

Abstract
Purpose
The genetics underlying inherited color vision deficiencies is well understood: causative mutations change the copy number or sequence of the long (L), middle (M), or short (S) wavelength sensitive cone opsin genes. This study evaluated the potential of opsin gene analyses for use in clinical diagnosis of color vision defects.
Methods
We tested 1872 human subjects using direct sequencing of opsin genes and a novel genetic assay that characterizes single nucleotide polymorphisms (SNPs) using the MassArray system. Of the subjects, 1074 also were given standard psychophysical color vision tests for a direct comparison with current clinical methods.
Results
Protan and deutan deficiencies were classified correctly in all subjects identified by MassArray as having red–green defects. Estimates of defect severity based on SNPs that control photopigment spectral tuning correlated with estimates derived from Nagel anomaloscopy.
Conclusions
The MassArray assay provides genetic information that can be useful in the diagnosis of inherited color vision deficiency including presence versus absence, type, and severity, and it provides information to patients about the underlying pathobiology of their disease.
Translational Relevance
The MassArray assay provides a method that directly analyzes the molecular substrates of color vision that could be used in combination with, or as an alternative to current clinical diagnosis of color defects.
Keywords: color vision testing, color blindness, genetic testing, opsin genes
Introduction
Color vision deficiency encompasses a broad range of disorders with a similarly large spectrum of consequences for those affected. An ideal color vision test should accomplish three aims: separate people with normal color vision from those with a color vision deficiency, classify the type of defect, and quantify its severity. However, current tests often fail in at least one of these objectives. The standard for research on color vision defects for more than 20 years has been analysis of the cone opsin genes to identify deleterious mutations and gene rearrangements, a more direct method of diagnosis that determines the underlying pathophysiology of the disorder. The purpose of this study was to evaluate the potential of analysis of the photopigment genes as a clinical tool for diagnosing color vision deficiency.
A great deal about the genetic underpinnings of human color vison is understood. The genes OPN1LW and OPN1MW encode the long (L) and middle (M) wavelength sensitive cone opsins, respectively. They are arranged in a tandem array on the long arm of the X-chromosome at Xq28 and the arrays vary in the number of genes.1 As shown in Figure 1, the ancestral L and M opsin gene array presumably contained one L opsin gene followed by one M opsin gene. Gene rearrangements associated with common color vision deficiencies occur during meiosis; the L opsin gene of one X chromosome can pair with the M opsin gene of the other and an “unequal” crossover occurs, resulting in two new arrays, both of which confer color vision deficiency in males. One contains a single opsin gene, a configuration that results in dichromacy. The other array has three opsin genes: an ancestral L, an ancestral M and, between them, an extra gene produced by the unequal crossover that encodes an L pigment, which may or may not be green-shifted depending on the location of the crossover. This array confers a color vision defect in males because only the first two genes in the array usually are expressed, and, thus, the M opsin gene has been displaced to a nonexpressed position.2 Over generations, this intermixing has continued, and because of relaxed selection against colorblindness in modern humans it has given rise to the large variability seen in the X chromosome cone opsin gene array.3–5
Figure 1.
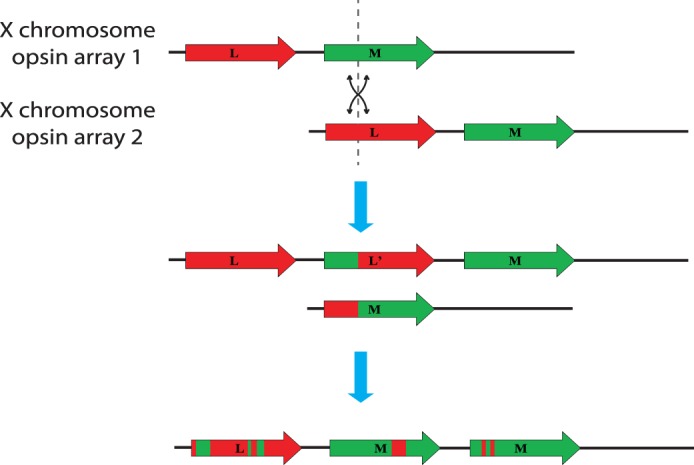
Schematic of the generation of hybrid cone opsins. During meiosis, homologous chromosomes engage in crossing over, where equivalent regions of DNA are swapped as a mechanism to enhance genetic diversity in the population. Because of the high degree of similarity and the proximity of the L and M opsin genes, the X chromosome is prone to misalignment at this locus. When crossovers occur at this point, the result is intermixing of the L and M opsin genes. Over generations, this intermixing produced the enormous variability seen today in opsin gene sequence and copy number. Many of these opsin gene arrays confer color vision defects.
The amino acids encoded by positions 277 and 285 together produce a spectral shift of approximately 20 nm in the photopigment, and, thus, define the difference between genes specifying an L versus M class pigment.5,6 The amino acids encoded by positions 116, 180, and 230/233 each shift the photopigment spectra by 2 to 4 nm and define the differences in spectral sensitivity of photopigments within a class.3–5 These differences determine the severity of the color vision defect in males in which the first two genes in the array specify a pigment of the same class.7 If the encoded pigments do not differ at any sites involved in spectral tuning or at sites that affect optical density,8 the array confers dichromacy. If the encoded pigments differ in peak sensitivity, the array confers anomalous trichromacy, and the severity of the defect is determined by the magnitude of the spectral difference.
The genetics of the S cone opsin are simpler. The OPN1SW gene is autosomal with a locus at 7q32 and lacks the variability of the L and M opsin genes. As with rhodopsin, almost any missense mutation is expected to cause disease. Currently six known mutations of the gene have been linked to S cone dysfunction and result in tritanopia.9–13 The color vision loss in these individuals appears progressive.12 Normal trichromatic color vision in childhood gradually succumbs to tritan defects as S cones slowly die.
We used knowledge of the cone opsin genes to examine the molecular substrates for color vision in 1872 subjects and to evaluate the potential of such genetic techniques for use in the clinical diagnosis of color vision deficiencies.
Subjects and Methods
Subject Recruitment
We recruited 1074 subjects for psychophysical and genetic color vision testing. Subjects were assessed using the fourth edition HRR pseudoisochromatic plate test and the Lanthony D-15 arrangement test. Those whose performance indicated the presence of a color vision deficiency were given additional color vision tests, including the second edition Dvorine pseudoisochromatic plate test and the Nagel anomaloscope. DNA was extracted from blood or saliva from each subject for cone opsin gene analysis.
DNA samples from 798 participants in the Age Related Eye Disease Study (AREDS) were purchased from Coriell Institute for Medical Research (Camden, NJ). DNA samples were from approximately equal numbers of individuals with age-related macular degeneration and matched controls. These patients were not selected for color vision and, thus, are assumed to be representative of the population as a whole in terms of the frequency of inherited color vision defects. The cone opsin genes of these samples were amplified and sequenced. No psychophysical color vision assessment was possible.
Informed consent was obtained from the subjects after explanation of the nature and possible consequences of the study. Experiments involving human subjects were conducted in accordance with the principles embodied in the Declaration of Helsinki, and were approved by Institutional Review Boards at the University of Washington.
Gene Sequencing
The sequencing technique was adapted from methods described previously.14 For each sample, the L and M opsin genes were separately and specifically amplified. In addition, the first gene, all downstream genes, and the last gene in the array were separately and specifically amplified. See Supplementary Table S1 for a list of the primer pairs, the PCR thermal cycling parameters for each primer pair and the details of how they were used.
The spectral class of the pigment encoded by the last gene in the array was identified by selectively amplifying the last gene with a reverse primer that lies outside the repeat unit of the array, then amplifying exon 5 from the last gene using primer pair 8, and finally subjecting the PCR product to Rsa I restriction enzyme digestion. The Rsa I site is created when codon 277 specifies tyrosine and the third position of codon 276 is a G residue. These are both present in L class pigment genes and absent in M class pigment genes; thus, digestion of PCR-amplified exon 5 with Rsa I followed by gel electrophoresis will yield two bands for L pigments and a single band for M pigments.15
Estimates of Photopigment Spectral Peak
To estimate the photopigment spectral peak, the identities of the amino acids at positions 116, 180, and 230 were determined. We estimated the longest L photopigment to have maximal sensitivity to 559 nm light and the spectral peak to be shifted 2.5 nm shorter when tyrosine is present at amino acid position 116, 3.5 nm shorter when alanine is present at amino acid position 180, and 4 nm shorter when threonine is present at position 230. We estimated the shortest M photopigment to have maximal sensitivity to 530 nm light and the spectral peak to be shifted 3 nm longer when serine is present at amino acid position 180 or when isoleucine is at amino acid position 230.3
Analysis of X-Chromosome Opsin Array Composition
We conducted a novel genetic color vision assay using the MassArray system (Agena Bioscience, Inc., San Diego, CA), which allows genotyping of multiple single nucleotide polymorphisms (SNPs) simultaneously using a primer extension process followed by mass spectrometry. An initial round of PCR amplifies from genomic DNA a short length of DNA surrounding each SNP. This is followed by single-base extensions of a primer that anneals directly adjacent to the SNP such that the extended primer carries the dideoxynucleotide that is the complement of the SNP. The nucleotides used for this extension are mass modified such that all four possible extension products are distinguishable by MALDI-TOF mass spectrometry.
A first version of the MassArray color vision assay was comprised of five SNPs that include: three sites that control spectral tuning differences within L and M genes (in codons 116, 180, and 230), one site that distinguishes L and M opsin (in codon 309), and a final site in the promoter region (+1) that differs between the first gene in the array and all downstream genes. Codon 309 was chosen to distinguish L from M opsin genes because it is closely linked with positions 277 and 285 and, unlike those important spectral tuning sites, is not surrounded by silent polymorphisms that make an SNP difficult to assay in the MassArray format. The assay reports opsin gene copy number, the proportion of L genes, and an estimate of spectral sensitivity differences between opsins encoded by the genes present. A second iteration of the test added sites of known mutations in L/M cone opsin genes as well as the S cone opsin gene. These mutations are C203R for L/M opsin and L56P, G79R, T190I, S214P, P264S, and R283Q for S opsin. All PCR and extension primers are listed in Supplementary Table S2.
For samples with MassArray results indicating the presence of extra L opsin genes, the long range PCR of the final gene in the array, described above, was performed to determine if the extra L opsin gene had displaced the M opsin gene from an expressed position.
Severity of Deficiency in Deuteranomalous Trichromats
For a subset of subjects, a highly-specialized two-alternative forced-choice procedure on the Nagel anomaloscope was used to measure the severity of color deficiencies. We argued that this procedure provides a valid measure of severity that can be used as a gold standard of comparison for evaluating clinical color vision tests and estimates of severity from genetics. While the anomaloscope is used as a clinical instrument, the procedure used here is too demanding and too time-consuming (usually requiring more than 30 minutes to complete) for clinical use. For each ratio of the 547 and 658 nm mixture lights set by the experimenter, the subject was asked to adjust the brightness of the 589 nm light to make the closest possible match. When this match was achieved, the subject was asked to describe the mixture light as either too red or too green compared to the 589 nm light. This procedure was repeated until two 547 nm/658 nm ratios were found: one that the subject always described as red and one as green across 10 trials. Ten measurements were taken at intermediate ratios between the too-red and too-green ratios to generate a psychometric function describing the ability of the subject to discriminate mixed red and green lights from monochromatic yellow. The data for each subject were fitted with a cumulative Gaussian weighted three times more heavily on the tails, the points that were always too red or too green. An estimate of the match range was calculated from the fitted curve; it was defined to be the range of values for which we were <95% confident that the 547 nm/658 nm mixture light was indistinguishable from the 589 nm standard light. Larger matching ranges are indicative of poorer red–green color vision.
Color Sensitivity of Dichromats
The trivector Cambridge Color Test was used with coordinates designed to test neutral points from dominant wavelengths 492 to 508 in 2-nm increments. The 498, 500, and 502 nm lines were tested three times and the other six were each tested twice. For each subject, the thresholds were averaged for each wavelength and then normalized by dividing by the subject's maximum threshold to determine the axis of lowest sensitivity.
Results
Detection and Categorization of Color Vision Defects
We analyzed the composition of the opsin gene array using the MassArray assay for two subject pools: a group of male subjects unselected for color vision status who participated in the AREDS study and a group of subjects for whom we performed standard color vision tests in our laboratory. The results for both subject pools are shown in Figure 2, where the percentage of genes in the array that are in the first position (the inverse of this proportion is opsin gene copy number) is compared to the percentage of the total genes that encode an L opsin. Subjects clustered into groups based on the characteristics of their arrays, and this provided a first pass diagnosis of the best possible color vision each subject can have.
Figure 2.

MassArray results for the AREDs subject pool and the psychophysically-tested subject pool. The MassArray color vision test uses two SNPs to characterize the composition of the L and M opsin gene array. The x-axis is the percentage of the total X-linked opsin genes that have the “first gene” promoter allele estimated from the areas under the mass spectrometer peaks for the first gene and down-stream gene promoter fragments. Example raw MassArray spectrometer data are shown in Supplemental Figure S1. The y-axis is an estimate of the percentage of the total X-linked genes that are L genes as measured by the relative areas under peaks in the MassArray spectrum for the SNP in codon 309. (A) Illustration of the theoretical polar angles corresponding to different numbers of L genes, labeled L = 0, L = 0.5, L = 1, L = 2. Subjects with polar angles along the 0° line (L = 0) have protan color vision deficiencies. Most normal subjects, have one L gene (L = 1) and fall near the 45° line. Individuals with more than one L gene have deutan type color deficiencies or they are “deutan suspects” and they fall near the line marked L = 2 genes or above it. Women who are carriers of protan color vision defects are predicted to fall near the line marked L = 0.5 genes because they typically have an L gene on one of their two X-chromosomes. Theoretical locations of different numbers of total numbers of genes also are marked. Individuals with one gene T = 1 are dichromats. Individuals with more than one gene can be colorblind or normal depending on the L:M gene ratio. (B) The classification of 798 males is based solely on genetic analysis and was accomplished using the first version of the test. (C) The classification of 1043 males and 31 females is based on genetic and behavioral diagnosis. Some subjects were tested using an earlier version of the MassArray test with fewer SNPs; results from the first version were normalized to allow plotting on the same graph.
Males with normal color vision typically have one L gene followed by one or more M genes,1 and they fall along the unity line below approximately [60%, 60%] in the MassArray assay. Rarely, males with dichromatic color vision (protanopic or deuteranopic) also will fall in this area because they have inactivating mutations in one of the first two genes in the array that are not detected by the MassArray assay.2,16 Subjects who lack L genes fall along the x-axis and all of these have, at best, protan color vision deficiencies, with single-gene protanopes clustering at [100%, 0%]. Subjects who lack M genes fall along the y = 100% line and all of these can have no better than deutan color vision deficiencies, with single-gene deuteranopes clustering at [100%, 0%]. Individuals whose results plot above the unity line possess at least one extra L opsin gene and may either be color normal or have a deutan-type color vision deficiency, depending on whether the first two genes in the array encode an L and an M pigment, or whether both encode an L pigment. Females who are likely to be carriers of red–green color vision deficiency also are distinguishable from color anomalous individuals and noncarrier normals: carriers of protan defects tend to have a paucity of L opsin genes compared to normal and, thus, should fall between the normal (one L gene) line and the x-axis, while carriers of deutan defects tend to have extra L genes compared to normal and, thus, should fall between the normal line and the deutan range.
The MassArray measurements are subject to a certain amount of error and it is necessary to set mathematical criteria for distinguishing genotypes and determine the likelihood of classification errors. With regard to providing information that is useful in diagnosing color vision defects the MassArray assay involves making the following distinctions: (1) Discriminate people with one X-chromosome photopigment gene from those with more than one gene. Everyone with only one photopigment gene on the X-chromosome is color blind and at best a dichromat. (2) The ability to distinguish between people who have L and M photopigment genes from those who have only L or only M genes. Those people with only L genes have deutan color vision deficiencies and those with only M genes have protan color vision deficiencies. (3) Approximately 90% of men with normal color vision have either two, three, or four X-linked photopigment genes. Men with two genes, both of which are not L, cannot have a deutan color vision defect; conversely, men with three or four genes in which two are of the L pigment class have a high probability of having a deutan-type color vision defect and, thus, can be classified as deutans or “deutan suspects.” The precision of the MassArray is sufficient to easily separate all the above classifications. For men with three or four genes, separating normals from deutan suspects is straightforward because the proportion of genes that are L is greatly different in these cases. For three genes, normals have 33% L and deutan suspects have 67% L genes. For four genes, normals have 25% L and deutans have 50%. However, the precision of the MassArray is near its limits of predictive accuracy for people with more than four genes on the X-chromosome, which constitute approximately 10% of the population. For these rarer cases, distinguishing between a person who has five genes, four M and one L, from a person with four M and two L, that is, between 20% and 33% L becomes statistical and there is theoretically a probability that a four M and two L deutan suspect could be misclassified as four M and one L color normal person. Thus, the following procedure was used to determine a mathematical criterion for distinguishing arrays with one L gene and multiple M genes, from arrays with more than one L gene indicating that the subject is a deutan or deutan suspect. The polar angle of each data point in the scatterplot (Fig. 2) was calculated. The polar angle indicates the number of L genes in the array for men and the average number of L genes per array for women (women have one array on each of their X chromosomes). As shown in Supplemental Figure S2, the polar angle distribution was fit to a Gaussian function representing arrays with one L gene and a sum of Gaussians representing individuals with two and three L genes (normals with extra downstream L genes and deutans). The angle at which the one L gene Gaussian intersected with the sum of the multiple L gene Gaussian was taken as the criterion dividing point for segregating subject into classes. We calculated the percentage of arrays expected to be misidentified from the overlap of the one L gene function and the sum of the two and three L gene Gaussian functions. The accuracy of separating normal individuals with one L gene and multiple M genes from protans, deutans, and deutan suspects estimated in this way is 99.6%.
To examine the same question of the possibility of misclassification another way, we looked specifically at the 1° bins on each side of the cutoff between normal and deutan suspects (1 vs. >1 L genes) for subjects whose color vision was tested. Nine males fell into this category. Six of the nine fell in the normal bin adjacent to the cutoff and not one was diagnosed as deutan in the color vision testing. For the three that fell on the deutan side of the cutoff, one was deutan by color vision tests and the other two had normal color vision in the tests, and were classified as normal with extra L genes. Thus, our results did not indicate that anyone was misclassified in our sample, although we cannot be sure that the normal men classified as having an extra L gene actually had the extra L. However, it is clear that there is a statistical probability that people with a large number of genes, in particular, could be misclassified. If for the nine people on either side of the cutoff, there is approximately a 50% to 50% chance of an error, then we estimated the error rate to be approximately 0.04%, very close to what was estimated from the Gaussian overlap.
We noted that the problem of avoiding misclassification errors could be easily solved. A histogram for polar angle examining just the approximately 10% of subjects with the highest estimated gene number shows that 7 of the 9 people falling in the uncertain zone were estimated to have more than 4 genes. A remedy that should almost eliminate any possibility of misclassification is to put people who are estimated to have high gene number and fall near the gap in a separate category. This would constitute <1% of all subjects. Something like the following could be appropriate “the genetic assay indicates that the patient has more than four pigment genes; the assay places the subject near the cutoff between normal and deutan subjects.” Being absolutely clear about when the classification could be ambiguous is a way of avoiding misclassifying patients with the MassArray assay.
Data from the pool of male AREDS subjects is shown in Figure 2A. Of 798 men, 708 fell along the unity line and are predicted to have normal color vision. The color coding for diagnoses in Figure 2A is the same as that indicated in Figure 2B. In Figure 2A, the diagnosis of each subject indicated by the color code was determined entirely from genetic data, including estimated %L and %downstream genes indicated in the plot, plus the estimate from the MassArray tuning site SNP data of spectral separation for deutan subjects with multiple L genes and protans with multiple M genes. The final piece of data used for the diagnosis indicated by the color code was the identity of the last gene in the array as L or M where applicable. Thus, in Figure 2A, eight males were identified as single-gene deuteranopes based on clustering at [100%, 100%] and two multigene deuteranopes were identified based on clustering along the y = 100% line along with absence of differences in SNPs in codons that influence spectral tuning. One person whose results fell along the y = 100% line was determined to be (at best) deuteranomalous with multiple L genes that differed in spectral tuning. Nine males were identified as protan: one single-gene protanope at [100%, 0%], two multigene protanopes with no differences in their M genes at SNPs that influence spectral tuning, and three protan-type individuals were identified whose M genes did encode spectral differences. The other three protan males had M genes that differed only in exon 2, which encodes amino acid differences that may affect optical density and give rise to protanomalous behavior in the anomaloscope test.8 The final 70 of the 798 males fell above the unity line but below the y = 100% line, indicative of arrays with at least two L opsin genes and at least one M opsin gene. These individuals may either be color normal or deutan, and distinguishing the two requires identification of the last gene in the array to deduce the order of the genes. For 20 of these subjects, there was a single extra L gene and it was at the 3′ end of the array; they are predicted to have normal color vision. For 21 of the subjects, the extra L gene was deduced to be in the second position; their arrays had only one M gene and it was found in the last position. Their L genes were different at spectral tuning sites so they are predicted to be, at best, deuteranomalous. The remaining 29 subjects had too many genes to determine whether the first and second genes both encoded L pigments and their diagnosis remains ambiguous. They are labeled as “Mild DA\Normal extra L” and they can be considered to be “deutan suspects.”
We also performed some direct gene sequencing for these subjects and we found two previously unreported missense mutations in expressed L opsin genes, L232V and L55V, and one silent mutation, GTC>GTT in codon 97. Additionally, one subject was homozygous and four were heterozygous for the deleterious C203R mutation in M genes. One of the heterozygous subjects had two M genes and the C203R mutation was found in the gene in the last gene position, meaning the expressed M did not have the mutation. The other three subjects had more than two M genes and so it could not be determined if the C203R mutation was in an expressed gene. Therefore, point mutations, all of which except the C203R are not detected by the MassArray assay, could render the subject dichromatic depending on the position of the gene encoding C203R and on the effects of the L232V and L55V mutations (which could affect protein folding or function) and the synonymous mutation (which could affect splicing). The L232V substitution is predicted by Mutation Taster17 to be disease-causing, while the L55V is not, and none of the three is predicted by Human Splicing Finder18 to affect splicing.
In the second subject pool, we used MassArray data, gene order, gene sequencing, and psychophysical tests to evaluate color vision status. The MassArray data for these subjects is shown in Figure 2B. All subjects are summarized in Table 1. Our subject pool included 31 females, 29 of whom passed the HRR and D15, one of whom made protan errors on those tests, and one of whom made deutan errors. The MassArray test classified all 29 women who were color normal in psychophysical assessments as having genes required for normal color vision. Additionally, the genetic test identified four of the women as probable deutan carriers and two as probable protan carriers based on established characteristics of opsin genes in carriers.14,19 The protan female had five spectrally identical M opsin genes in her array. The deuteranomalous female had an extra L opsin on one chromosome, displacing the M opsin gene from an expressed position, and a missense mutation (V63M) in the expressed M gene of the other chromosome, leaving her with three L opsins and a presumably nonfunctional M opsin. The V63M substitution is predicted by Mutation Taster to be disease-causing.
Table 1.
Summary of Psychophysically Tested Subjects

Among the male subjects (n = 1043), the MassArray identified 22 as having arrays comprised of only a single L gene ([100%, 0%]), making them deuteranopes; another 5 had arrays comprised of only a single M gene ([100%, 100%]), making them protanopes. All of these obligate dichromats failed the psychophysical tests.
The MassArray identified 9 males who had L opsin genes with distinct spectral tuning and no M opsin gene, making them, at best, deuteranomalous. However, only four made deutan errors on the psychophysical tests; the others, who we call “minimal deutans,” made no errors at all on either test. Examination of their L opsin sequences revealed that these subjects all had L photopigments with spectral separations of 6.5 to 10 nm. Three of these five agreed to return for testing with the Nagel anomaloscope, which confirmed that they were very mild deutans.
There were 18 males who had no L opsin gene and multiple M opsin genes. Five of them had arrays comprised solely of multiple identical M genes, making them obligate protanopes. Seven subjects had M opsin genes with differences only in exon 2, which does not affect spectral tuning but may create optical density differences; another 5 had M opsins with differences in both spectral tuning sites and in exon 2, making them, at best, protanomalous. The 18th protan male had three M opsin genes, but direct sequencing showed that one of them encoded a tyrosine at 309 (normally found in L pigments) making him appear normal in the MassArray assay. Seventeen of these 18 protans made protan errors on the color vision tests, but one (who had differences in spectral tuning and optical density controlling sites) was misdiagnosed by the HRR and D15 as a deutan.
Of the psychophysically tested male subjects, 887 had one L gene followed by a variable number of M opsin genes, a configuration normally associated with normal color vision. In 7 of those subjects, direct sequencing revealed the presence of mutations in the opsin genes that led to red–green color vision defects. One had a Cys203Arg mutation in his M gene, one had a Cys203Opal mutation in his M gene, one had a single base insertion in his M gene introducing a frame shift in exon 3, one had a gene rearrangement where his L was displaced to the third position leaving two spectrally identical M genes in expressed positons, and three had toxic opsin LIAVA or LVAVA variants.20 These seven subjects all failed the psychophysical color vision tests.
Sequencing found that 5 of the 888 subjects with normal arrays had mutations in the SNPs that the MassArray assays uses to characterize the array. One of those subjects was outside all diagnosis clusters at [100%, 50%] and appeared to have an impossible array – 50% of his genes were of the L class, but he seemed to have only a single gene in his array. Sequencing of the promoter region revealed that he had in all genes the “first gene version” of the SNP which the MassArray assay uses to determine gene number (i.e., a G at nucleotide +1) but was heterozygous at other positions, meaning he had an LM array sufficient for normal color vision. The other four subjects had mutations in the codon 309 SNP that the MassArray uses to distinguish L and M opsin genes. One had an LM array in which the L had a phenylalanine at 309 (typically associated with M genes), making him appear protan to the MassArray. Similarly, three subjects who appeared to have deutan LLM arrays actually had normal LMM arrays because one M opsin gene encoded a tyrosine at 309, normally found in L pigments. All of these five subjects passed the color vision tests.
Direct sequencing of the opsin genes for the other 875 of the 888 subjects with normal numbers of L and M genes showed no deleterious mutations, together indicating the genetic basis for normal color vision. Of these 875 subjects, 869 passed the HRR and D15 color vision tests. The remaining six showed mild defects in protan, deutan, or, in two cases, tritan stimuli on psychophysical color vision tests. No genetic cause for color vision deficiency was ultimately found for these subjects.
The final group of psychophysically-tested subjects were 102 men with at least two L opsin genes and at least one M opsin gene. In 26 of those subjects, the extra L opsin gene was in the last position of the array, not displacing the M gene. These arrays (LML or LMML) are consistent with normal color vision, and, indeed, 25 of these men passed the psychophysical color vision tests. The last man had an LMML array but made mild errors on the HRR and D15, had a Rayleigh match range of 44 to 59, and was of East Asian descent. We sequenced his promoter region and found he was heterozygous for an A-71C mutation, a common cause of red–green defects in Asian populations.21 In another 28 men, two L opsin genes were in expressed positions by deduction from last gene sequencing (LLM or LLLM arrays). L opsin sequences indicated that there was no spectral difference between the L opsins for two of these 28, making them multigene deuteranopes, and there was a spectral difference for the other 26, making them, at best, deuteranomalous. Of the 26 deuteranomals, 22 were classified as mild or medium deutans by the HRR and D15. However, two were minimal deutans who made no errors on either test. The last two deuteranomals had a 2.5 nm spectral separation but behaved more poorly on color vision tests than we would predict from having two pigments with that separation. One had very poor acuity and amblyopia, which likely led him to perform worse on the psychophysical tests than his color vision defect would have allowed; the other was not tested for visual acuity and reported no other eye disorders.
The remaining 48 of the 102 subjects with extra L opsin genes had long arrays containing four or more opsin genes. While we can specifically analyze the first and last genes in the array, we cannot access the second gene. For longer arrays, we cannot always deduce the identity of the second expressed gene. Of the subjects with extra L opsin genes and a long array, 11 had spectrally identical L genes. If their extra L was in an expressed position, these men would be deuteranopes, otherwise, they would have normal color vision. All of them passed the HRR and D15, consistent with normal color vision. Another 19 failed the psychophysical tests so we assume the extra L gene is in the second position. The final 18 subjects with long arrays and extra L genes passed the HRR and D15 but had a spectral separation between their Ls of at least 6.5 nm; these men may be normal with the extra L genes downstream of the expressed positions or be minimal deutans able to pass the psychophysical tests. Four of these agreed to return for testing with the Nagel anomaloscope, which revealed that three were normal trichromats and one was a minimal deutan.
Estimates of Severity of the Deficit in Anomalous Trichromats
The SNPs at amino acid positions 116, 180, and 230/233 each encode a shift of 2 to 4 nm in spectral sensitivity. By determining the identity of these amino acids, one can estimate the spectral separation between two opsins of the same class and thus predict the severity of the deficiency in anomalous trichromats. It is possible to determine the spectral peaks of the expressed opsins for almost all subjects using direct sequencing of relevant exons specifically amplified from first genes, last genes, L genes, and M genes. The MassArray also provides an estimate of the spectral separation of photopigments by identifying the proportion of genes with each allele present at codons 116, 180, and 230. However, it does not give information about which SNPs are present specifically in L versus M genes or about the SNPs associated with genes in a particular order in the array. When using only information from the MassArray we estimated spectral sensitivities under the assumption that ancestral L opsin amino acids would tend to be in genes more upstream in the array and ancestral M opsin amino acids would be in more downstream genes. Results using this assumption agreed for 19 of 22 subjects diagnosed as deuteranomalous with estimates obtained from gene sequencing.
To test the accuracy of genetic estimates of color sensitivity from spectral separation, the severity of the deficiency in 20 deutan subjects and nine normal controls was assessed by a forced choice procedure on the Nagel anomaloscope (see methods) and compared to estimates of spectral separation derived from gene sequence information.
Figure 3A shows example results from a deuteranomalous male with L pigments separated by 2.5 nm. His midpoint is approximately 21 and his match range is 2 Nagel units. Data from subjects with similar spectral separations were averaged to generate Figure 3B to show the relation between spectral separations predicted from genetics and defect severity assessed by the Nagel anomaloscope. The genetic estimates agree well with the anomaloscope assessment. Subjects with a single X chromosome opsin gene or no separation between multiple opsin genes, perform far worse than all anomalous trichromats and cannot even distinguish the pure red or green lights from the yellow light. We measured the spectra of the lights in the Nagel anomaloscope with a Konica Minolta CS-2000 spectroradiometer and found that the average Nagel range of 0.345 units for our normal subjects equates to a cone contrast of only 0.174%. With such impressive sensitivity, even the 10-fold higher thresholds of moderate anomalous trichromats still represent a considerable color discrimination ability compared to a dichromat.
Figure 3.

Results of forced choice Rayleigh match procedure. (A) Example results from a deuteranomalous male with 2.5 nm separating the spectral peaks of his L opsin photopigments. (B) An estimate of the spectral separation between X chromosome photopigments was derived from gene sequence information for deuteranopic, deuteranomalous, and normal trichromatic subjects (deuteranopes were given a separation of 0 nm). This estimate was compared to the width of the Rayleigh match range obtained from the forced choice protocol.
Color Discrimination Thresholds in Dichromats
As described earlier, rather than a single L and a single M photopigment, there exists a family of each, and members of each family can differ in their spectral sensitivity. The peak absorbance of L pigments can range from 549 to 559 nm, while the peak absorbance of M pigments can range from 530 to 536 nm. This means that not all dichromats will have the same color confusion lines. This variability presents a problem for discriminating dichromats from anomalous trichromats using cone contrast type tests, such as the HRR. Because color discrimination thresholds of red–green color defectives peak at a single confusion line and decrease with distance from that axis, stimuli designed to produce confusions along a given color axis will fail to correctly diagnosis people with confusion lines that differ from the standard chosen for a particular cone contrast type test. To evaluate this, we measured color discrimination thresholds for 12 dichromats – three protanopes and nine deuteranopes using the Cambridge Color Test (Fig. 4) which allows measurement of thresholds along a variety of hypothetical confusion lines. All subjects had only a single X chromosome cone opsin gene or two genes with the same amino acids at spectral tuning sites, and were categorized as protanopes or deuteranopes based on the identity of those genes. Deuteranopes varied greatly in their color discrimination for different confusion lines, with some displaying their worst performance along a line through a dominant wavelength of 506 nm and others of 500 nm. For a stimulus designed to test a confusion line for the deuteranope whose threshold peaks at 500 nm, the deuteranope whose threshold peaks at 506 nm would have a threshold of approximately 50% below his maximum and, thus, would be indistinguishable from an anomalous trichromat whose confusion line is at 500 nm.
Figure 4.

Finding dichromat confusion lines using the Cambridge Color Test. The thresholds of five sample dichromats across a range of confusion lines are shown in (A). All thresholds are normalized to the neutral point, the wavelength where the subject performed worst. Two protanopes are shown in green and three deuteranopes in red. The two vertical dotted lines show the standard neutral points for protans and deutans with circles to show how well the individual subjects perform at that point. The neutral points are shown for all 12 dichromats in (B). Dots represent individuals who had a single worst performance and bars represent individuals who performed equally badly on multiple wavelengths. The five individuals from (A) are numbered and shown again in (B).
Testing along a single standard confusion line for protan, deutan, and tritan defects is predicted to produce errors in determining the severity of color vision defects and, indeed, they were found to occur quite frequently. Using the fourth edition HRR plate test we assessed the color vision of 22 subjects with only a single opsin gene on the X chromosome, multiple genes with the same sequence, or genes with inactivating mutations. Even though such subjects are necessarily dichromats, seven (32%) were categorized as “medium” by the HRR plate test.
Discussion
Separating Normal Color Vision from Congenital Red–Green Deficiency Using an Assay of the Number and Ratio of L and M Opsin Genes
Red–green color vision deficiency is by far the most common single locus disorder in humans. Unlike many other common genetic polymorphisms that have been driven to high frequency by selective pressure, the high frequency of red–green color vison defects is the result of “mutation pressure” from the extremely high frequency of rearrangements at this locus. Relaxation of selection against colorblindness in humans in the presence of the high mutation rate accounts for its high frequency.3
Taking advantage of the fact that almost all common color vision defects are the result of gene rearrangements, we developed a MassArray assay designed to separate people with normal color vision from those with different forms of red–green colorblindness by determining the number and ratio of L and M genes. The assay also included a quantitation of the spectral tuning sites allowing multigene protanopes and deuteranopes to be differentiated from individuals with the genetic potential to be anomalous trichromats and allowing us to predict their relative severity. In addition, the MassArray assay included a probe for a C203R mutation that interrupts photopigment function and occurs at a relatively high frequency in the population.
The sample of subjects that we studied from the Seattle area (discussed below) who underwent color vision testing included individuals who volunteered because they knew (or suspected) that they might have color vision defects, so it cannot be considered to represent a random sample from the population. Thus, a sample of 798 male AREDS subjects, who were unselected for color vision phenotype, shown in Figure 2A, were run to give an estimate of how results for the assay might be distributed for the general population unselected for color vision phenotype.
Some large studies have used psychophysical color vision testing methods that were designed to have a sensitivity for detecting congenital red–green color vision deficiencies that is very close to 100%. For example, Waaler22 screened his subjects using the Ishihara test, but knowing that some mild deuteranomalous individuals can pass the Ishihara making zero errors, he used a criterion of selecting subjects who made errors or even showed difficulty on the Ishihara for additional testing using the anomaloscope. Subjects chosen for anomaloscope testing were classified into 5 categories based on the results with that test: normal, protanope, deuteranope, protanomalous, or deuteranomalous. Most clinics do not have an anomaloscope or personnel trained to use one, so this is not a practical clinical testing method; however, results from this approach do represent close to a gold standard for sensitivity in detecting red–green color vision defects. Waaler22 found that 8.01% of males had red–green color vision deficiency in which 0.88% were protanopes, 1.03% were deuteranopes, 1.04% were protanomalous, and 5.06% were deuteranomalous.
For the AREDS sample (Fig. 2A), we diagnosed people based solely on the MassArray plus one extra step of assaying the last gene for 71 subjects (9%) who had extra L genes and four with a C203R mutation. Opsin gene arrays conferring color vision defects were found in 42 subjects (5.3%) including obligate single-gene and multigene-dichromats, obligate protans with no L genes, and 21 people with an extra L gene that was deduced to be in the second position, making their best possible color vision deuteranomalous. In 724 subjects (90.7%), no gene rearrangements were detected that would cause a red–green color vision defect; thus, they were classified by this test as having normal red–green color vision. However, from sequencing of exons 2, 3, and 4 of the L and M genes of all subjects, three previously unreported point mutations in expressed L opsin genes -–L55V, L232V, and a silent substitution in codon 97 – were discovered. If any of these changes cause the loss of L-cone function, these people could be protanopes that were not detected by the MassArray assay (up to 0.3% of the population). There also may have been deleterious mutations in exons 1, 5, and 6 or the regulatory regions which were not sequenced but, except for the A-71C (discussed below) that occurs at high frequency in some Asian populations, people with such mutations are expected to be exceedingly rare.
Finally, in 29 subjects with extra L genes and in three who were heterozygous for the C203R mutation there were too many genes to determine whether the first and second genes encoded functional L and M pigments or not, so all 32 (4.0% of the sample) were categorized as “deutan suspects.” To compare with prior studies, we can estimate how many of these men had color vision defects by assuming a similar proportion of deutans as found in men with shorter arrays. For men in this sample with arrays that could be resolved by last gene analysis, approximately 50% (21/41) had two L genes in expressed positions and, thus, are deutans. If a similar percentage of longer arrays with multiple L genes are deutan then 7.2% of AREDS males had red/green color vision deficiencies of which 0.5% were protanopes, 1.3% deuteranopes, 0.8% protanomals, and 4.6% deuteranomals. These rates are very similar to the previously published population estimates such as obtained in the classic work of Waaler,22 mentioned above, and a more recent study of a Greek population.23 Thus, a purely genetic population survey is consistent with the classic results using the anomaloscope.
Detection of Color Vision Deficiencies with the Massarray Assay
The MassArray test separates subjects into three categories (normal red–green color vision, red–green color vision defects, and deutan suspects) instead of two, normal and color defective. Nonetheless, the following statements can be made about the detection of genetic mutations that cause red–green color vision deficiency with the assay. It missed two new point missense mutations; if these disrupt cone function then 61 of an estimated 63 males with defects known to cause color vision deficiency were identified with the assay. Thus, 97% of the males with mutations that could cause red–green color vision deficiency in the AREDS sample were correctly identified with the assay as either having a color vision deficiency or being a deutan suspect. All point mutations that cause color vision defects are expected to interrupt photopigment function and be associated with dichromacy rather than anomalous trichromacy. Earlier, we examined the genes of 128 red–green dichromats and, retrospectively, we can say that 6 (4.7%) dichromats had mutations that would not be caught by the MassArray assay.16 Estimating that 25% of all males with red–green color vision deficiencies are dichromats provides an estimate of 1.4% of all color defects would not be detected by the assay. Conversely, we estimated that approximately 98.6% of individuals with genes known to cause vision deficiency would be identified with a purely genetic MassArray test.
Everyone identified as having a color vision deficiency had a gene defect that positively prevents either the normal L or normal M pigment from being produced. However, in comparing genetics with psychophysical tests of color vision in our Seattle sample (discussed below), of 942 men and women who passed color vision tests, five had mutations in SNPs used by the MassArray to determine the number and ratio of L and M genes and they would have been misidentified as red–green color blind. Thus, as presently implemented, 0.5% of people with normal color vision would be diagnosed incorrectly as having a color vision defect with this assay.
As an alternative genetic testing method, sequencing the entirety of the L and M genes and the promoter region for all subjects would almost completely avoid false-positives. With the technology presently available; however, it would be impractical for use as part of a routine clinical assessment for red–green color vision deficiency. As a diagnostic tool, ability of the MassArray to separate deutan defects from protan is perfect, and its ability to identify a subset of people as obligate dichromats is very valuable since separating dichromats from anomalous trichromats is one of the worst failings of current psychophysical color vision tests used in the clinic. The two weaknesses of the MassArray test are that it can miss the small faction of people with previously undiscovered point mutations and that diagnosis is ambiguous for the 4% of the population identified as “deutan suspects.” Nonetheless, as a much more direct measure of the underlying pathophysiology, the genetic test described here has many advantages over conventional clinical color vision testing methods.
The ability to discriminate colors in the middle-to-long wavelengths is determined by the spectral proximity of photopigments encoded on the X-chromosome.24 Here, as shown in Figure 3, we have examined the relationship between the photopigment spectral separation and red–green color vision thresholds with a high degree of precision using a forced choice anomaloscope task. Two aspects of this relationship are striking. First, a reduction in spectral separation from the normal approximately 30 nm to only 10 nm results in a very modest elevation in thresholds of only two to three times normal. Second, even deuteranomalous individuals with very small spectral separations of approximately 2.5 nm experience color vision threshold elevations that are only approximately 10 times higher than normal, which is dramatically better than dichromats that are more than 100 times worse than normal. Thus, dichromats have color vision that is approximately 10 times worse than the common more severe anomalous trichromats. This highlights the great significance in clinical color vision testing of separating dichromats from anomalous trichromats.
As shown in Figure 5, testing along a single standard confusion line for protan, deutan, and tritan defects as is done in some of the most advanced conventional psychophysical diagnostic tests, such as the HRR, is predicted to produce errors in determining the severity of color vision defects. Thus, it is not surprising that neither the widely used Ishihara pseudoisochromatic plate test nor the HRR can reliably distinguish the two categories. The ability to estimate levels of severity among anomalous trichromats through the deduced spectral separation between pigments (Fig. 3) and to correctly discriminate anomalous trichromats from dichromats is a very strong reason in favor of incorporating genetic testing into the standard diagnosis protocol for clinical color vision testing.
Figure 5.

Sample psychophysical color vision test to accompany genetic assay. A simple color vision test that can be administered quickly and with no experimenter training. Each group of dots contains one colored dot whose chromaticity falls along a protan, deutan, or tritan confusion line. The subject must identify the colored dot in each group and a single mistake constitutes failure.
Combining Genetic and Psychophysical Approaches
The extremely high frequency of color vision defects, the fact that they usually appear in children for whom neither parent is affected, and the damage that can result from being undiagnosed or misdiagnosed, are reasons for everyone to have an accurate diagnosis of color vision deficiency before entering preschool. Colors are used extensively in the early school years in teaching subjects, such as reading and math, and colorblind children, particularly dichromats, can be seriously disadvantaged if they are not accurately diagnosed and appropriate accommodations are not made. The administration of a genetic test can be done at a very early age, even at birth, and from the discussion above, it may correctly diagnose an estimated 98.6% of everyone tested, as either having the genetic basis for normal color vision, or genes associated with color defective vision or those of a deutan suspect.
Information from psychophysical color vision testing complements information gained from the practical genetic test described here. People with missense mutations that are not picked up in the MassArray assay will be dichromats. Such people would appear normal in the genetic screen but be diagnosed as having a moderate to severe red–green defect on a psychophysical test, as was the case for 7 of 887 male subjects in our psychophysically-tested group. The combination of a normal appearing gene array coupled with obvious failure on a psychophysical color vision test could be used as a criterion indicating the need for sequencing the L and M genes and their regulatory regions in such individuals, which would both detect and correctly identify the type of all red–green dichromats. Sequencing the L and M genes of people who fail a psychophysical screening test also would identify toxic sequence combinations, such as “LIAVA” and “LVAVA,” that result in splicing errors,25 and it would identify the small percentage of colorblind people who appear normal in the MassArray assay because of mutations in the SNPs used by the assay to determine array configurations.
The ideal protocol for clinical color vision diagnosis combines a basic psychophysical color vision screening test with the genetic test described here. It is most efficient to use a simple screening test focused on separating normals from deutans among the “deutan suspects” and identifying subjects with rare mutations that require further genetic analysis including complete sequencing of the opsin genes. With this in mind, we developed the disposable minimalist “paper and pencil” color vision screening test shown in Figure 5. This is designed to detect blue–yellow and red–green color vision deficiencies.
The minimalist test correctly identified 99% of 260 color normal subjects. People with minimal deutan deficiencies are a challenge for all screening tests. With a failure criterion of a single missed dot, the minimalist test shown in Figure 5 correctly identified 89% of 55 subjects diagnosed with red–green deficiencies from testing with the desaturated D15 and HRR. While the minimalist test did correctly fail two of five minimal deutans who passed the HRR and desaturated D15, there were still minimal deutans and a few mild deutans who passed, accounting for why only 89% of color defects were detected. However, as summarized in Table 1, a fraction of deuteranomalous subjects has only L genes and no M genes, and well over half of subjects with multiple L genes have only three genes total so deutans among them can be separated from normal with the last gene assay. Thus, the combination of the minimalist screening test with the MassArray test could comprise a very practical protocol that we estimate would identify more than 96% of all individuals with color vision deficiencies, missing only a fraction of mild deutans. However, the small fraction of people who pass the screening test but have multiple L genes could be referred for additional screening to insure that everyone with a color vision deficiency is identified.
Summary
Overall, the MassArray test described here is capable of directly identifying the underlying pathobiology of color vision deficiencies and offers many important advantages over traditional psychophysical color vision tests. When combined with a simplified screening test, the MassArray opsin gene assay provides a powerful tool that can accomplish all the aims of an ideal color vision test. It is the best way, clinically, to separate protans from deutans and dichromats from anomalous trichromats, and it performs well in stratifying different levels of severity among anomalous trichromats. It can distinguish inherited color vision deficiencies from those that arise from other causes. An additional benefit of genetic testing is the ability to distinguish probable female carriers of color vision deficiencies from normal trichromatic women, a feat impossible for psychophysical tests. The test can even predict future color vision loss in younger people with known S cone mutations. Moreover, as gene therapies for color blindness become available, knowledge of the causal mutations leading to color defects will identify subjects who are good candidates and determine which type of therapy they would require.
Supplementary Material
Acknowledgments
The authors thank Jessica Rowland, Netta Smith, Toni Haun, Katharina Foote, and Sally Chu for help with psychophysical color vision testing and with PCR and DNA sequencing.
Supported by Neurobiology Training Grant T32GM007108; National Institutes of Health (NIH; Bethesda, MD) Grants P30EY001730, R01EY009620, and R01EY021242; Macular Vision Research Foundation; and unrestricted funds from Research to Prevent blindness. J. Neitz is the Bishop Professor, and M. Neitz is the Ray H. Hill Professor in Ophthalmology.
References
- 1. Nathans J,, Thomas D,, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986. ; 232: 193–202. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi T,, Motulsky AG,, Deeb SS. Position of a 'green-red' hybrid gene in the visual pigment array determines colour-vision phenotype. Nat Genets. 1999. ; 22: 90–93. [DOI] [PubMed] [Google Scholar]
- 3. Neitz J,, Neitz M. The genetics of normal and defective color vision. Vision Res. 2011. ; 51: 633–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merbs SL,, Nathans J. Absorption spectra of the hybrid pigments responsible for anomalous color vision. Science. 1992. ; 258: 464–466. [DOI] [PubMed] [Google Scholar]
- 5. Asenjo AB,, Rim J,, Oprian DD. Molecular determinants of human red/green color discrimination. Neuron. 1994. ; 12: 1131–1138. [DOI] [PubMed] [Google Scholar]
- 6. Neitz M,, Neitz J,, Jacobs GH. Spectral tuning of pigments underlying red–green color vision. Science. 1991. 252: 971–974. [DOI] [PubMed] [Google Scholar]
- 7. Neitz J,, Neitz M,, Kainz PM. Visual pigment gene structure and the severity of human color vision defects. Science. 1996. ; 274: 801–804. [DOI] [PubMed] [Google Scholar]
- 8. Neitz J,, Neitz M,, He JC,, Shevell SK. Trichromatic color vision with only two spectrally distinct photopigments. Nat Neurosci. 1999. ; 2: 884–888. [DOI] [PubMed] [Google Scholar]
- 9. Weitz CJ,, Miyake Y,, Shinzato K,, et al. Human tritanopia associated with two amino acid substitutions in the blue sensitive opsin. Am J HumGenet. 1992. ; 50: 498–507. [PMC free article] [PubMed] [Google Scholar]
- 10. Weitz CJ,, Went LN,, Nathans J. Human tritanopia associated with a third amino acid substitution in the blue sensitive visual pigment. Am J HumGenet. 1992. ; 51: 444–446. [PMC free article] [PubMed] [Google Scholar]
- 11. Gunther KL,, Neitz J,, Neitz M. A novel mutation in the short-wavelength sensitive cone pigment gene associated with a tritan color vision defect. VisNeurosci. 2006. ; 23: 403–409. [DOI] [PubMed] [Google Scholar]
- 12. Baraas RC,, Carroll J,, Gunther KL,, et al. Adaptive optics retinal imaging reveals S-cone dystrophy in tritan color vision deficiency. J Opt Soc AmA. 2007. ; 1438–1447. [DOI] [PMC free article] [PubMed]
- 13. Baraas RC,, Hagen LA,, Dees EW,, Neitz M. Substitution of isoleucine for threonine at position 190 of S-opsin causes S-cone-function abnormalities. Vision Res. 2012. ; 73: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oda S,, Ueyama H,, Tanabe S,, Tanaka Y,, Yamade S,, Kani K. Detection of female carriers of congenital color-vision deficiencies by visual pigment gene analysis. Curr Eye Res 2000;21: 767–773. [DOI] [PubMed]
- 15. Nathans J,, Piantanida TP,, Eddy RL,, Shows TB,, Hogness DS. Molecular genetics of inherited variation in human color vision. Science. 1986. ; 232: 203–210. [DOI] [PubMed] [Google Scholar]
- 16. Neitz M,, Carroll J,, Renner A,, Knau H,, Werner JS,, Neitz J. Variety of genotypes in males diagnosed as dichromatic on a conventional clinical anomaloscope. Vis Neurosci. 2004. ; 21: 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwarz JM,, Cooper DN,, Schuelke M,, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014. ; 11: 361–362. [DOI] [PubMed] [Google Scholar]
- 18. Desmet FO,, Hamroun D,, Lalande M,, Collod-Béroud G,, Claustres M,, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37: e67. [DOI] [PMC free article] [PubMed]
- 19. Kainz PM,, Neitz M,, Neitz J. Molecular genetic detection of female carriers of protan defects. Vision Res. 1998; 38: 3365–3369. [DOI] [PubMed] [Google Scholar]
- 20. Carroll J,, Dubra A,, Gardner JC,, et al. The effect of cone opsin mutations on retinal structure and the integrity of the photoreceptor mosaic. Invest Ophthalmol Vis Sci. 2012. ; 53: 8006–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ueyama H,, Muraki S,, Tanabe S,, Yamade S,, Ogita H. A new subset of deutan color-vision defect associated with an L/M visual pigment gene array of normal order and −71C substitution in the Japanese population. J Biochem. 2015; 158: 197–204. [DOI] [PubMed] [Google Scholar]
- 22. Waaler GHM. Über die ErblichkeitsverhaItnisse der verschiedenen Arten von angeborener Rotgrünblindheit. Zeit Indukt Abstamm Vererbungs. 1927; 45: 279–233. [Google Scholar]
- 23. Koliopoulos J,, Iordanides P,, Palimeris G,, Chimonidou E. Data concerning colour vision deficiencies amongst 29,985 young Greeks. Mod Probl Ophthalmol. 1976. ; 17: 161–164. [PubMed] [Google Scholar]
- 24. Neitz J,, Neitz M,, Kainz PM. Visual pigment gene structure and the severity of color vision defects. Science 1996. ; 274: 801–804. [DOI] [PubMed] [Google Scholar]
- 25. Ueyama H,, Muraki-Oda S,, Yamade S,, et al. Unique haplotype in exon 3 of cone opsin mRNA affects splicing of its precursor, leading to congenital color vision defect. Biochem Biophys Res Commun. 2012. ; 424: 152–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.