tissue extracts. A. Example response curves for the peptide GLQSLPTHDPSPLQR (ERBB2) measured by LC-MRM. Responses measured in FFPE and frozen tissues are overlaid, and curves are plotted on a log10 scale. Error bars are the standard deviation of complete process triplicate measurements. B. Example response curves for the peptide GLQSLPTHDPSPLQR (ERBB2) measured by immuno-MRM. Responses measured in FFPE and frozen tissues are overlaid, and curves are plotted on a log10 scale. Error bars are the standard deviation of complete process triplicate measurements. C. Distribution of LLOQs and CVs measured in FFPE and frozen tissue for all analytes detected by LC-MRM. D. Distribution of LLOQs and CVs measured in FFPE and frozen tissues for all analytes detected by immuno-MRM. Box plots show the median (bar), inner quartiles (box), 5-95th percentiles (line), and outliers (points).
Overall, performance of the assays is conserved in FFPE tissue compared with frozen tissue. 227 LC-MRM assays were characterized with endogenous signal in both frozen and FFPE samples, with a median LLOQ of 0.78 fmol/μg and median CV of 12.1% (ranging <1% to 160%) in frozen curves and median LLOQ of 0.28 fmol/μg and median CV of 11.3% (ranging <1% to 157%) in FFPE curves (Figure 3C). There were 31 immuno-MRM assays characterized in both FFPE and frozen tissue, showing effective retrieval and preservation of the epitope. Again, the assay performances for immuno-MRM assays were comparable, with median LLOQ of 0.014 fmol/μg and median CV of 16.9% (ranging <1% to 43%) in frozen curves and median LLOQ of 0.0046 fmol/μg and median CV of 11.6% (ranging <1% to 48%) in FFPE curves (Figure 3D). The similarity of analytical characteristics indicates the assays can be consistently applied in either matrix with similar performance. Furthermore, the LLOQs of immuno-MRM demonstrate the ability to analyze low abundance targets from FFPE.
Comparison of quantitative MRM measurements in matched FFPE and frozen breast cancer tumor tissue samples
We next asked if analyte concentrations measured in FFPE tissues are consistent with measurements of the same analytes from matched, frozen specimens. Three individual ER+/Her2+ breast cancer tissues were collected and analyzed in process triplicate (i.e. one process on each of three separate days), using both the multiplex direct MRM35 and immuno-MRM63 assays described above. To minimize variability due to heterogeneity of the sample, we alternated pieces of the tissue prepared as FFPE samples or fresh frozen, and evenly distributed three 10 μm tissue sections from each sample across the assay replicates (see also Supplemental Figure S3). FFPE samples were extracted by the RapiGest/urea protocol, and frozen samples were processed using a previously reported urea-based method.41
Protein recovery was measured by BCA assay, and sensitivity and repeatability of the direct LC-MRM and immuno-MRM multiplex assays were compared in the matched FFPE and frozen biospecimens. While there is a significant difference in the amount of protein extracted in each of the three individual samples (due to differences in the size of the individual tumor samples and the heterogeneity of tissue), the protein recovery was comparable between the FFPE and frozen samples for a given sample in all three individual tumor samples (p-value > 0.04 in all three samples) (Figure 4A). Previous reports suggested that incomplete reversal of protein cross-linking in fixed tissues could lead to depressed levels of protein measured by BCA, due to the unavailability of amino acids for the BCA reaction.46 However, the comparability of measured protein in our results indicates high reversal of cross-links and efficient extraction of protein from the FFPE samples. Estimation of the extraction yield indicates similar yields in both tissue types. Tumor tissue sample #3 had a mass of 88 mg, of which 60% was FFPE-treated and 40% was frozen. Three 10 μm sections from this sample would contain roughly 0.60 mg wet tissue weight, (0.36 mg FFPE-treated and 0.24 mg frozen). Based on the average protein recovery reported in Figure 4A, we achieved an approximate extraction yield of 14% for both the FFPE and frozen samples.
Figure 4. Comparison of protein quantification in matched frozen and FFPE biospecimens.

Three individual breast cancer tissues were sectioned and analyzed by multiplexed LC-MRM (targeting 512 peptide analytes) and immuno-MRM (targeting 42 peptide analytes) assays. Each sample was analyzed in process triplicate, and each replicate consisted of extraction, digestion, and enrichment performed on three separate days. A. Protein recovery from matched FFPE and frozen samples measured by BCA assay. B. Overlap in number of peptides detected in quantitative LC-MRM and immuno-MRM assays in matched FFPE and frozen tissue specimens. C. Scatter plot of endogenous peptide concentrations (n= 356) determined by MRM measurements made in the matched FFPE and frozen tissue. D. Repeatability (CV) of immuno-MRM and LC-MRM assays applied to FFPE and frozen tissue, each box plot represents three samples measured on three days (n = 9). E. Scatter plot of endogenous peptides (n= 49) with overlapping immuno-MRM and LC-MRM measurements in FFPE (yellow) and frozen (blue) tissues. Error bars are the standard deviation of triplicate measurements.
We next compared the endogenous peptide concentrations determined in FFPE and fresh frozen tissue. In total, 249 peptides were quantified in all of the samples (236 by direct LC-MRM and 23 by immuno-MRM), with a high overlap of about two-thirds of peptides quantified in both tissues (Figure 4B), indicating good overall sensitivity for peptide detection. The correlation of peptide concentrations measured in the FFPE and frozen samples is plotted in Figure 4C. Overall, there is very good correlation for the LC-MRM (R2 = 0.94) and immuno-MRM (R2 = 0.89) assays, supporting the preservation of peptide quantification in FFPE. As expected, there are more low abundance targets detected following enrichment by immuno-MRM. Also, based on previous results64, it is expected that increasing the input amount will further enable analysis of very low abundance species by immuno-MRM.
To examine if any potential bias exists in peptide retrieval as a function of amino acid content, we separated results for lysine- and arginine- containing peptides. Correlation of these subsets of peptides (correlation plots are shown in Supplemental Figure S4) show equivalent quantities in the fresh and fixed specimens, further supporting robust quantification for tryptic peptides using the optimized protocol.
Precision of the assays was also equivalent in the two sample types. The distribution of CVs for the targeted assays are plotted in Figure 4D. Median CVs for immuno-MRM (11.6% in FFPE versus 16.6% in frozen) and LC-MRM (11.3% in FFPE versus 12.0% in frozen) are comparable for the assays measured in the matched FFPE and frozen samples, demonstrating that the assays are highly reproducible for measurements made across different days.
To determine whether the optimized FFPE processing method affected the immunoaffinity enrichment step of the immuno-MRM assays, we compared the measurements of 10 peptides whose endogenous levels were quantified by both LC-MRM and immuno-MRM. Figure 4E shows a very high correlation (R2 > 0.98) of the measurements for the overlapping peptides between the two sample types.
The results demonstrate robust quantification of protein in FFPE (compared with matched frozen tissues) using the optimized process coupled with MRM. This establishes a standardized protocol for protein quantification by MRM in fixed tissues, supporting the ability to investigate large numbers of analytes in retrospective studies using the multiplexed MRM technology. While the results clearly demonstrate precise, relative quantification, the measured concentration of any peptide may not accurately reflect the amount of endogenous protein present in the undigested sample, since protein extraction and trypsin digestion may not quantitatively recover all proteins and peptides equally. The presence of intact protein isoforms or post-translational modifications near cleavage sites can affect the inferred protein quantitation, and incomplete cross-link reversal can adversely affect trypsin digestion by blocking access to cleavage sites, affecting recovery. Despite these caveats, the data show that the optimized protocol is capable of precisely quantifying reproducibly recovered peptides, and that equivalent quantification can be achieved in FFPE samples compared to fresh frozen specimens.
CONCLUSION
The optimized process presented herein enables robust, MRM-based quantification of peptide analytes from FFPE biospecimens, comparable to quantification performed in matched fresh frozen tissues. This enables large numbers of biomarker candidates to be quantified in multiplex in archival sample sets. The ability to perform retrospective analyses on archived samples with clinical annotation will significantly improve understanding of disease processes and help usher in personalized medicine through novel tissue-based protein biomarkers.
Supplementary Material
ACKNOWLEDGMENT
Research was supported by the National Cancer Institute of the National Institutes of Health (R33CA173300) and the Clinical Proteomics Tumor Analysis Consortium Initiative (U24CA160034). NW BioTrust and NWBioSpecimen services are supported by the FHCRC/UW Cancer Consortium Cancer Center Support Grant P30 CA015704 (D. Gilliland, principal investigator) from the National Institutes of Health (NIH) National Cancer Center, State of Washington Life Sciences Discovery Fund (LSDF) Washington Phenotyped Biospecimen Resource grant (J. Slattery, principal investigator), LSDF Consortium Biospecimen Program grant (P. Porter, principal investigator), Fred Hutchinson Cancer Research Center, the University of Washington School of Medicine, the University of Washington Department of Pathology, and the Institute of Translational Health Sciences which is supported by the Clinical and Translational Science Awards Program grant UL1 TR000423 (M. Disis, principal investigator) from the NIH National Center for Advancing Translational Sciences. Histology and Image Analysis was done by Fred Hutchinson Experimental Histopathology Shared Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- FFPE
formalin fixed paraffin embedded
- BCA
bicinchoninic acid
- MRM
multiple reaction monitoring
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no conflict of interest.
REFERENCES
- 1.Khleif SN, Doroshow JH, Hait WN, AACR-FDA-NCI Cancer Biomarkers Collaborative AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010;16(13):3299–3318. doi: 10.1158/1078-0432.CCR-10-0880. [DOI] [PubMed] [Google Scholar]
- 2.La Thangue NB, Kerr DJ. Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat. Rev. Clin. Oncol. 2011;8(10):587–596. doi: 10.1038/nrclinonc.2011.121. [DOI] [PubMed] [Google Scholar]
- 3.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 4.Paulovich AG, Whiteaker JR, Hoofnagle AN, Wang P. The interface between biomarker discovery and clinical validation: The tar pit of the protein biomarker pipeline. PROTEOMICS Clin. Appl. 2008;2(10–11):1386–1402. doi: 10.1002/prca.200780174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, Yan P, Schoenherr RM, Zhao L, Voytovich UJ, et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 2011;29(7):625–634. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anagnostou VK, Dimou AT, Botsis T, Killiam EJ, Gustavson MD, Homer RJ, Boffa D, Zolota V, Dougenis D, Tanoue L, et al. Molecular classification of nonsmall cell lung cancer using a 4-protein quantitative assay. Cancer. 2012;118(6):1607–1618. doi: 10.1002/cncr.26450. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein NS, Ferkowicz M, Odish E, Mani A, Hastah F. Minimum formalin fixation time for consistent estrogen receptor immunohistochemical staining of invasive breast carcinoma. Am. J. Clin. Pathol. 2003;120(1):86–92. doi: 10.1309/QPHD-RB00-QXGM-UQ9N. [DOI] [PubMed] [Google Scholar]
- 8.Werner M, Chott A, Fabiano A, Battifora H. Effect of formalin tissue fixation and processing on immunohistochemistry. Am. J. Surg. Pathol. 2000;24(7):1016–1019. doi: 10.1097/00000478-200007000-00014. [DOI] [PubMed] [Google Scholar]
- 9.Sauter G, Lee J, Bartlett JMS, Slamon DJ, Press MF. Guidelines for human epidermal growth factor receptor 2 testing: biologic and methodologic considerations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009;27(8):1323–1333. doi: 10.1200/JCO.2007.14.8197. [DOI] [PubMed] [Google Scholar]
- 10.Taylor CR. Standardization in immunohistochemistry: the role of antigen retrieval in molecular morphology. Biotech. Histochem. Off. Publ. Biol. Stain Comm. 2006;81(1):3–12. doi: 10.1080/10520290600667866. [DOI] [PubMed] [Google Scholar]
- 11.Vani K, Sompuram SR, Fitzgibbons P, Bogen SA. National HER2 proficiency test results using standardized quantitative controls: characterization of laboratory failures. Arch. Pathol. Lab. Med. 2008;132(2):211–216. doi: 10.5858/2008-132-211-NHPTRU. [DOI] [PubMed] [Google Scholar]
- 12.Press M, Spaulding B, Groshen S, Kaminsky D, Hagerty M, Sherman L, Christensen K, Edwards DP. Comparison of different antibodies for detection of progesterone receptor in breast cancer. Steroids. 2002;67(9):799–813. doi: 10.1016/s0039-128x(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 13.Moeder CB, Giltnane JM, Harigopal M, Molinaro A, Robinson A, Gelmon K, Huntsman D, Camp RL, Rimm DL, American Society of Clinical Oncology et al. Quantitative justification of the change from 10% to 30% for human epidermal growth factor receptor 2 scoring in the American Society of Clinical Oncology/College of American Pathologists guidelines: tumor heterogeneity in breast cancer and its implications for tissue microarray based assessment of outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007;25(34):5418–5425. doi: 10.1200/JCO.2007.12.8033. [DOI] [PubMed] [Google Scholar]
- 14.Cregger M, Berger AJ, Rimm DL. Immunohistochemistry and quantitative analysis of protein expression. Arch. Pathol. Lab. Med. 2006;130(7):1026–1030. doi: 10.5858/2006-130-1026-IAQAOP. [DOI] [PubMed] [Google Scholar]
- 15.Blackler AR, Morgan NY, Gao B, Olano LR, Armani MD, Romantseva E, Kakareka JW, Bonner RF, Mukherjee S, Xiao B, et al. Proteomic analysis of nuclei dissected from fixed rat brain tissue using expression microdissection. Anal. Chem. 2013;85(15):7139–7145. doi: 10.1021/ac400691k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeSouza LV, Krakovska O, Darfler MM, Krizman DB, Romaschin AD, Colgan TJ, Siu KWM. mTRAQ-based quantification of potential endometrial carcinoma biomarkers from archived formalin-fixed paraffin-embedded tissues. Proteomics. 2010;10(17):3108–3116. doi: 10.1002/pmic.201000082. [DOI] [PubMed] [Google Scholar]
- 17.Fu Z, Yan K, Rosenberg A, Jin Z, Crain B, Athas G, Heide RSV, Howard T, Everett AD, Herrington D, et al. Improved protein extraction and protein identification from archival formalin-fixed paraffin-embedded human aortas. Proteomics Clin. Appl. 2013;7(3–4):217–224. doi: 10.1002/prca.201200064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gámez-Pozo A, Sánchez-Navarro I, Calvo E, Díaz E, Miguel-Martín M, López R, Agulló T, Camafeita E, Espinosa E, López JA, et al. Protein phosphorylation analysis in archival clinical cancer samples by shotgun and targeted proteomics approaches. Mol. Biosyst. 2011;7(8):2368–2374. doi: 10.1039/c1mb05113j. [DOI] [PubMed] [Google Scholar]
- 19.Güzel C, Ursem NTC, Dekker LJ, Derkx P, Joore J, van Dijk E, Ligtvoet G, Steegers EAP, Luider TM. Multiple reaction monitoring assay for pre-eclampsia related calcyclin peptides in formalin fixed paraffin embedded placenta. J. Proteome Res. 2011;10(7):3274–3282. doi: 10.1021/pr1010795. [DOI] [PubMed] [Google Scholar]
- 20.Hembrough T, Thyparambil S, Liao W-L, Darfler MM, Abdo J, Bengali KM, Taylor P, Tong J, Lara-Guerra H, Waddell TK, et al. Selected Reaction Monitoring (SRM) Analysis of Epidermal Growth Factor Receptor (EGFR) in Formalin Fixed Tumor Tissue. Clin. Proteomics. 2012;9(1):5. doi: 10.1186/1559-0275-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hembrough T, Thyparambil S, Liao W-L, Darfler MM, Abdo J, Bengali KM, Hewitt SM, Bender RA, Krizman DB, Burrows J. Application of selected reaction monitoring for multiplex quantification of clinically validated biomarkers in formalin-fixed, paraffin-embedded tumor tissue. J. Mol. Diagn. JMD. 2013;15(4):454–465. doi: 10.1016/j.jmoldx.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Hembrough T, Liao W-L, Hartley CP, Ma PC, Velcheti V, Lanigan C, Thyparambil S, An E, Monga M, Krizman D, et al. Quantification of Anaplastic Lymphoma Kinase Protein Expression in Non-Small Cell Lung Cancer Tissues from Patients Treated with Crizotinib. Clin. Chem. 2016;62(1):252–261. doi: 10.1373/clinchem.2015.245860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nuciforo P, Thyparambil S, Aura C, Garrido-Castro A, Vilaro M, Peg V, Jimenez J, Vicario R, Cecchi F, Hoos W, et al. High HER2 protein levels correlate with increased survival in breast cancer patients treated with anti-HER2 therapy. Mol. Oncol. 2016;10(1):138–147. doi: 10.1016/j.molonc.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catenacci DVT, Liao W-L, Thyparambil S, Henderson L, Xu P, Zhao L, Rambo B, Hart J, Xiao S-Y, Bengali K, et al. Absolute quantitation of Met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PloS One. 2014;9(7):e100586. doi: 10.1371/journal.pone.0100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang S-I, Thumar J, Lundgren DH, Rezaul K, Mayya V, Wu L, Eng J, Wright ME, Han DK. Direct cancer tissue proteomics: a method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene. 2007;26(1):65–76. doi: 10.1038/sj.onc.1209755. [DOI] [PubMed] [Google Scholar]
- 26.Myers MV, Manning HC, Coffey RJ, Liebler DC. Protein expression signatures for inhibition of epidermal growth factor receptor-mediated signaling. Mol. Cell. Proteomics MCP. 2012;11(2):M111.015222. doi: 10.1074/mcp.M111.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimura T, Nomura M, Tojo H, Hamasaki H, Fukuda T, Fujii K, Mikami S, Bando Y, Kato H. Proteomic analysis of laser-microdissected paraffin-embedded tissues: (2) MRM assay for stage-related proteins upon non-metastatic lung adenocarcinoma. J. Proteomics. 2010;73(6):1100–1110. doi: 10.1016/j.jprot.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Takadate T, Onogawa T, Fukuda T, Motoi F, Suzuki T, Fujii K, Kihara M, Mikami S, Bando Y, Maeda S, et al. Novel prognostic protein markers of resectable pancreatic cancer identified by coupled shotgun and targeted proteomics using formalin-fixed paraffin-embedded tissues. Int. J. Cancer J. Int. Cancer. 2013;132(6):1368–1382. doi: 10.1002/ijc.27797. [DOI] [PubMed] [Google Scholar]
- 29.Sprung RW, Martinez MA, Carpenter KL, Ham A-JL, Washington MK, Arteaga CL, Sanders ME, Liebler DC. Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed, paraffin-embedded tissue. J. Proteome Res. 2012;11(6):3498–3505. doi: 10.1021/pr300130t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steiner C, Tille J-C, Lamerz J, Kux van Geijtenbeek S, McKee TA, Venturi M, Rubbia-Brandt L, Hochstrasser D, Cutler P, Lescuyer P, et al. Quantification of HER2 by Targeted Mass Spectrometry in Formalin-Fixed Paraffin-Embedded (FFPE) Breast Cancer Tissues. Mol. Cell. Proteomics MCP. 2015;14(10):2786–2799. doi: 10.1074/mcp.O115.049049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Want EJ, Cravatt BF, Siuzdak G. The expanding role of mass spectrometry in metabolite profiling and characterization. Chembiochem. 2005;6(11):1941–1951. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 32.Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38(4):296–309. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 33.Netzel BC, Grant RP, Hoofnagle AN, Rockwood AL, Shuford CM, Grebe SKG. First Steps toward Harmonization of LC-MS/MS Thyroglobulin Assays. Clin. Chem. 2016;62(1):297–299. doi: 10.1373/clinchem.2015.245266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoofnagle AN, Whiteaker JR, Carr SA, Kuhn E, Liu T, Massoni SA, Thomas SN, Townsend RR, Zimmerman LJ, Boja E, et al. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin. Chem. 2016;62(1):48–69. doi: 10.1373/clinchem.2015.250563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, Kim JS, Zhang Y, Wang X, Ivey RG, et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods. 2014;11(2):149–155. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009;27(7):633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prakash A, Rezai T, Krastins B, Sarracino D, Athanas M, Russo P, Zhang H, Tian Y, Li Y, Kulasingam V, et al. Interlaboratory reproducibility of selective reaction monitoring assays using multiple upfront analyte enrichment strategies. J. Proteome Res. 2012;11(8):3986–3995. doi: 10.1021/pr300014s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, et al. Large-Scale Interlaboratory Study to Develop, Analytically Validate and Apply Highly Multiplexed, Quantitative Peptide Assays to Measure Cancer-Relevant Proteins in Plasma. Mol. Cell. Proteomics MCP. 2015;14(9):2357–2374. doi: 10.1074/mcp.M114.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3(2):235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 40.Whiteaker JR, Zhao L, Yan P, Ivey RG, Voytovich UJ, Moore HD, Lin C, Paulovich AG. Peptide Immunoaffinity Enrichment and Targeted Mass Spectrometry Enables Multiplex, Quantitative Pharmacodynamic Studies of Phospho-Signaling. Mol. Cell. Proteomics MCP. 2015;14(8):2261–2273. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoenherr RM, Whiteaker JR, Zhao L, Ivey RG, Trute M, Kennedy J, Voytovich UJ, Yan P, Lin C, Paulovich AG. Multiplexed quantification of estrogen receptor and HER2/Neu in tissue and cell lysates by peptide immunoaffinity enrichment mass spectrometry. Proteomics. 2012;12(8):1253–1260. doi: 10.1002/pmic.201100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Razavi M, Frick LE, LaMarr WA, Pope ME, Miller CA, Anderson NL, Pearson TW. High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J. Proteome Res. 2012;11(12):5642–5649. doi: 10.1021/pr300652v. [DOI] [PubMed] [Google Scholar]
- 43.Berna M, Ott L, Engle S, Watson D, Solter P, Ackermann B. Quantification of NTproBNP in rat serum using immunoprecipitation and LC/MS/MS: a biomarker of drug-induced cardiac hypertrophy. Anal. Chem. 2008;80(3):561–566. doi: 10.1021/ac702311m. [DOI] [PubMed] [Google Scholar]
- 44.Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, et al. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol. Cell. Proteomics MCP. 2012;11(6):M111.013854. doi: 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoofnagle AN, Becker JO, Wener MH, Heinecke JW. Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin Chem. 2008;54(11):1796–1804. doi: 10.1373/clinchem.2008.109652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sprung RW, Brock JWC, Tanksley JP, Li M, Washington MK, Slebos RJC, Liebler DC. Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell. Proteomics MCP. 2009;8(8):1988–1998. doi: 10.1074/mcp.M800518-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hood BL, Darfler MM, Guiel TG, Furusato B, Lucas DA, Ringeisen BR, Sesterhenn IA, Conrads TP, Veenstra TD, Krizman DB. Proteomic analysis of formalin-fixed prostate cancer tissue. Mol. Cell. Proteomics MCP. 2005;4(11):1741–1753. doi: 10.1074/mcp.M500102-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Crockett DK, Lin Z, Vaughn CP, Lim MS, Elenitoba-Johnson KSJ. Identification of proteins from formalin-fixed paraffin-embedded cells by LC-MS/MS. Lab. Investig. J. Tech. Methods Pathol. 2005;85(11):1405–1415. doi: 10.1038/labinvest.3700343. [DOI] [PubMed] [Google Scholar]
- 49.Palmer-Toy DE, Krastins B, Sarracino DA, Nadol JB, Merchant SN. Efficient method for the proteomic analysis of fixed and embedded tissues. J. Proteome Res. 2005;4(6):2404–2411. doi: 10.1021/pr050208p. [DOI] [PubMed] [Google Scholar]
- 50.Xu H, Yang L, Wang W, Shi S-R, Liu C, Liu Y, Fang X, Taylor CR, Lee CS, Balgley BM. Antigen retrieval for proteomic characterization of formalin-fixed and paraffin-embedded tissues. J. Proteome Res. 2008;7(3):1098–1108. doi: 10.1021/pr7006768. [DOI] [PubMed] [Google Scholar]
- 51.Prieto DA, Hood BL, Darfler MM, Guiel TG, Lucas DA, Conrads TP, Veenstra TD, Krizman DB. Liquid Tissue: proteomic profiling of formalin-fixed tissues. BioTechniques. 2005;Suppl:32–35. doi: 10.2144/05386su06. [DOI] [PubMed] [Google Scholar]
- 52.Groseclose MR, Massion PP, Chaurand P, Caprioli RM. High-throughput proteomic analysis of formalin-fixed paraffin-embedded tissue microarrays using MALDI imaging mass spectrometry. Proteomics. 2008;8(18):3715–3724. doi: 10.1002/pmic.200800495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang S-I, Thumar J, Lundgren DH, Rezaul K, Mayya V, Wu L, Eng J, Wright ME, Han DK. Direct cancer tissue proteomics: a method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene. 2007;26(1):65–76. doi: 10.1038/sj.onc.1209755. [DOI] [PubMed] [Google Scholar]
- 54.Gustafsson OJR, Arentz G, Hoffmann P. Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta. 2015;1854(6):559–580. doi: 10.1016/j.bbapap.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 55.Steiner C, Ducret A, Tille J-C, Thomas M, McKee TA, Rubbia-Brandt L, Scherl A, Lescuyer P, Cutler P. Applications of mass spectrometry for quantitative protein analysis in formalin-fixed paraffin-embedded tissues. Proteomics. 2014;14(4–5):441–451. doi: 10.1002/pmic.201300311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schoenherr RM, Saul RG, Whiteaker JR, Yan P, Whiteley GR, Paulovich AG. Anti-peptide monoclonal antibodies generated for immuno-MRM assays have a high probability of supporting Western blot and ELISA. Mol. Cell. Proteomics MCP. 2014 doi: 10.1074/mcp.O114.043133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schoenherr RM, Zhao L, Whiteaker JR, Feng LC, Li L, Liu L, Liu X, Paulovich AG. Automated screening of monoclonal antibodies for SISCAPA assays using a magnetic bead processor and liquid chromatography-selected reaction monitoring-mass spectrometry. J. Immunol. Methods. 2010;353(1–2):49–61. doi: 10.1016/j.jim.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolff AC, Hammond MEH, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 2007;131(1):18–43. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- 59.Hammond MEH, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010;28(16):2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinforma. Oxf. Engl. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi SR, Cote RJ, Yang C, Chen C, Xu HJ, Benedict WF, Taylor CR. Development of an optimal protocol for antigen retrieval: a “test battery” approach exemplified with reference to the staining of retinoblastoma protein (pRB) in formalin-fixed paraffin sections. J. Pathol. 1996;179(3):347–352. doi: 10.1002/(SICI)1096-9896(199607)179:3<347::AID-PATH559>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 62.Nirmalan NJ, Hughes C, Peng J, McKenna T, Langridge J, Cairns DA, Harnden P, Selby PJ, Banks RE. Initial development and validation of a novel extraction method for quantitative mining of the formalin-fixed, paraffin-embedded tissue proteome for biomarker investigations. J. Proteome Res. 2011;10(2):896–906. doi: 10.1021/pr100812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schoenherr RM, Saul RG, Whiteaker JR, Yan P, Whiteley GR, Paulovich AG. Anti-peptide monoclonal antibodies generated for immuno-multiple reaction monitoring-mass spectrometry assays have a high probability of supporting Western blot and ELISA. Mol. Cell. Proteomics MCP. 2015;14(2):382–398. doi: 10.1074/mcp.O114.043133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics MCP. 2010;9(1):184–196. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.